Introduction
In Japan, >90% of esophageal cancer is squamous
cell carcinoma, which prognosis is poorer than adenocarcinoma of
esophagus (1). Patients with
advanced stage of esophageal cancer have a poor prognosis with a
lethal outcome, despite efforts to improve diagnostic procedures
and treatment modalities (2).
Dendritic cells (DCs), which are potent
antigen-presenting cells, could coordinate innate and adaptive
immune responses. Hence, DC-based tumor immunotherapy was
introduced in the patients with prostate cancer, melanoma, renal
cell carcinoma, glioma, gastric cancer, colon cancer, and pediatric
solid tumor (3). In 2010,
monocyte-derived dendritic cells (moDCs) pulsed with fusion antigen
protein consisting of full-length prostatic acid phosphatase (PAP)
and full-length GM-CSF were approved by U.S. Food and Drug
Administration for the treatment of men with hormone refractory
prostate cancer. Cellular immunotherapy using these moDCs prolonged
overall survival among men enrolled in the phase III clinical study
compared with placebo group (4).
As demonstrated in phase III trials of this prostate cancer
immunotherapy, moDC-based cellular immunotherapy was shown to be
effective in cancer patients when the candidate is selected
properly. However, with regard to DC-based cellular immunotherapy
for carcinoma of esophagus, only few studies (5–7) have
been carried out. One of the studies dealt with WT1 peptide-pulsed
DC therapy with activated T lymphocyte therapy for advanced cancers
including two patients with esophageal cancer (5). The study showed that there is
beneficial effect to some extent. The other two reports dealt with
DC therapy for primary malignant melanoma of the esophagus
(6,7). On this note, Asakage et al
showed that tumor lysate-pulsed DC therapy is a safe and promising
approach as adjuvant therapy for primary malignant melanoma of the
esophagus (6). Likewise, Ueda
et al reported that peptide-specific immune response could
be induced in patients with primary malignant melanoma of the
esophagus after immunotherapy using DCs pulsed with MAGE peptides
(7).
We performed phase I/II clinical trial of moDCs
pulsed with SART1 (8) peptide for
patients with advanced squamous cell carcinoma of esophagus. In
addition, we performed in vitro studies concerning
cytotoxicity of patient’s lymphocytes cultured with SART1
peptide-pulsed moDCs or exosomes secreted from these moDCs against
esophageal carcinoma cell line. We also performed an IFN-γ ELISPOT
assay using patient lymphocytes obtained before and after SART1
peptide-pulsed moDC vaccination. Although clinical benefit was not
clearly demonstrated, in vitro and in vivo immune
responses caused by SART1 peptide-pulsed moDC vaccination was
revealed in the present study.
Materials and methods
Study design
The study was carried out according to a protocol
approved by the Institutional Review Board of Niigata University
School of Medicine and conducted in accordance with the Helsinki
Declaration. Written informed consent was obtained from all
patients. Seven patients were enrolled in this open-labeled,
non-randomized phase I/II clinical trial. The primary aim of the
study was to evaluate feasibility and safety, whereas the secondary
aim was to evaluate immunological and clinical responses to DC
vaccination (Table I).
 | Table IPatient profile, mode of DC therapy
and clinical response. |
Table I
Patient profile, mode of DC therapy
and clinical response.
| Patient no. | Age/Gender | Tumor
progression | Previous
therapy | Cytokines for DC
culture | Antigen
peptides | Mode of
infusion | No. of
infusion | Average no. of
infused cells (x108) | DTH | Effect of DC
therapy | Tumor marker | Outcome | Adverse effects
(association) |
|---|
| 1 | 58/M | Liver
metastasis | NAC, esohagectomy,
hepatectomy | GM-CSF, IL-4 | SART-1 | IV | 3 | 3.08 | ND | PD | Elevated | Died (10 M) | No |
| 2 | 71/M | Lung
metastasis | Esophagectomy, Cx,
Rx | GM-CSF, IL-4 | SART-1 | IV | 5 | 1.28 | ND | NC | Negative for 20 M
after DC therapy | Survived for 20 M
then died | Hypophosphatemia
(possible) |
| 3 | 61/M | Aortic invasion,
liver and lung metastasis | Cx | GM-CSF | SART-1
IL-4 | IV | 3 | 1.79 | ND | PD | Elevated | Died (2 M) | No |
| 4 | 59/M | Mediastinal LNs
matastasis, pleural infiltration | Esophagectomy, Cx,
Rx | GM-CSF, IL-4 | SART-1 | IV | 2 | 0.54 | ND | PD | Elevated | Died (1 M) | No |
| 5 | 66/M | Liver, kidney and
skin involvement | NAC,
esohagectomy | GM-CSF, IL-4,
TNF-α | SART-1/KLH | SC | 3 | 1.09 | Negative | PD | Elevated | Died (2 M) | No |
| 6 | 67/M | Liver and neck LNs
metastasis | Esophagectomy,
Rx | GM-CSF, IL-4,
TNF-α | SART-1/KLH | SC | 3 | 0.31 | KLH positive | PD | Elevated | Died (4 M) | No |
| 7 | 53/M | Liver and abdominal
LNs metastasis | NAC, esohagectomy,
Cx | GM-CSF, IL-4,
TNF-α | SART-1/KLH | SC | 2 | 0.77 | KLH positive | PD | Elevated | Died (3 M) | No |
Patients and treatment
Eligibility criteria for the present study were as
follows: i) advanced or relapsed squamous cell carcinoma of
esophagus, which had been treated with standard therapy; ii)
presence of HLA-A*24:02; and iii) performance status (PS) ≤1
(ECOG-scale).
The buffy coat cells of patients were collected by
leukapheresis (in 14 out of 21 times DC preparation) or bag method
(in seven times) with written informed concent. Peripheral blood
mononuclear cells (PBMCs) were separated by Ficoll-Hypaque
(Lymphoprep; Axis-Shield Poc AS, Oslo, Norway) density
centrifugation. Monocytes were isolated by culturing PBMNCs in
plastic culture dish (BD Biosciences, San Jose, CA, USA) at a cell
concentration of 3–5×106/ml and removing non-adherent
cells from the dish. Immature moDCs were induced from monocytes by
culturing plastic adherent cells in the same culture dish
containing RPMI-1640 (Invitrogen Life Technologies, Carlsbad, CA,
USA) with 5% autologous serum, 100 ng/ml GM-CSF (Kirin Brewer y
Co., Ltd., Gunma, Japan) and 10 ng/ml IL-4 (Schering-Plough
Research Institute, Kenilworth, NJ, USA) for 7 days. Immature moDCs
were matured by adding 10 ng/ml TNF-α (PeproTech, Inc., Rocky Hill,
NJ, USA) on day 6. Mature moDCs were collected from culture dish by
pipetting and occasionally using cell scraper (Corning Life
Sciences, Tewksbury, MA, USA).
Tumor antigen peptide used for the study was
SART1690–698 (EYRGFTQDF, HLA-A*24:02 restricted, GMP
grade), which was produced by Multiple Peptide Systems (San Diego,
CA, USA) and donated by Prof. Kyogo Itoh (Kurume University,
Fukuoka, Japan). SART1 peptide was added to the moDC culture at a
concentration of 50 μg/ml during the last 24 h. In the last three
patients, keyhole limpet hemocyanin (KLH: carrier protein for
peptide antigen) (Calbiochem, La Jolla, CA, USA) was pulsed at a
concentration of 50 μg/ml together with SART1 peptide for the last
24 h of moDC culture (Table I). On
occasion, PBMNCs and peptide-pulsed moDCs were cryopreserved for
later in vitro study.
On the day of vaccination, peptide-pulsed moDCs were
washed and re-suspended in 500 μl saline with 5% autologous serum
and transferred to a 1 ml syringe for injection. The moDC
suspension was injected intravenously (IV) in the first four
patients (patients 1–4) and subcutaneously (SC) in the upper arm in
the last three patients (patients 5–7). Infusions with
peptide-pulsed moDCs were repeated every three weeks up to five
times depending on the patient.
Clinical evaluation
Evaluation with CT scan and clinical examinations
were performed before, during and after vaccinations. A skin test
for delayed-type hypersensitivity (DTH) reaction was performed
using an intradermal injection of 100 μl of the peptide or KLH (5
mg/ml each) on the palmar side of the forearm. Saline solution (100
μl) was used as a negative control. More than 2 mm red induration
area after 48 h was defined as a positive DTH skin test
reaction.
