Introduction
Colorectal carcinoma (CRC) is the most frequent type
of cancer in Germany. Death is mostly due to development of distant
metastases in liver and/or lung. Surgical resection of metastases
with curative intent is possible only in a small proportion
(10–15%) of patients (1). If this
is not possible, such patients normally receive palliative
chemotherapy. They die on average within a period of 2 years. The
development of new therapeutic strategies is therefore urgently
needed for patients with metastasized CRC.
Immunotherapy is a new strategy which looks
promising for CRC because this tumor is immunogenic and patients
with distinct tumor-infiltrating T cells have a higher survival
probability than those without such immune cells (2). Active vaccination aims at supporting
the generation of a polyclonal T-cell response to multiple
tumor-associated antigens (TAAs). In the past, we developed a live
tumor cell vaccine for CRC and other cancers. This ATV-NDV vaccine
is composed of 10 million autologous tumor cells which are first
infected by a bird paramyxovirus, Newcastle disease virus (NDV) and
then become irradiated with 200 Gy (3). The advantages of the use of
autologous tumor cells and of NDV as adjuvans to introduce
so-called danger signals have been described (4). A randomized prospective study
revealed a significant long-term survival benefit for colon cancer
patients upon vaccination with ATV-NDV following resection of liver
metastases (5).
Vaccination of patients in late-stage disease has in
general been rather ineffective (6), possibly due to dysfunction of the
immune system. Tumor-reactive T cells from such patients are likely
anergized (7) because of long-term
confrontation with TAAs in the absence of costimulatory signals
(8). We hypothesized that it may
perhaps be possible to partially reactivate such T cells by
providing strong costimulatory signals in combination with TAAs. To
this end and to further increase the effectivity of the vaccine
ATV-NDV, we developed in the past bispecific single-chain fusion
proteins (bs-scFvs) based on antibodies directed against NDV and
against CD28 (9). The construct
bsHN-CD28 binds directly to the ATV-NDV vaccine cells while the
second arm is directed against CD28, an important molecule on T
cells to deliver costimulatory signals (10).
This report describes the study design, the
production of the vaccine ATV-NDV-bsHN-CD28 and the results. The
latter are primarily based on the T-cell responses of patients as
analyzed by interferon-γ ELISPOT assays.
Materials and methods
Study design
The study was initiated in 2004 after a positive
response from the Ethics Committee of the University of Heidelberg
(no. L-149/2004). Included were only patients who suffered from CRC
UICC stage IV (with distant metastases). Tumor tissue was obtained
either from operation specimens or from interventional punction of
liver metastases. After generation of the vaccine (see below), the
patients received five intracutanous vaccinations in the thigh. The
first four vaccines were applied at 2-week intervals and the fifth
after 4 weeks. Blood samples were taken regularly immediately
before and 72 h after each vaccination for immunological monitoring
of the patients T-cell response (see below).
In this phase I study, the dose of the bispecific
fusion protein bsHN-CD28 to be added to the ten million ATV-NDV
vaccine cells was stepwise increased according to the Fibonacci
scheme: D0, D1, D2, D3 (=Dm). D0=0.199 Dm; D1=0.398 Dm; D2=0.658
Dm; D3=Dm (=1 μg protein).
Patient characteristics
Included were 14 patients, five women and nine men,
median age 55 years (range, 39–71). The primary tumor was either
from rectum (n=7), sigma (n=3), colon (n=1) or coecum (n=3).
Metastases were localized in liver (n=13), lung (n=5) or in the
peritoneum (n=4). Before inclusion into the study, the tumors were
progressive under chemotherapy (n=6), stable under chemotherapy
(n=1) or there was progress without chemotherapy of synchronous
metastases (n=7).
Generation of the vaccine
ATV-NDV-bsHN-CD28
The vaccine was generated in the cell culture
laboratory of the Department of Surgical Oncology at the Heidelberg
University Hospital. After enzymatic digestion of mechanically
dissected tumor samples, the single cells were either used directly
for further modification or they were first expanded in primary
cell cultures to reach the necessary number of cells (at least 50
million). The cells were then modified by infection with NDV
(strain Ulster) as described (3)
and the bsHN-CD28 protein added at the required amount.
The construction, production and purification of
bsHN-CD28 from the plasmid pERdhfr (αHN-αCD28) was described before
(11). Transfection of the
plasmids into dCHO cells was carried out by electroporation. Clones
that were stably expressing the desired fusion protein were
selected by limiting dilution. The production was conducted in high
density cell culture systems (Integra Biosciences AG, Zizers,
Switzerland). The E tag containing fusion protein was purified by
means of an anti-E tag immunoaffinity chromatography procedure
(Amersham Pharmacia Biotech, Amersham, UK). The protein
concentrations were determined with the CB-Protein Assay™ reagent
(Calbiochem-Merck Co., Schawalbach, Germany). Optimization studies
were performed earlier to find the best protocol for its coupling
to viral hemagglutinin-neuraminidase (HN) anchor molecules of the
vaccine ATV-NDV (12). Before
application, the modified vaccine cells were inactivated by gamma
irradiation (200 Gy). In addition, the absence of pathogens and
fungi had been confirmed by cultivation on blood-agar or on
Sabouraud-agar plates.
ELISPOT assay
The isolation of T cells, the generation of
dendritic cells (DCs) and the performance of the ELISPOT assay was
done essentially as described before (13,14).
