Introduction
Hepatocellular carcinoma (HCC) is one of the most
common cancers, and the third leading cause of cancer-related
mortality worldwide (1). The main
risk factors of HCC in humans include infection with hepatitis B
and/or hepatitis C virus, and alcoholic liver disease (2). The outcome of HCC treatment is
dependent on the tumor stage at the time of diagnosis. A good
curability can be achieved if HCC were detected in an early stage.
Unfortunately, most HCC patients with symptoms have progress to
advanced stage when they are diagnosed. The poor prognostic
outcomes are mainly due to cancer metastasis and tumor recurrence
after surgery. Various factors are involved in the regulation of
cancer metastasis, including the epithelial-mesenchymal transition
(EMT) (3). Nevertheless, the
molecular mechanisms underlying EMT in HCC tumor metastasis are
poorly characterized.
Glypican-3 (GPC3) is one of the cell surface heparan
sulfate proteoglycans that bind to the cell membrane via a
glycosyl-phosphatidylinositol (GPI) anchor. GPC3 is released from
cell membranes by the extracellular lipase Notum at the GPI anchor
(4). Functionally, GPC3 regulates
signaling pathways associated with the Wnt, Sonic hedgehog (Shh),
fibroblast growth factor, and bone morphogenetic protein (5–7).
Previous studies indicated that GPC3 plays an important role in
regulating cancer progression in a stage- and tissue-specific
manner; e.g. GPC3 acted as a tumor suppressor in breast cancer
(7,8). Interestingly, overexpression of GPC3
presented in lung squamous cell carcinoma, but not in lung
adenocarcinoma (9).
GPC3 is involved in the migration, proliferation,
and modulation of cell survival in different tissues (8). It has been shown that GPC3 is highly
expressed in patients with HCC, either in serum or liver tissues,
but is absent in benign hepatic lesions, hepatic cirrhosis and
hepatitis (10–12). Furthermore, GPC3 has been reported
to be used to discriminate α-fetoprotein (AFP)-negative HCC from
cirrhotic nodules (13). Thus,
GPC3 might be a more reliable tumor marker that could allow for an
early diagnosis of HCC (14,15).
In addition, several studies have shown that GPC3 promoted growth
of HCC in vivo and in vitro (6,16,17).
However, the clinical function of GPC3 in HCC progression remains
obscure. To date, few studies elucidate the potential functions of
GPC3 in HCC EMT and metastasis.
The present study examined the effects of GPC3 in
HCC progression and metastasis. We focused on the role of GPC3 in
HCC EMT and underlying molecular mechanisms.
Materials and methods
Patients
We obtained paired liver tissues (tumor vs.
non-tumor) from 45 patients with HCC who received hepatic resection
between July 2008 and January 2009 at Beijing You’an Hospital. One
part of resected tissues was fixed in formalin and embedded in
paraffin for histological examination. The remained liver tissues
were snap-frozen and stored at −80°C for real-time PCR and western
blot analyses. Clinical characteristics of these patients are shown
in Table I. HCC was confirmed by
histological examinations according to the AASLD guidelines
proposed in 2005 (18). This study
was approved by Research Ethics Committee, You’an Hospital
affiliated with Capital Medical University. Written informed
consent was obtained from each patient.
 | Table IClinical characteristics of the study
population (n=45). |
Table I
Clinical characteristics of the study
population (n=45).
| Age (mean/range)
(years) | 48.9 (22–67) |
| Gender
(male/female) | 38/7 |
| Primary liver
disease (HBV/HCV/alcohol/others) | 39/3/4/2 |
| Patients with
cirrhosis | 45 |
| Child-Pugh class
(A/B/C) | 27/18/0 |
| Tumor diameter
(<3 cm/≥3 cm) | 14/31 |
| Tumor number
(≤3/>3) | 40/5 |
| Portal vein
thrombosis | 14 |
| Vascular invasion
(present/absent) | 14/31 |
| Serum AFP (≤20
μg/l/>20 μg/l) | 19/16 |
| Histological
differentiation (good/moderate and poor) | 4/18/23 |
| BCLC stage
(A/B/C/D) | 13/14/18/0 |
Immunohistochemistry analyses for
GPC3
Four-μm paraffin-embedded sections were
deparaffinized and rehydrated. Antigen was retrieved using
heat-induced epitopes in 10 mM citrate buffer, pH 6.0 and
endogenous peroxidases were blocked for 5 min using 0.3%
H2O2. Then, the sections were incubated with
mouse monoclonal anti-GPC3 antibody (diluted 1:100, Santa Cruz
Biotechnology, Santa Cruz, CA, USA) overnight in a cool room. Next
day, the slides were applied for horseradish peroxidase-labeled
secondary antibody (Zhongshan Golden Bridge, Beijing, China) after
three PBS washes. To evaluate GPC3 immunostaining, the percentage
of positive cells and intensity of staining were analyzed. The
intensity of immunostaining was semi-quantitatively estimated
according to the following scores: 0, no positive staining; 1,
positive staining was weak yellow; 2, positive staining was yellow;
and 3, strong positive staining was brown. The mean percentage of
positive tumor cells was determined after counting positive GPC3
cells in five fields under ×400 magnification. The scores were
defined as follows: 0, ≤10%; 1, 10%–25%; 2, 25%–50%; 3, 50–75%; and
4, >75%. The final GPC3 immune score was calculated as staining
intensity × mean percentage of positive tumor cells. We defined the
scores ≤4 as low expression of GCP3 and 5–12 as high expression of
GPC3.
RNA extraction and quantitative real-time
PCR
Total RNA was purified from frozen HCC tissues using
an RNeasy Mini kit (Qiagen, Hilden, Germany). Total RNA (1 μg) was
reverse transcribed using a Reverse-Transcription system (Promega,
Madison, WI, USA). Transcript levels of GPC3 cDNA were
quantified using real-time PCR (Rotor-Gene Q, Qiagen, Germany). The
primers used to amplify GPC3 were: forward,
5′-TTCTCAACAACGCCAATA-3′, and reverse, 5′-GATGTAGCCAGGCAAAGC-3′.
Amplicon expression in each sample was normalized to
β-actin. The primers used to amplify β-actin were:
forward, 5′-ACCCACACTGTGCCCATCTA-3′, and reverse,
5′-GCCACAGGATTCCATACCCA-3′. After normalization, expression of
GPC3 was quantified using a 2−ΔΔCt calculation.
PCR was performed in duplicate and three independent experiments
were performed.
Cell culture
The human HCC cell lines HepG2, Hep3B and Huh7 were
obtained from the Cell Bank of the Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). HepG2, Hep3B and
Huh7 were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco Life Technology, Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum (FBS; Hyclone, Rockford, IL, USA), 100 μg/ml
streptomycin, and 100 U/ml penicillin (Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China) in a humidified
atmosphere of 5% CO2 at 37°C. For experiments, cells at
70% confluence were serum-starved 24 h before treatment. To block
the ERK pathway, cells were preincubated with PD98059
(Sigma-Aldrich, St. Louis, MO, USA) for 3 h before GPC3 (Sino
Biological Inc., Beijing, China) treatment.