Analysis of surface phenotypes of moDCs
prepared for infusion
Immediately before injection, antigen-pulsed moDCs
were spared and subjected to phenotypic analysis as previously
described (9). The cells were
stained by incubation with monoclonal antibodies against CD1a
(Immunotech, Marseille, France), CD14, CD80, CD86 and HLA-DR (BD
Biosciences) together with the relevant isotype controls to analyze
the expression of cell surface antigens.
Preparation of moDC-derived exosomes
moDC-derived exosomes were prepared using the method
described by Zitvogel et al (10). The whole culture medium of
SART1-pulsed moDCs was harvested at the time of preparing moDCs for
injection. moDC culture medium was centrifuged at 300 × g for 20
min and the supernatant was collected. The supernatant was
centrifuged at 10,000 × g for 30 min and the supernatant was
collected again for eliminating cell debris. Then the supernatant
was ultra-centrifuged at 100,000 × g for 60 min and the pellet was
collected and washed once in a large volume of medium. Exosome
pellet was finally dissolved at the concentration of exosomes
derived from 107 moDCs in 1 ml RPMI-1640 medium. The
protein concentration in the exosome preparation was measured and
used for SART1 peptide-specific cytotoxic T lymphocyte (CTL)
induction assay.
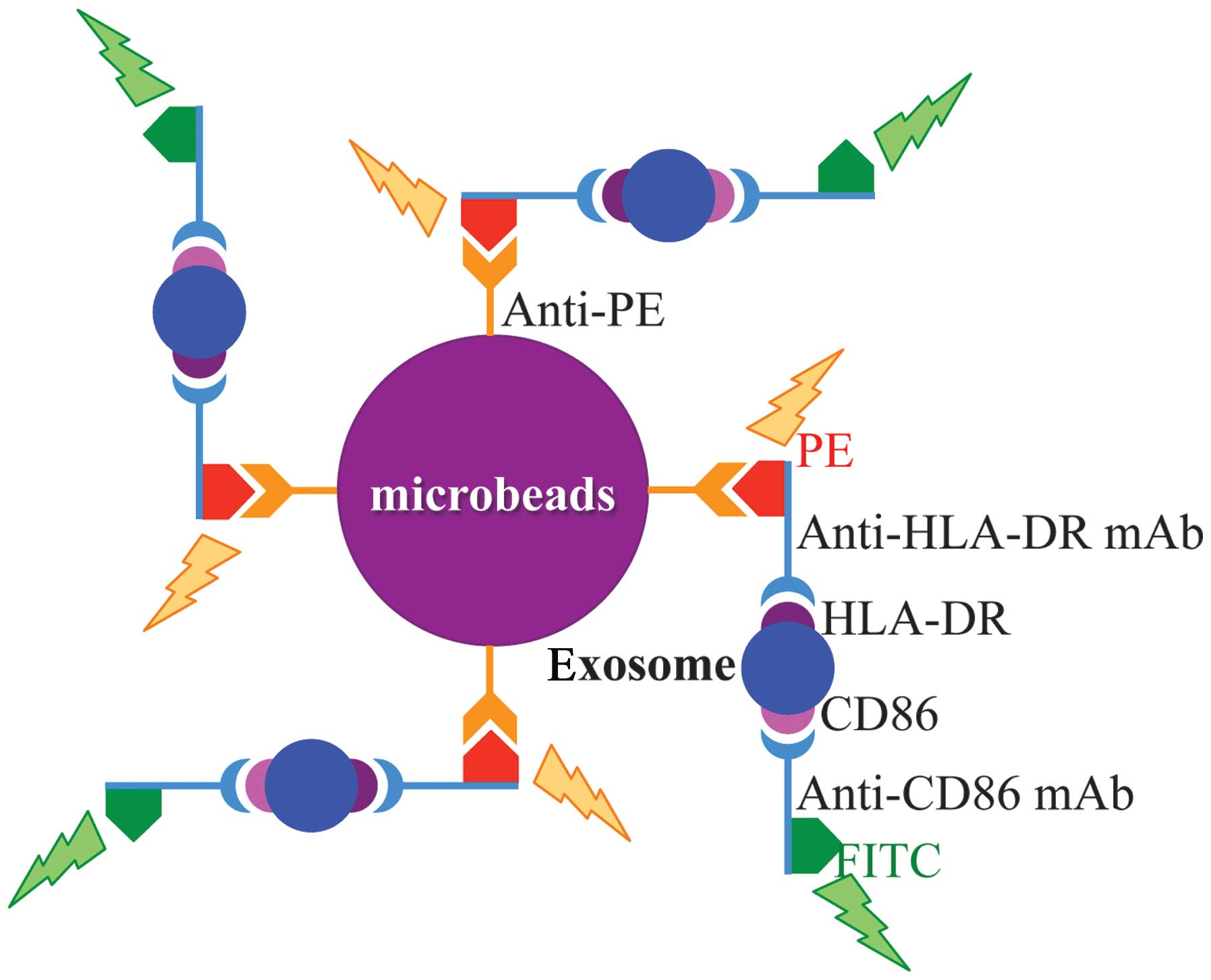
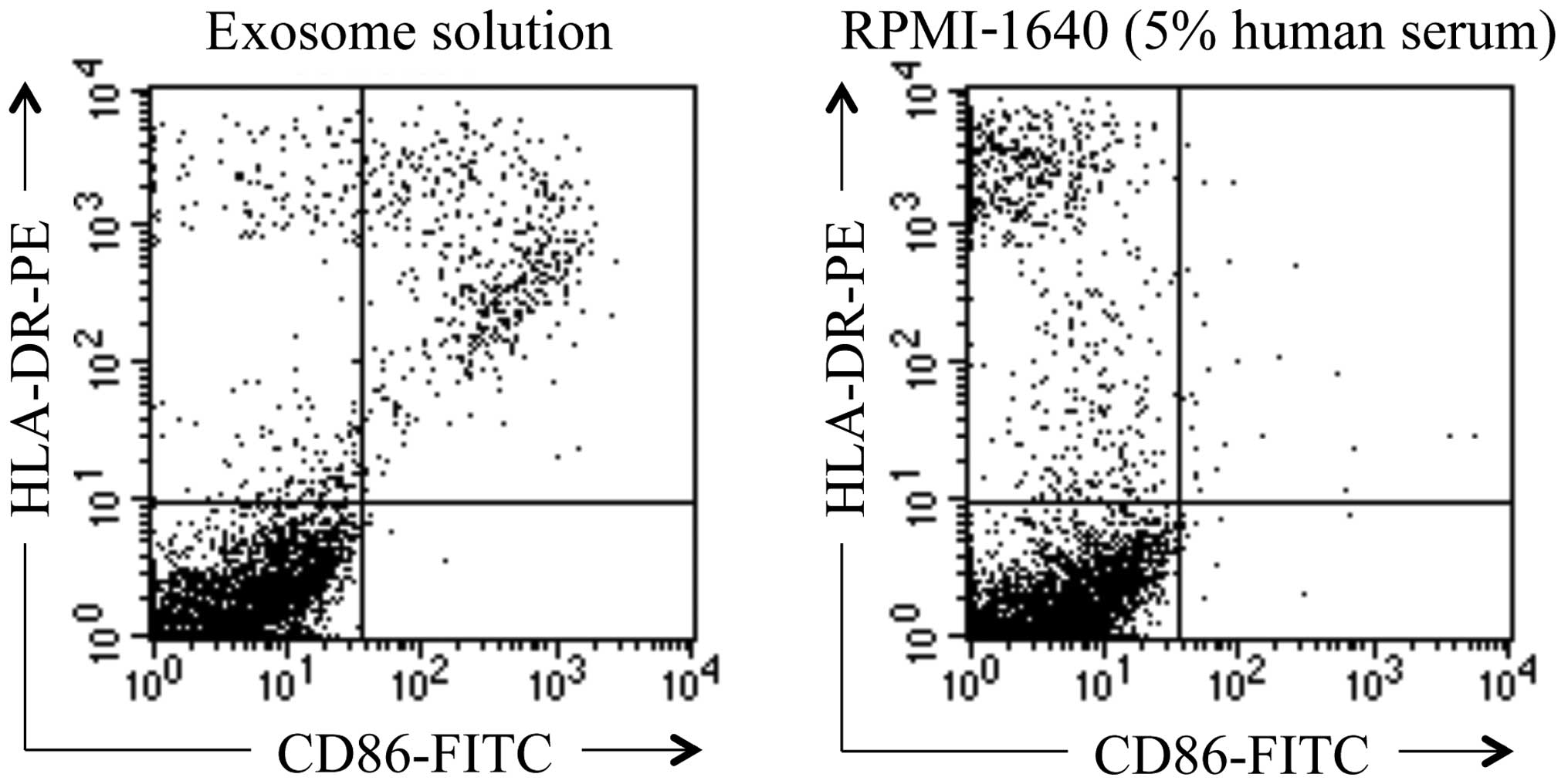
Identification of exosome
Exosome was identified by demonstrating the
expressions of both HLA-DR and CD86 on the surface of the nanoscale
vesicle (Fig. 1) (11). Anti-PE micro-beads (Miltenyi Biotec
GmbH, Bergisch Gladbach Germany) were incubated with PE labeled
anti-HLA-DR monoclonal antibody (BD Biosciences) for 1 h at 4°C
with tapping every 10 min. The mixture was washed with PBS by
centrifuging at 9,100 × g (10,000 rpm) for 10 min at 4°C twice in
order to eliminate free monoclonal antibody. Microbeads pellet was
suspended with Fc receptor blocking solution (PBS with 0.5% human
γ-globulin and 0.1% sodium azide) (for blocking the Fc receptors
possibly expressed on the surface of exosome) and mixed with
exosome solution, and then incubated for 1.5 h at 4°C with tapping
every 20 min. The mixture was washed with PBS by centrifuging at
9,100 × g (10,000 rpm) for 10 min at 4°C twice in order to
eliminate free exosomes. Microbeads bound with exosomes were
incubated with FITC-labeled anti-CD86 monoclonal antibody for 1 h
at 4°C with tapping every 10 min. The mixture was washed with PBS
by centrifuging at 9,100 × g (10,000 rpm) for 10 min at 4°C twice.
