Introduction
Myelodysplastic syndrome (MDS), as a heterogeneous
group of related clonal diseases, is typified by monolineage or
multilineage dysplasia, ineffective hematopoiesis, or high risk of
transformation to acute myelocytic leukemia (AML) (1). MDS has been associated with aberrant
methylation of relevant gene promoters that can facilitate tumor
onset by silencing anti-oncogenes and by changing the expressions
of tumor-related genes (2,3). Unlike genetic mutation, these
epigenetic changes can be reverted by drugs such as DNA
methyltransferase inhibitor 5-azacytidine (AZA) that targets the
treatment of AML/MDS. Compared with traditional chemotherapeutic
agents, AZA is able to significantly increase the overall survival
rates of medium-risk II, high-risk MDS and WHO-AML patients
(4).
As a cytosine nucleoside analogue, AZA results in
demethylation at low concentration while endows cytotoxicity at
high concentration (5).
Low-concentration AZA weakens the methylation of CpG islands in
anti-oncogene promoter regions by inhibiting methyltransferase,
thus promoting the expressions of anti-oncogenes (e.g., p15, p16
and other negative cell cycle regulatory genes). However, it barely
toxifies normal cells (6).
Although AZA evidently raises the overall survival rates of
medium-risk II, high-risk MDS and WHO-AML patients, the rate of
complete remission remains low (7,8).
Heme oxygenase-1 (HO-1), which is an isozyme of heme oxygenase, has
been observed in many solid tumors including melanoma, brain tumor
and lymphosarcoma (9). Previous
studies also revealed that HO-1 affects the response of leukemic
cells to chemotherapy as well as their proliferation and apoptosis
via the TNF and NF-κB pathways (10). High HO-1 expression may indicate
progression, poor prognosis and chemotherapy resistance of leukemia
(11), which protects normal bone
marrow cells from the side effects of chemotherapy but allows tumor
cells to resist drugs (12).
Recent studies have reported that demethylation of Nrf2 induced by
certain drugs led to an increase in the protein expression and
activity of heme oxygenase-1, including 5-fluorouracil and dietary
phytochemicals (13,14). Nevertheless, neither the role of
HO-1 in MDS nor in the demethylating effect of AZA has been well
studied hitherto. Our study found that after being treated with low
concentration AZA (0.5 μM), SKM-1 cells expressed more HO-1, and
the bone marrow mononuclear cells (MNCs) from high-risk and very
high-risk MDS patients also expressed more HO-1 than those from
low-risk and very low-risk MDS patients did. Considering the role
of HO-1, we hypothesized that high HO-1 expression may weaken the
therapeutic effects of AZA and promote MDS malignant
progression.
Therefore, we analyzed the influence of HO-1 on
AZA-induced proliferation inhibition, apoptosis, cell cycle arrest
and demethylation in SKM-1 cells by silencing HO-1 gene with
lentivirus-mediated siRNA and by upregulating it with Hemin (15
μmol/l). Furthermore, the possible mechanism was explored.
Materials and methods
Cell lines and cell culture
conditions
Human AML cell lines, including HEL, U937 and THP-1,
were propagated in a monolayer culture in RPMI-1640 medium. MDS
cell line SKM-1 was purchased from the Japanese Collection of
Research Bioresources. RPMI-1640 medium was supplemented with 15%
fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml
streptomycin. The medium and antibiotics were bought from
Invitrogen (Carlsbad, CA, USA). All cells were maintained in a 37°C
incubator with 95% humidity and 5% CO2.
Patient samples
Bone marrow samples were collected during routine
diagnostic assessment after written informed consent had been
obtained. The patients were diagnosed by using WHO classification.
Patients’ bone marrow MNCs were separated by Ficoll-Hypaque (Sigma
Chemical Co.) density-gradient centrifugation and used immediately.
All participants provided written informed consent prior to
entering the study. The study was approved by the Institutional
Review Board of the Affiliated Hospital of Guiyang Medical
College.
Chemicals and antibodies
AZA (99.0% purity) was obtained from the Shanghai
Huilun Life Science and Technology Corp. Antibodies for western
blot analysis were obtained from Cell Signaling Technology
(Beverly, MA, USA) and secondary antibodies were purchased from
Li-Cor Corp. (Lincoln, NE, USA).
Cell proliferation assay
Cells were seeded at a density of 1,000 cells/well
in a 96-well plate. After overnight incubation at 37°C, serial
dilutions of test compounds were added and the cells were further
incubated in 5% CO2 for 24 h at 37°C. The inhibitory
effects were determined using colorimetric
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (MTT; Sigma, USA).
Apoptosis analysis
Apoptotic cells were analyzed by flow cytometry with
propidium iodide (PI) staining (BD Biosciences, San Jose, CA, USA).
Cells were treated with fresh drug preparations and medium for 24
h, washed in PBS and resuspended in 100 μl of binding buffer
containing 5 μl of Annexin V (BD Pharmingen, San Diego, CA, USA).
The cells were analyzed by flow cytometry after adding 5 μl of PI.
Annexin V binds to cells that express phosphatidylserine on the
outer layer of the cell membrane, while PI stains the cellular DNA
of those cells with a compromised cell membrane. This allows viable
cells (unstained with either fluorochrome) to be distinguished from
apoptotic cells (stained only with Annexin V) and necrotic cells
(stained with both Annexin V and PI). After being stained at room
temperature for 15 min in dark, the cells were measured by flow
cytometry and Cell Quest software (BD Biosciences). All experiments
were conducted at least three times.
Cell cycle analysis
Cell cycle was determined by flow cytometry using
PI. Cells (105/ml) were washed with PBS and fixed with
70% ice-cold ethanol. After 2 h, the cells were washed twice in PBS
and resuspended in PBS containing 50 mg/ml PI (Sigma), 200 mg/ml
DNase free RNase A (Citomed), and 0.1% Triton X-100 for 1 h at room
temperature. Acquisition was performed on a FACS Calibur flow
cytometer (Becton-Dickinson). Data were analyzed with the cell
cycle program from FlowJo software (Tree Star, Inc. Ashland, OR,
USA).
Quantitative real-time PCR
Total RNA was isolated and purified from cells using
the RNeasy kit (Qiagen, Hilden, Germany) and reverse-transcribed
using the Omniscript Reverse Transcription kit (Qiagen). cDNAs were
analyzed by quantitative real-time PCR using primers provided by
Airui Technology Corp. (Guiyang, China) and iQ SYBR Green supermix
(Bio-Rad, Hercules, CA, USA).
Methylation-specific PCR
Genomic DNA was prepared from cells and then
subjected to bisulfite conversion. The methylation status of CpG
islands in the p16 gene promoter was determined by
methylation-specific PCR (MSP). The primers used for unmethylated
p16 were: sense, 5′-TTTTTGGTGTTAAAGGGTGGTGTACT-3′ and antisense,
5′-CACAAAAACCCTCACTCACAACAA-3′, which yielded a fragment of 132 bp.
The primers used for methylated p16 were: sense,
5′-GTGTTAAAGGGCGGCGTAGC-3′ and antisense,
5′-AAAACCCTCACTCGCGACGA-3′, which yielded a PCR product of 122 bp.
DNA was amplified according to the following protocol: 95°C for 5
min, followed by 40 cycles at 95°C for 1 min, 60°C for 30 sec, 72°C
for 1 min, and a final extension step at 72°C for 10 min. Amplified
products were resolved on 3% agarose gels and visualized under
ultraviolet light after staining with ethidium bromide. Results
were confirmed by repeating MSP assays after an independently
performed bisulfite treatment.
Western blot analysis
Western blot analysis was carried out to analyze
protein expression and activation after cells were treated with AZA
alone or AZA plus Hemin, or after specific HO-1 knockdown. Briefly,
cells were washed in PBS, collected, and then lysed in
radioimmunoprecipitation assay buffer (50 mmol/l Tris-HCl; 150
mmol/l NaCl; 0.1% SDS; 0.5% Na-deoxycholate; 1% NP40) containing
proteinase inhibitor cocktail and phosphatase inhibitor cocktail
(Roche Applied Science, Indianapolis, IN, USA). The lysate was
centrifuged at 10000 × g at 4°C for 10 min. The supernatant (50–100
mg protein) was fractioned by SDS-PAGE using 10% gels and was
transferred electrophoretically to Hybond-enhanced
chemiluminescence membranes (GE Healthcare Life Sciences,
Piscataway, NJ, USA). The membrane was blocked with blocking buffer
(Li-Cor Corp.) at room temperature for 1 h and then incubated with
the primary antibody at 4°C overnight. After being washed by PBS
with 0.1% Tween-20 (PBST), the membrane was incubated with the
IRDye infrared secondary antibody (Li-Cor Corp.) for 1 h at room
temperature, washed with PBST again and detected with enhanced
chemiluminescence.