Western blotting
Western blotting was performed as described
previously (19). Briefly, total
cellular proteins were extracted using cell lysis buffer (20 mM
Tris/HCl, 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS and protease
inhibitor cocktail, pH 7.5). The protein concentrations of the
lysates were determined according to the bicinchoninic acid (BCA)
method using a protein assay kit (Pierce Biotechnology, Rockford,
IL, USA). Cell or liver tissue lysates containing 100 μg protein
were generated and total proteins were separated by standard
SDS-PAGE, followed by transfer to polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA). Membranes were incubated
with primary antibodies against GPC3 (diluted 1:500, Santa Cruz
Biotechnology), MMP9 (diluted 1:1,000, Cell Signaling Technology,
CA, USA), E-cadherin (diluted 1:1000, Santa Cruz Biotechnology),
α-SMA (diluted 1:1,000, Cell Signaling Technology), β-catenin
(diluted 1:1,000, Cell Signaling Technology), Snail (diluted
1:1,000, Cell Signaling Technology), p-ERK (diluted 1:1,000, Cell
Signaling Technology), ERK (diluted 1:1,000, Cell Signaling
Technology), or Sonic hedgehog (Shh; diluted 1:500, Santa Cruz
Biotechnology). Next, membranes were incubated with an appropriate
horseradish peroxidase-conjugated secondary antibody. Reactive
bands were detected using enhanced chemiluminescence reagents
(Applygen Technologies Inc., Beijing, China). To ensure equal
loading of samples in each lane, membranes were stripped and
reprobed with an anti-glyceraldehyde 3-phosphate dehydrogenase
antibody (GAPDH; diluted 1:10,000; Kang Chen, Shanghai, China). The
relative densities of the protein bands were quantitatively
determined using ImageJ software (National Institutes of Health,
Bethesda, MD, USA).
Scratch assay
The migration of cells was examined with scratch
assays. When cells grew to 90% confluency, a scratch wound in the
monolayer was made using a pipette tip. After washing away all
detached cells with PBS, the remaining cells were treated with 0.1
μg/ml GPC3. The wound distances were measured by microscope at 0,
24, and 48 h after treatment. Each test was performed in
triplicate.
Migration and invasion assays
Serum-starved cells were resuspended in DMEM free of
FBS and seeded in a 24-well plate at a concentration of
105 cells/well on inserts with 8-μm filter pores, which
were either uncoated (migration assay; BD Falcon™ Cell Culture
Insert, San Jose, CA, USA) or Matrigel-coated (invasion assay; BD
Biocoat™ Matrigel™ Invasion Chamber). As a chemoattractant, 1% FBS
was added to the lower chamber while FBS-free DMEM with 0, 0.05,
0.1 or 0.2 μg/ml GPC3 or 0, 25, 50 or 100 nM PD98059 was put into
the upper chamber, respectively. After 24 h, cells were fixed and
stained with methanol and hematoxylin. Migrated or invaded cells
were counted at ten random fields per triplicate filter under an
inverted microscope whereas non-migrating and non-invading cells
were removed from the upper surface by wiping with a cotton swab.
Each test was performed in triplicate.
Statistical analysis
Clinicopathological parameters in patients with GPC3