Microbeads pellet was suspended with PBS and processed for flow
cytometry analysis. Anti-PE microbeads, anti-PE microbeads bound
with PE-labeled anti-HLA-DR monoclonal antibody (in excess of
microbeads), and anti-FITC microbeads bound with FITC-labeled
anti-HLA-DR monoclonal antibody (in excess of microbeads) were used
for compensation of flow cytometry analysis. RPMI-1640 medium with
5% of human serum was used as control for exosomes.
Proliferation assay
Proliferation assay was performed for evaluating an
antigen-specific proliferative capacity of lymphocytes in
moDC-treated patients. Briefly, SART1 peptide-pulsed moDCs, which
were used for vaccination, were irradiated with 30 Gy of
l37Cs generated from gamma irradiation apparatus
(PS-3000SB Cs-137; Pony Industry Co., Ltd., Osaka, Japan)
immediately before MLC. One hundred thousand allogeneic or
patient’s autologous PBMCs, which were collected before or after
3rd vaccination then cryopreserved, were co-cultured in a 96-well
flat-bottom microtiter plate (BD Biosciences) with graded numbers
of moDCs. Co-cultured cells were pulsed with 0.5 μCi (18.5
kBq)/well [methyl-3H]-thymidine (PerkinElmer, Boston,
MA, USA) on day 5 of culture and harvested after overnight culture
with a cell harvester (Labo Mash; Futaba Medical Inc., Tokyo,
Japan). Cellular proliferation was evaluated by measuring
3H-thymidine incorporation with a liquid scintillation
counter (LSC-5100; Aloka Co., Ltd., Tokyo, Japan). The experiments
were performed in triplicate.
SART1 peptide-specific CTL induction
using peptide-pulsed moDCs
Patient’s PBMCs were co-cultured with SART1
peptide-pulsed and irradiated moDCs at a cell ratio of 10:1 in
24-well plate containing 2 ml of 5% autologous serum-containing
RPMI-1640 medium as described previously (12). IL-2 (Shionogi & Co., Ltd.,
Osaka, Japan) and IL-7 (Cytheris S.A., Vanves, France) were added
to the co-culture on day 3 at the final concentration of 50 U/ml
and 10 ng/ml, respectively. Two thirds of the medium with IL-2 and
-7 were replenished every 2–3 days throughout the culture period.
Patient’s MNCs were stimulated repeatedly every week with the same
cryopreserved and thawed peptide-pulsed moDCs and the co-culture
was maintained for 4 weeks. For CTL induction by exosomes derived
from SART1-pulsed moDCs, autologous PBMCs were cultured in 2 ml
autologous serum-containing medium with 500 μl of exosome solution,
which contains exosomes derived from 5×106 moDCs.
Addition of IL-2 and -7, and replenishment with fresh medium were
undertaken in the same manner as the co-culturing with SART1
peptide-pulsed moDCs.
Cytotoxicity assay
Patient lymphocytes, which were cultured with SART1
peptide-pulsed autologous moDCs or their exosomes for 4 weeks, were
used as effector cells in 51Cr-release cytotoxicity
assay by the method described previously (9). Esophageal cancer cell line, KE4 cells
(expressing of H LA-A*24 a nd SA RT1), a nd ch ronic myelogenous
leukemia-blastic crisis (CML-BC) cell line, C2F8 cells (expressing
HLA-A*24 but not SART1) (13) were
used as target cells for the cytotoxicity assay. Target cells
(1×106), were labeled with 100 μCi (100 μl) of
Na51CrO4 (NEN Life Sciences Inc., Boston, MA,
USA) and cultured with effector cells in a 96-well round bottom
plate (BD Biosciences) at 37°C in a fully humidified 5%
CO2 atmosphere. Cytotoxicity of effector cells was
determined at various effector-target cell ratios after incubation
for 4 h. The supernatants of the co-culture were then harvested and
analyzed for 51Cr release in an auto-well gamma system
ARC-300 (Aloka Co., Ltd.). Maximum and spontaneous 51Cr
release was measured after incubation of labeled target cells with
1 N HCL or medium alone, respectively. Percentages of cytotoxicity
of the effector cells were calculated using the following formula:
% cytotoxicity = [(51Cr release of sample wells −
spontaneous 51Cr release)/(maximum 51Cr
release − spontaneous 51Cr release)] ×100.
ELISPOT assay
Cryopreserved patients’ PB-MNCs were plated in 2
ml/well at a concentration of 2×106 cells in 24-well
plates (BD Biosciences) in 5% human serum-containing RPMI-1640
medium with 10 μg/ml of SART1 peptide. Two days later, 300 IU/ml
IL-2 was added to the cultures. The cultured cells were tested for
reactivity in the ELISPOT on day 12. The ELISPOT assay for
quantifying SART1 peptide-specific IFN-γ-releasing cells was
performed using ELISpotPLUS for Human IFN-γ kit (Mabtech
AB, Nacka Strand, Sweden). The cultured cells and SART1 peptides
were added to the ELISPOT plates, which had been coated with
anti-IFN-γ antibody (1-D1K), and the plates were incubated
overnight. The following day, biotinylated detection antibody
(7-B6-1-biotin) was added to the washed wells. The plates were
incubated for 2 h and washed, and the streptavidin-ALP was added to
each well. Plates were incubated at room temperature for 1 h, and
the enzyme substrate 5-bromo-4-chloro-3-indolyl phosphate/nitroblue
tetrazolium (BCIP/NBT-plus) was added to each well and incubated at
room temperature for 15 min. The spots were counted using
stereomicroscope imaging system (Olympus Corp., Tokyo, Japan), and
the peptide-specific CTL frequency could be calculated from the
numbers of spot-forming cells.
Statistical analysis
The statistical relevance of differences in
3H-thymidine incorporation in proliferation assay and
51Cr release in cytotoxicity assay was evaluated with a
two-way ANOVA, applying GraphPad Prism Software (GraphPad Software,
Inc., San Diego, CA, USA). Differences were considered as
significant at p<0.05 and markedly significant at p<0.01.
