Introduction
Hepatocellular carcinoma (HCC) accounts for 80–90%
of primary liver cancers, and is the sixth leading cause of death
from cancer worldwide (1). Today,
developments in imaging have enabled the detection of HCC at an
early stage and new therapeutic options for HCC have improved
survival rates (2–5). However, HCC still has high recurrence
rates after surgical resection, with most recurrences being
confined to the remnant liver (6,7).
Therefore, it is important to identify genetic markers relevant to
the oncogenesis of HCC (8).
We reported a novel method named double-combination
array that combines a gene expression array and a single nucleotide
polymorphism (SNP) array, and have developed this method and
reported a number of cancer-related genes in HCC (9–14).
Through these studies, we hypothesized that the decrease in
expression of these genes could be caused by DNA methylation of the
CpG islands in the promoter region, and would lead to HCC
progression. In addition to the double-combination array analysis,
we obtained further data from the same specimens using methylation
array analysis to make this association with DNA methylation more
conclusive. Thus, this triple combination array analysis seems to
be an efficient procedure for the detection of cancer-related genes
of HCC (15–17).
From our triple combination array analysis, cyclin J
(CCNJ) was identified as one of the new candidate
cancer-related genes of HCC. Cyclin family members, known as cell
cycle regulators, play important roles in cell proliferation and
are linked to cell carcinogenesis. The function of most cyclins in
human cancer, including HCC, has been reported to be oncogenic.
However, some cyclins play suppressive roles in carcinogenesis or
cancer progression (18–23). The function of CCNJ in
humans is still unclear and there are only a few reports of a
relationship between CCNJ and cancer, although there have
been no previous studies on the role of CCNJ in HCC
(24,25). We predicted CCNJ might be
involved in the cell cycle in human HCC and evaluated the
expression and methylation status and investigated
clinicopathological features of CCNJ in HCC cases.
Materials and methods
Ethics
This study was conducted in accordance with the
ethical guidelines of the World Medical Association Declaration of
Helsinki-Ethical Principles for Medical Research Involving Human
Subjects and has been approved by the Institutional Review Board of
Nagoya University, Japan. All patients gave written informed
consent for usage of clinical samples and data, as required by the
institutional review board.
Sample collection and DNA
preparation
Nine HCC cell lines (HepG2, Hep3B, HLE, HLF, HuH1,
HuH2, HuH7, PLC/PRF-5 and SK-Hep1) were obtained from the American
Type Culture Collection (Manassas, VA, USA). The cell lines were
cultured in RPMI-1640 supplemented with 10% fetal bovine serum and
incubated in 5% CO2 at 37°C.
A 68-year-old woman with chronic hepatitis C was
diagnosed with HCC in the right lobe and underwent a liver
resection. Specimens of her tumor and adjacent non-cancerous tissue
were excised, and total RNA and DNA were extracted. Total RNA was
sent to the manufacturer of Affymetrix to prepare it for expression
array analysis. Genomic DNA was used in the SNP-Chip array, and
bisulfite-converted DNA was used in the Illumina Infinium
HumanMethylation27 BeadChip array (Illumina, San Diego, CA, USA).
The tumor was pathologically confirmed as HCC. RNA and DNA were
extracted from an area composed of >80% cancerous cells in the
tumor sample.
HCC tissue (HTs) and non-cancerous tissue (NTs)
samples were obtained from 85 patients (72 males and 13 females)
who underwent liver resection at Nagoya University Hospital,
Nagoya, Japan between 1994 and 2001. Patient ages ranged from 21–77
years (mean ± SD, 61.6±10.1 years). Sixty-one patients had
hepatitis C and 16 had hepatitis B. The median duration of
follow-up was 63.4 months (range, 2.3–208.9 months). All tissues
were reviewed pathologically to confirm the diagnosis of HCC.
Written informed consent, as required by the institutional review
board, was obtained from all patients. Tissue samples were
immediately frozen in liquid nitrogen and stored at −80°C until
required. Genomic DNA was obtained from tissue samples using
proteinase K digestion followed by phenol/chloroform
extraction.
RNA isolation and microarray
procedure
Total RNA was isolated from each of the frozen
samples with RNeasy Mini kit (Qiagen, Chatsworth, CA, USA)
according to the manufacturer’s protocol. RNA quality was assessed
as an RNA integrity number ≥8 using an Agilent 2100 Bioanalyzer
(Missisauga, ON, Canada). Total RNA was labeled with cyanine-3 dye
using a Quick Amp Labeling kit (Agilent) and hybridized to Agilent
Whole Human Genome (4×44 K) Microarrays for 17 h in a rotating
SciGene model 700 oven (Sunnyvale, CA, USA). Arrays were scanned
using an Agilent Technologies DNA Microarray Scanner, and data were
feature-extracted using Feature Extraction Software 10.5.1.1
(Agilent) and statistically analyzed using default setting on
GeneSpring GX 12.1.0 software (Agilent) (26).
GeneChip Affymetrix platform
SNP chip array experiments were done according to
the standard protocol for Affymetrix GeneChip Mapping 500 K arrays
(Affimetrix). Briefly, total genomic DNA was digested with a
restriction enzyme (XbaI or HindIII), ligated to an
appropriate adapter for each enzyme, and subjected to PCR
amplification using a single primer. After digestion with DNase I,
the PCR products were labeled with a biotinylated nucleotide
analogue using terminal deoxynucleo-tidyl transferase and
hybridized to the microarray. Hybridized probes were captured by
streptavidin-phycoerythrin conjugates, and the array was scanned
and genotypes called as previously described (27).
All the examples of copy number analysis with
Affymetrix GeneChip Mapping 500 K arrays were done using Copy
Number Analyzer for Affymetrix GeneChip Mapping 500 K arrays (CNAG)
version 2.0 (28).
Methylation array platform
Methylation arrays were performed using DNA
extracted from tissue samples obtained from the 68-year-old female
patient, as described above. The Illumina Infinium
HumanMethylation27 BeadChip protocol required 0.5–1 μg of
bisulfite-converted DNA (29). Of
the approximately 28 million CpG sites found throughout the haploid
human genome, Illumina initially designed Infinium methylation
probes for 27,578 CpG sites located in promoter regions (up to 1 kb
upstream or 500 bp downstream of transcription start sites). Of
these, 27,324 CpG sites relate to 14,475 consensus coding
sequences, including around 1,000 cancer-associated genes, and 254
CpG sites relate to approximately 100 microRNA genes. The probes
were preferentially selected to occur within CpG islands using the
NCBI ‘relaxed’ definition of a CpG island: CpG islands identified
bioinformatically with a CpG content of >50% and an
observed/expected ratio of >0.6 (30).