Results
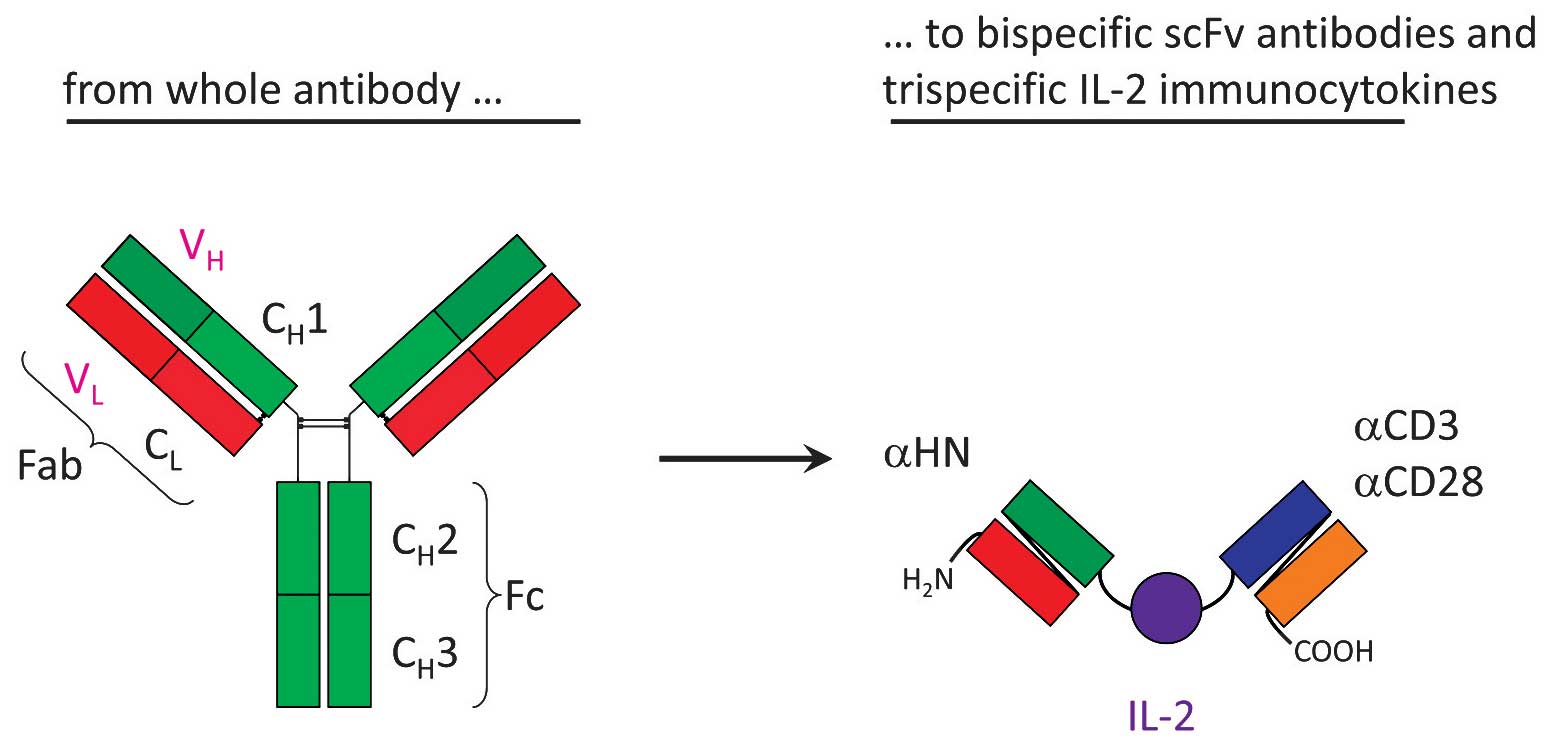
Construction of bsHN-CD28
Fig. 1 shows a
scheme of the construction of NDV-specific T-cell stimulatory
fusion proteins. Single-chain (scFv) antibodies specific for NDV HN
were derived from the hybridoma HN.B (Dr R.M. Iorio, University of
Massachusetts, Worcester, MA, USA). The CD28-specific scFv was
derived from the hybridoma 9.3 (Dr J.D. Hansen, US Geological
Survey-Western Fisheries Research Center, Seattle, WA, USA). The
plasmid that encoded the fusion protein bsHN-CD28 (no. 290) had the
structure
Flag-VH-L-VL-L-L-VH-VL-E
tag. Other plasmids encoded anti-CD3 instead of anti-CD28 and human
IL-2 instead of the linker poly-l-lysine (L) (10–12).
The highest dose of bsHN-CD28 that was added to the
vaccine ATV-NDV in the patient group D3 (n=4) was 1 μg purified
protein. The group D2 (n=4) received 1/3 of this amount, the group
D1 (n=3) 1/9 and the group D0 (n=3) 1/27 μg.
Side-effects
The vaccinations were well tolerated. Severe adverse
events were not observed. At the injection site swellings often
occurred with indurations which caused itching. Systemic symptoms
were subfebrile temperature (37–38°C) (n=8), fever (>38°C)
(n=2), vomiting (n=2), tiredness (n=2) and abdominal pain
(n=2).
Immune response monitoring
The ELISPOT assay was adjusted to measure
selectively the reactivity of memory T cells (MTCs). Purified T
cells from the peripheral blood were co-incubated for 40 h with
antigen-presenting DCs and the number of IFN-γ secreting T cells
per 75,000 or 100,000 T cells enumerated. Per patient there were
eight time points at which blood probes were taken.
Response to autologous tumor lysate
The vaccinations were performed with viable but
irradiated autologous tumor cells whose T cell costimulatory
function had been enhanced in two ways: i) by infection with NDV
(15); and ii) by attachment of
bsHN-CD28 (10–12). The T cells thereby received very
proper costimulatory signals.