The recombinant lentiviral vector and
transfection
Self-prepared recombinant
lentivirus-V5-D-TOPO-HO-1-vector and control vector
lentivirus-V5-D-TOPO-vector were cotransfected into the 293FT
packaging cell line. The supernatant was collected 48 and 72 h
after infection to harvest the recombinant virus.
Lentivirus-V5-DTOPO-vector and lentivirus-V5-D-TOPO- vector -HO-1
were cotransfected into SKM-1 cells. The transfection rate was
determined by western blot analysis.
Intravenous MDS model of NOD/SCID
mice
NOD/SCID mice, purchased from Beijing laboratory
animal center, were injected intraperitoneally with 150 mg/kg
cyclophosphamide (Wako Pure Chemical Industries, Kyoto, Japan) on
each of two consecutive days to repress residual immunity. The day
after the third cyclophosphamide injection, mice were randomized
into three groups, SKM-1 (without treatment), SKM-1-vector
(transfected with empty vector) and SKM-1-siHO-1 (transfected with
siRNA targeting HO-1) cells (3×107 cells per animal)
were separately injected intravenously into the mice tail vein of
the three groups. On the sixth day after inoculation (day 0), the
three groups were further grouped into six: SKM-1, SKM-1-vector,
SKM-1-siHO-1, SKM-1 (AZA), SKM-1-vector (AZA), SKM-1-siHO-1 (AZA).
The mice were administered azacitidine (2.5 mg/kg) or NS once a day
from day 1 for seven consecutive days. The untreated control
received NS, drugs and NS were injected intraperitoneally. The
weight loss and survival times of the mice were recorded and
analyzed. The status of injected SKM-1 cells in peripheral blood
was confirmed by Wright staining. All procedures were conducted in
accordance with Guidelines for the Care and Use of Laboratory
Animals. The protocol was approved by the Committee on the Ethics
of Animal Experiments of Guiyang Medical College.
Statistical analysis
Each experiment or assay was performed at least
three times, and representative examples are shown. Data are
reported as means ± SEM. Statistically significant differences
between the treated groups were calculated using Student’s t-test.
Differences were considered statistically significant at
P<0.05.
Results
AZA treatment increases HO-1 expression
in SKM-1 cells
MDS cell line SKM-1 and AML cell lines U937, HEL and
THP-1 were treated with AZA (0.5 μM) for 24 h, the HO-1 expression
was detected by real-time PCR. AZA treatment obviously increased
HO-1 expression in SKM-1 cells, but not in the three AML cell lines
(Fig. 1A). Bone marrow MNCs of 48
MDS patients were collected and divided into a very-low-risk group,
a low-risk group, a high-risk group and a very high-risk group
according to WHO risk classification criteria (Table I). Real-time PCR results showed
that the HO-1 expression level of bone marrow MNCs from high-risk
and very high-risk MDS patients exceeded that of very low-risk and
low-risk patients (Fig. 1B).
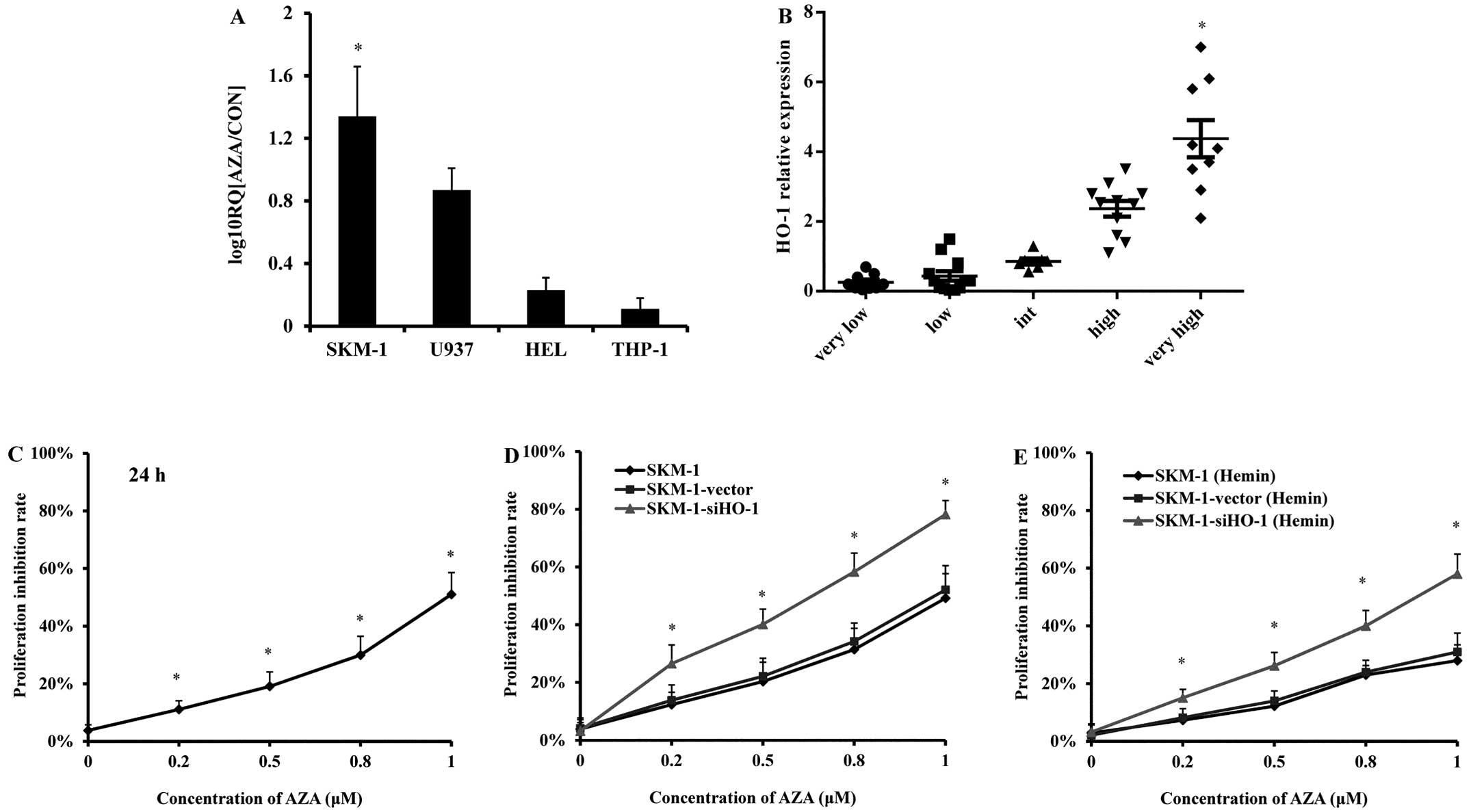 | Figure 1HO-1 relative expression in MDS/AML
cell lines and in MDS patients and the inhibitory effects of AZA on
SKM-1 cell proliferation. (A) HO-1 expressions in MDS cell line
SKM-1 and AML cell line U937, HEL, THP-1 treated with AZA (1 μM) by
qPCR. (B) HO-1 expressions in MDS patients by qPCR, classified
according to the WHO classification. Characteristics of MDS
patients are shown in Table I. (C)
Effects of AZA (0.2, 0.5, 0.8 and 1 μM) on cell growth in SKM-1
(untreated) cells. Cells were treated with AZA for 24 h. (D)
Effects of AZA (0.2, 0.5, 0.8 and 1 μM) on cell growth in SKM-1
(control), SKM-1-vector (transfected with empty vector),
SKM-1-siHO-1 (transfected with lentivirus-mediated HO-1 siRNA)
cells. Cells were treated with AZA for 24 h. (E) Effects of AZA
combined with Hemin on cell growth in SKM-1 (control),
SKM-1-vector, SKM-1-siHO-1. Cells were treated with Hemin for 24 h,
and then treated with AZA for 24 h. Cell viability was measured by
MTT. The MTT uptake and the effect were analyzed by Prism V5.0
(GraphPad Software, San Diego, CA, USA). Statistical differences
were calculated by one-way ANOVA. *P<0.05. |
 | Table IPatient characteristics. |
Table I
Patient characteristics.