Bisulfite-converted DNA was then whole-genome
amplified, enzymatically fragmented and hybridized to the array.
During hybridization, the bisulfite-converted DNA annealed to
methylation-specific probes on the chip. Each CpG locus was
represented by two bead types, one of which was specific to the
methylated state and the other specific to the unmethylated state,
which was directly related to the underlying sequence change
catalyzed during bisulfite conversion. Therefore, for each CpG
site, a possible C/T variant could be assayed through the
single-base extension step, which was possible because of the
ability to hybridize to either the ‘protected’ methylated cytosine
or the converted (unmethylated) thymine.
After hybridization, a single-base extension step
was carried out using a multi-layer staining process, as described
below. The BeadChip was then scanned on the Illumina iScan and the
resulting ‘idat’ files were analyzed using BeadStudio software. The
output of the BeadStudio analysis was a β-value for each CpG site.
This was a continuous value between 0 and 1 where 0=0% methylation
and 1=100% methylation at a given CpG site. Therefore, this assay
enables quantitative analysis of methylation at individual CpG
sites.
The data were extensively analyzed and CCNJ
was eventually identified as a potential cancer-related gene. Since
the gene was identified from a specimen from a single patient, the
result could have been applicable only to that particular patient.
We therefore conducted additional experiments to verify the
clinical relevance of CCNJ.
Reverse transcription-polymerase chain
reaction (RT-PCR)
CCNJ mRNA expression was analyzed using
semi-quantitative RT-PCR and real-time RT-PCR. Total RNA (10 μg)
isolated from nine HCC cell lines, primary HTs and NTs were used to
generate cDNAs. The resulting cDNAs were then amplified using PCR
primers for CCNJ (sense, 5′-TGC GCT TTC CTG CCT GCT TC-3′ in
exon 3; antisense 5′-AGG CTG TTG AGC TGC TCC AG-3′ in exon 4),
which amplified an 81-bp product. Initial denaturation at 94°C for
5 min was followed by amplification consisting of 37 cycles of 94°C
for 10 sec, 60°C for 8 sec and 72°C for 6 sec. RT-PCR of β-actin
was performed to confirm that equal amounts of cDNA were used as
templates. Each PCR product was loaded directly onto 3% agarose
gels, stained with ethidium bromide and visualized under UV
illumination.
Quantitative RT-PCR analysis
PCR was performed with the SYBR Green PCR Core
Reagents kit (Perkin-Elmer Applied Biosystems, Foster City, CA,
USA) under the following conditions: one cycle at 95°C for 10 sec,
followed by 40 cycles at 95°C for 5 sec and at 60°C for 30 sec.
SYBR Green emission was detected in real-time with an ABI prism
7000 Sequence Detector (Perkin-Elmer Applied Biosystems). Primers
used in the PCR were the same as those described above for RT-PCR.
For standardization, the expression of GAPDH was quantified
in each sample. Quantitative RT-PCR was performed at least three
times, including negative controls without template. Expression of
CCNJ was normalized to that of GAPDH in each
sample.
Methylation-specific PCR (MSP)
DNA from HCC cell lines, HTs and NTs were subjected
to bisulfite treatment. Briefly, 2 μg of DNA were denatured using
NaOH and modified using sodium bisulfite. DNA samples were then
purified using Wizard purification resin (Promega Corp., Madison,
WI, USA), treated with NaOH, precipitated with ethanol and
resuspended in water. Primer pairs were used to detect methylation
(sense, 5′-GGC GGT TGA GCG TTT CGG-3′; antisense, 5′-AAA CGA CGC
GAC GAC TC-3′; 104-bp product) and non-methylation (sense, 5′-GTT
GTG TGG GAT GTT GAT TG-3′; antisense, 5′-CCA AAC CCA AAT ACA ACA
GC-3′; 70-bp product) of the CCNJ promoter region near exon
1. MSP and unmethylated-specific PCR (UNMSP) amplification
consisted of denaturation at 94°C for 5 min followed by 35 cycles
of 94°C for 8 sec, 60°C for 5 sec, and 72°C for 3 sec. PCR products
were loaded directly onto 3% agarose gels, stained with ethidium
bromide and visualized under UV illumination.
Sequence analysis
Bisulfite-treated genomic DNA obtained from HCC cell
lines was sequenced and PCR was performed in all cases. Semi-nested
PCR was performed to obtain adequate products for TA cloning. PCR
amplification consisted of denaturation at 94°C for 3 min followed
by 35 cycles of 94°C for 10 sec, 52°C for 10 sec and 72°C for 20
sec with the following primer pair: sense 5′-GGA TTG GGT ATT GTTA
GTG AT-3′; antisense, 5′-ACC TCA ACT AAA ACC CAA C-3′; yielding a
378-bp product. PCR products were subcloned into a TA cloning
vector (Invitrogen, Carlsbad, CA, USA). Six cloned samples that
originated from two HCC cell lines (SK-Hep1 and Hep3B) were picked
out. Each DNA clone was mixed with 3 μl of the specific primer
(M13) and 4 μl of Cycle Sequence Mix (ABI PRISM Terminator v1.1
Cycle Sequencing kit; Applied Biosystems, Foster City, CA, USA).
Samples were then subjected to the following cycling conditions:
95°C for 30 sec followed by 25 cycles of 96°C for 10 sec, 50°C for
5 sec, and 60°C for 4 min, and then purified using ethanol
precipitation. Sequence analysis was carried out using an Applied
Biosystems ABI310, and sequence electropherograms were generated
using ABI Sequence Analysis software version 3.0.
5-Aza-2′-deoxycytidine (5-aza-dC)
treatment
To confirm that promoter hypermethylation was
responsible for silencing of gene expression, the nine HCC cell
lines were treated with 1 μM 5-aza-dC (Sigma-Aldrich, St. Louis,
MO, USA) to inhibit DNA methylation. Cells (1.5×106)
were cultured for 6 days with medium changes on days 1, 3 and 5. On
day 6, cells were harvested, RNA was extracted, and RT-PCR was
performed as described above.
Statistical analysis
Continuous variables were expressed as medians
(range) and comparisons were made using the Mann-Whitney U test.
Categorical variables were compared using χ2 or Fisher’s
exact tests, where appropriate. Overall survival rates were
analyzed using Kaplan-Meier and log-rank tests. All statistical
analyses were performed using JMP software version 9.0.2 (SAS
International Inc., Cary, NC, USA). The level of statistical
significance was set at P<0.05.