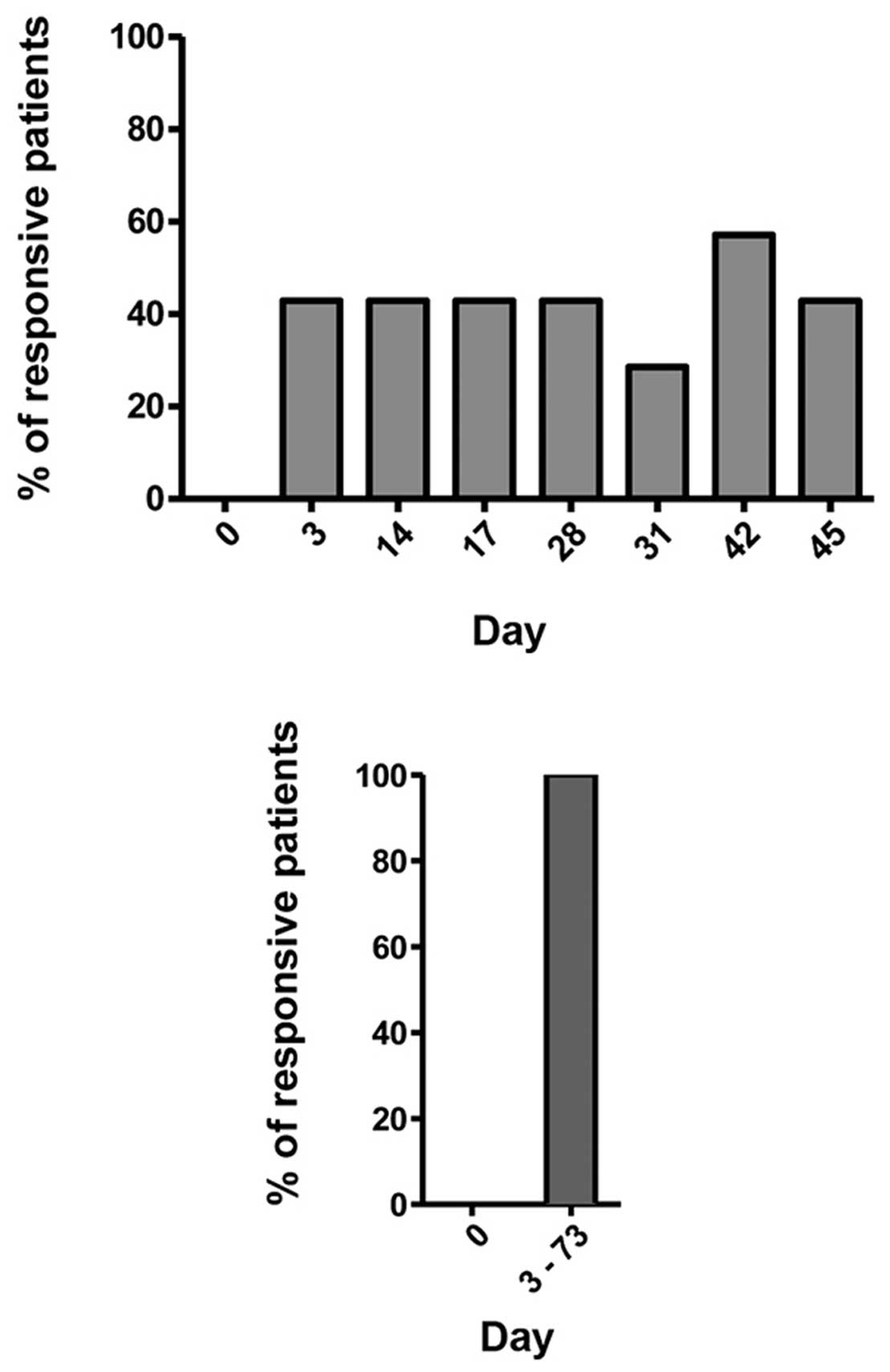
As can be seen from Fig. 2, none of the 14 patients showed a
response at day 0, that is before the first vaccination. In
contrast, at the seven time points after vaccination, always ~40%
of the patients were responsive. These were not always the same
patients whose T cells responded. When analyzing the response rate
of each patient either before vaccination or after vaccination
during the whole time period of 3–73 days, 100% of them showed at
least once a response after vaccination. This is remarkable
considering the fact that they were all late-stage metastasized
patients. This result is significant and based on ~500
ELISPOTs.
Response to defined tumor antigens
Since no information exists on the relationship
between the vaccination with whole autologous tumor cells and the
response to defined TAAs we investigated this further.
The patient DCs were pulsed either with autologous
tumor lysate (16) or loaded with
known 20mer peptides from defined common TAAs as described
(13,14) and used for MTC stimulation.
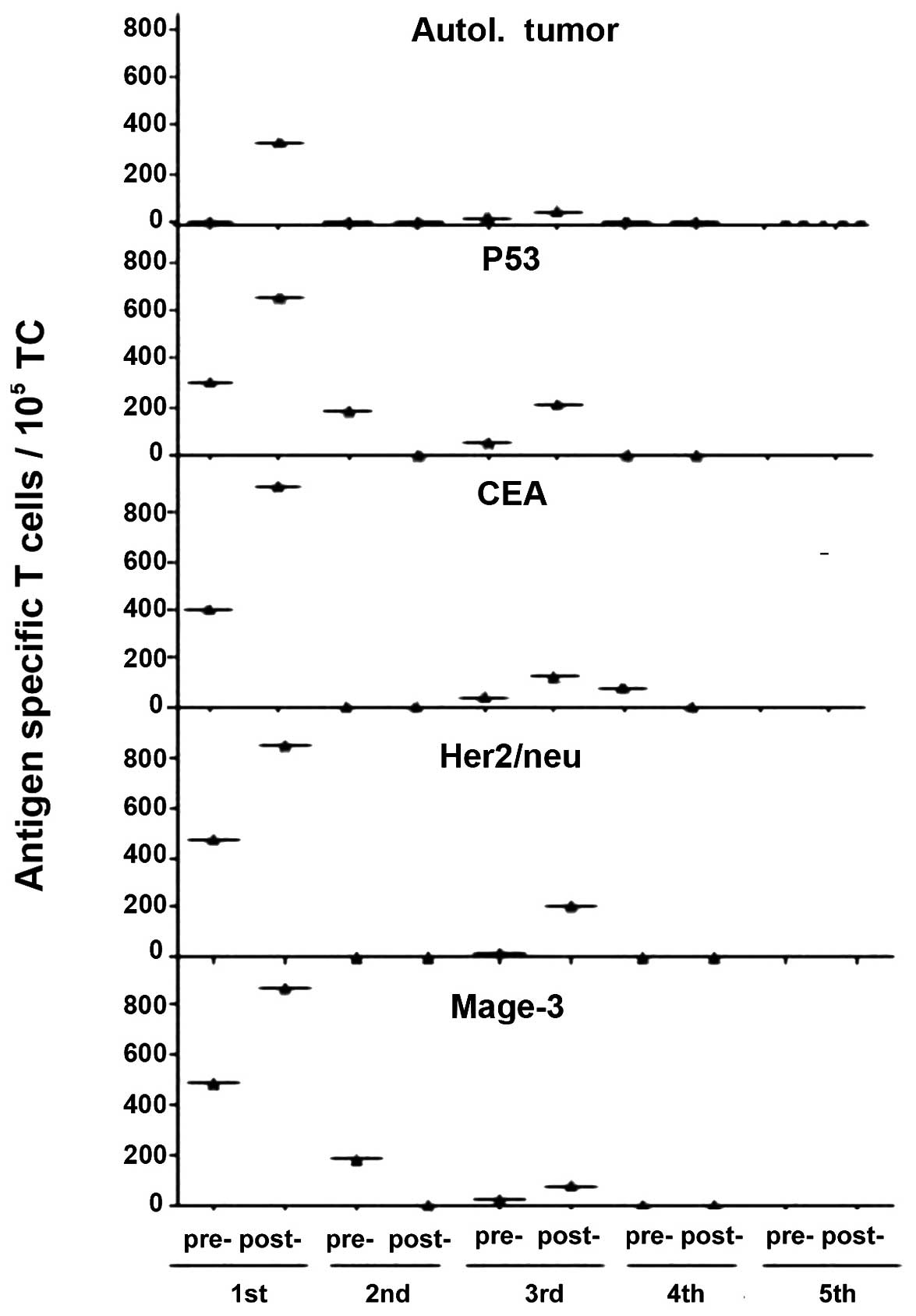
Fig. 3 shows results from one
patient (no. 1622). As can be seen, there existed a response
already before vaccination to the TAAs CEA, Her2/neu, Mage-3 and
p53. This pre-existing memory pool was apparently not sufficient to
control the tumor of the patient. Three days after the first
vaccination, this patient had responded by a strong expansion of
these TAA-specific T cells. This suggests that the vaccine
contained all of these common TAAs. Through combination with
powerful costimulatory signals the vaccine cells became immunogenic
and caused a polyvalent augmentation of TAA-specific T cells. This
result corroborates our concept of the design of this vaccine.