high/low were compared using Pearson’s χ2 test or
Fisher’s exact test. Continuous variables were analyzed with
one-way analysis of variance. The results of multiple observations
are presented as the means ± SDs of at least three independent
experiments. The Kaplan-Meier method was used to determine survival
probability and differences between groups were assessed using the
log-rank test. Overall survival (OS) was defined as the interval
between surgery and death or the last follow-up time-point.
Statistical analyses were conducted using SPSS version 17.0
software (SPSS Inc., Chicago, IL, USA).
Results
High expression of GPC3 in cancer cells
is associated with HCC progression
To examine whether GPC3 involved in HCC progression,
GPC3 mRNA and protein levels were assessed in liver tissues from
patients with different HCC stages. The Barcelona Clinic Liver
Cancer (BCLC) system was applied to classify the disease stage
(20). Among the 45 pathologically
diagnosed HCC patients, 13 had BCLC-stage A, 14 BCLC-stage B, and
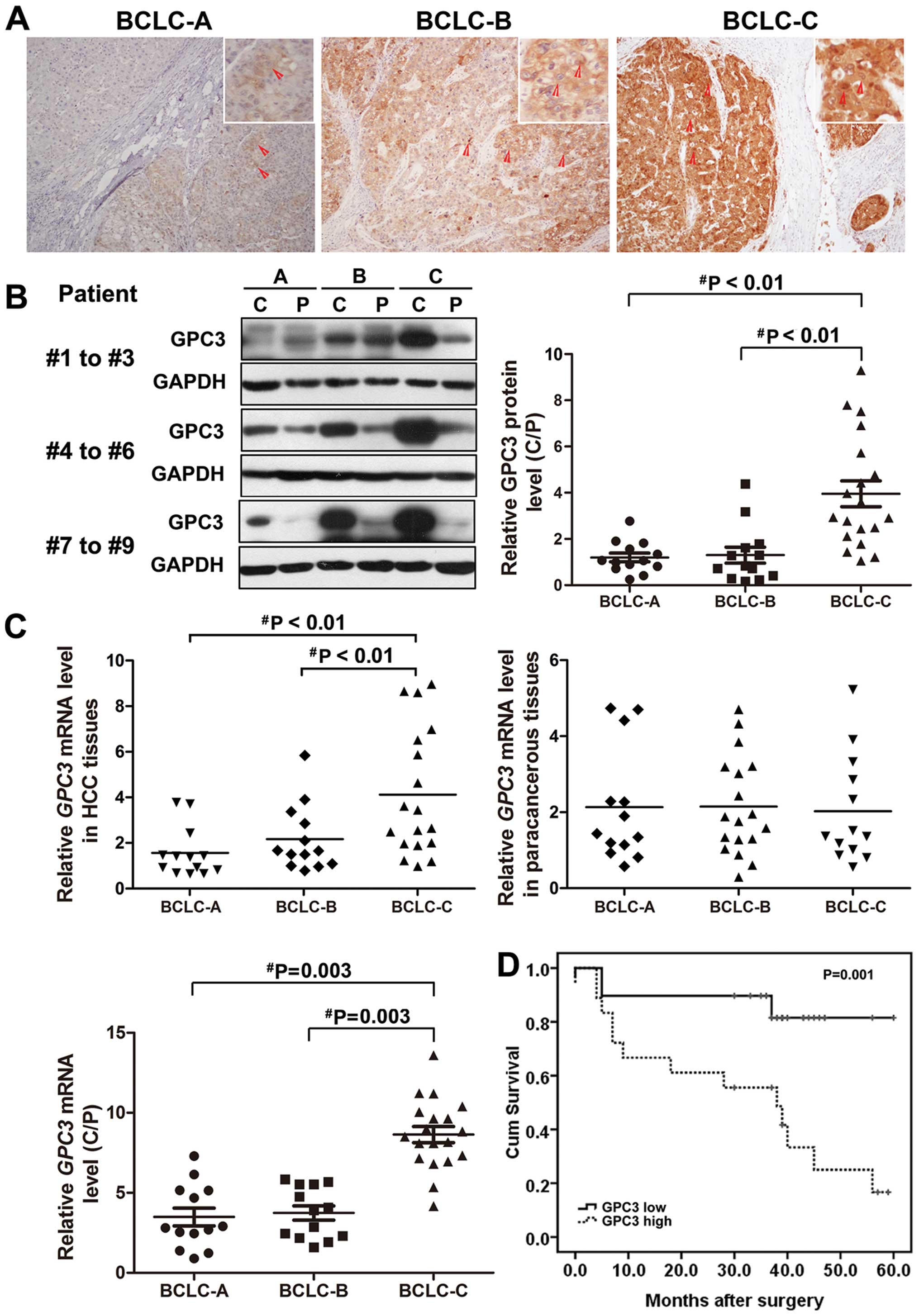
18 BCLC-stage C. The expression of GPC3 was examined in all HCC
tissues using an anti-GPC3 antibody. Immunostaining of GPC3 was
detected in the majority of hepatocyte membranes and cytoplasms of
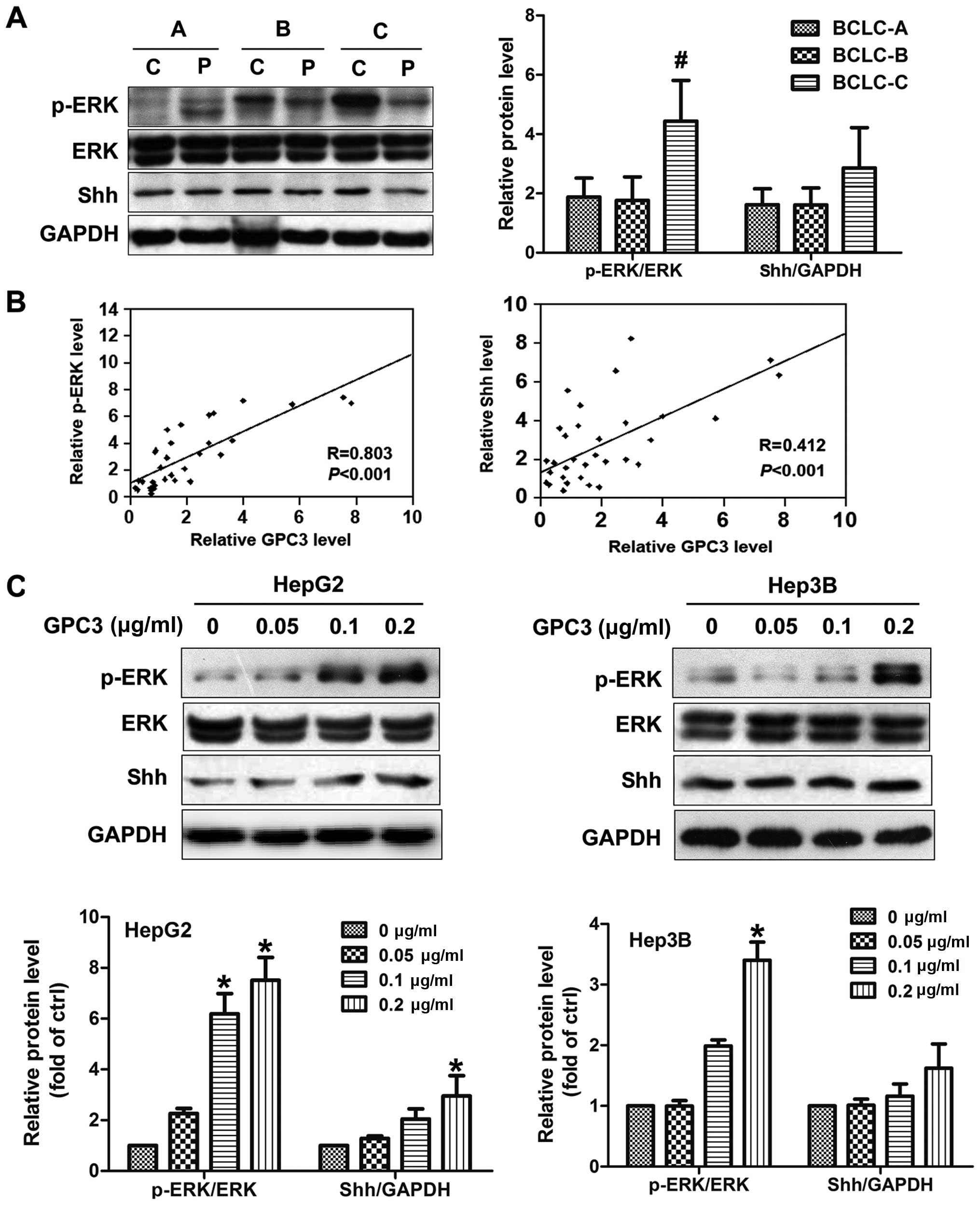
most HCC cancer cells (Fig. 1A).
Notably, both mRNA and protein expression of GPC3 was remarkably
elevated in HCC patients with BCLC-stage C compared to those with
BCLC-stage A or B (Fig. 1B and C).
Real-time PCR and western blot analyses showed that GPC3 was
upregulated on average ≤2.4-fold in patients with BCLC-stage C
compared to those with BCLC-stage A or B (P=0.003, Fig. 1B and C). There were no significant
difference of GPC3 expression between patients with BCLC-stages A
and B (P>0.05; Fig. 1B and C),
or the paracancerous tissues of different stages (P>0.05;
Fig. 1B and C).
As mentioned in Materials and methods, we divided 45
patients into GPC3high and GPC3low groups
based on GPC3 immune positivity. Table II shows that 22 patients (48.9%)
belonged to GPC3high group and 23 (51.1%)
GPC3low group, respectively. Correlations between GPC3
expression and clinicopathological features were further analyzed.
Consistent with the findings based on real-time PCR and western
blotting, there was a significant difference in GPC3 expression
levels between BCLC-stages C and A (P=0.002) or B (P=0.001), while
there was no significant difference between BCLC-stages A and B. In
addition, high levels of GPC3 expression were strongly correlated
with the rate of vascular invasion (P<0.001). No other clinical
characteristics, including age, gender, hepatitis B virus
infection, serum AFP level, tumor size, tumor number, and
Child-Pugh class, were related to the expression of GPC3. We then
investigated whether the GPC3 expression is associated with the
prognosis of HCC patients. The 5-year overall survival rate of HCC
patients with low GPC3 expression levels reached 80.8% whereas only
31.8% of the HCC patients with high GPC3 expression were still
surviving at the end of the follow-up (P=0.042, Mantel-Cox test;
Fig. 1D).