Results
Expression, SNP and methylation
arrays
To identify novel tumor-related genes in HCC, we
used expression arrays and found genes whose expression was
decreased in HCC samples compared with corresponding non-cancerous
tissues. The result of the expression profiling showed that the
expression of CCNJ was strongly decreased with a −2.3
(log2 ratio) value in the HCC expression array (Table I).
 | Table IExpression array analysis of the
CCNJ gene. |
Table I
Expression array analysis of the
CCNJ gene.
| Probe set ID | Gene symbol | Log2
ratio | Non-cancer
signal | Detection | Tumor signal | Detection | Probe ID | Chromosomal
location |
|---|
| 229091_s_at | CCNJ | −2.3 | 36.9 | P | 10.3 | P | HU133p2_38346 | 10q24.1 |
| 219670_x_a | CCNJ | −1.3 | 31.6 | P | 11.2 | P | HU133p2_28755 | 10q24.1 |
We then confirmed the reduction of expression of
CCNJ in tumor tissues using semi-quantitative RT-PCR for
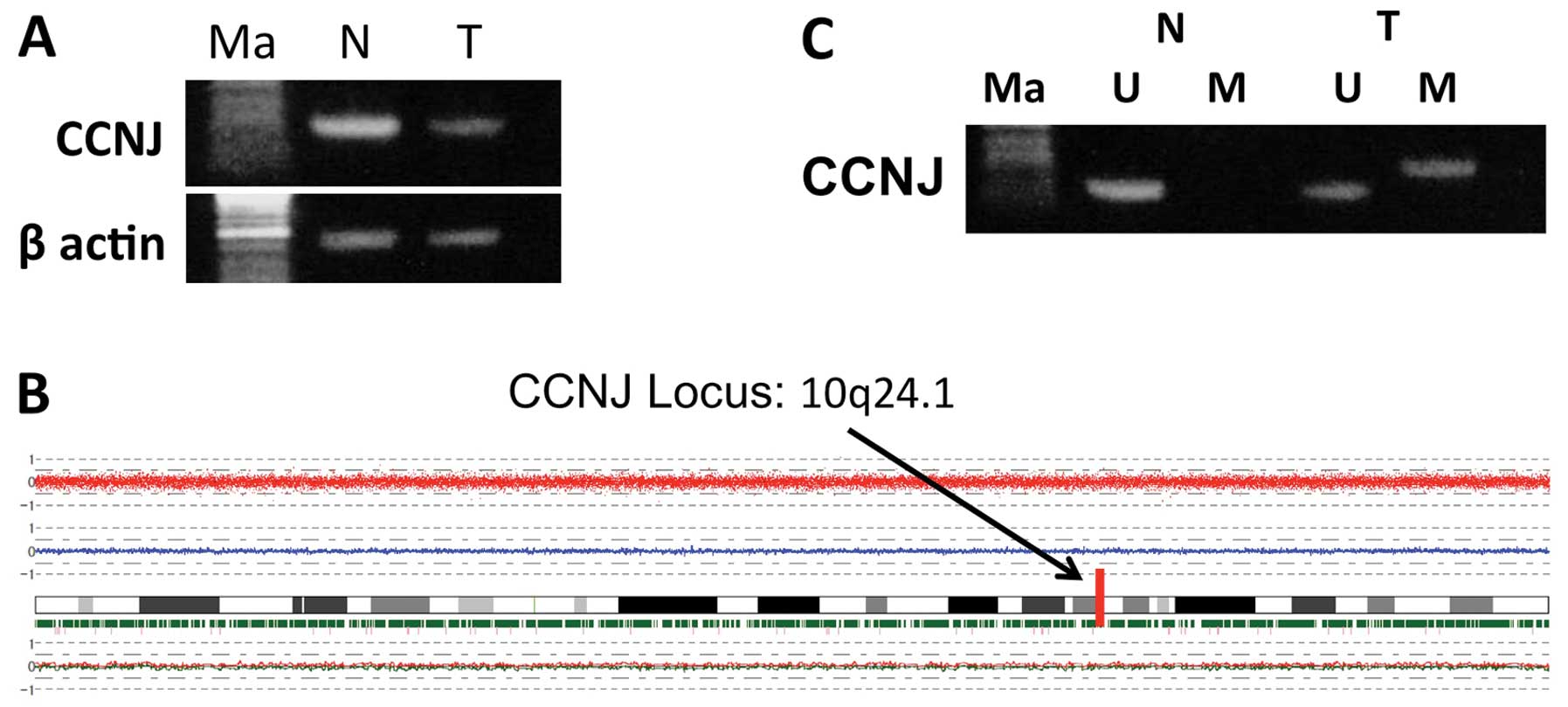
case samples used in the array analysis (Fig. 1A). Next, we analyzed the SNP
arrays. We observed deletions in 3q, 8p, 11q, 12p, 12q, 16p, 17p,
19p and X chromosomes, and chromosomal gains in 1q, 3q, 11q, 12p
and 12q. Chromosome 10, which contains CCNJ, did not show
any deletions or amplifications (Fig.
1B). We also looked for detailed data of the SNP array at the
CCNJ gene locus at 10q24.1, and found four SNPs. Two of
these four SNPs showed a heterozygous AB allele in both tumor and
non-tumor tissue samples (Table
II). These results suggested that the expression of CCNJ
decreased without loss of heterozygosity (LOH) or gene
deletion.
| Table IIMethylation array analysis of the HCC
samples. |
Table II
Methylation array analysis of the HCC
samples.
| Probe set ID | Chromosome | Physical
position | Non-cancer | 1_Confidence | Tumor | 2_Confidence |
|---|
| SNP_A_4268671 | 10 | 97804751 | AB | 0.003906 | AB | 0.002441 |
| SNP_A_2004209 | 10 | 97804806 | AA | 0.25 | AA | 0.039063 |
| SNP_A-4289679 | 10 | 97807640 | AB | 0.011719 | AB | 0.088379 |
| SNP_A-1922485 | 10 | 97808330 | AA | 0.0625 | AA | 0.007813 |
We subsequently checked the results of the
methylation array: the continuous β-values were 0.906 for tumor
tissue vs. 0.112 for corresponding non-cancerous tissue, indicating
a high degree of methylation in HCC samples (Table III). Using MSP, we confirmed the
hypermethylation of the CCNJ promoter in the tumor tissue
obtained from the 68-year-old woman whose DNA was used for the
methylation array (Fig. 1C). These
results implied that CCNJ expression decreased without LOH,
possibly because of hypermethylation of the promoter region.
| Table IIIMethylation array analysis of the
CCNJ gene. |
Table III
Methylation array analysis of the
CCNJ gene.