|
Characteristics | (n, %) |
|---|
| Age(years), n | 48 |
| <70 | 34 (71) |
| ≥70 | 14 (29) |
| Sex, n | 48 |
| Male | 37 (77) |
| Female | 11 (23) |
| WHO Classification,
n | 48 |
| RA/RAS | 10 (21) |
| RCMD/RSCMD | 14 (29) |
| RAEB-I | 4 (8) |
| RAEB-II | 17 (36) |
| RAEB-t/AML | 3 (6) |
| WPSS risk
group | 48 |
| Very low | 9 (19) |
| Low | 12 (25) |
| Intermediate | 7 (14) |
| High | 11 (23) |
| Very high | 9 (19) |
Silencing HO-1 enhances the inhibitory
effects of AZA on SKM-1 cell growth
As detected by MTT assay, the growth inhibition
rates of SKM-1 cells at 24 h increased (11, 19, 30 and 51%) with
rising AZA concentration (0.2, 0.5, 0.8 and 1 μM) (Fig. 1C). Thus, AZA inhibited the growth
of SKM-1 cells in a concentration-dependent manner. After being
treated with AZA (0.2, 0.5, 0.8 and 1 μM) for 24 h, the
SKM-1-siHO-1 cells (transduced with HO-1 siRNA) were more prone to
inhibition (growth inhibition rates: 26.50, 40, 58 and 78%). In
contrast, the growth inhibition rates of the blank SKM-1 cells
(without any treatment) and SKM-1-vector cells (transduced with
empty vector) remained almost unchanged (Fig. 1D). When HO-1 expression was
upregulated first by Hemin (15 μmol/l), the growth inhibition
rates, especially those of the blank SKM-1 and SKM-1-vector cells,
were apparently decreased after treatment with AZA at the
concentrations mentioned above for 24 h (Fig. 1E). Therefore, the growth inhibitory
effect of AZA on SKM-1 cells was attenuated by HO-1 expression and
boosted by silencing HO-1 with siRNA.
HO-1 silencing sensitizes SKM-1 cells to
AZA-induced apoptosis through the caspase-3-dependent pathway
Given that HO-1 expression resisted AZA-induced
inhibition of SKM-1 cell growth, the apoptosis of SKM-1 cells in
which HO-1 was silenced by siRNA or upregulated by Hemin was
detected by flow cytometry after 24 h of treatment with 1 μM AZA.
Compared with the blank control and empty vector group (25.90 and
28.08%), the apoptotic rate of the HO-1 silencing SKM-1 cells
increased remarkably (48.64%), which, however, plummeted due to
HO-1 expression induced by Hemin (Fig.
2A). Hence, HO-1 expression protected SKM-1 cells from
AZA-induced apoptosis. To further analyze the mechanism for
enhanced apoptosis, the expressions of caspase-3 and −9, cleaved
caspase-3 and −9, BCL-2 and Bax were detected by western blot
analysis. After treatment with AZA for 24 h, the expression levels
of cleaved caspase-3 and −9 and Bax increased in the HO-1 silencing
SKM-1 cells, whereas that of BCL-2 decreased (Fig. 2B). To prove that increase of SKM-1
cell apoptosis was associated with caspase-3-dependent apoptotic
pathways, we treated SKM-1 cells with the caspase-3 inhibitor
Z-DEVE-FMK as well as AZA for 24 h, and then we evaluated the cell
apoptosis by flow cytometry. SKM-1 cells, especially for the HO-1
silencing group, were dramatically less subjected to apoptosis when
Z-DEVE-FMK was used (Fig. 2C).
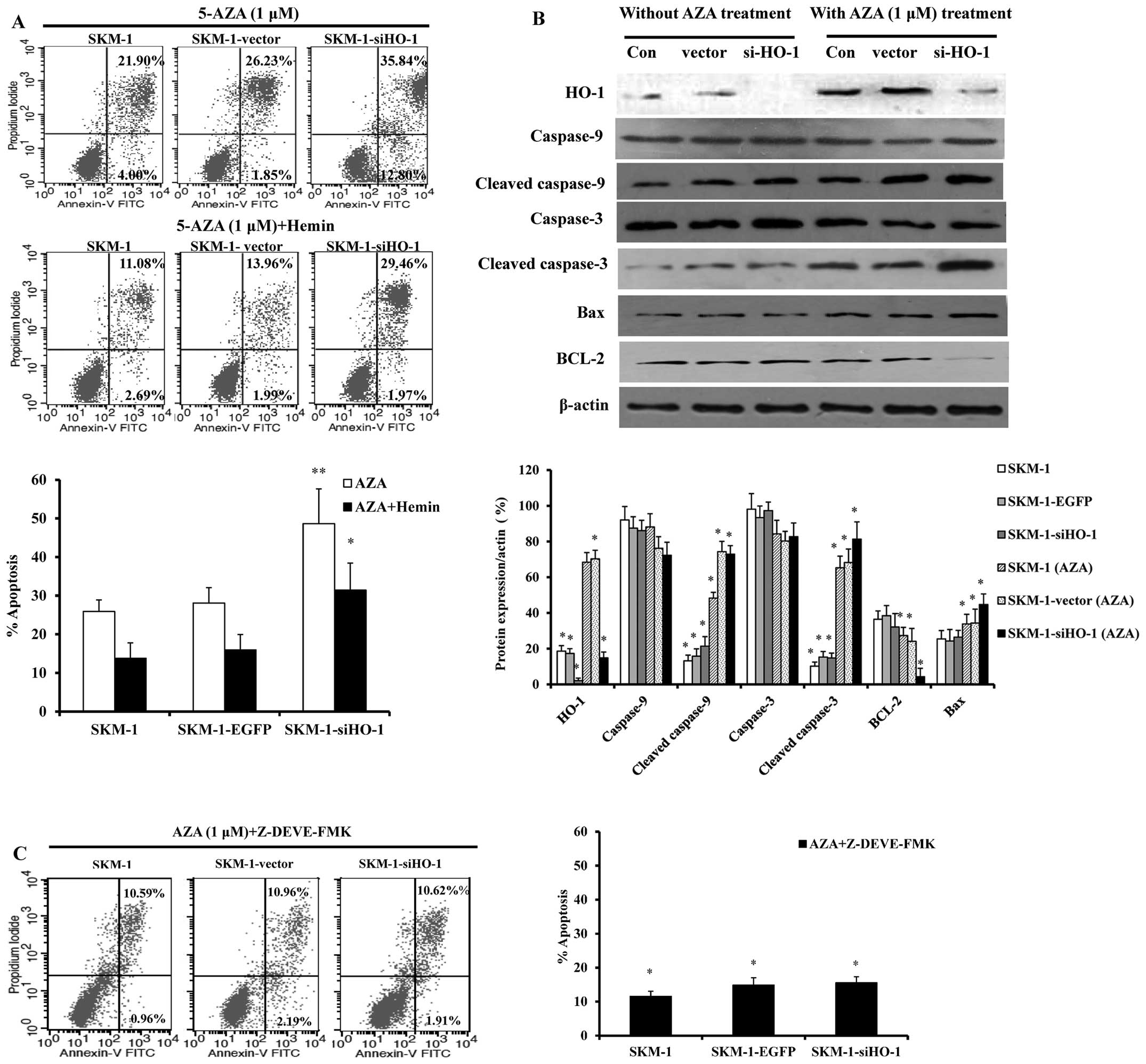 | Figure 2Silencing HO-1 sensitizes SKM-1 cells
to apoptosis induced by AZA. SKM-1 cells were grouped into SKM-1
(control), SKM-1-vector, SKM-1-siHO-1. (A) The cells were cultured
in the presence of AZA or AZA plus Hemin and apoptosis was analyzed
24 h thereafter by flow cytometry. Annexin-V-FITC/PI-positive cells
were determined by flow cytometry. Bar graph indicates the percent
of Annexin V-positive cells (apoptotic cells). (B) Western blot
analysis of apoptosis proteins in SKM-1 cells. Con, SKM-1
(untreated); vector, SKM-1-vector; siHO-1, SKM-1-siHO-1 with or
without AZA (1 μM) treatment. Protein expressions of HO-1,
caspase-9, −3, cleaved caspase-9, −3, Bcl-2 and BAX, are depicted.