Results
Patient characteristics and vaccination
with peptide-pulsed moDCs
Seven patients with advanced stage of squamous cell
carcinoma of esophagus were enrolled for the study. Patient
characteristics are shown in Table
I. The mean age of the patients was 62 years (range, 53–71
years) and all were males. All the patients, who had developed
metastases to liver, lung or kidney and so on, were treated with
operation, chemotherapy and/or radiotherapy before entering the
clinical trial. In the first four patients, moDCs were prepared by
culturing monocytes with GM-CSF/IL-4 and pulsing with SART1
peptide. In the last three patients, TNF-α was used for maturation
of moDCs generated by culturing monocytes with GM-CSF/IL-4 and
mature moDCs were pulsed with KLH in combination with SART1.
Infusions of moDCs were undertaken two to five times, IV or SC in
the first four patients, and in the last three patients,
respectively.
The average number of infused cells in each infusion
varied (0.31–3.08×108 cells) from patient to patient
depending on the method of blood drawing. Pre-culture number of
MNCs, number of all infused cells, and percentage of large cells
estimated by forward scatter and side scatter dot plots of flow
cytometry, as well as the number of DCs (large cells) in each
infusion of individual patient are shown in Table II. Comparison of these values
among leukapheresis and bag method for blood drawing is shown in
Table III. Mean and SD of moDCs
in a single infusion procedure was 0.37±0.32×108 cells
in whole with 0.50±0.31×108 cells in leukapheresis and
0.12±0.09×108 cells in bag method (Table III).
| Table IINumber of infused cells each time of
cell processing in patients with DC treatment. |
Table II
Number of infused cells each time of
cell processing in patients with DC treatment.
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 |
|---|
|
|
|
|
|
|
|
|---|
| DC therapy no. | 1 | 2 | 3 | 1 | 2 | 3 | 4a | 5a | 1 | 2 | 3 | 1a | 2a | 1 | 2 | 3b | 1a | 2a | 3a |
|---|
| Pre-culture no. of
MNCs (×108) | 12.0 | 19.0 | 14.3 | 7.8 | 7.9 | 5.7 | 1.6 | 1.4 | 11.3 | 6.3 | 15.8 | 2.4 | 2.1 | 11.5 | 9.4 | 2.3 | 1.2 | 1.5 | 3.1 |
| No. of all infused
cells (×108) | 2.45 | 3.94 | 2.85 | 2.44 | 1.40 | 1.83 | 0.49 | 0.22 | 2.52 | 1.37 | 1.48 | 0.56 | 0.52 | 1.58 | 1.47 | 0.21 | 0.16 | 0.12 | 0.64 |
| % of large cells
estimated by flow cytometry | 30.2 | 7.0 | 25.2 | 42.9 | 19.2 | 46.7 | 27.4 | ND | 32.5 | 11.2 | 36.4 | 18.1 | 55.1 | 11.4 | 12.1 | 3.4 | 30.0 | ND | 7.5 |
| Probable no. of DCs
infused (×108) | 0.74 | 0.28 | 0.72 | 1.05 | 0.27 | 0.85 | 0.13 | | 0.82 | 0.15 | 0.54 | 0.10 | 0.29 | 0.18 | 0.18 | 0.01 | 0.05 | | 0.05 |
| Table IIIComparison of number (mean ± SD) of
infused cells among leukapheresis and the bag method. |
Table III
Comparison of number (mean ± SD) of
infused cells among leukapheresis and the bag method.
| Blood drawing | All (n=21) | Leukapheresis
(n=13) | Bag (n=7) | Insufficient
leukapheresis (n=1) |
|---|
| Pre-culture no. of
MNCs (x108) | 7.42±5.19 | 10.78±3.68 | 1.90±0.62 | 2.30 |
| No. of all infused
cells (x108) | 1.32±1.02 | 1.91±0.86 | 0.39±0.20 | 0.21 |
| % of large cells
estimated by flow cytometry | 24.9±14.36 | 25.6±12.8 | 27.6±15.9 | 3.40 |
| Probable no. of DCs
infused (x108) | 0.37±0.32 | 0.50±0.31 | 0.12±0.09 | 0.01 |
Surface phenotypes and allogeneic antigen
presenting abilities of injected moDCs
In the first four patients, immature moDCs were
generated by culture with GM-CSF and IL-4 and pulsed with SART1
peptide only. In these patients, immature moDCs were used for the
therapy since immature DCs were presumed to mature physiologically
in the process of interaction with T cells in vivo. In the
last three patients, mature moDCs were generated by culture with
TNF-α in addition to GM-CSF and IL-4 and pulsed with SART1 peptide
and KLH. These prepared moDCs were analyzed for surface phenotypes
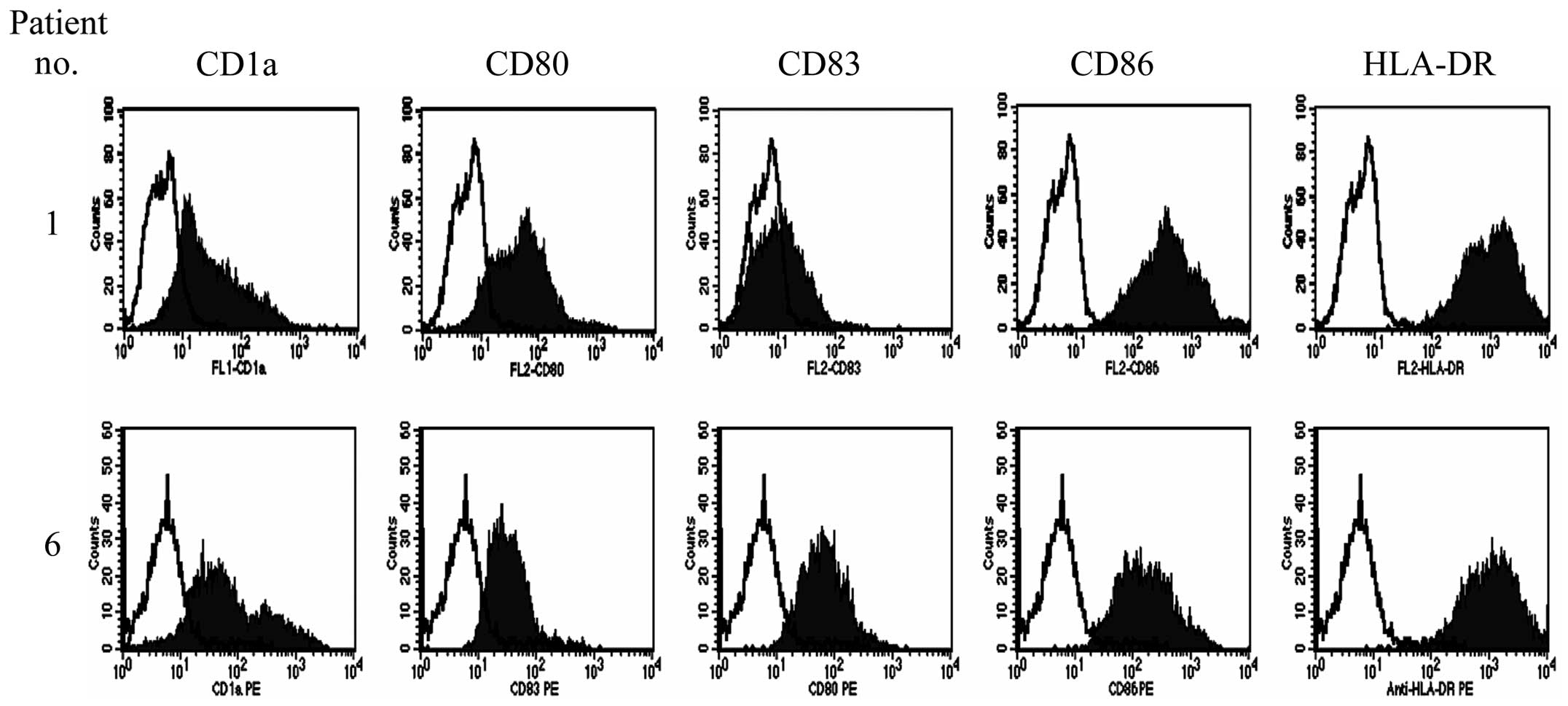
relating to antigen presentation. Although moDCs prepared from all
the enrolled patients were positive for CD1a, CD80 CD86 and HLA-DR,
the expression of CD83 was much higher in moDCs generated by
culture with TNF-α in addition to GM-CSF and IL-4 compared with
those generated by culture with GM-CSF and IL-4 only (Fig. 2). Therefore, the surface phenotypes
of infused moDCs were characteristic for immature moDCs in the
first four patients and mature moDCs in the last three
patients.