| Probe ID | Gene symbol | Sample | Methylation
value | Total | Status | Confidence | Chromosomal
location |
|---|
|
|---|
| Unmethylated | Methylated |
|---|
| 229091_s_at | CCNJ | Normal | 0.112033 | 1346 | 1184 | 162 | 3.678E-38 | 10q24.1 |
| 219670_x_a | CCNJ | Tumor | 0.906054 | 3732 | 260 | 3472 | 3.678E-38 | |
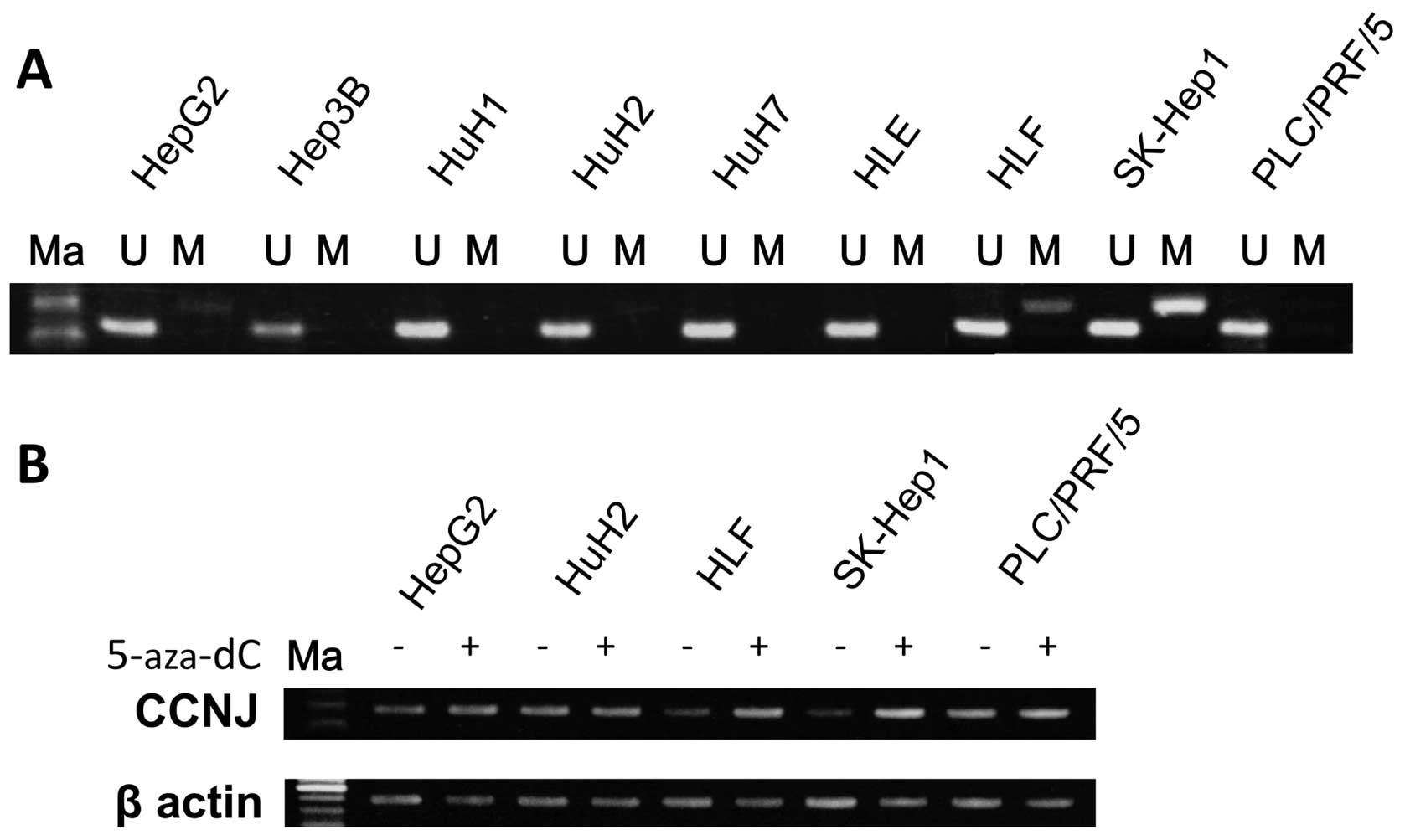
MSP and UNMSP of nine HCC cell lines
We designed primers for methylation-specific MSP and
UNMSP and checked the methylation status of nine HCC cell lines.
For MSP, we obtained bands of appropriate size in lanes containing
HLF, HepG2 and SKHep1 samples, while in UNMSP, appropriate bands
were identified in lanes for all cell lines (Fig. 2A). We subsequently identified
partial methylation in HLF, HepG2 and SKHep1, and no methylation in
HuH1, HuH2, HuH7, Hep3B, HLE and PLC/PRF/5 cells.
5-Aza-dC treatment of nine HCC cell
lines
To confirm that hypermethylation of the promoter in
the lesion inhibited CCNJ expression, we checked mRNA
expression of the gene before and after 5-aza-dC treatment in the
nine HCC cell lines. After 5-Aza-dC treatment, expression of
CCNJ in HLF, HepG2 and SKHep1 cells was clearly reactivated,
as shown using semi-quantitative RT-PCR (Fig. 2B).