Also, the number of cells in the vaccine and of other components
seems to be physiological and capable of activating a fast MTC
response from patients with metastasized CRC.
While in patient no. 1622 the vaccine was
immunogenic towards all TAAs used in the ELISPOT assay, the T cells
from another patient (no. 1630) showed a dominant response only
against Her2/neu and p53 and not to CEA or Mage-3 (data not shown).
We showed before for breast cancer patients that the tumor-reactive
MTC repertoire is highly individual (14).
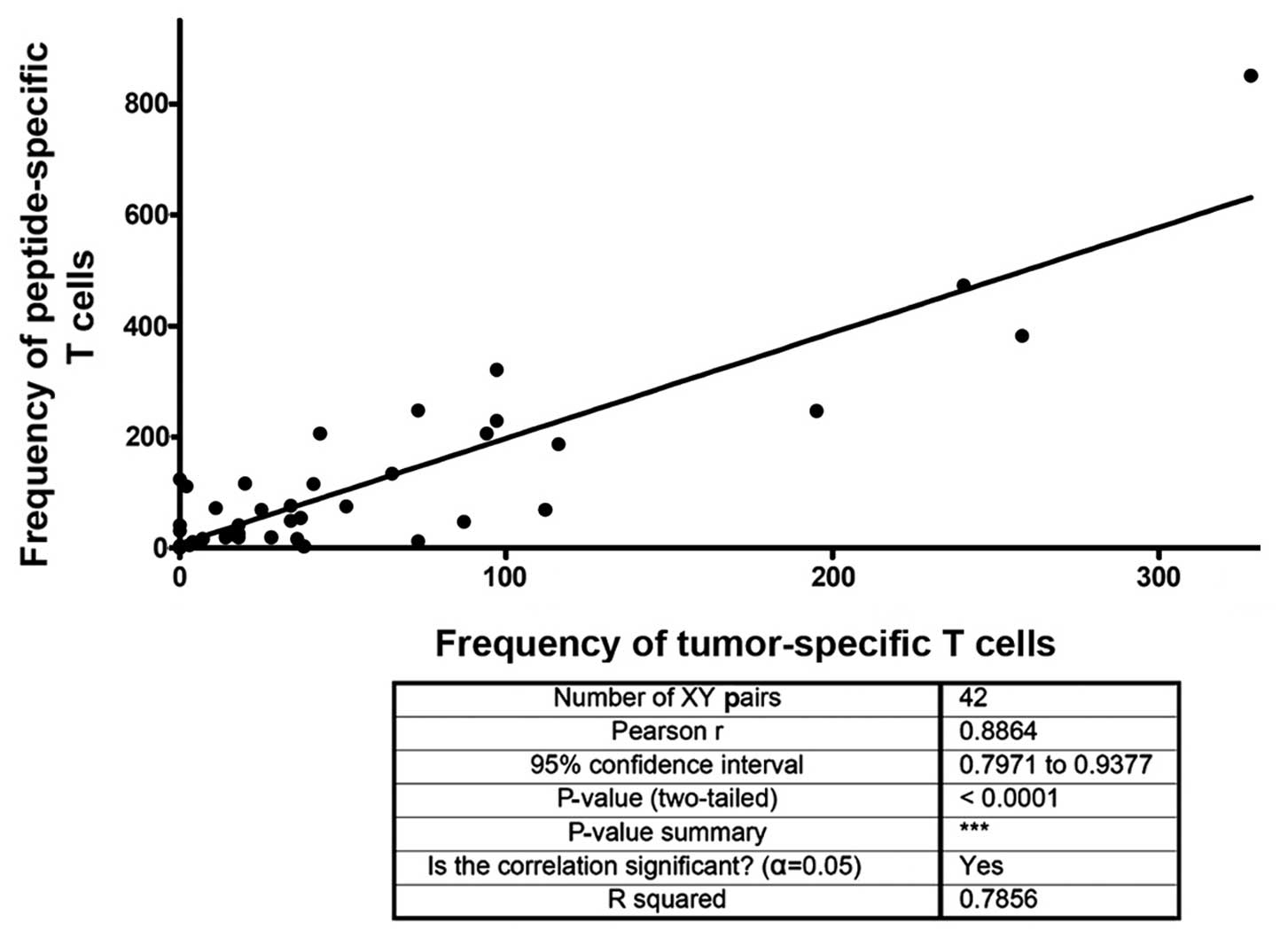
Fig. 4 shows
results from a correlation analysis of the frequency of patients T
cells responding to autologous tumor lysate and the frequency of
TAA peptide-specific T cells. The analysis revealed a significant
correlation. This result also means that our protocol of pulsing
DCs with tumor lysate leads to presentation of TAA-derived
MHC-peptide complexes in similarity to external loading with
defined peptides.