| Table IIThe correlation between GPC3
expression and clinicopathologic variables of patients with
HCC.a |
Table II
The correlation between GPC3
expression and clinicopathologic variables of patients with
HCC.a
| Feature | Low expression of
GPC3 (n=23) | High expression of
GPC3 (n=22) | P-value |
|---|
| Gender |
| Male | 19 | 19 | 1.000 |
| Female | 4 | 3 | |
| Age (years) |
| ≤50 | 10 | 13 | 0.295 |
| >50 | 13 | 9 | |
| HBsAg |
| Positive | 21 | 18 | 0.619 |
| Negative | 2 | 4 | |
| Child-Pugh
class |
| A | 15 | 12 | 0.465 |
| B | 8 | 10 | |
| Tumor size
(cm) |
| ≤3 | 7 | 7 | 0.920 |
| >3 | 16 | 15 | |
| Tumor number |
| Single | 14 | 17 | 0.235 |
| Multiple | 9 | 5 | |
| Vascular
invasion |
| No | 22 | 9 | 0.001 |
| Yes | 1 | 13 | |
| Serum AFP
(μg/l) |
| ≤20 | 10 | 9 | 0.862 |
| >20 | 13 | 13 | |
| BCLC HCC stage |
| A | 9 | 4 | 0.002b |
| B | 11 | 3 | 0.001b |
| C | 3 | 15 | |
HCC patients with high GPC3 expression
demonstrated the EMT phenotype
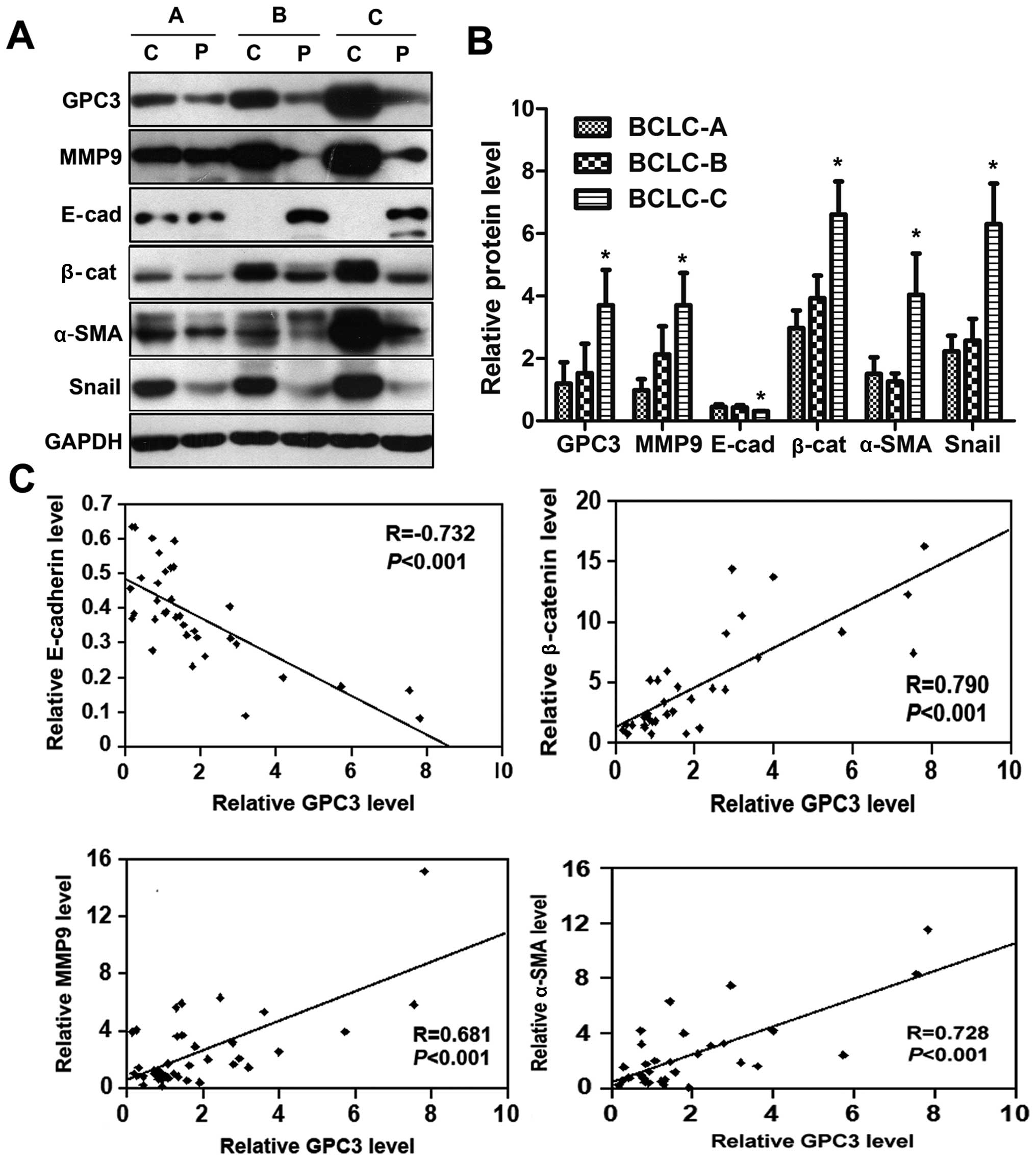
HCC progression and metastasis were highly
associated with phenotypic alterations of cancer cells,
particularly EMT (21,22). In this study, we measured the
expression of the epithelial cell markers, e.g., E-cadherin and
β-catenin, and the mesenchymal markers such as MMP9 and α-SMA, as
well as Snail, a transcription factors associated with EMT. Western
blot analyses showed that the expression of Snail, MMP9, β-catenin
and α-SMA in cancerous tissues with BCLC-stage C were significantly
increased compared to those with BCLC-stages A or B (P<0.05,
Fig. 2A). By contrast, expression
of E-cadherin in cancerous tissues with BCLC-stage C was lower than
in those with BCLC-stages A or B (P<0.05; Fig. 2A). There was a positive correlation
between the expression levels of GPC3 and MMP9, β-catenin, and
α-SMA, but a negative correlation between the expression of GPC3
and E-cadherin (Fig. 2B). These
results strongly suggest that GPC3 might contribute to EMT of
HCC.