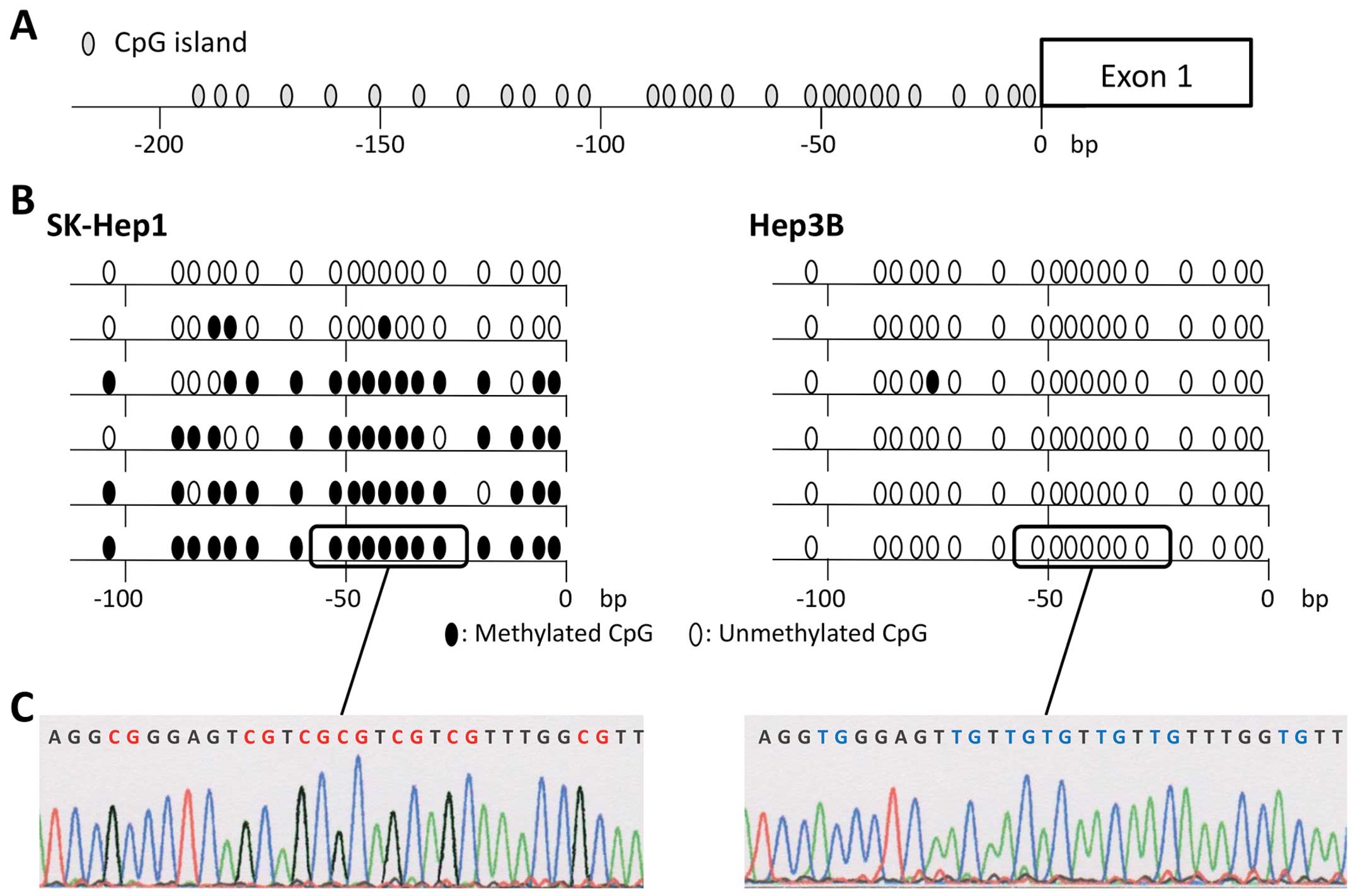
Sequence analysis
To confirm that the amplification of MSP was done
correctly, we performed sequence analysis of bisulfite-treated DNA
of the CCNJ promoter region cloned into TA cloning vectors
with cDNAs from both SK-Hep1 and Hep3B cells. Almost all CpG
islands in cDNA fragments isolated from SKHep1 cells were
methylated, while those from Hep3B were unmethylated (Fig. 3). These results showed the high
degree of accuracy of MSP and UNMSP.
MSP and UNMSP of adjacent non-cancerous
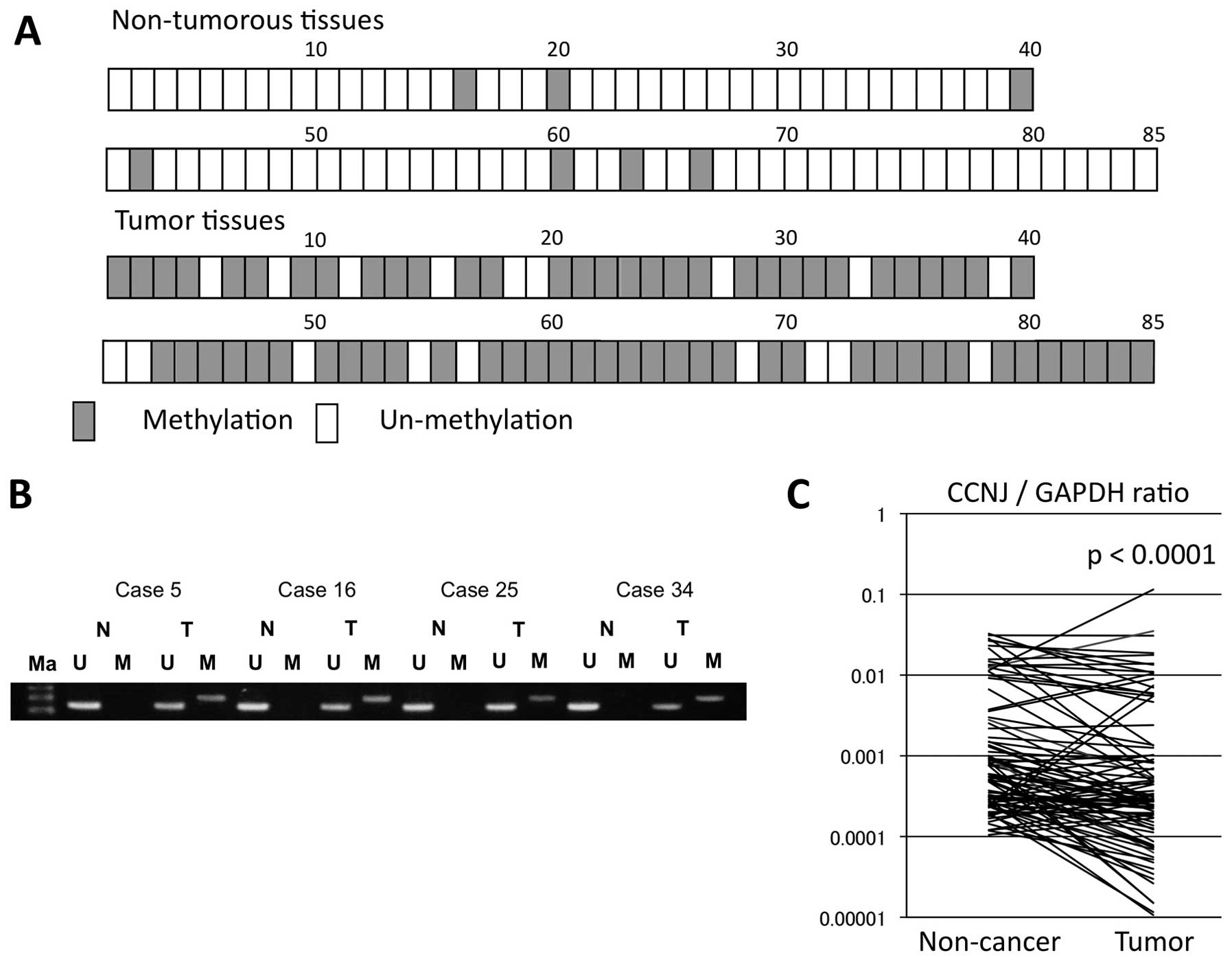
and tumor tissues from 85 HCC patients
We assessed CCNJ promoter hypermethylation in
adjacent non-cancerous and tumor tissues from 85 HCC patients.