Western blot bands were quantified with Quantity One software. Each
sample was normalized by related β-actin expression. (C) SKM-1
(untreated), SKM-1-vector, SKM-1-siHO-1 cells were added to
caspase-3 inhibitor combined with AZA, apoptosis was analyzed 24 h
thereafter by flow cytometry. Bar graph indicates the percent of
Annexin V-positive cells (apoptotic cells). All measurements were
conducted in triplicate. *P<0.05,
**P<0.01. |
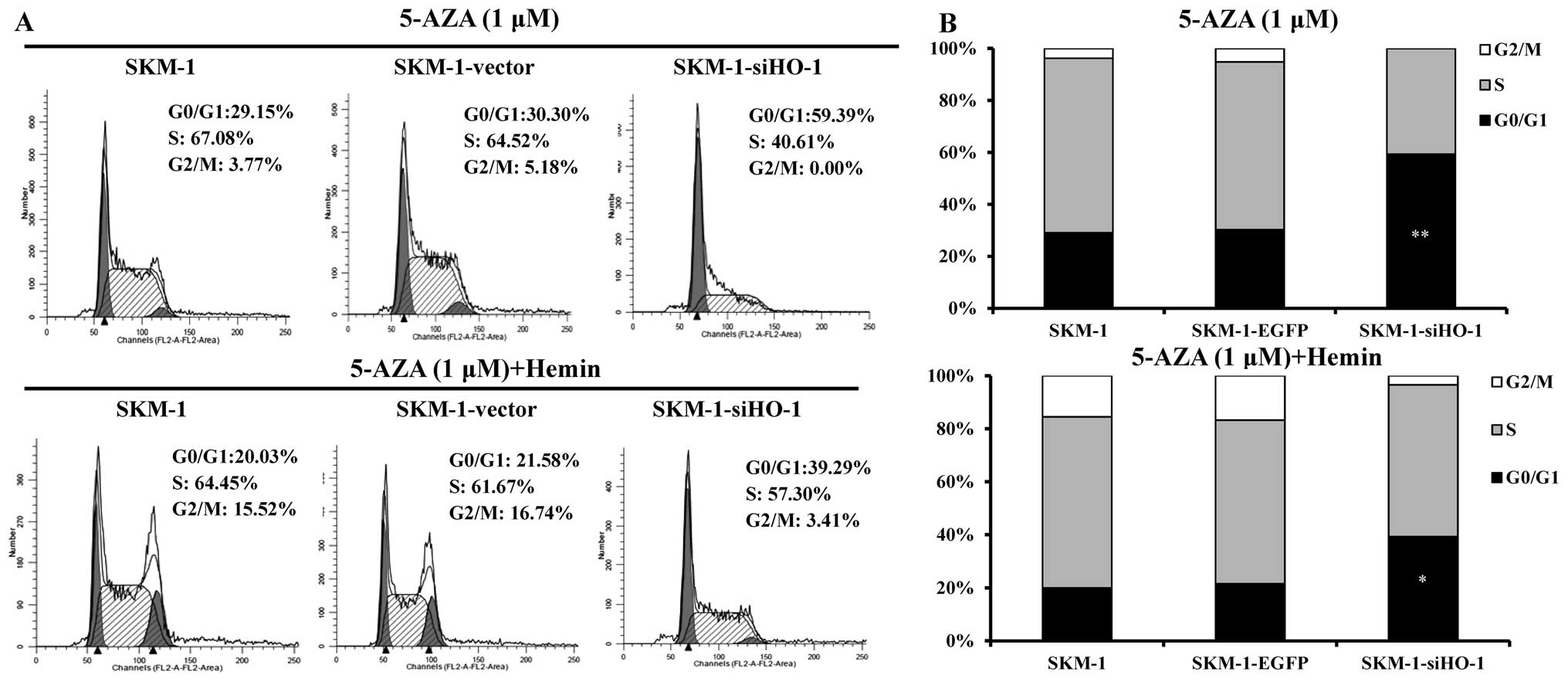
AZA arrests more SKM-1 cells in the G0/G1
phase by silencing HO-1
By regulating the methylation levels of promoters of
cell cycle negative regulatory genes such as p15 and p16, AZA
facilitates the expressions of p15 and p16 genes and influences the
regulation of cell cycle as a result. To explore whether HO-1
affected the cell cycle, flow cytometry was used for the blank
control, empty vector and HO-1 silencing SKM-1 cells before and
after treatment with AZA or AZA plus Hemin. The results revealed
that AZA-induced SKM-1 cell apoptosis was accompanied by G0/G1
arrest, particularly when HO-1 was silenced. On the contrary, HO-1
expression induced by Hemin accelerated the cell cycle progression
to G2/M phase (Fig. 3), probably
by promoting cell proliferation and differentiation (15,16).
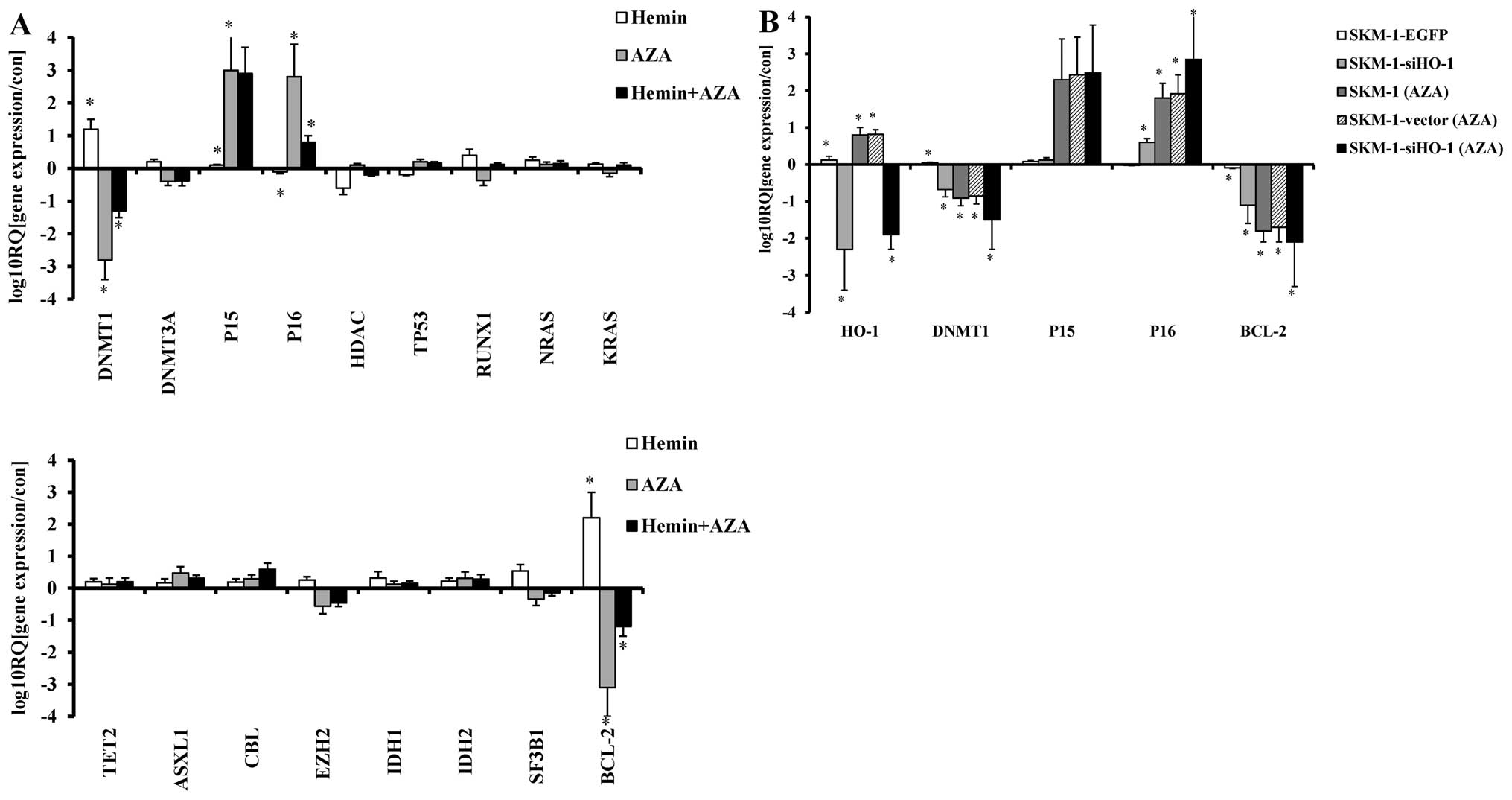
P16 overexpression mediates G0/G1 arrest
of SKM-1 cells
Decrease of cell apoptosis and malignant
proliferation, which are bound to occur during malignant
progression of MDS, are associated with silencing of anti-oncogenes
and activation of oncogenes that are dominantly controlled by
aberrant methylation (17,18). To clarify whether AZA-induced
significant HO-1-silencing SKM-1 cell proliferation inhibition,
apoptosis increase and G0/G1 arrest were associated with regulation