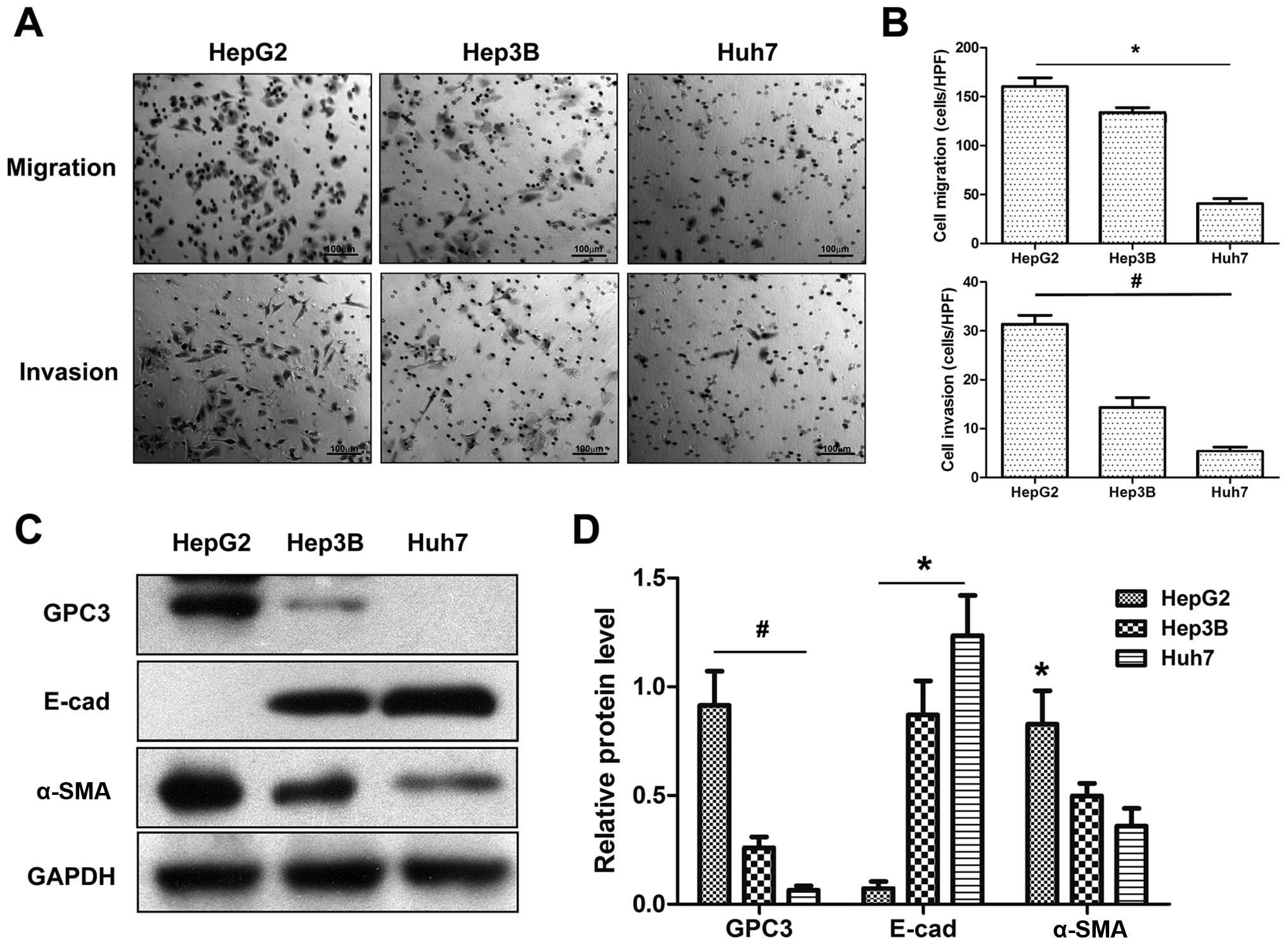
GPC3 participates in EMT in vitro
To further clarify the potential association between
GPC3 expression and HCC metastasis, we investigated the expression
of GPC3 and the metastatic capacity in HCC cell lines e.g., HepG2,
Hep3B and Huh7. Migration and invasion assays indicated that HepG2
cells had the highest and Huh7 cells had the lowest metastatic
capacity among three HCC cell lines (P<0.05; Fig. 3A). Western blotting showed that
HepG2 cells expressed the highest levels of GPC3, whereas Huh7
cells expressed the lowest (Fig.
3C). Alternatively, HepG2 cells expressed higher levels of
α-SMA and lower levels of E-cadherin compared to Hep3B and Huh7
cells (Fig. 3C and D).
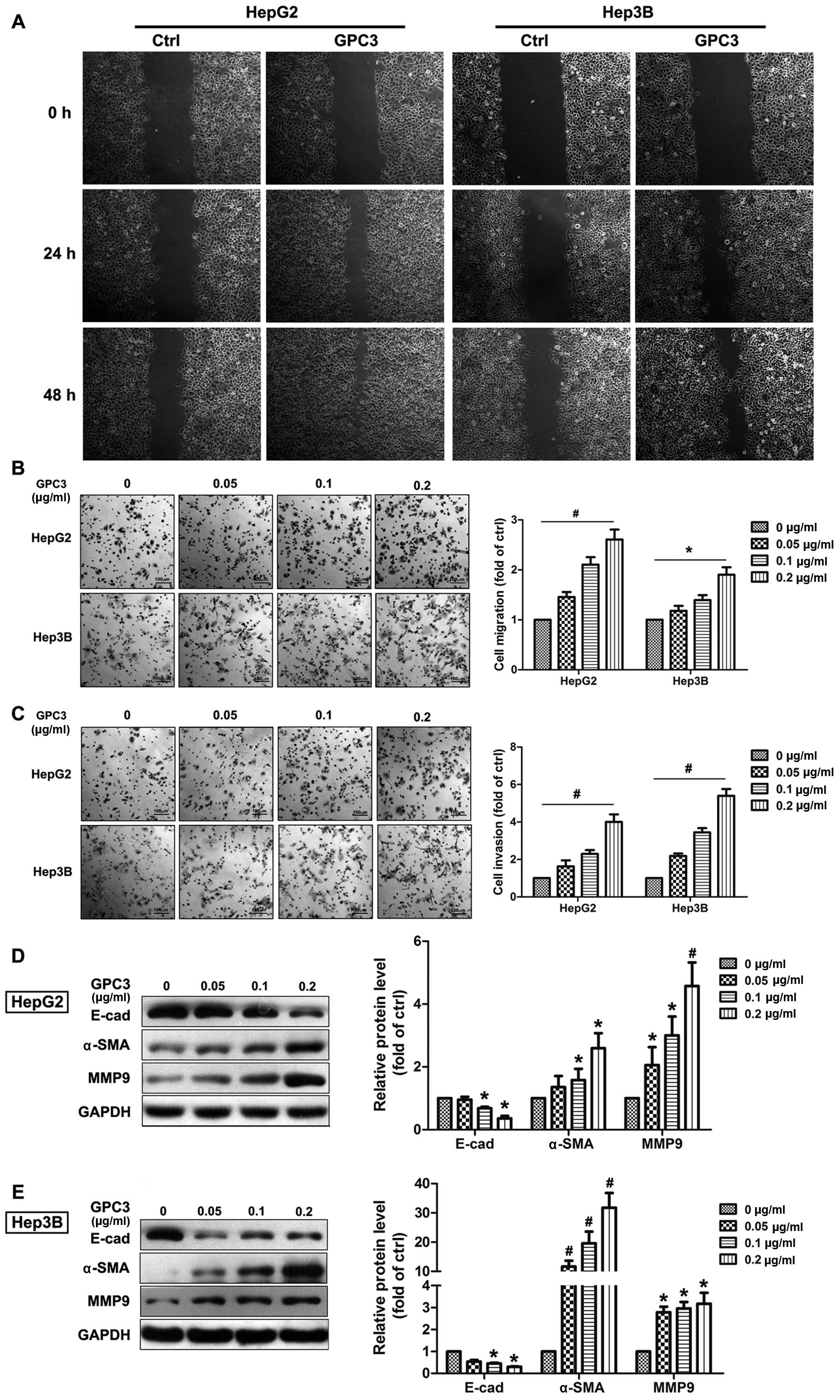
In addition to the cell surface expression, GPC3 can
also be detected in the extracellular environment after being
released from the cell membrane by Notum, a lipase that cleaves GPI
anchors (4). Next, the impact of
soluble exogenous GPC3 on HCC cellular motility was studied.