Overall, 67 of the 85 (78.8%) displayed CCNJ promoter
hypermethylation in HCC tissues, compared with eight of 85 (9.4%)
in adjacent non-cancerous tissues (Fig. 4A). Hypermethylation of the
CCNJ promoter occurred statistically significantly more
frequently in tumor tissues (P<0.001).
Quantitative RT-PCR analysis of specimens
from 85 HCC patients
We evaluated expression levels of CCNJ mRNA
using real-time RT-PCR in the 85 cases, and by calculating the
expression index (the value of CCNJ mRNA expression divided
by that of GAPDH for each sample). Total CCNJ
expression in tumor tissues was significantly lower compared with
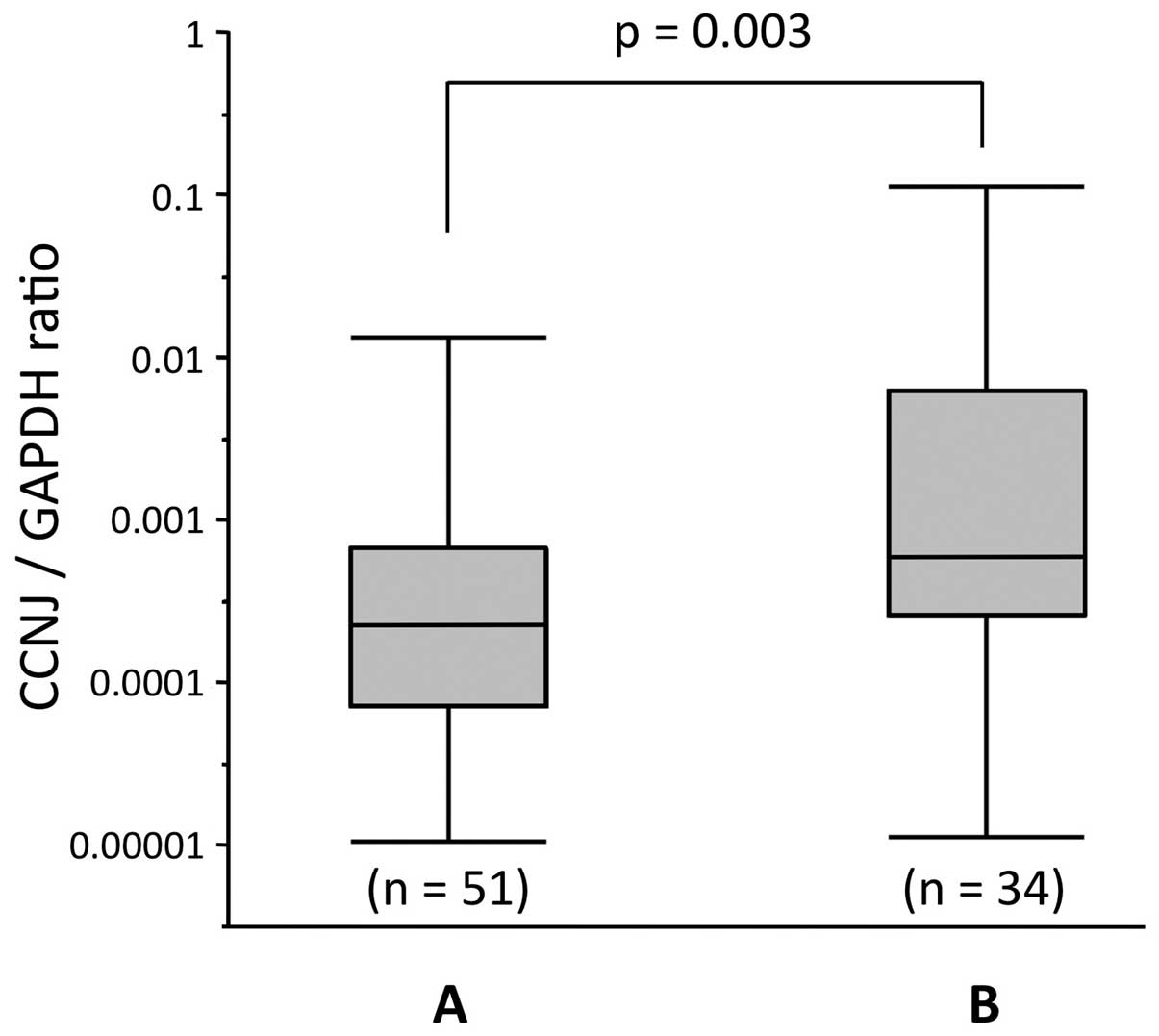
that in adjacent non-cancerous tissues (P<0.001; Fig. 4C). Furthermore, the cases with
CCNJ promoter hypermethylation showed significantly lower
expression of CCNJ in tumor tissues than those without
promoter hypermethylation (Fig.
5).