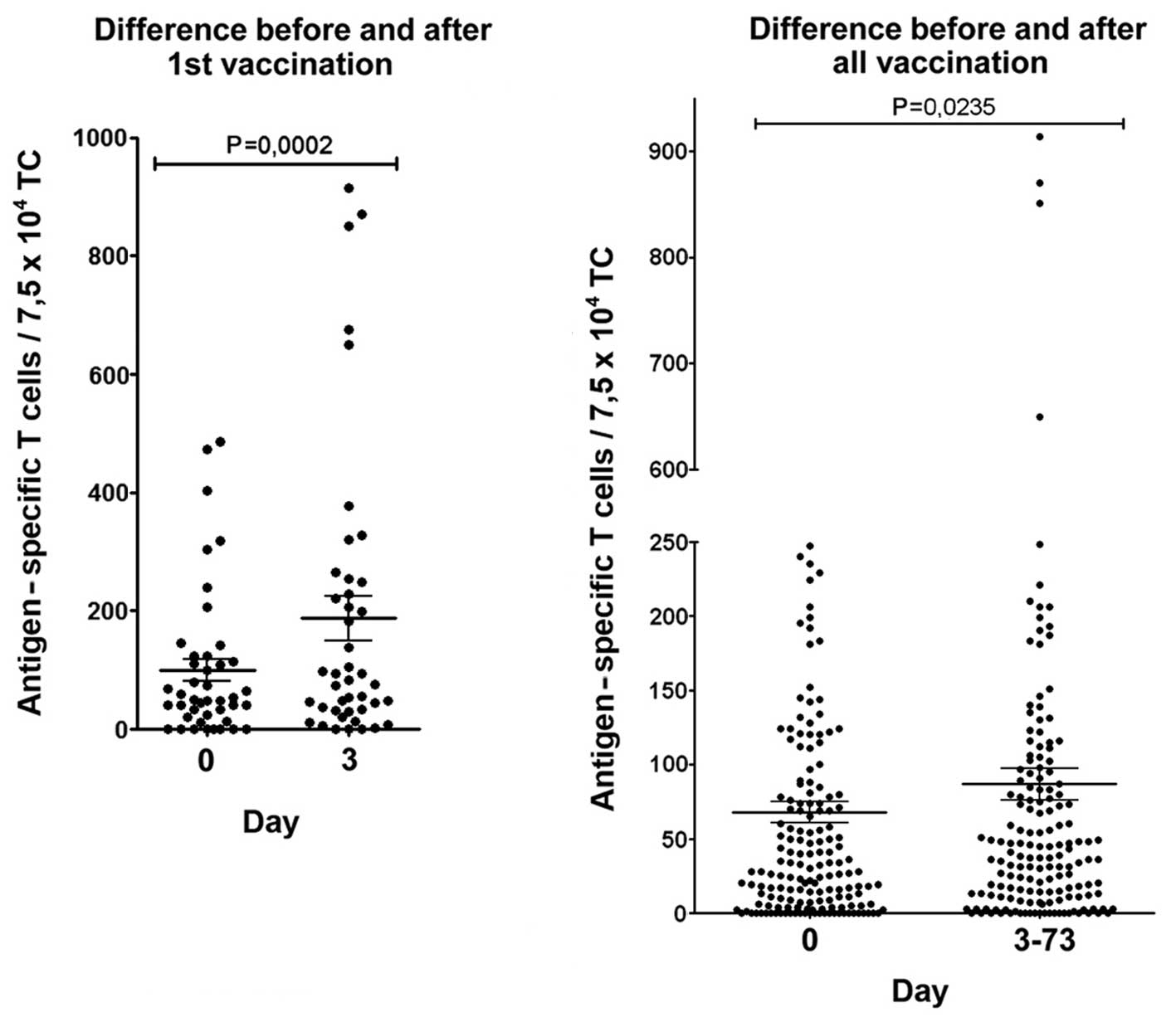
Cumulative analysis of TAA
peptide-specific T cells
Within the first 3 days after the first vaccination
there was in this cumulative analysis of the data from all patients
a strong and highly significant (P=0.0002) increase of TAA
peptide-specific T cells (Fig. 5,
left side). Also, the difference before and after all vaccinations
(Fig. 5, right side) was highly
significant. This suggests a functional reactivation of the
pre-existing repertoire (4,14) of
tumor-reactive MTCs by this vaccine in spite of the advanced
disease stage in these patients.
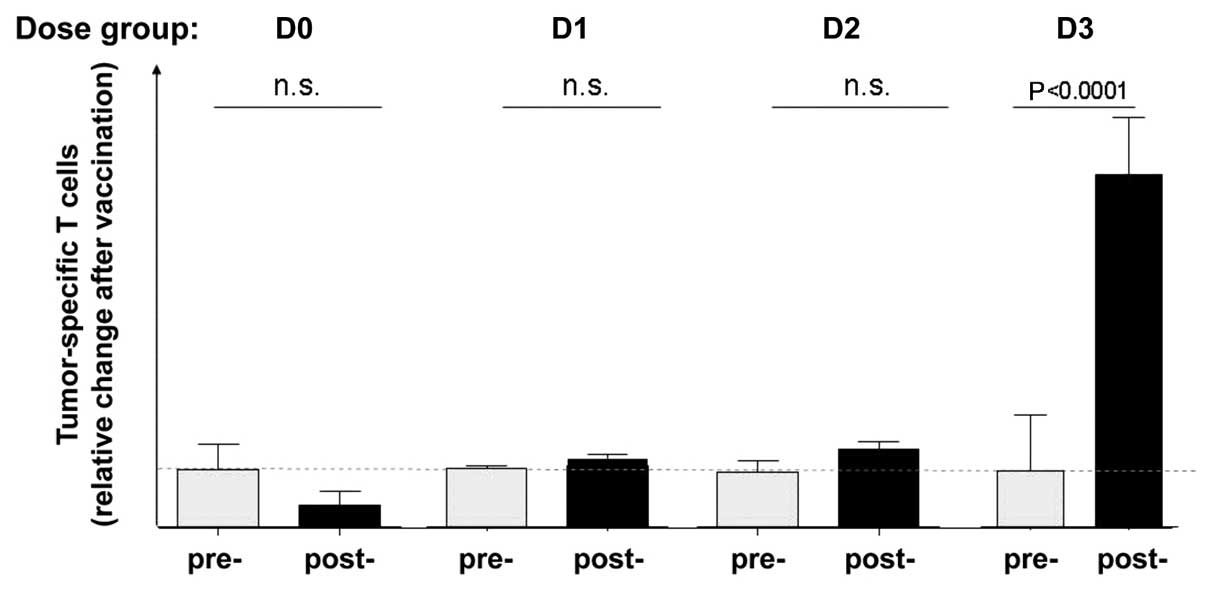
Dose-response analysis of bsHN-CD28
An important aspect of the immune response
monitoring relates to a possible relationship between the dose of
the applied fusion protein in the vaccine and the T-cell response
observed. The cumulative analysis of all T-cell responses before
and after vaccination, including all the dose groups, had revealed
a significant increase of TAA-specific T cells (Fig. 5). The differentiation between the
four-dosis groups D0–D3 revealed, however, a clear increase of
T-cell responses with increasing dose of the bispecific fusion
protein (Fig. 6). This result
shows that there was an influence of the added bsHN-CD28 protein on
the observed response and corroborates the importance of optimal
T-cell costimulation by a tumor vaccine to reactivate pre-existing,
perhaps already partially anergic TAA-specific T cells. That 1 μg
protein in the vaccine can exert such an effect in late-stage CRC
is a remarkable biological activity.