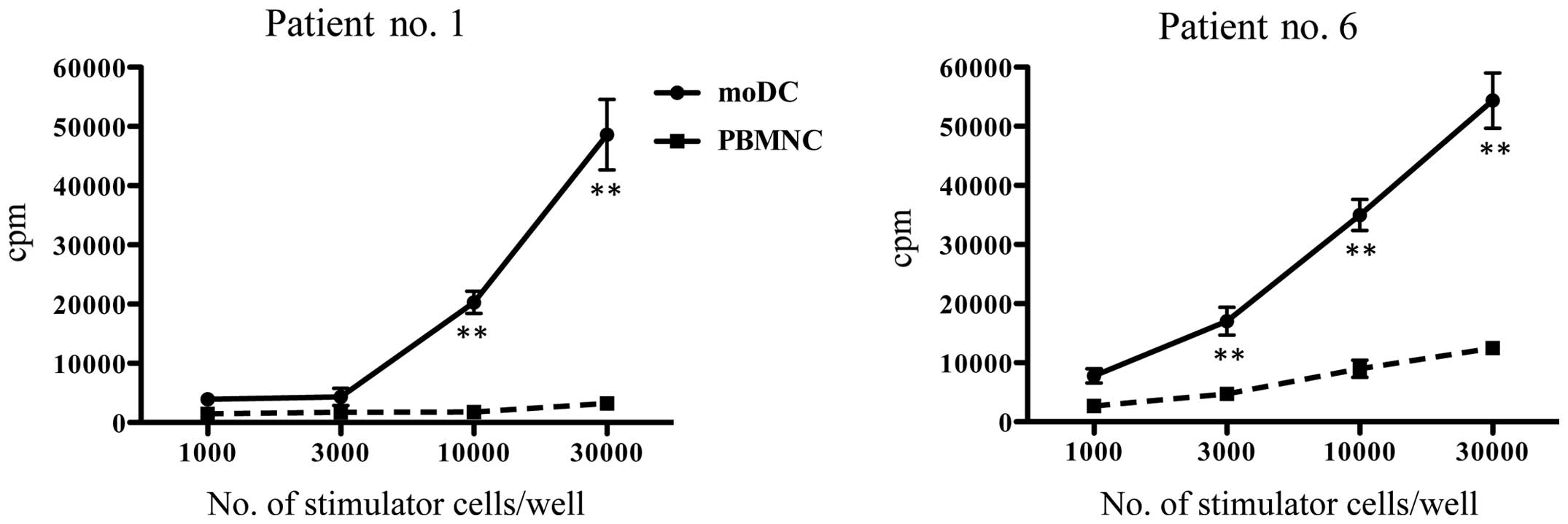
These moDCs were analyzed for antigen presenting
ability by using allogeneic proliferation assay. Although mature
moDCs showed slightly higher 3H-thymidine incorporation
than immature moDCs in low stimulator/responder ratio of the
proliferation assay, immature and mature moDCs were demonstrated to
possess a considerable potent ability of antigen presentation
(Fig. 3).
DTH and effects of DC therapy
Although skin DTH reactions against KLH were
detected in two out of three patients vaccinated with moDCs pulsed
with SART1 and KLH, DTH reaction against SART1 peptide was not
observed in all the seven patients (Table I). One patient who received SART1
peptide-pulsed moDC vaccine (patient no. 2) remained stable for 20
months after moDC therapy judging from tumor marker and CT findings
and he was categorized as no change (NC). But thereafter he
developed lung metastasis, for which the operation was undertaken.
The remaining six patients had progressive disease (PD) with the
median survival of 3.7 months and no favorable response was
observed during and after the vaccination course (Table I).
Toxicity
The vaccination was generally well-tolerated and no
allergic reaction to the vaccine was observed. One patient who
received SART1 peptide-pulsed moDCs showed a moderate
hypophosphatemia, although the relationship with moDC vaccination
was not definite (Table I).
Induction of SART1-specific CTLs by using
SART1 peptide-pulsed moDCs
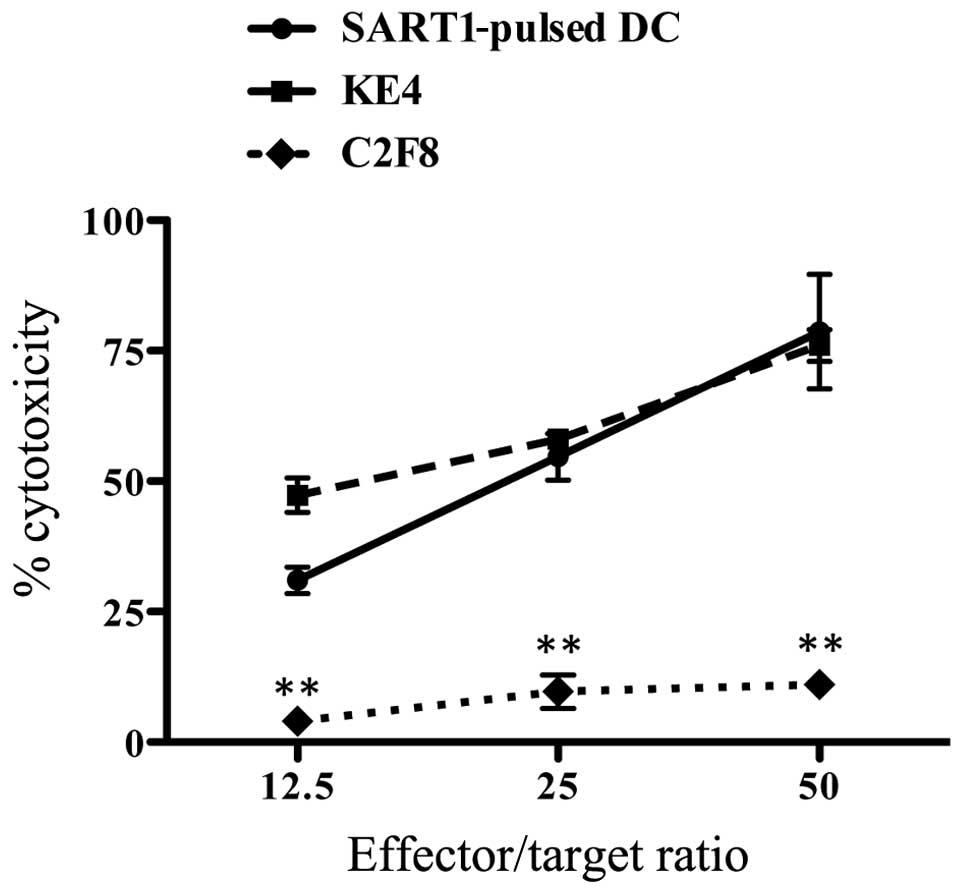
Lymphocytes of patient no. 7 primed with autologous
SART1 peptide/KLH-pulsed moDCs three times showed a significant
cytotoxic ability against SART1 peptide-pulsed moDCs and KE4 cells,
which were positive for the expression of both HLA-A24 and SART1,
in an effector-to-target ratio dependent manner. However, CML-BC
cell line C2F8 cells (13), which
did not express SART1, were not killed by lymphocytes primed with
SART1/KLH peptide-pulsed moDCs (Fig.
4).