Scratch-assays displayed an obviously greater migration capacity of
both HepG2 and Hep3B cells when they were treated by exogenous GPC3
compared to untreated cells (Fig.
4A). Additionally, in Boyden chamber and Matrigel invasion
assays, exogenous GPC3 induced dynamic migration and invasion of
both HepG2 and Hep3B cells in a dose-dependent manner (Fig. 4B and C). Furthermore, GPC3
treatment significantly reduced expression of the epithelial cell
markers such as E-cadherin and increased the mesenchymal markers
MMP9 and α-SMA in a dose-dependent manner in both HepG2 and Hep3B
cells.
GPC3 induces EMT via increasing ERK
activation
Given the critical roles of the mitogen-activated
protein kinase (MAPK) (23) and
Sonic hedgehog (Shh) signaling pathways in EMT (24), we investigated the impact of GPC3
in the two pathways. We found that ERK activity was strongly
elevated in HCC patients with BCLC-stage C (P<0.05; Fig. 5A). A positive correlation between
GPC3 expression and phospho-ERK was observed in HCC tissues by
regression analysis, with a coefficient (R) of 0.803 (P<0.001;
Fig. 5B). In vitro, the
exposure of cells to soluble GPC3 promoted the phosphorylation of
ERK in a dose-dependent manner in both HepG2 and Hep3B cells
(Fig. 5C). To further confirm
whether GPC3-induced cell migration and invasion is via the ERK
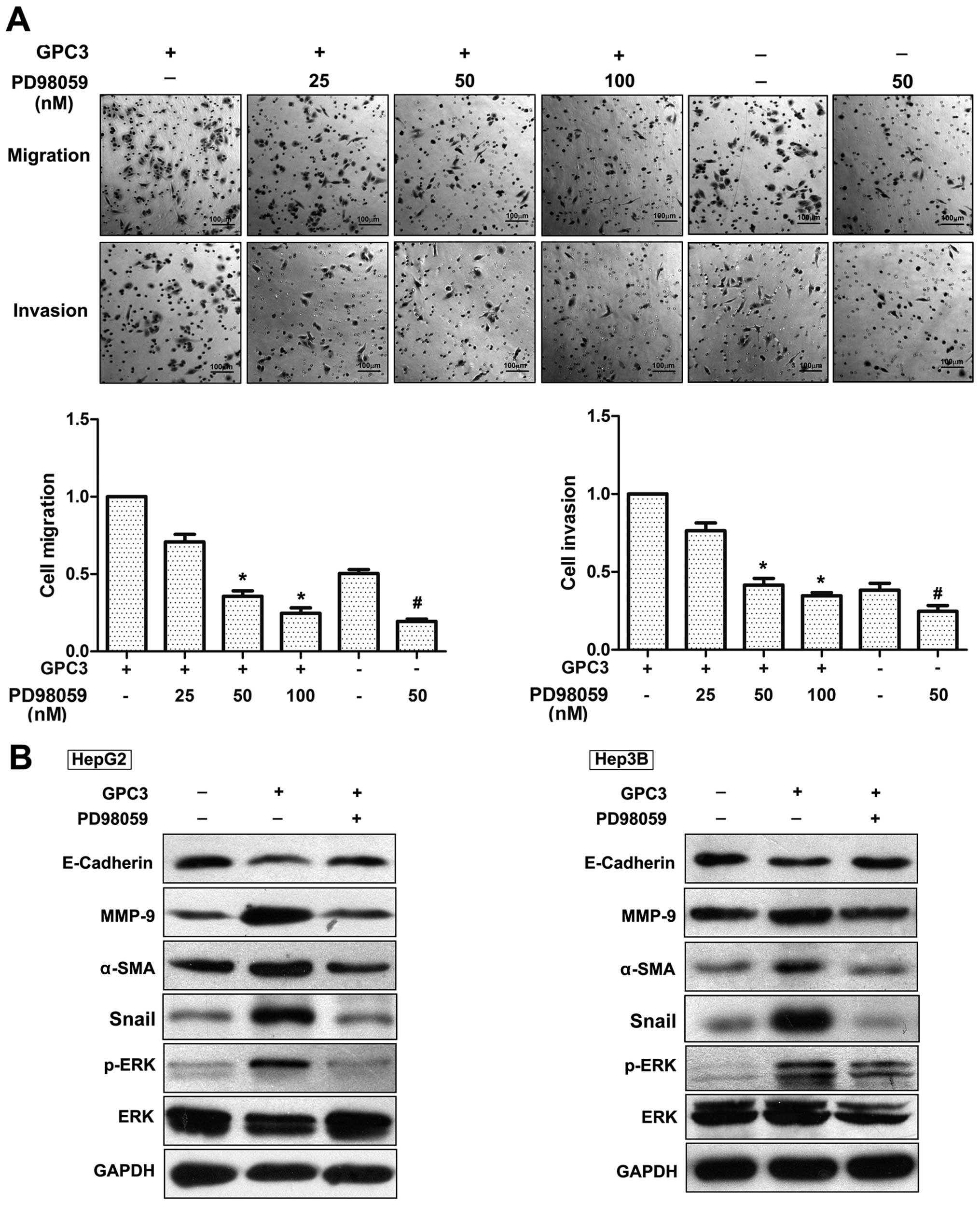
pathway, we applied PD98059, an inhibitor of MEK (a kinase that
activates ERK), to HepG2 cells prior to GPC3 treatment. We observed
that PD98059 could decrease HepG2 cell migration and invasion.
Furthermore, this inhibitor significantly attenuated the
GPC3-induced migration of HepG2 cells in a dose-dependent manner
(25 nM, 82.80±16%; 50 nM, 35.19±6%; and 100 nM, 24.32±6%). In an
invasion assay, PD98059 reduced the GPC3-induced invasion capacity
of HepG2 cells in a dose-dependent manner (25 nM, 73.00±6.74%; 50
nM, 41.70±3.07%; and 100 nM, 35.54±2.13%; Fig. 6A). Notably, PD98059 significantly
attenuated GPC3-induced EMT. As shown in Fig. 6B, E-cadherin was increased whereas
MMP-9, α-SMA and Snail were decreased under PD98059 treatment.
Besides the ERK pathway, we also investigated whether the Shh
pathway was involved in GPC3-induced EMT. Unexpectedly, no
significance was obtained on Shh expression among different stages
(P>0.05; Fig. 5A). The
correlation of Shh expression with GPC3 was weaker compared to ERK
(Fig. 5B). In vitro, GPC3
treatment could not markedly increase the Shh expression (Fig. 5C).
Discussion
The expression levels of GPC3 mRNA and protein were
markedly increased in primary tissues and serum from HCC patients
compared to those from healthy people or patients with benign liver
lesions (10,12,13).