DTH reactions
The local delayed-type hypersensitivity skin
reaction (DTH) was evaluated always 24 and 72 h after vaccination
(3). To distinguish TAA-specific
DTH reactions from those against NDV virus or the fusion protein,
the skin challenge was performed only with ATV, the inactivated
autologous tumor cells. In seven patients an increase of the DTH
reaction (>100%) in comparison to the response before
vaccination was observed.
Tumor marker
The marker CEA, which was followed during the course
of vaccinations, either decreased (n=4), remained where it was
(n=3) or it increased (n=7).
Metastases (RECIST criteria)
By radiological means the following processes were
observed: Partial response (>30% decrease) (n=4), stable disease
(n=4), progress (>20% increase) (n=5). No data exist for one
patient who withdrew from the study.
Survival
In 2009, 5 years after initiation of the study,
seven of the 14 patients were still alive, two patients had been
further operated after a good first response and four patients had
died. One patient withdrew from the study.
Discussion
In this clinical phase I dose escalation study we
tested the T-cell costimulatory activity of a bispecific scFv
fusion protein, bsHN-CD28. If found to be active in vivo,
this new immunostimulatory protein could become attached to any
tumor cell infectable by NDV, because the cell surface exposed
viral HN molecules would serve as universal anchor molecule for
this reagent.
Other bispecific antibodies which are being
developed for tumor targeting of immune cells generally use a
distinct tumor target structure such as EpCAM (17), HER2 (18), FAP (19) or others. This means that for each
tumor type respective reagents need to be developed and tested
separately. The advantage of a universal reagent that could be
coupled through an adaptor like NDV to any type of tumor appears
obvious. Also, this tumor-targeting approach does not interfere
with the presentation of TAAs by the tumor cells.
We described that NDV can infect virtually any human
tumor cell type and replicate within it, thereby expressing at the
tumor cell surface a high density of viral HN and F molecules
(20). Normal cells, in contrast,
are resistant to replication of NDV (21). NDV infection of tumor cells was
found to introduce a CD80 (B7.1)-independent costimulatory function
which was capable of breaking tolerance in vitro (15). It is therefore likely that the
vaccine cells in this study can also break tolerance and reverse
states of anergy of T cells in situ in cancer patients,
especially after augmentation of the costimulatory signals by the
bsHN-CD28.
Our concept of introducing into a tumor vaccine a
costimulatory function via activation of CD28 on T cells avoids the
use of CD28 ligands such as CD80 or CD86. These ligands can also
bind to CTLA-4, a receptor mediating negative signals, even with
higher affinity. Such interaction would thus downregulate the
T-cell response. Instead, we decided to use an agonistic anti-CD28
hybridoma and to clone and select a functional scFv. This was then
fused with an anti-HN scFv to create the bispecific product we
aimed at.
In preclinical studies the dual binding specificity
of the reagent had been demonstrated (11). In further studies, the combination
of this reagent with the vaccine ATV-NDV was optimized (10). Once bound, the reagent was stable
at the cell surface for a time period of 24–48 h, a time sufficient
for T-cell activation (12).
Before discussing the results of this study, it is
important to understand how the bsHN-CD28 reagent functions. Both
binding sites are monovalent. By itself the reagent can thus not
costimulate a T cell because this requires cross-linking of CD28
molecules at its plasma membrane. Only when multiple bsHN-CD28
molecules become attached to a platform such as the cell surface of
the vaccine ATV-NDV they are capable of aggregating CD28 molecules
on co-cultured T cells.
Infection of tumor cells by NDV was shown to
upregulate expression of MHC and ICAM-1 molecules (22). Similar to professional APCs, the
tumor vaccine ATV-NDV-bsHN-CD28 is capable of co-presenting
MHC-peptide complexes (including TAAs) to be recognized by the
antigen-specific T cell receptor complex (TCR) to deliver signal 1
and anti-CD28 scFvs to interact with CD28 to deliver signal 2. In
addition, ICAM-1 molecules will interact with LFA-1 molecules on T
cells thereby stabilizing the cell-cell interaction. It is
conceivable that the vaccine cells of our study can form an
immunological synapse with T cells, similar to APCs. Such synapse
consists of two concentric rings: the central supramolecular
activation cluster (c-SMAC) enriched with TCRs and CD28 molecules
and the surrounding peripheral supramolecular activation cluster
(p-SMAC) enriched with LFA-1. The p-SMAC provides adhesive
anchoring of the T cell to the APC, while the c-SMAC represents a
protected zone for sustained signaling via TCR and CD28 (23). It is likely that the strength of
positive costimulatory signaling is decisive in a situation of
late-stage cancer in which negative signals are likely to
dominate.
When signal 2 is lacking, for instance on a
TAA-presenting tumor cell, interactions with TAA-specific T cells
render the T cell anergic: the T cell becomes refractory to signals
even when the TCR interacts with TAA. This situation has been
described in cancer patients, especially in chronic situations of
advanced disease (24), like the
patients of this study. T-cell anergy (7) may exist in different states, not all
of them being irreversible. For instance, tumor-reactive MTCs from
draining lymph nodes of carcinoma patients could be reactivated in
a short-term ELISPOT assay using as vaccine ATV-NDV with optimized
signals 1 and 2, but not with a similarly modified vaccine from an
unrelated tumor cell line (10).