Association between promoter methylation
status of the CCNJ gene and clinicopathological characteristics in
85 HCC patients
We analyzed the association between the methylation
status of CCNJ and clinicopathological features in the 85
HCC patients. There was no notable association between methylation
status and clinicopathological factors (data not shown). However,
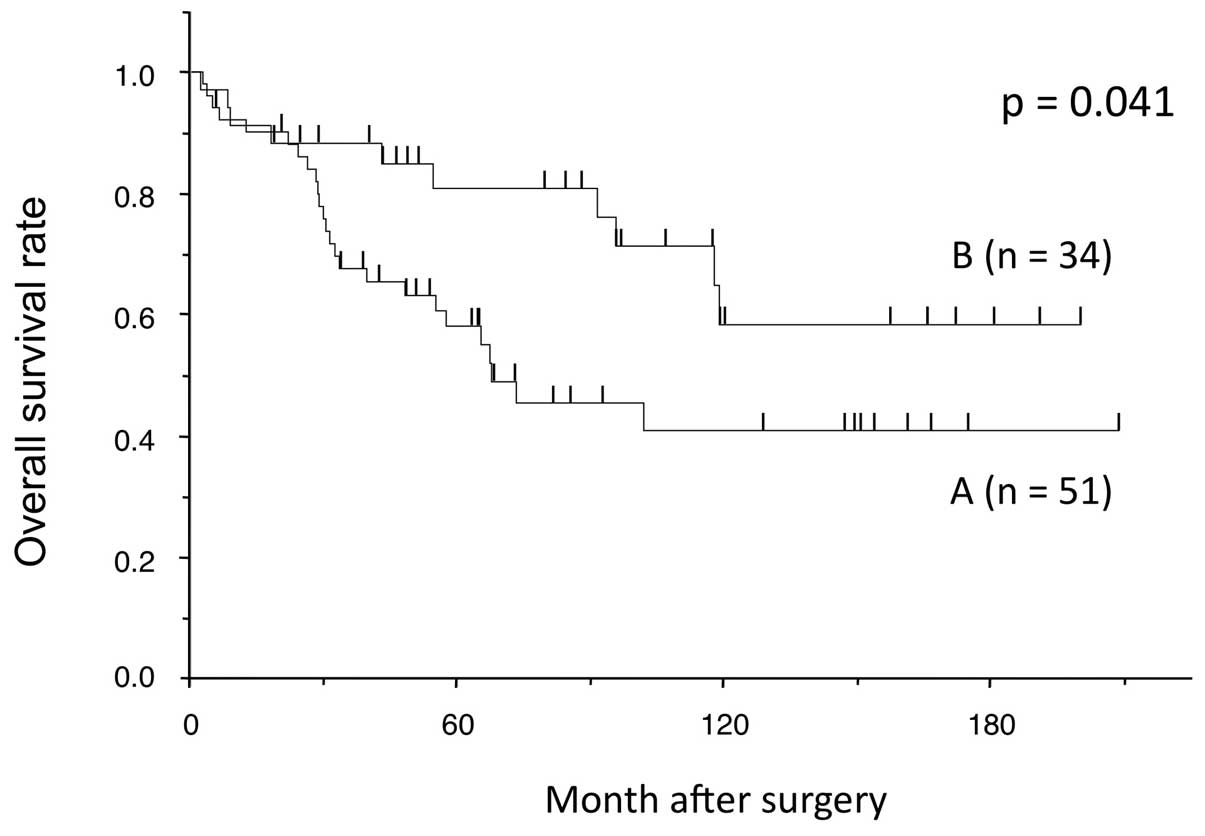
cases with both hypermethylation and a lower expression value of
CCNJ in tumor tissue than in adjacent non-cancerous tissue
had a poorer prognosis in terms of overall survival than the other
cases (P=0.038; Fig. 6).
Discussion
The cell cycle is controlled by a highly conserved
family of proteins called cyclin-dependent kinases (CDKs) and their
regulatory subunits, the cyclins (31). In mammals, approximately 20
subtypes of cyclin family members have been detected. The
progression of the cell cycle requires the cyclic activity of
several distinct CDK/cyclin complexes. The transition from the G2
to the mitotic (M) phase requires active complexes consisting of
CDK1/cyclin, and from the G1 to the synthesis (S) phase requires
active CDK2/cyclin complexes (32).
CCNJ was previously identified in a yeast
two-hybrid screen for embryonic proteins that interact with CDK2
(33). The biological function of
CCNJ in humans is still unclear, but in Drosophila
early embryogenesis, a role for CCNJ in nuclear division has
been reported. In the early embryo of Drosophila, CCNJ is
present in the S phase in an active complex with CDK2. Thus,
CDK2/CCNJ complexes may be functioning during the S phase in the
cell cycle (34).
In humans, the function of several cyclin family
members has been linked to the process of carcinogenesis or
proliferation of cancer, and many of them are considered to be
oncogenic. Also, in human HCC, the correlation between tumor
progression and overexpression of some cyclin family members has
been reported. Nishida et al reported that overexpression of
CCND1 resulted in the rapid growth of HCC (35). Gramantieri et al reported
that CCNG1 was inversely correlated with miR-122, which
influences p53 stability and transcriptional activity, and higher
CCNG1 expression was related to decreased survival (36,37).
However, our results showed that downregulation with promoter
hypermethylation of CCNJ in tumor tissues correlated with a
significantly poorer prognosis.
Similar to our results, cyclin G2 (CCNG2) has
been reported to be a tumor suppressor gene, and it has been shown
that the level of CCNG2 was downregulated in human gastric
adeno-carcinomas (18), oral
squamous carcinomas (19),
colorectal carcinomas (20) and
bladder metastatic carcinomas (21), when compared with adjacent
non-cancerous tissues. Moreover, low levels of CCNG2
expression correlated significantly with poor overall survival
(22).
The biological function of CCNG2 is obviously
different from conventional cell cycle proteins, since CCNG2
negatively regulates cell cycle progression (23). DNA damage and other growth
inhibitory signals may upregulate the activity of the CCNG2
gene, and activated CCNG2 interacts with protein phosphatase
2A (PP2A) and the CCNG2-PP2A complex stops cell cycle
progression and regulates cell proliferation by inhibiting CDK2
(38). As indicated by our
results, CCNJ behaved similarly to CCNG2 in that it
had a suppressive role against tumor progression by regulating the
G1 to S phase transition of the cell cycle, because CCNJ is
considered to interact with CDK2 in the cell cycle.
In conclusion, our results indicated that
CCNJ could be a novel prognostic marker of HCC. The
mechanism of CCNJ silencing might be related to promoter
hypermethylation. The method of triple combination array analysis,
which we devised, may be potentially useful in detecting new
tumor-related genes and their mechanisms.
Acknowledgements
This work was supported by Japan Society for the
Promotion of Science (JSPS) KAKENHI Grant-in-Aid for Scientific
Research (C) number 22591427.
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Livraghi T, Goldberg SN, Lazzaroni S,
Meloni F, Solbiati L and Gazelle GS: Small hepatocellular
carcinoma: Treatment with radio-frequency ablation versus ethanol
injection. Radiology. 210:655–661. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takayasu K, Arii S, Ikai I, et al: Liver
Cancer Study Group of Japan: Prospective cohort study of
transarterial chemo-embolization for unresectable hepatocellular
carcinoma in 8510 patients. Gastroenterology. 131:461–469. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Llovet JM, Ricci S, Mazzaferro V, et al:
SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu MC and Yuan JM: Environmental factors
and risk for hepatocellular carcinoma. Gastroenterology. 127(Suppl
1): S72–S78. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cusnir M and Patt YZ: Novel systemic
therapy options for hepatocellular carcinoma. Cancer J. 10:97–103.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kanda M, Nomoto S, Okamura Y, Nishikawa Y,
Sugimoto H, Kanazumi N, Takeda S and Nakao A: Detection of
metallothionein 1G as a methylated tumor suppressor gene in human
hepatocellular carcinoma using a novel method of double combination
array analysis. Int J Oncol. 35:477–483. 2009.PubMed/NCBI
|
|
10
|
Nomoto S, Kanda M, Okamura Y, Nishikawa Y,
Qiyong L, Fujii T, Sugimoto H, Takeda S and Nakao A: Epidermal
growth factor-containing fibulin-like extracellular matrix protein
1, EFEMP1, a novel tumor-suppressor gene detected in hepatocellular
carcinoma using double combination array analysis. Ann Surg Oncol.