However, the clinical role of GPC3 in HCC progression and the
underlying molecular mechanisms have remained unclear. Here, we
showed that GPC3 expression was correlated with the clinical
features of HCC, including BCLC staging and invasive ability.
Furthermore, we found that exogenous GPC3 could regulate the
migration, invasion capacity and EMT of HCC cells by activating the
ERK signaling pathway.
GPC3 plays an important role in regulation of HCC
tumorigenesis and progression. However, the effect of GPC3 on HCC
proliferation is controversial. Depending on different cellular
environments, GPC3 may either promote or inhibit cell growth. For
example, transfection of Huh7 and HepG2 cells with GPC3-specific
siRNA effectively inhibited cell proliferation (25). Cheng et al (26) reported that NIH3T3 cells
transfected with a GPC3 expression vector revealed overexpression
of GPC3 and excessive cell proliferation. On the other hand, Pan
et al revealed that overexpression of GPC3 in Huh7 and
SK-HEP-1 cells effectively inhibited cell proliferation and cell
invasion through induction of apoptosis (27). Sung et al (28) revealed that antisense-mediated
knockdown of GPC3 in HepG2 cells signifcantly promoted cell growth
through pathways independent of IGF2. Furthermore, Lin et al
(29) reported that hepatocyte
proliferation and hepatomegaly induced by phenobarbital and 1,4-bis
[2-(3,5-dichloropyridyloxy)] benzene is suppressed in
hepatocyte-targeted glypican-3 transgenic mice. Our laboratory is
in the process of confirming the relationship between GPC3
expression and HCC cell proliferation.
Metastasis is an important aspect of HCC
progression. Several studies have reported that GPC3 can stimulate
the migration, and invasion (16,17)
of HCC cell lines. Here, we focused on the clinical importance of
GPC3 in HCC, and noted that there was a positive association
between GPC3 expression and HCC metastasis progression. As
metastasis or vascular invasion are major factors in the
progression from BCLC-stages A and B to C, we proposed that GPC3
might be involved in HCC metastasis. Furthermore, we showed that
both endogenous and exogenous GPC3 expression were correlated with
metastasis and invasion. First, we found endogenous GPC3 expression
was associate with HCC metastasis in HCC tissues and cells, which
was consistent with the report from Miao et al that the
knockdown of GPC3 resulted in the inhibition of cell migration and
invasion in Huh7 cells (16). The
migration and invasion capacities of HCC cell lines were also
increased by exogenous GPC3 treatment, suggesting that in addition
to endogenous GPC3, soluble/exogenous GPC3 may also function to
promote HCC metastasis in patients. So far, few studies have
explored how endogenous and exogenous GPC3 regulate HCC metastasis.
Capurro et al reported that GPC3 was needed for attachment
to the cell surface by a GPI anchor to stimulate growth of HCC
cells (6). Thus, we suspected that
the GPI anchor is necessary in endogenous and exogenous
GPC3-promoting HCC metastasis. Further study will be carried out
using genetic approaches in the cell models such as loss of
function studies.
The importance of the EMT in cancer metastasis and
other human diseases has been recognized (30). A key step in the EMT is the
downregulation of E-cadherin (31). The E-cadherin-catenin complex plays
an important role in the process of cell adhesion. Its dysfunction
has been associated with a reduction in cell differentiation and
increased cell invasiveness and metastasis. Snail, a negative
regulator of E-cadherin, is one of the key transcription factors
promoting EMT (32). Furthermore,
the mesenchymal markers MMP9 and α-SMA have been particularly
associated with tumor progression and metastatic dissemination in
different human cancers (33,34).
Here, we demonstrated that GPC3 promoted tumor metastasis through
the induction of EMT and could potentially be used as a biomarker.
Unexpectedly, β-catenin, an epithelial cell marker, was positively
correlated with GPC3 expression levels. We believe that these
contradictory results are a consequence of the dual function of
β-catenin in HCC. It acts in both cadherin-based adhesion and Wnt
signaling. Catenins can associate with cadherins at the membrane
level, forming adhesion complexes. Additionally, in response to a
Wnt stimulus or specific gene mutations, β-catenin is stabilized
and translocates to the nucleus where it binds TCF/LEF-1
transcription factors to trans-activate genes that drive tumor
formation. Here, we measured the total levels of β-catenin.
Additional studies will be needed to confirm the localization and
function of β-catenin in GPC3-induced metastasis.
Many reports have shown that GPCs regulated various
signaling pathways, including ERK, Shh, Wnt, bone morphogenetic
proteins, and fibroblast growth factors (5–7,26).
We found GPC3 regulates EMT and metastasis through the ERK
signaling pathway. Recent studies indicate that ERK signaling
pathway is involved in Claudin-1 (35) and αB-Crystallinζ (36) induces EMT in HCC. Yoshida et
al also reported that sublethal heat treatment skewed HCC cells
toward EMT transforming them to progenitor-like by the ERK pathway
(37). Thus, we believe that ERK
pathway may play an important role in GPC3-induced EMT of HCC. Chen
et al reported that the EMT and enhanced hedgehog signaling
activity may be responsible for HCC cell chemoresistance and
invasion (24). In this study,
although Shh expression had an increasing trend in BCLC stage C, no
significance was obtained among different stages. In addition, GPC3
treatment did not markedly increase the Shh expression in
vitro. Therefore, we speculate the Shh pathway may be not
involved in GPC3-induced EMT in HCC cell. Nevertheless, we cannot
exclude the possibility that alternative pathways are involved in
the promotion of HCC progression by GPC3. For example, GPC3 could
also stimulate the in vitro and in vivo growth of HCC
cells by increasing autocrine/paracrine canonical Wnt signaling
(6). So identifying the precise
mechanisms and signal pathways involved in these processes remains
to be addressed.
In conclusion, our findings suggest that GPC3
stimulates the invasiveness and metastasis of HCC cells. GPC3 may
promote the EMT of HCC cells through ERK signaling pathways. Thus,
GPC3 might be a potential indicator of HCC EMT and metastasis, and
our findings provide a theoretical basis for therapeutically
targeting GPC3 for the treatment of HCC.
Acknowledgements
This study was supported by the Major Projects on
Infectious Disease (2012ZX1002-008-05), as well as the Capital
Science and Technology Development Fund (2014-1-1824) and the
Beijing High-Level Talent Academic Leader/Personnel Training
Programs awarded to Hui-Guo Ding (2011-2-19).
Abbreviations:
|
GPC3
|
glypican-3
|
|
HCC
|
hepatocellular carcinoma
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
GPI
|
glycosyl-phosphatidylinositol
|
|
AFP
|
α-fetoprotein
|
|
BCLC
|
Barcelona Clinic Liver Cancer
|
|
MAPK
|
mitogen-activated protein kinas
|
|
Shh
|
Sonic hedgehog
|
References
|
1
|
He J, Gu D, Wu X, et al: Major causes of
death among men and women in China. N Engl J Med. 353:1124–1134.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kreuger J, Perez L, Giraldez AJ and Cohen
SM: Opposing activities of Dally-like glypican at high and low
levels of Wingless morphogen activity. Dev Cell. 7:503–512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Filmus J, Capurro M and Rast J: Glypicans.