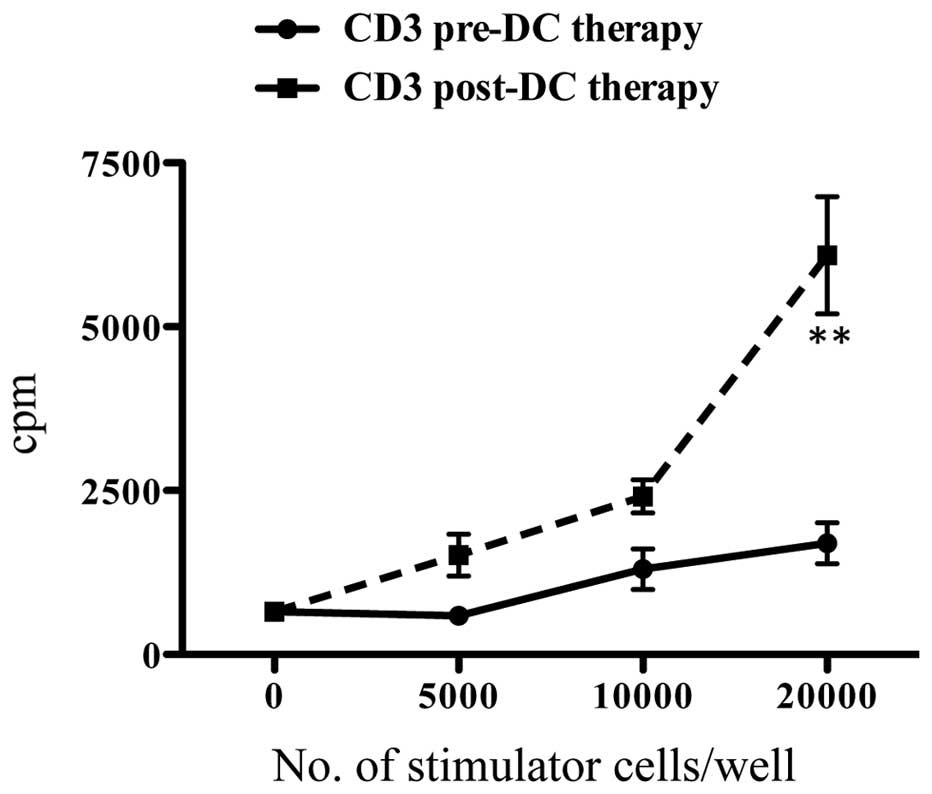
Increased reactivity of vaccinated
patient’s lymphocytes against SART1 peptide/KLH-pulsed moDCs
Reactivity of CD3+ T cells of patient no.
6 against moDCs pulsed with SART1 peptide/KLH was compared between
CD3+ T cells in pre-treatment phase and those in
post-vaccination phase (after three times infusion of SART1
peptide/KLH-pulsed moDCs). CD3+ T cells in
post-vaccination phase showed a much higher reactivity against
SART1 peptide/KLH-pulsed moDCs in autologous MLC compared with
those in pre-treatment phase (Fig.
5). This enhancement of CD3+ T-cell reactivity was
thought to be mainly caused by an increased reactivity against
KLH.
Production of antigen-presenting exosomes
by moDCs
Ultra-centrifuged preparation from moDC supernatant
of patient no. 1 was demonstrated to possess microvesicles
expressing both HLA-DR and CD86, which were presumed to be
exosomes. However, HLA-DR-bound microbeads were negative for CD86
in RPMI-1640 with 5% human serum (Fig.
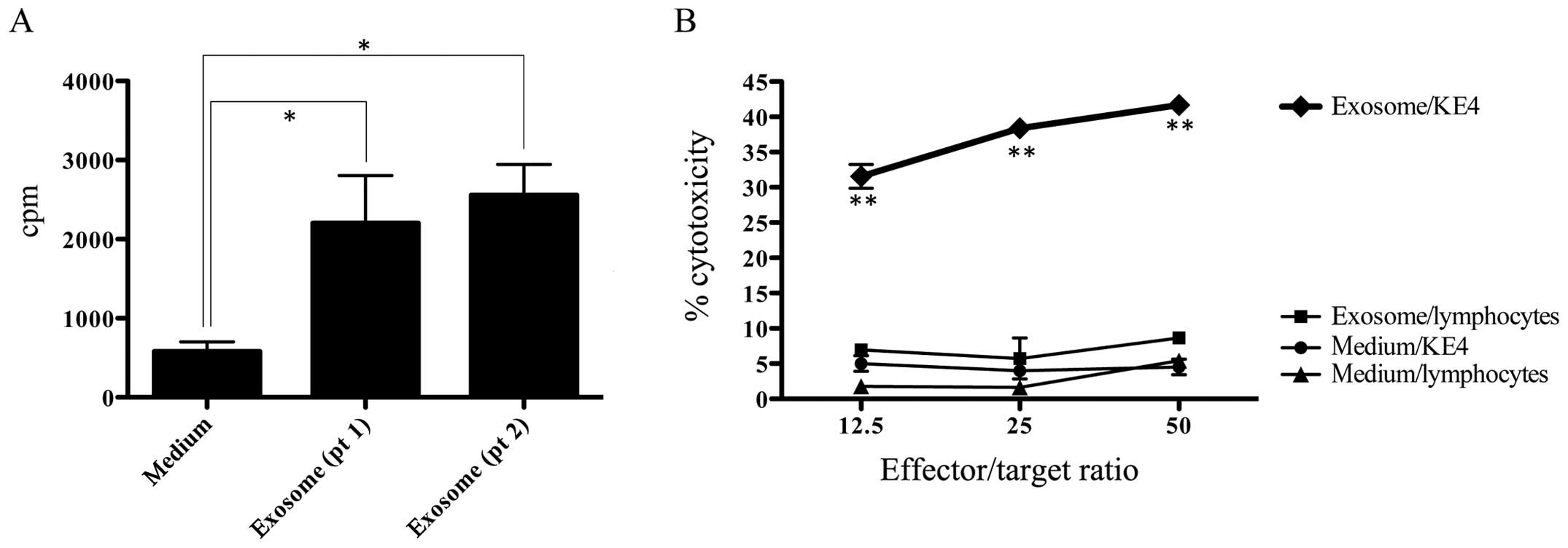
6). Exosome solutions prepared from moDC cultures of patient
no. 1 and 2 were used as substitute for stimulator cells in MLC
using allogeneic PBMNCs as responder cells. Exosome solutions from
both patients were demonstrated to possess a weak but definite
antigen presenting ability to allogeneic lymphocytes (Fig. 7A). Exosome solution prepared from
patient no. 1 was shown to induce SART1-specific CTLs in 4
weeks-culture of autologous PBMNCs when stimulated three times with
exosome solution derived from moDCs (Fig. 7B).
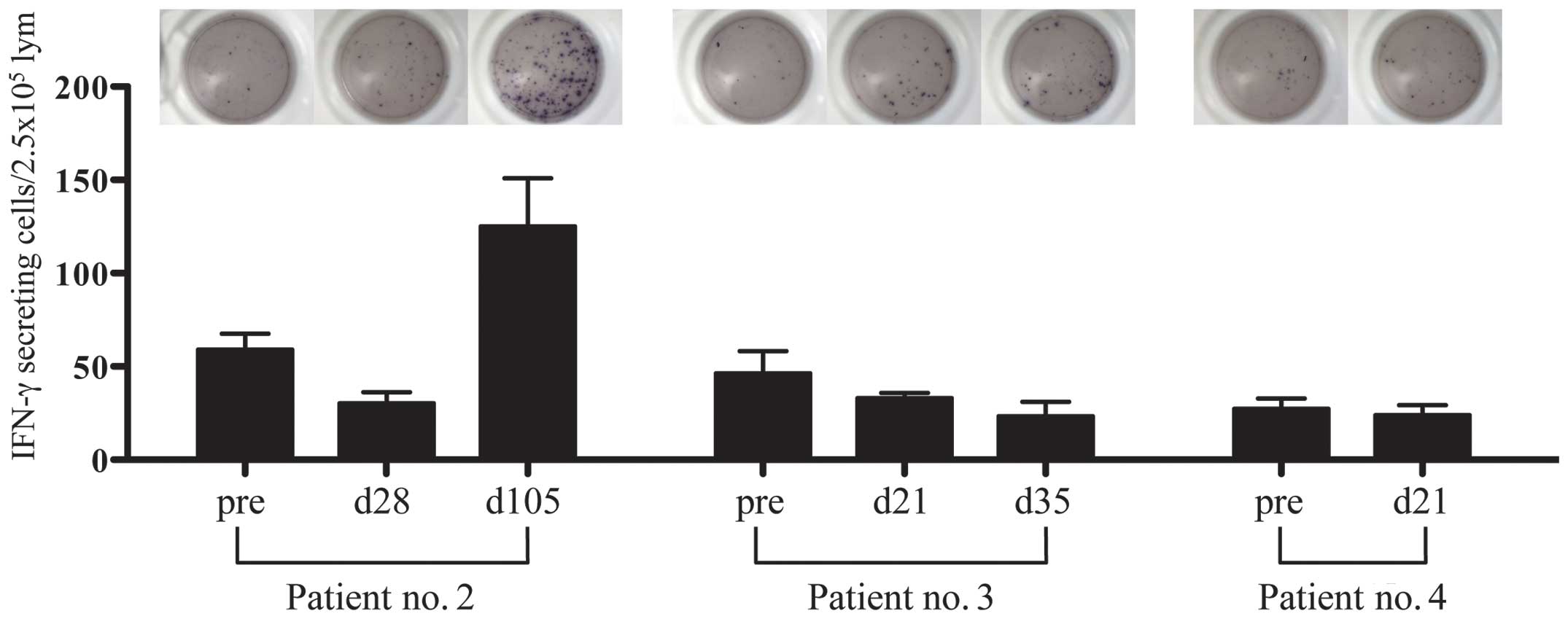
SART1-specific T-cell response by
ELISPOT
PBMNCs obtained from three patients (patient no. 2,
3 and 4) before moDC vaccination and at time points during the
vaccinations were analyzed for quantifying SART1 peptide-specific
IFN-γ-releasing cells. One (patient no. 2) of three patients had a
SART1-specific immune response in ELISPOT assay of lymphocytes at
day 84 from the initiation of the vaccination (after four times of
moDC vaccination) (Fig. 8). In the
other two patients (patient nos. 3 and 4), SART1-specific
IFN-γ-releasing cells did not increase probably due to the short
period after the vaccination (not >42 days from the initiation
of moDC vaccination). Patient no. 2, who showed a definite increase
of IFN-γ ELISPOT after moDC vaccination, is the case identified to
be stable disease after infusion of SART1 peptide-pulsed moDCs.