The main findings of this study are the
following:
The new fusion protein bsHN-CD28 did not cause major
adverse events. A superagonistic anti-CD28 antibody (TGN1412) which
has in the past been applied to human beings caused unexpected
severe adverse events (25). These
were mainly due to non-specific polyclonal T-cell activation and
initiation of a so-called cytokine storm reaction. This did not
happen with bsHN-CD28. As a monovalent reagent this scFv can only
activate T cells when it is bound on a cell surface to its ligand
HN. Even then, it will only activate T cells in combination with
signal 1 (10–12). Since NDV is a bird virus, it does
not exist in man. Monovalency and dependency on a foreign agent
thus explain the high safety profile of this costimulatory
molecule.
Vaccination with ATV-NDV-bsHN-CD28 caused a strong
significant increase of tumor-reactive T cells in the peripheral
blood of many patients. The fast appearance of tumor-reactive T
cells after the first vaccination is likely due to reactivation and
expansion of pre-existing but inactive MTCs. De novo
generation of antigen-specific T cells from the pool of naïve T
cells would require a time period longer than 3 days.
Reactivity of the T cells was directed against DCs
pulsed with autologous tumor lysate and also against DCs loaded
with peptides from defined common TAAs. Autologous tumor cells
express individually distinct (unique) as well as common TAAs and
also normal self antigens. The simultaneous analysis of responses
to autologous tumor lysate and to peptides from common TAAs
revealed similarities, both in terms of frequencies and in terms of
kinetics of their appearance in - and disappearance from - the
peripheral blood. Responses to autologous tumor lysate correlated
with responses to common TAAs. These findings corroborate the
assumed presence of TAAs in the vaccine in the form of TAA-derived
MHC-peptide complexes. From these data it can be deduced that
autologous tumor cells express a broad spectrum of relevant TAAs
and that vaccination with modified autologous tumor cells is an
effective means to re-enforce a polyvalent TAA-specific repertoire
of MTCs (4).
A cumulative analysis of the data from all 14
patients revealed a significant increase of the frequencies of
tumor-reactive MTCs in the blood, especially 3 days after the first
vaccination. The increase of frequencies of tumor-reactive T cells
suggests expansion of a pre-existing polyclonal pool of
TAA-specific MTCs. Some of these may be derived from the bone
marrow where they reside long-term in distinct niches (26–28).
During the course of multiple vaccinations, the frequencies found
in the blood were not always as high as after the first
vaccination. This finding could be explained by extravasation of
MTCs from the blood into tissues, including tumor tissue. More
detailed information on the biology and dynamics of MTC responses
in general and about those in cancer patients are required before
the phenomena observed can be definitely explained.
With increasing dose of the reagent bsHN-CD28 there
was a clear-cut increase of tumor-reactive T cells. A dose-response
relationship is perhaps the best proof of the functionality of a
reagent such as the new biological product bsHN-CD28. This seems to
be the case in this study. A summary of T-cell analyses from all
patients before and after vaccination revealed a significant
increase of tumor-reactive T cells in the whole group (Fig. 5). A differentiation between the
different dosis groups revealed nevertheless an increase of T-cell
reactivity with increasing dose. The highest and only significant
increase was observed with the dose of 1 μg protein that was added
to 10 million ATV-NDV tumor vaccine cells. This result underlines
the importance of the additional costimulation via CD28 and
corroborates the assumption that only strong costimulatory
signaling can override negative signals on T cells in a chronic
disease situation.
All patients showed reactivity to the vaccination,
at least once during the time course of immune monitoring. The fact
that 100% of the 14 late-stage cancer patients showed a response,
at least once, to this type of vaccine is rather exceptional. Of
course, response in this case means T-cell mediated cancer-reactive
response which is not equal to clinical response but nevertheless
impressive.
With regard to clinical effects, the main
observations were decrease of CEA in four patients and partial
response of metastases in four patients. Seven patients were still
alive in 2009. Unfortunately, we have no further follow-up.
In July 2009, the PI of this study (P.B.) was
informed by the Regulatory Office of Karlsruhe (Baden-Württemberg,
Germany) that the production of the vaccine for a clinical study is
now considered as a somatic cellular therapeutic and that this
requires a permit according to legal code no. 13 Arzneim
ittelgesetz (AMG). According to study protocol, this phase I
clinical study, which generated interesting new information and pro
mising results without having caused severe harm to any patient,
should have been followed by a phase II study. Although the study
had been approved before, the new regulations via the European
Medicines Agency (EMA) do not allow a continuation. The
prerequisite for a new study are a GMP status for each of the drug
components, tumor cells, NDV and bispecific fusion protein. The
time and the costs for this go beyond that what a PI doing research
at a public institution can afford. It requires huge investments
which only a pharmaceutical company can do. The new regulations are
said to be in the interest of the safety of patients but an
interest of the pharmaceutical industry in this development can not
be disputed. As a consequence of the new legislation, the
proportion of PI guided objective non-biased clinical studies among
all clinical studies in Europe is dramatically decreasing.
Nevertheless, we hope that the new ideas behing this
study will survive the new regulations to the benefit of future
cancer patients.
Acknowledgements
We are gratefull to the Dietmar Hopp Foundation for
financial support of this study. It is based on three decades of
intensive research by the first author (V.S.) and his team at the
German Cancer Research Center, Heidelberg, which paved the way for
the concept of active-specific antitumor vaccination using
patient-derived tumor cells, infecting them by NDV and finally
attaching bispecific antibodies. We thank all the research people,
co-operation partners and clinicians involved in the past. With
regard to the bispecific antibodies we acknowledge in particular C.
Haas, M. Lulei and P. Fournier.