of methylation-related genes, the expressions of such genes (TP53,
RUNX1, NRAS, KRAS, TET2, ASXL1, CBL, EZH2, IDH1, IDH2, DNMT1,
DNMT3A, SF3B1, BCL-2, p15, p16 and HDAC) in AZA- or/and
Hemin-treated SKM-1 cells were detected by real-time PCR. We found
AZA-treated SKM-1 cells showed decreased DNMT1, BCL-2 expression
and increased p16, p15 expression, of these genes, DNMT1, BCL-2 and
p16 showed significant changes. However, HO-1 expression induced by
Hemin evidently decreased such effects (Fig. 4A). Moreover, after
lentivirus-mediated HO-1 knockdown and treatment with 1 μM AZA for
24 h, compared with treatment with AZA alone, the expression of
DNMT1 and BCL-2 was further reduced and p16 further increased
(Fig. 4B). We also found that HO-1
silenced SKM-1 cells tended to arrest in the G0/G1 phase after
treatment with AZA, for which increased expression of p16 may be
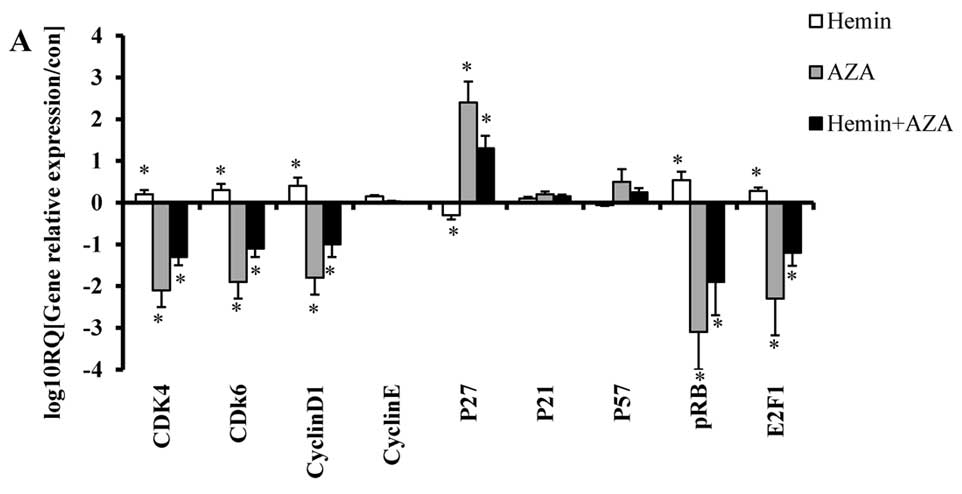
responsible. Next, we detected the relative expression of cell
cycle-related genes (CDK4, CDK6, Cyclin D1, Cyclin E, p21, p27,
P57, pRB and E2F1) by real-time PCR, silencing HO-1 further reduced
CDK4, CDK6, pRB, E2F1 expressions induced by AZA, whereas p27
expression further increased (Fig.
5A). Western blot analysis showed the protein expressions of
p16, CDK4, CDK6, pRB, E2F1 and p27 changed in accordance with the
above real-time PCR results (Fig.
5B).
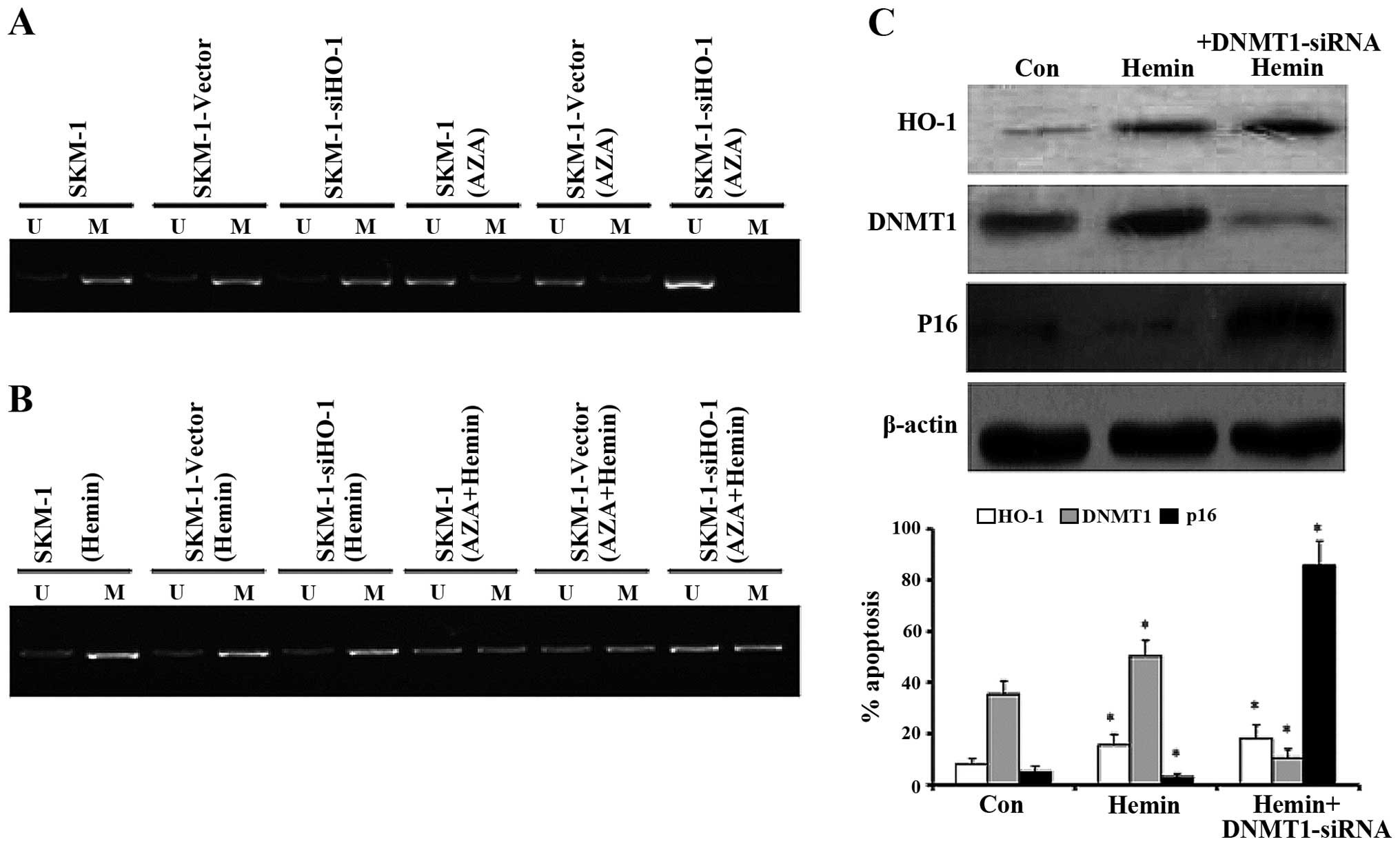
HO-1 silencing enhances the demethylating
effect of AZA on p16 gene promoter and subsequently promoted p16
expression through inhibiting DNMT1
Given that HO-1 silencing increases p16 expression
induced by AZA in SKM-1 cells, we analyzed whether HO-1 promoted
p16 expression by influencing its demethylation level through MSP.