Discussion
DC-based antitumor immunotherapy has been
demonstrated to be feasible without side-effects and bring clinical
benefits with immune responses (14), but esophageal cancers have been
rarely enrolled in DC therapy so far. There have been several in
vitro studies indicating DC immunotherapy as a promising
strategy for esophageal cancer. Milano and Krishnadath reported a
patient-specific autologous readout assay for pre-clinical testing
of DC-mediated cytotoxic immune responses. They demonstrated that
the use of DCs transfected with autologous total tumor RNA could be
effective for treating esophageal cancer (16). While in the migration study of
administered DCs, Fujiwara et al performed an intratumoral
administration of in-labeled DCs in combination with preoperative
chemotherapy in esophageal cancer patients. Their study revealed
that the intratumoral administration of DCs during chemotherapy
does not give rise to DC migration from the tumor to the draining
lymph nodes, and suggested that an impairment of DC migration may
be associated with difficulty in achieving an optimal clinical
response in DC therapy (15). It
is now generally recognized that clinical outcomes of patients
receiving DC vaccination alone for advanced stage cancer have not
been satisfactory. Therefore, the treatment strategy to combine DC
therapy with another treatment modality to improve clinical
outcomes is considered (16). With
regard to antigen peptide-based immunotherapy for esophageal
squamous cell carcinoma, Kono et al reported that the immune
response induced by multiple-peptides vaccination could make the
prognosis better by analyzing the data of a multicenter phase II
clinical trial consisting of 60 patients with advanced stage of
esophageal cancer (17).
Exosomes, which are nanoscale (50–100 nm) vesicles,
can mediate an immune response by activating T lymphocytes (through
antigen presentation), natural killer (NK) cells (through NKG2D
ligand binding), and DCs (through antigen transfer) (18). Exosomes secreted by DCs loaded with
tumor antigen have been shown to generate potent immune responses
against cancer cells by inducing antigen-specific CD8+ T
cells (19) and abolishing the
suppressive function of regulatory T cells (20). Until now, only three clinical
trials have been undertaken, on the application of exosomes for
antitumor immunotherapy. Dai et al reported that autologous
ascites-derived exosomes combined with GM-CSF could induce tumor
antigen-specific CTL responses in phase I clinical trial for
patients with colorectal cancer, with no to minimal adverse effects
(21). Escudier et al
disclosed that phase I clinical trial of autologous DC
derived-exosomes was feasible and safe in patients with melanoma
and minor to stable clinical responses were observed in skin and
lymph node sites (22).
Furthermore, Morse et al demonstrated a MAGE-specific T-cell
response and increased NK lytic activity in patients with non-small
cell lung carcinoma treated with autologous DC derived-exosomes
loaded with multiple MAGE peptides (23).
Safety and efficacy were explored in the current
phase I/II vaccination for patients with esophageal cancer. The
vaccination was well-tolerated and no side-effect except for
possible hypophosphatemia was observed, similar to those reported
in other vaccination studies (24–26).
One patient (patient no. 2) treated with SART1-pulsed moDCs
remained stable for 20 months after moDC therapy, although
thereafter he developed lung metastasis, for which surgery was
undertaken. In patient no. 2, we could observe that the number of
IFN-γ-producing cells increased after four times of SART1-pulsed
moDC vaccination by IFN-γ ELISPOT assay. The other six patients
died after 1–10 months from vaccination with PD. Although clinical
and survival benefits were not observed in this vaccination
treatment for the enrolled patients with advanced stage of squamous
cell carcinoma of esophagus, feasibility of tumor antigen
peptide-pulsed moDC therapy was demonstrated. In the present
clinical trial, DTH against antigen peptide was negative, although
positive for KLH in some patients. We used peptide itself for
priming in DTH. Instead of antigen peptides, antigen peptide-pulsed
DCs should have been used for priming in DTH. On this note,
Ellebaek et al reported that antigen-pulsed DCs should be
used as antigen in DTH test in order to present antigens to obtain
the highest local immune reactivity (27). Also in vitro, the reactivity
of patient’s CD3+ T cells against SART1
peptide/KLH-pulsed moDCs increased after three times DC
vaccination. This enhancement of CD3+ T-cell reactivity
was presumed to be due to an increased reactivity against KLH but
not against SART1 peptide as shown in vivo of DTH. On the
contrary, moDCs prepared from each patient expressed molecules
associated with antigen presentation, such as CD1a, CD80, CD83,
CD86 and HLA-DR, although the expression of CD83 among them was
influenced by the culture method with or without TNF-α. Patient’s
lymphocytes primed with SART1 peptide-pulsed moDCs were
demonstrated to have a significant cytotoxic ability against
HLA-A24+/SART1+ esophageal carcinoma cell
line and SART1 peptide-pulsed autologous moDCs. These SART1
peptide-pulsed moDCs prepared from enrolled cancer patients were
shown to produce antigen-presenting exosomes, which could generate
SART1-specific CTLs in culture of autologous lymphocytes being
stimulated with exosome preparation. In addition, ELISPOT assay
using cryopreserved lymphocytes of the patients demonstrated that
IFN-γ ELISPOTs were increased after four times of moDC vaccinations
in one patient. These findings suggest that injected moDCs had an
ability to induce antigen-specific CTLs and the patient lymphocytes
acquired antigen-specific reactivity when primed with antigens
presented by injected moDCs. In the present clinical application of
antigen peptide-pulsed moDCs for advanced stage of squamous cell
carcinoma of esophagus and related in vitro and in
vivo studies, it was shown that DC-based cellular immunotherapy
for these cancer patients was feasible, functional DCs could be
generated from these patients, and patient’s immunity is elevated
by the infusion of DCs prepared from monocytes. In order to
establish a clinically effective DC-based immunotherapy, the
patient indication criteria for these therapies and the manner of
preparing highly qualified DCs for injection were presumed to be
the principle issues.
References
|
1
|
Siewert JR and Ott K: Are squamous and
adenocarcinomas of the esophagus the same disease? Semin Radiat
Oncol. 17:38–44. 2007. View Article : Google Scholar
|
|
2
|
Lagergren J and Lagergren P: Oesophageal
cancer. BMJ. 341:c62802010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Palucka K and Banchereau J: Cancer
immunotherapy via dendritic cells. Nat Rev Cancer. 12:265–277.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kantoff PW, Higano CS, Shore ND, Berger
ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims
RB, Xu Y, Frohlich MW and Schellhammer PF; IMPACT Study
Investigators. Sipuleucel-T immunotherapy for castration-resistant
prostate cancer. N Engl J Med. 363:411–422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kato Y: WT1 peptide pulsed dendritic cell
therapy with activated T lymphocytes therapy for advanced cancers.