References
|
1
|
Fong Y, Fortner J, Sun RL, Brennan MF and
Blumgart LH: Clinical score for predicting recurrence after hepatic
resection of metastatic colorectal cancer: analysis of 1001
consecutive cases. Ann Surg. 230:309–318. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galon J, Costes A, Sanchez-Cabo F, et al:
Type, density, and location of immune cells within colorectal
tumors predict clinical outcome. Science. 313:1960–1964. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ockert D, Schirrmacher V, Beck N, et al:
Newcastle disease virus-infected intact autologous tumor cell
vaccine for adjuvant active specific immunotherapy of resected
colorectal carcinoma. Clin Cancer Res. 2:21–28. 1996.
|
|
4
|
Schirrmacher V, Fournier P and Schlag P:
Autologous tumor cell vaccines for post-operative active-specific
immunotherapy of colorectal carcinoma: long-term patient survival
and mechanism of function. Exp Rev Vaccines. 13:117–130. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schulze T, Kemmner W, Weitz J, et al:
Efficiency of adjuvant active specific immunization with Newcastle
disease virus modified tumor cells in colorectal cancer patients
following resection of liver metastases: results of a prospective
randomized trial. Cancer Immunol Immunother. 58:61–69. 2009.
View Article : Google Scholar
|
|
6
|
Rosenberg SA, Yang JC and Restifo NP:
Cancer immunotherapy: moving beyond current vaccines. Nat Med.
10:909–915. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schwartz RH: T cell anergy. Annu Rev
Immunol. 21:305–334. 2003. View Article : Google Scholar
|
|
8
|
Greenfield EA, Nguyen KA and Kuchroo VK:
CD28/B7 costimulation: a review. Crit Rev Immunol. 18:389–418.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fournier P and Schirrmacher V: Bispecific
antibodies and trispecific immunocytokines for targeting the immune
system against cancer: preparing for the future. BioDrugs.
27:35–53. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aigner M, Janke M, Lulei M, Beckhove P,
Fournier P and Schirrmacher V: An effective tumor vaccine optimized
for costimulation via bispecific and trispecific fusion proteins.
Int J Oncol. 32:777–789. 2008.PubMed/NCBI
|
|
11
|
Haas C, Lulei M, Fournier P, Arnold A and
Schirrmacher V: T-cell triggering by CD3- and CD28-binding
molecules linked to a human virus-modified tumor cell vaccine.
Vaccine. 23:2439–2453. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Haas C, Lulei M, Fournier P, Arnold A and
Schirrmacher V: A tumor vaccine containing anti-CD3 and anti-CD28
bispecific antibodies triggers strong and durable antitumor
activity in human lymphocytes. Int J Cancer. 118:658–667. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Koch M, Beckhove P, Op den Winkel J, et
al: Tumor infiltrating T lymphocytes in colorectal cancer:
Tumor-selective activation and cytotoxic activity in situ. Ann
Surg. 244:986–992. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sommerfeldt N, Schütz F, Sohn C, Förster
J, Schirrmacher V and Beckhove P: The shaping of a polyvalent and
highly individual T-cell repertoire in the bone-marrow of breast
cancer patients. Cancer Res. 66:8258–8265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Termeer CC, Schirrmacher V, Bröcker EB and
Becker JC: Newcastle disease virus infection induces
B7-1/B7-2-independent T-cell costimulatory activity in human
melanoma cells. Cancer Gene Ther. 7:316–323. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai L, Koopmann J, Fiola C, Fournier P and
Schirrmacher V: Dendritic cells pulsed with viral oncolysates
potently stimulate autologous T cells from cancer patients. Int J
Oncol. 21:685–694. 2002.PubMed/NCBI
|
|
17
|
Brischwein K, Schlereth B, Guller B, et
al: MT110: a novel bispecific single-chain antibody construct with
high efficacy in eradicating established tumors. Mol Immunol.
43:1129–1143. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Biburger M, Weth R and Wels WS: A novel
bispecific tetravalent antibody fusion protein to target
costimulatory activity for T-cell activation to tumor cells
overexpressing ErbB2/HER2. J Mol Biol. 346:1299–1311. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Müller D, Frey K and Kontermann RE: A
novel antibody-4-1BBL fusion protein for targeted costimulation in
cancer immunotherapy. J Immunother. 31:714–722. 2008.PubMed/NCBI
|
|
20
|
Schirrmacher V, Haas C, Bonifer R, Ahlert
T, Gerhards R and Ertel C: Human tumor cell modification by virus
infection: an efficient and safe way to produce cancer vaccine with
pleiotropic immune stimulatory properties when using Newcastle
disease virus. Gene Ther. 6:63–73. 1999. View Article : Google Scholar
|
|
21
|
Fournier P, Wilden H and Schirrmacher V:
Importance of retinoic acid-inducible gene I and of receptor for
Type I interferon for cellular resistance to infection by Newcastle
disease virus. Int J Oncol. 40:287–298. 2012.PubMed/NCBI
|
|
22
|
Washburn B and Schirrmacher V: Human tumor
cell infection by Newcastle Disease Virus leads to upregulation of
HLA and cell adhesion molecules and to induction of interferons,
chemokines and finally apoptosis. Int J Oncol. 21:85–93. 2002.
|
|
23
|
Grakoui A, Bromley SK, Sumen C, et al: The
immunological synapse: a molecular machine controlling T cell
activation. Science. 285:221–227. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zitvogel L, Tesniere A and Kroemer G:
Cancer despite immunosurveillance: immunoselection and
immunosubversion. Nat Rev Immunol. 6:715–727. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
St Clair EW: The calm after the cytokine
storm: lessons from the TGN1412 trial. J Clin Invest.
118:1344–1347. 2008.PubMed/NCBI
|
|
26
|
Feuerer M, Beckhove P, Bai L, Solomayer
EF, Bastert G, Diehl IJ, Pedain C, Oberniedermayr M, Schirrmacher V
and Umansky V: Therapy of human tumors in NOD/SCID mice with
patient-derived reactivated memory T cells from bone marrow. Nat
Med. 7:452–458. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Feuerer M, Beckhove P, Garbi N, Mahnke Y,
Limmer A, Hommel M, Hämmerling GJ, Kyewsky B, Hamann A, Umansly V
and Schirrmacher V: Bone marrow as a priming site for T-cell
responses to blood-borne antigen. Nat Med. 9:1151–1157. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schirrmacher V, Feuerer M, Fournier P,
Ahlert T, Umansky V and Beckhove P: T-cell priming in bone marrow:
the potential for long-lasting protective anti-tumor immunity.
Trends Mol Med. 9:526–534. 2003. View Article : Google Scholar : PubMed/NCBI
|