Compared with SKM-1 (without treatment) and SKM-1-vector cells
(transfected with empty vector), the demethylation level of p16 in
SKM-1-siHO-1 cells (transfected with HO-1 siRNA) did not change
obviously, which was then augmented significantly after treatment
with AZA for 24 h (Fig. 6A).
However, when HO-1 was upregulated by Hemin, the demethylating
effects of AZA, as suggested by MSP, were diminished (Fig. 6B). We have found that silencing
HO-1 facilitated AZA-induced p16 expression, then we blocked DNMT1
gene by DNMT1 siRNA and upregulated HO-1 expression by Hemin at the
same time, western blot analysis confirmed HO-1 induction promoted
DNMT1 expression, however, when blocking DNMT1 by siRNA at the same
time, DNMT1 expression significantly decreased, and p16 showed a
relatively high protein expression level (Fig. 6C).
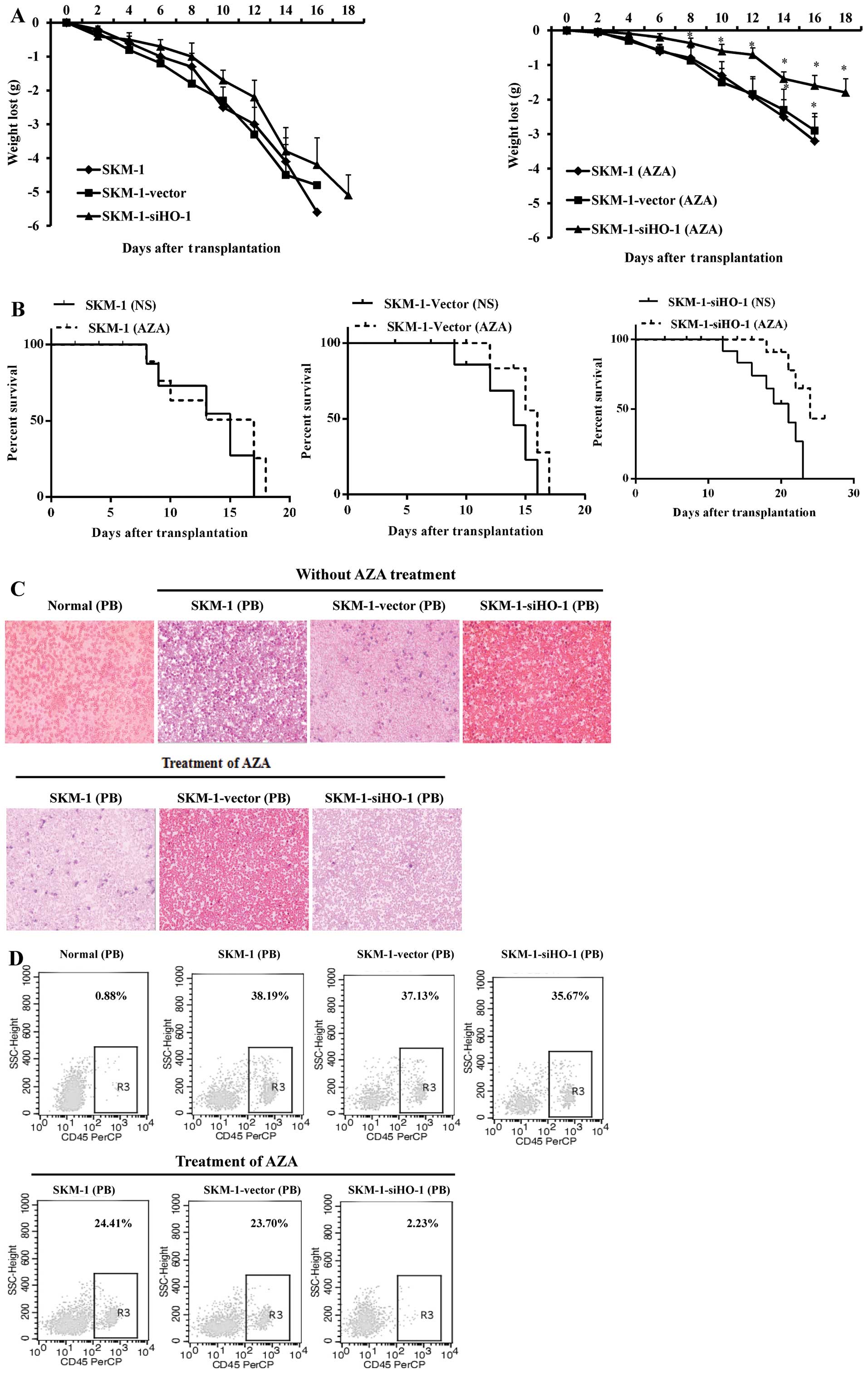
Silencing HO-1 efficiently enhances the
effects of AZA on inhibiting SKM-1 cells in intravenous MDS mouse
model
In vivo, we established the intravenous MDS
mouse model through injecting SKM-1 (without any treatment),
SKM-1-vector (transfected with empty vector), SKM-1-siHO-1
(transfected with lentivirus-mediated HO-1 siRNA) cells. Treatment
of the MDS mice with 2.5 mg/kg azacitidine resulted in significant
suppression of SKM-1 cell growth, especially for the HO-1 silenced
SKM-1 cells. On day 14, the AZA-treated groups: SKM-1 (AZA),
SKM-1-vector (AZA), SKM-1-siHO-1 (AZA) weighed, respectively 92,
91.4 and 93% of the mean body weight of the normal NOD/SCID mice,
the NS-treated groups: SKM-1, SKM-1-vector and SKM-1-siHO-1
weighed, respectively 56, 57.4 and 60% of the mean body weight of
the normal NOD/SCID mice (Fig.
7A). We observed that the overall survival of AZA-treated MDS
mice was prolonged, and in NS-treated MDS mice it shortened
(Fig. 7B). Wright-staining
analysis revealed a relatively lower number of primitive SKM-1
cells in AZA-treated MDS mice compared with the NS-treated in
peripheral blood, and we found that the AZA-treated MDS mice
injected the SKM-1-siHO-1 cells showed the lowest number of the
primitive SKM-1 cells (Fig. 7C).
In addition, flow cytometry showed decreased human CD45+
SKM-1 cells in the peripheral blood of AZA-treated MDS mice, the
AZA-treated SKM-1-siHO-1 mice, however, showed the most significant
decrease of human CD45+ cells (Fig. 7D).
Discussion
Epigenetic changes, in addition to genetic mutation,
predominantly control the onset of MDS/AML. As a common epigenetic
inhibitor, AZA, when inserted into newly synthesized DNA, can
demethylate DNA by binding DNA methyltransferase irreversibly. It
inhibits the synthesis of proteins after being inserted into RNA
(5). Besides, AZA is able to
regulate cell differentiation and proliferation (19). AZA was approved by FDA to treat all
MDS subtypes in 2004, and by using them Kumode et al
successfully treated MDS-derived AML (20). Despite the significantly elevated
overall survival rates of medium-risk II and high-risk MDS
patients, AZA usually gives unsatisfactory complete remission
rates. Some patients are insensitive to AZA, and the mechanism
remains unknown. Moreover, the therapeutic effects of AZA may
depend on gender, and females tend to have higher plasma AZA
concentrations than those of males and thus enjoy slower disease
progression (21). If found and
regulated in time, the latent adverse factors no longer hinder AZA
to exert effects, which are beneficial to the treatment of
high-risk and very high-risk MDS. Recently, HO-1 has been
implicated in many solid tumor-related studies, and high HO-1
expression may suggest progression, poor prognosis and chemotherapy
resistance of leukemia (11). For
patients receiving chemotherapy, high HO-1 expression can both
protect normal bone marrow cells and result in resistance of tumor
cells (12). By regulating
transcription factors Nrf2, NF-κB and AP-1 and by decreasing ROS
accumulation in AML cells, HO-1 shields the cells from TNF-induced
apoptosis (22). Rushworth et
al and Rushworth and MacEwan reported that Fas-associated death
domain-like interleukin 1β-converting enzyme-like inhibitory
protein (FLIP) promoted AML cells to resist apoptosis by regulating
HO-1 expression (10,11). There are no research reports on the
role of HO-1 in AZA treatment. HO-1 counteracts the therapeutic
effects of AZA on MDS via a yet unknown mechanism. In this study,
we silenced HO-1 by lentivirus-mediated siRNA or upregulated HO-1
by Hemin to evaluate the influences of AZA on the proliferation,
apoptosis, and cell cycle of SKM-1 cells in vitro and in
vivo, and to explore the possible mechanism.