Genome Biol. 9:2242008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Capurro MI, Xiang YY, Lobe C and Filmus J:
Glypican-3 promotes the growth of hepatocellular carcinoma by
stimulating canonical Wnt signaling. Cancer Res. 65:6245–6254.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stigliano I, Puricelli L, Filmus J,
Sogayar MC, Bal de Kier Joffe E and Peters MG: Glypican-3 regulates
migration, adhesion and actin cytoskeleton organization in mammary
tumor cells through Wnt signaling modulation. Breast Cancer Res
Treat. 114:251–262. 2009. View Article : Google Scholar
|
|
8
|
Peters MG, Farias E, Colombo L, Filmus J,
Puricelli L and Bal de Kier Joffe E: Inhibition of invasion and
metastasis by glypican-3 in a syngeneic breast cancer model. Breast
Cancer Res Treat. 80:221–232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Aviel-Ronen S, Lau SK, Pintilie M, et al:
Glypican-3 is overexpressed in lung squamous cell carcinoma, but
not in adenocarcinoma. Mod Pathol. 21:817–825. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Capurro M, Wanless IR, Sherman M, et al:
Glypican-3: a novel serum and histochemical marker for
hepatocellular carcinoma. Gastroenterology. 125:89–97. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Filmus J and Capurro M: Glypican-3 and
alphafetoprotein as diagnostic tests for hepatocellular carcinoma.
Mol Diagn. 8:207–212. 2004. View Article : Google Scholar
|
|
12
|
Liu H, Li P, Zhai Y, et al: Diagnostic
value of glypican-3 in serum and liver for primary hepatocellular
carcinoma. World J Gastroenterol. 16:4410–4415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li B, Liu H, Shang HW, Li P, Li N and Ding
HG: Diagnostic value of glypican-3 in alpha fetoprotein negative
hepatocellular carcinoma patients. Afr Health Sci. 13:703–709.
2013.PubMed/NCBI
|
|
14
|
Llovet JM, Chen Y, Wurmbach E, et al: A
molecular signature to discriminate dysplastic nodules from early
hepatocellular carcinoma in HCV cirrhosis. Gastroenterology.
131:1758–1767. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang XY, Degos F, Dubois S, et al:
Glypican-3 expression in hepatocellular tumors: diagnostic value
for preneoplastic lesions and hepatocellular carcinomas. Hum
Pathol. 37:1435–1441. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miao HL, Pan ZJ, Lei CJ, et al: Knockdown
of GPC3 inhibits the proliferation of Huh7 hepatocellular carcinoma
cells through down-regulation of YAP. J Cell Biochem. 114:625–631.
2013. View Article : Google Scholar
|
|
17
|
Zittermann SI, Capurro MI, Shi W and
Filmus J: Soluble glypican 3 inhibits the growth of hepatocellular
carcinoma in vitro and in vivo. Int J Cancer. 126:1291–1301.
2010.
|
|
18
|
Bruix J and Sherman M; Practice Guidelines
Committee AAftSoLD. Management of hepatocellular carcinoma.
Hepatology. 42:1208–1236. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu YL, Wang NN, Gu L, Yang HM, Xia N and
Zhang H: The suppressive effect of metabotropic glutamate receptor
5 (mGlu5) inhibition on hepatocarcinogenesis. Biochimie.
94:2366–2375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vauthey JN, Lauwers GY, Esnaola NF, et al:
Simplified staging for hepatocellular carcinoma. J Clin Oncol.
20:1527–1536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding W, You H, Dang H, et al:
Epithelial-to-mesenchymal transition of murine liver tumor cells
promotes invasion. Hepatology. 52:945–953. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang W, Mendoza MC, Pei X, et al:
Down-regulation of CMTM8 induces epithelial-to-mesenchymal
transition-like changes via c-MET/extracellular signal-regulated
kinase (ERK) signaling. J Biol Chem. 287:11850–11858. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen X, Lingala S, Khoobyari S, Nolta J,
Zern MA and Wu J: Epithelial mesenchymal transition and hedgehog
signaling activation are associated with chemoresistance and
invasion of hepatoma subpopulations. J Hepatol. 55:838–845. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun CK, Chua MS, He J and So SK:
Suppression of glypican 3 inhibits growth of hepatocellular
carcinoma cells through up-regulation of TGF-beta2. Neoplasia.
13:735–747. 2011.PubMed/NCBI
|
|
26
|
Cheng W, Tseng CJ, Lin TT, et al:
Glypican-3-mediated oncogenesis involves the Insulin-like growth
factor-signaling pathway. Carcinogenesis. 29:1319–1326. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pan Z, Chen C, Long H, et al:
Overexpression of GPC3 inhibits hepatocellular carcinoma cell
proliferation and invasion through induction of apoptosis. Mol Med
Rep. 7:969–974. 2013.PubMed/NCBI
|
|
28
|
Sung YK, Hwang SY, Farooq M, Kim JC and
Kim MK: Growth promotion of HepG2 hepatoma cells by
antisense-mediated knockdown of glypican-3 is independent of
insulin-like growth factor 2 signaling. Exp Mol Med. 35:257–262.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin CW, Mars WM, Paranjpe S, et al:
Hepatocyte proliferation and hepatomegaly induced by phenobarbital
and 1,4-bis [2-(3,5-dichloropyridyloxy)] benzene is suppressed in
hepatocyte-targeted glypican 3 transgenic mice. Hepatology.
54:620–630. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
the importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu M, Yin F, Fan X, et al: Decreased
TIP30 promotes Snail-mediated epithelial-mesenchymal transition and
tumor-initiating properties in hepatocellular carcinoma. Oncogene.
Mar 31–2014.(Epub ahead of print). View Article : Google Scholar
|
|
33
|
Cho NH, Shim HS, Rha SY, et al: Increased
expression of matrix metalloproteinase 9 correlates with poor
prognostic variables in renal cell carcinoma. Eur Urol. 44:560–566.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li H, Wang H, Wang F, Gu Q and Xu X: Snail
involves in the transforming growth factor beta1-mediated
epithelial-mesenchymal transition of retinal pigment epithelial
cells. PLoS One. 6:e233222011. View Article : Google Scholar
|
|
35
|
Suh Y, Yoon CH, Kim RK, et al: Claudin-1
induces epithelial-mesenchymal transition through activation of the
c-Abl-ERK signaling pathway in human liver cells. Oncogene.
32:4873–4882. 2013. View Article : Google Scholar
|
|
36
|
Huang XY, Ke AW, Shi GM, et al:
alphaB-crystallin complexes with 14-3-3zeta to induce
epithelial-mesenchymal transition and resistance to sorafenib in
hepatocellular carcinoma. Hepatology. 57:2235–2247. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yoshida S, Kornek M, Ikenaga N, et al:
Sublethal heat treatment promotes epithelial-mesenchymal transition
and enhances the malignant potential of hepatocellular carcinoma.
Hepatology. 58:1667–1680. 2013. View Article : Google Scholar : PubMed/NCBI
|