17:923–932. 2010. View Article : Google Scholar
|
|
11
|
Okamura Y, Nomoto S, Kanda M, Li Q,
Nishikawa Y, Sugimoto H, Kanazumi N, Takeda S and Nakao A: Leukemia
inhibitory factor receptor (LIFR) is detected as a novel suppressor
gene of hepatocellular carcinoma using double-combination array.
Cancer Lett. 289:170–177. 2010. View Article : Google Scholar
|
|
12
|
Okamura Y, Nomoto S, Kanda M, Hayashi M,
Nishikawa Y, Fujii T, Sugimoto H, Takeda S and Nakao A: Reduced
expression of reelin (RELN) gene is associated with high recurrence
rate of hepatocellular carcinoma. Ann Surg Oncol. 18:572–579. 2011.
View Article : Google Scholar
|
|
13
|
Kanda M, Nomoto S, Okamura Y, Hayashi M,
Hishida M, Fujii T, Nishikawa Y, Sugimoto H, Takeda S and Nakao A:
Promoter hypermethylation of fibulin 1 gene is associated with
tumor progression in hepatocellular carcinoma. Mol Carcinog.
50:571–579. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayashi M, Nomoto S, Kanda M, Okamura Y,
Nishikawa Y, Yamada S, Fujii T, Sugimoto H, Takeda S and Kodera Y:
Identification of the A kinase anchor protein 12 (AKAP12) gene as a
candidate tumor suppressor of hepatocellular carcinoma. J Surg
Oncol. 105:381–386. 2012. View Article : Google Scholar
|
|
15
|
Okamura Y, Nomoto S, Hayashi M, et al:
Identification of the bleomycin hydrolase gene as a methylated
tumor suppressor gene in hepatocellular carcinoma using a novel
triple-combination array method. Cancer Lett. 312:150–157. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hishida M, Nomoto S, Inokawa Y, et al:
Estrogen receptor 1 gene as a tumor suppressor gene in
hepatocellular carcinoma detected by triple-combination array
analysis. Int J Oncol. 43:88–94. 2013.PubMed/NCBI
|
|
17
|
Inokawa Y, Nomoto S, Hishida M, et al:
Dynamin 3: A new candidate tumor suppressor gene in hepatocellular
carcinoma detected by triple combination array analysis. Onco
Targets Ther. 6:1417–1424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi W, Yu KR, Wu GY and Zhang XB:
Expression of CCNG in gastric carcinoma and its relationship with
prognosis. Chin J Cell Biol. 33:994–997. 2011.
|
|
19
|
Kim Y, Shintani S, Kohno Y, Zhang R and
Wong DT: Cyclin G2 dysregulation in human oral cancer. Cancer Res.
64:8980–8986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun GG, Zhang J and Hu WN: CCNG2
expression is down-regulated in colorectal carcinoma and its
clinical significance. Tumour Biol. 35:3339–3346. 2014. View Article : Google Scholar
|
|
21
|
Shan G, Shan SG and Zhang XB: Expression
of Cyclin G1 and Cyclin G2 in transitional cell carcinoma of
bladder. Chin J Histochem Cytochem. 18:267–272. 2009.
|
|
22
|
Sun GG, Hu WN, Cui DW and Zhang J:
Decreased expression of CCNG2 is significantly linked to the
malignant transformation of gastric carcinoma. Tumour Biol.
35:2631–2639. 2014. View Article : Google Scholar
|
|
23
|
Bates S, Rowan S and Vousden KH:
Characterisation of human cyclin G1 and G2: DNA damage inducible
genes. Oncogene. 13:1103–1109. 1996.PubMed/NCBI
|
|
24
|
Harvey RC, Mullighan CG, Wang X, et al:
Identification of novel cluster groups in pediatric high-risk
B-precursor acute lymphoblastic leukemia with gene expression
profiling: Correlation with genome-wide DNA copy number
alterations, clinical characteristics, and outcome. Blood.
116:4874–4884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Feliciano A, Castellvi J, Artero-Castro A,
et al: miR-125b acts as a tumor suppressor in breast tumorigenesis
via its novel direct targets ENPEP, CK2-α, CCNJ, and MEGF9. PLoS
One. 8:e762472013. View Article : Google Scholar
|
|
26
|
Stangegaard M: Gene expression analysis
using agilent DNA microarrays. Methods Mol Biol. 529:133–145. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kennedy GC, Matsuzaki H, Dong S, et al:
Large-scale genotyping of complex DNA. Nat Biotechnol.
21:1233–1237. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nannya Y, Sanada M, Nakazaki K, et al: A
robust algorithm for copy number detection using high-density
oligonucleotide single nucleotide polymorphism genotyping arrays.
Cancer Res. 65:6071–6079. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Takai D and Jones PA: The CpG island
searcher: A new WWW resource. In Silico Biol. 3:235–240.
2003.PubMed/NCBI
|
|
30
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
31
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: Several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Finley RL Jr, Thomas BJ, Zipursky SL and
Brent R: Isolation of Drosophila cyclin D, a protein expressed in
the morphogenetic furrow before entry into S phase. Proc Natl Acad
Sci USA. 93:3011–3015. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kolonin MG and Finley RL Jr: A role for
cyclin J in the rapid nuclear division cycles of early Drosophila
embryogenesis. Dev Biol. 227:661–672. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nishida N, Fukuda Y, Komeda T, et al:
Amplification and overexpression of the cyclin D1 gene in
aggressive human hepatocellular carcinoma. Cancer Res.
54:3107–3110. 1994.PubMed/NCBI
|
|
36
|
Gramantieri L, Ferracin M, Fornari F, et
al: Cyclin G1 is a target of miR-122a, a microRNA frequently
down-regulated in human hepatocellular carcinoma. Cancer Res.
67:6092–6099. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fornari F, Gramantieri L, Giovannini C, et
al: MiR-122/cyclin G1 interaction modulates p53 activity and
affects doxorubicin sensitivity of human hepatocarcinoma cells.
Cancer Res. 69:5761–5767. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bennin DA, Don AS, Brake T, McKenzie JL,
Rosenbaum H, Ortiz L, DePaoli-Roach AA and Horne MC: Cyclin G2
associates with protein phosphatase 2A catalytic and regulatory B′
subunits in active complexes and induces nuclear aberrations and a
G1/S phase cell cycle arrest. J Biol Chem. 277:27449–27467. 2002.
View Article : Google Scholar : PubMed/NCBI
|