MDS/AML cell lines SKM-1, HEL, U937 and THP-1 were
treated with AZA (0.5 μM) for 24 h, and bone marrow MNCs from 48
MDS patients were collected. HO-1 expressions were then detected by
real-time PCR. Compared with AML cell lines HEL, U937 and THP-1,
SKM-1 cells expressed higher level of HO-1, which was further
increased by AZA treatment. In contrast, AZA treatment barely
varied the expression levels of HO-1 in AML cells. In addition, the
HO-1 expression levels of bone marrow MNCs from high-risk and very
high-risk MDS patients exceeded those of low-risk and very low-risk
patients, implying that HO-1 may be associated with MDS malignant
progression.
Subsequently, we silenced HO-1 by
lentivirus-mediated siRNA or upregulated HO-1 by Hemin to evaluate
the roles of HO-1 in the AZA-induced growth inhibition, apoptosis
and cell cycle arrest of SKM-1 cells. MTT assay showed that AZA
suppressed the proliferation of SKM-1 cells
concentration-dependently, and that the inhibitory effects were
drastically boosted by silencing HO-1 but were diminished by Hemin.
However, the HO-1 silencing SKM-1 cells were less prone to Hemin
treatment, probably because Hemin could not induce HO-1 expression
as effectively as it usually did when HO-1 was silenced by siRNA.
After HO-1 was silenced, flow cytometry exhibited that AZA induced
significant apoptosis of SKM-1 cells. When HO-1 was upregulated by
Hemin, however, we found the apoptosis effects induced by AZA were
weaken, the blank control and empty vector SKM-1 cells underwent
significantly less apoptosis than the HO-1 silencing SKM-1 cells
did, similar outcomes to those of MTT assay were obtained.
Furthermore, flow cytometry showed that AZA arrested SKM-1 cells in
the G0/G1 phase, particularly when HO-1 was silenced. Upregulating
HO-1 by Hemin accelerated cell cycle progression to the G2/M phase,
which may be ascribed to the facilitated cell proliferation and
differentiation by HO-1 (15). By
using real-time PCR, the expressions of methylation-related genes
in AZA- or/and Hemin-treated SKM-1 cells [RNA splicing gene
(SF3B1), DNA methylation genes (TET2, DNMT1, DNMT3A, IDH1 and
IDH2), chromatin-modifying genes (ASXL1 and EZH2), transcriptional
regulatory gene (RUNX1), DNA repair gene (TP53), signal
transduction genes (CBL, NRAS and KRAS), histone acetylation gene
(HDAC), negative cell cycle regulatory genes (p15 and p16), and
apoptosis regulatory gene (BCL-2)] were detected (23). Of all the genes, only DNMT1, p16
and BCL-2 were subjected to expression changes. With HO-1
upregulation, the expression of DNMT1 and BCL-2 increased, while
that of p16 decreased. Based on the above experiments, the
expressions of HO-1, DNMT1, p16 and BCL-2 genes were detected again
in HO-1 silencing SKM-1 cells with or without AZA, the expression
of p16 further increased, and DNMT1, BCL-2 further decreased. BCL-2
is a target for sensitizing AZA, specific inhibition of BCL-2
contributes greatly to the growth inhibitory effects of AZA on AML
cell lines TF-1 and ML-2 (24).
BCL2L10 is a predictive factor which can predict whether or not a
patient will become resistant to AZA (25), so we considered the increased
apoptosis of SKM-1 cells was related to BCL-2 downregulation. p16
gene is methylated upon MDS or AML (26). Its high expression in low-risk MDS
patients may be associated with ineffective hematopoiesis that
results from p16-mediated cell senescence, while its low
expressions in high-risk MDS patients may be related with MDS
malignant progression (27).
Moreover, p16 arrests the T-ALL cell line CCRF-CEM in the G0/G1
phase via the pRB-E2F1 pathway (28). Therefore, we largely attributed
HO-1 silencing-enhanced sensitivity of AZA to increase in p16
expression.
In gastric cancer, restoration of p16 suppressed
CDK4, Cyclin D1 and delayed cell cycle transition (29). Real-time PCR showed that of all the
cell cycle-related genes (CDK4, CDK6, Cyclin D1, Cyclin E, p21,
p27, p57, pRB and E2F1), the levels of AZA-induced CDK4, CDK6, pRB
and E2F1 expression plummeted after HO-1 was silenced, whereas,
that of p27 expression increased and those of Cyclin E and p21
remained almost unchanged. Moreover, silencing HO-1 and treating
SKM-1 cells with AZA for 24 h simultaneously, as indicated by
western blot analysis, augmented p16, p27 expression and reduced
the expression of CDK4, CDK6, pRB and E2F1, suggesting that G0/G1
arrest was related with p16 expression. In our study, however, we
did not use specific p16 inhibitor or p16 siRNA to intervene with
its expression, so, it remains further to be confirmed.
P27/CDKN1B is known to mediate the cell cycle exit
with differentiation (30), so p27
may be involved in G0/G1 exit due to AZA-induced differentiation.
By using MSP, we analyzed whether HO-1 promoted the p16 expression
by influencing its demethylation level. After HO-1 was silenced,
AZA demethylated p16 much more effectively, as evidenced by the
apparently increased demethylation products. It has been reported
that PM exposure increased ROS production, DNMT1 expression and
methylation of the p16 promoter, and finally increased the risk of
lung cancer (31). Our study
revealed that AZA suppressed DNMT1 more evidently and p16
expression further increased after silencing HO-1. In order to
determine how HO-1 affected p16 expression, we blocked the DNMT1 by
DNMT1 siRNA and found that hemin-induced HO-1 expression can
promote DNMT1 expression and inhibit p16 expression slightly, and
blocking DNMT1 at the same time, DNMT1 decreased and p16 increased
significantly, suggesting that HO-1 may inhibit p16 expression
through promoting DNMT1 expression. However, HO-1 alone showed
sight effects on p16 expression, silencing HO-1 just sensitized AZA
to facilitate p16 expression.
Furthermore, the expression levels of caspase-3,
cleaved caspase-3, caspase-9, cleaved caspase-9, BCL-2 and Bax
proteins in AZA-treated SKM-1-siHO-1 cells were detected. The
expressions of caspase-3 and −9 fluctuated mildly, and those of
cleaved caspase-3 and −9 increased, the ratio of BCL-2/BAX
decreased. To further determine the apoptosis-related mechanism,
caspase-3 inhibitor, Z-DEVE-FMK, was added, after which the
apoptotic rate was, as detected by flow cytometry, significantly
reduced. When caspase-3 was inhibited, its activated form, cleaved
caspase-3, also decreased. In myeloid (P39, HL60) and T cells
(Jurkat), AZA induced apoptosis via multiple and separately
regulated pathways, such as cleavage of Bcl-2 family proteins,
activation of caspase-2 and −3-like, and induction of
hypomethylation (32). Thus, we
concluded that the AZA-induced apoptosis may be correlated with
caspase-3-dependent pathway.
In vivo, silencing HO-1 efficiently enhances
the effects of AZA on inhibiting SKM-1 cells with no significant
weight decrease, prolonging survival time and reduction in SKM-1
cells in peripheral blood. These results further confirmed that
silencing HO-1 played a crucial role in enhancing the AZA-induced
apoptosis.
In conclusion, our study revealed that silencing
HO-1 sensitized SKM-1 cells to AZA in vitro and in
vivo. After being treated with AZA, SKM-1 cells expressed more
HO-1, and the bone marrow MNCs from high-risk and very high-risk
MDS patients had higher HO-1 expression than those from low-risk
and very low-risk patients. With HO-1 silenced, AZA began to
inhibit the proliferation of SKM-1 cells more potently, accompanied
by raised apoptotic rate and dominant arrest in the G0/G1 phase.