Gan To Kagaku Ryoho. 37:2240–2242. 2010.(In Japanese).
|
|
6
|
Asakage M, Kitayama J, Tsuno NH, Komuro Y,
Kaisaki S, Hori N, Nagawa H, Tsuno NH, Hori N and Takahashi K:
Primary malignant melanoma of the esophagus treated by
esophagectomy and adjuvant dendritic-cell therapy. J Gastroenterol.
40:545–546. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ueda Y, Shimizu K, Itoh T, Fuji N, Naito
K, Shiozaki A, Yamamoto Y, Shimizu T, Iwamoto A, Tamai H and
Yamagishi H: Induction of peptide-specific immune response in
patients with primary malignant melanoma of the esophagus after
immunotherapy using dendritic cells pulsed with MAGE peptides. Jpn
J Clin Oncol. 37:140–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kikuchi M, Nakao M, Inoue Y, Matsunaga K,
Shichijo S, Yamana H and Itoh K: Identification of a SART-1-derived
peptide capable of inducing HLA-A24-restricted and tumor-specific
cytotoxic T lymphocytes. Int J Cancer. 81:459–466. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Narita M, Tochiki N, Saitoh A, Watanabe N,
Kaji M, Satoh N, Yamahira A, Nakamura T, Masuko M, Furukawa T, Toba
K, Fuse I, Aizawa Y and Takahashi M: Induction of antigen-specific
cytotoxic T lymphocytes by using monocyte-derived DCs transfected
with in vitro-transcribed WT1 or SART1 mRNA. Med Oncol. 26:429–436.
2009. View Article : Google Scholar
|
|
10
|
Zitvogel L, Regnault A, Lozier A, Wolfers
J, Flament C, Tenza D, Ricciardi-Castagnoli P, Raposo G and
Amigorena S: Eradication of established murine tumors using a novel
cell-free vaccine: dendritic cell-derived exosomes. Nat Med.
4:594–600. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Théry C, Boussac M, Véron P,
Ricciardi-Castagnoli P, Raposo G, Garin J and Amigorena S:
Proteomic analysis of dendritic cell-derived exosomes: a secreted
subcellular compartment distinct from apoptotic vesicles. J
Immunol. 166:7309–7318. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamahira A, Narita M, Nakamura T, Watanabe
N, Kaji M, Taniguchi T, Hashimoto S, Furukawa T, Toba K, Aizawa Y,
Kuzushima K and Takahashi M: Generation of antigen-specific
cytotoxic T lymphocytes using a leukemic plasmacytoid dendritic
cell line as antigen presenting cells. Leuk Res. 35:793–799. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Furukawa T, Koike T, Ying W, Kishi K, Aoki
S, Gotoh T, Hashimoto S, Saitoh H, Hanano M, Shinada S, et al:
Establishment of a new cell line with the characteristics of a
multipotential progenitor from a patient with chronic myelogenous
leukemia in early erythroblastic crisis. Leukemia. 8:171–180.
1994.PubMed/NCBI
|
|
14
|
Shore ND, Mantz CA, Dosoretz DE, Fernandez
E, Myslicki FA, McCoy C, Finkelstein SE and Fishman MN: Building on
sipuleucel-T for immunologic treatment of castration-resistant
prostate cancer. Cancer Control. 20:7–16. 2013.PubMed/NCBI
|
|
15
|
Fujiwara S, Wada H, Miyata H, Kawada J,
Kawabata R, Nishikawa H, Gnjatic S, Sedrak C, Sato E, Nakamura Y,
Sakakibara M, Kanto T, Shimosegawa E, Hatazawa J, Takahashi T,
Kurokawa Y, Yamasaki M, Nakajima K, Takiguchi S, Nakayama E, Mori M
and Doki Y: Clinical trial of the intratumoral administration of
labeled DC combined with systemic chemotherapy for esophageal
cancer. J Immunother. 35:513–521. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Milano F and Krishnadath KK: Novel
therapeutic strategies for treating esophageal adenocarcinoma: the
potential of dendritic cell immunotherapy and combinatorial
regimens. Hum Immunol. 69:614–624. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kono K, Iinuma H, Akutsu Y, Tanaka H,
Hayashi N, Uchikado Y, Noguchi T, Fujii H, Okinaka K, Fukushima R,
Matsubara H, Ohira M, Baba H, Natsugoe S, Kitano S, Takeda K,
Yoshida K, Tsunoda T and Nakamura Y: Multicenter, phase II clinical
trial of cancer vaccination for advanced esophageal cancer with
three peptides derived from novel cancer-testis antigens. J Transl
Med. 10:1412012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Théry C, Ostrowski M and Segura E:
Membrane vesicles as conveyors of immune responses. Nat Rev
Immunol. 9:581–593. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hao S, Bai O, Li F, Yuan J, Laferte S and
Xiang J: Mature dendritic cells pulsed with exosomes stimulate
efficient cytotoxic T-lymphocyte responses and antitumour immunity.
Immunology. 120:90–102. 2007. View Article : Google Scholar
|
|
20
|
Taieb J, Chaput N, Schartz N, Roux S,
Novault S, Ménard C, Ghiringhelli F, Terme M, Carpentier AF,
Darrasse-Jèze G, Lemonnier F and Zitvogel L: Chemoimmunotherapy of
tumors: cyclophosphamide synergizes with exosome based vaccines. J
Immunol. 176:2722–2729. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H
and Li G: Phase I clinical trial of autologous ascites-derived
exosomes combined with GM-CSF for colorectal cancer. Mol Ther.
16:782–790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Escudier B, Dorval T, Chaput N, André F,
Caby MP, Novault S, Flament C, Leboulaire C, Borg C, Amigorena S,
Boccaccio C, Bonnerot C, Dhellin O, Movassagh M, Piperno S, Robert
C, Serra V, Valente N, Le Pecq JB, Spatz A, Lantz O, Tursz T,
Angevin E and Zitvogel L: Vaccination of metastatic melanoma
patients with autologous dendritic cell (DC) derived-exosomes:
results of the first phase I clinical trial. J Transl Med.
3:102005. View Article : Google Scholar
|
|
23
|
Morse MA, Garst J, Osada T, Khan S,
Hobeika A, Clay TM, Valente N, Shreeniwas R, Sutton MA, Delcayre A,
Hsu DH, Le Pecq JB and Lyerly HK: A phase I study of dexosome
immunotherapy in patients with advanced non-small cell lung cancer.
J Transl Med. 3:92005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Berntsen A, Trepiakas R, Wenandy L,
Geertsen PF, thor Straten P, Andersen MH, Pedersen AE, Claesson MH,
Lorentzen T, Johansen JS and Svane IM: Therapeutic dendritic cell
vaccination of patients with metastatic renal cell carcinoma: a
clinical phase 1/2 trial. J Immunother. 31:771–780. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trepiakas R, Berntsen A, Hadrup SR, Bjørn
J, Geertsen PF, Straten PT, Andersen MH, Pedersen AE, Soleimani A,
Lorentzen T, Johansen JS and Svane IM: Vaccination with autologous
dendritic cells pulsed with multiple tumor antigens for treatment
of patients with malignant melanoma: results from a phase I/II
trial. Cytotherapy. 12:721–734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Svane IM, Pedersen AE, Johnsen HE, Nielsen
D, Kamby C, Gaarsdal E, Nikolajsen K, Buus S and Claesson MH:
Vaccination with p53-peptide-pulsed dendritic cells, of patients
with advanced breast cancer: report from a phase I study. Cancer
Immunol Immunother. 53:633–641. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ellebaek E, Engell-Noerregaard L, Iversen
TZ, Froesig TM, Munir S, Hadrup SR, Andersen MH and Svane IM:
Metastatic melanoma patients treated with dendritic cell
vaccination, Interleukin-2 and metronomic cyclophosphamide: results
from a phase II trial. Cancer Immunol Immunother. 61:1791–1804.
2012. View Article : Google Scholar : PubMed/NCBI
|