The changes were related with increases in the expressions of p16,
cleaved caspase-3 and −9 as well as decrease in BCL-2/Bax ratio.
Hence, HO-1 may be one of the targets that sensitize AZA to fight
against MDS malignant progression, which paves the way for treating
high-risk and very high-risk MDS in clinical practice.
Acknowledgements
This study was supported, in part, by the National
Natural Science Foundation of China (nos. 81070444, 81270636,
81360501 and 81470006), International Cooperation Project of
Guizhou Province (no. 2011-7010), Social Project of Guizhou
Province (no. 2011-3012), Provincial Government Special Fund of
Guizhou Province (no. 2010-84), and Project of Science and
Technology Bureau of Guiyang City (no. [2012103-36]).
References
|
1
|
Sekeres MA and Cutler C: How we treat
higher-risk myelodysplastic syndromes. Blood. 123:829–836. 2014.
View Article : Google Scholar
|
|
2
|
Jiang Y, Dunbar A, Gondek LP, et al:
Aberrant DNA methylation is a dominant mechanism in MDS progression
to AML. Blood. 113:1315–1325. 2009. View Article : Google Scholar :
|
|
3
|
Stintzing S, Kemmerling R, Kiesslich T,
Alinger B, Ocker M and Neureiter D: Myelodysplastic syndrome and
histone deacetylase inhibitors: ‘to be or not to be acetylated’? J
Biomed Biotechnol. 2011:2141432011. View Article : Google Scholar
|
|
4
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
et al: Efficacy of azacitidine compared with that of conventional
care regimens in the treatment of higher-risk myelodysplastic
syndromes: a randomised, open-label, phase III study. Lancet Oncol.
10:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kimura S, Kuramoto K, Homan J, et al:
Antiproliferative and antitumor effects of azacitidine against the
human myelodysplastic syndrome cell line SKM-1. Anticancer Res.
32:795–798. 2012.PubMed/NCBI
|
|
6
|
Figueroa ME, Skrabanek L, Li Y, et al: MDS
and secondary AML display unique patterns and abundance of aberrant
DNA methylation. Blood. 114:3448–3458. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ades L and Santini V: Hypomethylating
agents and chemotherapy in MDS. Best Pract Res Cl Ha. 26:411–419.
2013. View Article : Google Scholar
|
|
8
|
Khan C, Pathe N, Fazal S, Lister J and
Rossetti JM: Azacitidine in the management of patients with
myelodysplastic syndromes. Ther Adv Hematol. 3:355–373. 2012.
View Article : Google Scholar
|
|
9
|
Jozkowicz A, Was H and Dulak J: Heme
oxygenase-1 in tumors: is it a false friend? Antioxid Redox Sign.
9:2099–2117. 2007. View Article : Google Scholar
|
|
10
|
Rushworth SA, Zaitseva L, Langa S, Bowles
KM and MacEwan DJ: FLIP regulation of HO-1 and TNF signalling in
human acute myeloid leukemia provides a unique secondary
anti-apoptotic mechanism. Oncotarget. 1:359–366. 2010.
|
|
11
|
Rushworth SA and MacEwan DJ: HO-1
underlies resistance of AML cells to TNF-induced apoptosis. Blood.
111:3793–3801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma D, Fang Q, Li Y, et al: Crucial role of
heme oxygenase-1 in the sensitivity of acute myeloid leukemia cell
line Kasumi-1 to ursolic acid. Anti-cancer Drug. 25:406–414. 2014.
View Article : Google Scholar
|
|
13
|
Su ZY, Shu L, Khor TO, Lee JH, Fuentes F
and Kong AN: A perspective on dietary phytochemicals and cancer
chemoprevention: oxidative stress, nrf2, and epigenomics. Topics
Curr Chem. 329:133–162. 2013. View Article : Google Scholar
|
|
14
|
Kang KA, Piao MJ, Kim KC, et al:
Epigenetic modification of Nrf2 in 5-fluorouracil-resistant colon
cancer cells: involvement of TET-dependent DNA demethylation. Cell
Death Dis. 5:e11832014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang LF, Qi J, Zuo G, et al:
Osteoblast-secreted factors promote proliferation and osteogenic
differentiation of bone marrow stromal cells via
VEGF/heme-oxygenase-1 pathway. PLoS One. 9:e999462014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wegiel B, Hedblom A, Li M, et al: Heme
oxygenase-1 derived carbon monoxide permits maturation of myeloid
cells. Cell Death Dis. 5:e11392014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parker JE, Mufti GJ, Rasool F, Mijovic A,
Devereux S and Pagliuca A: The role of apoptosis, proliferation,
and the Bcl-2-related proteins in the myelodysplastic syndromes and
acute myeloid leukemia secondary to MDS. Blood. 96:3932–3938.
2000.PubMed/NCBI
|
|
18
|
Issa JP: The myelodysplastic syndrome as a
prototypical epigenetic disease. Blood. 121:3811–3817. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Curik N, Burda P, Vargova K, et al:
5-azacitidine in aggressive myelodysplastic syndromes regulates
chromatin structure at PU.1 gene and cell differentiation capacity.
Leukemia. 26:1804–1811. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumode T, Fukui A, Eguchi G, Yamaguchi T
and Maeda Y: A case of secondary leukemia subsequent to
myelodysplastic syndromes successfully treated with azacitidine.
Case Rep Med. 2014:7939282014.PubMed/NCBI
|
|
21
|
Jasielec J, Saloura V and Godley LA: The
mechanistic role of DNA methylation in myeloid leukemogenesis.
Leukemia. 28:1765–1773. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heasman SA, Zaitseva L, Bowles KM,
Rushworth SA and Macewan DJ: Protection of acute myeloid leukaemia
cells from apoptosis induced by front-line chemotherapeutics is
mediated by haem oxygenase-1. Oncotarget. 2:658–668.
2011.PubMed/NCBI
|
|
23
|
Cazzola M, Della Porta mg and Malcovati L:
The genetic basis of myelodysplasia and its clinical relevance.
Blood. 122:4021–4034. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bogenberger JM, Kornblau SM, Pierceall WE,
et al: BCL-2 family proteins as 5-Azacytidine-sensitizing targets
and determinants of response in myeloid malignancies. Leukemia.
28:1657–1665. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cluzeau T, Robert G, Mounier N, et al:
BCL2L10 is a predictive factor for resistance to azacitidine in MDS
and AML patients. Oncotarget. 3:490–501. 2012.PubMed/NCBI
|
|
26
|
Karlic H, Herrmann H, Varga F, et al: The
role of epigenetics in the regulation of apoptosis in
myelodysplastic syndromes and acute myeloid leukemia. Crit Rev
Oncol Hemat. 90:1–16. 2014. View Article : Google Scholar
|
|
27
|
Wang YY, Cen JN, He J, et al: Accelerated
cellular senescence in myelodysplastic syndrome. Exp Hematol.
37:1310–1317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ausserlechner MJ, Obexer P, Geley S and
Kofler R: G1 arrest by p16INK4A uncouples growth from cell cycle
progression in leukemia cells with deregulated cyclin E and c-Myc
expression. Leukemia. 19:1051–1057. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim JK, Noh JH, Eun JW, et al: Targeted
inactivation of HDAC2 restores p16INK4a activity and exerts
antitumor effects on human gastric cancer. Mol Cancer Res.
11:62–73. 2013. View Article : Google Scholar
|
|
30
|
Ng KP, Ebrahem Q, Negrotto S, et al: p53
independent epigenetic-differentiation treatment in xenotransplant
models of acute myeloid leukemia. Leukemia. 25:1739–1750. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soberanes S, Gonzalez A, Urich D, et al:
Particulate matter Air Pollution induces hypermethylation of the
p16 promoter Via a mitochondrial ROS-JNK-DNMT1 pathway. Sci Rep-UK.
2:2752012.
|
|
32
|
Khan R, Schmidt-Mende J, Karimi M, et al:
Hypomethylation and apoptosis in 5-azacytidine-treated myeloid
cells. Exp Hematol. 36:149–157. 2008. View Article : Google Scholar : PubMed/NCBI
|