Introduction
Breast cancer remains the second leading cause of
cancer deaths among women. The troubling mortality rates of breast
cancer patients, along with the continued incidence of new breast
cancer diagnoses each year, illustrate a critical need for new
therapeutic targets and improved therapies in breast cancer
treatment. Emerging therapeutic targets potentially reside in the
transient receptor potential (TRP) superfamily of cation channels.
Recent studies have demonstrated important roles for TRP channels
in several types of human cancer (1–3).
However, little is known regarding the role of these cation
channels in breast cancer. Determining the role of TRPs in breast
cancer may help identify novel molecular targets for the treatment
of breast cancer patients and thus help reduce the mortality rates
of this devastating disease.
The TRP superfamily is a diverse set of cation
channels that facilitate a variety of cellular functions. The
largest TRP subfamily is the TRP melastatin (TRPM) set of cation
channels. TRPM channels are known to mediate sensory and adaptive
functions, such as taste, thermosensitivity, and touch (4,5).
TRPM2 is a unique member of the TRPM subfamily, a widely expressed,
non-selective cation channel that also possesses adenosine
diphosphoribose (ADP-ribose) pyrophosphatase activity (6). The binding of ADP-ribose leads to the
enzymatic activity and the opening of this ion channel. Thus, upon
activation of this ‘chanzyme’ by ADP-ribose, cations are gated into
the cell. Most notable of these cations is calcium, where the
influx of calcium in response to oxidative stress leads to the
calcium-mediated activation of pro-cell death apoptotic (7) and non-apoptotic proteins (8,9).
TRPM2 thus appears to facilitate the progression of
caspase-dependent and caspase-independent cell death mechanisms
after oxidative stress (10).
Accordingly, activation of TRPM2 has been shown to exacerbate the
injury that occurs in response to oxidative stress in noncancerous
cells, including neuronal (11),
pancreatic (12), and
hematopoietic cells (9).
Pharmacologic inhibition of TRPM2 was subsequently shown to
decrease cell death in these instances, as well as increase cell
survival in several other cell lines and tissues (13–15).
The rationale for pharmacologically inhibiting the activation of
TRPM2 is based upon the ability of TRPM2 inhibitors to decrease the
cell death and tissue injury that occurs due to debilitating
diseases and conditions. Taken together, the current knowledge of
TRPM2 has provided the basis for the development of pharmacologic
inhibitors of TRPM2 in order to treat debilitating conditions that
involve excessive cell death, including stroke, diabetes, immune
disorders and inflammation.
Since TRPM2 has mostly been investigated in
noncancerous cells, less is known about the function of the TRPM2
cation channel in cancer cells. Two TRPM2 mRNA transcripts, one
antisense transcript and one truncated TRPM2 transcript, were shown
to be increased in 80% of metastatic melanoma cell lines (16). Functional analysis of the protein
products of these transcripts demonstrated that overexpression of
wild-type TRPM2 or knockout of the truncated TRPM2 transcript
increased cytotoxicity in melanoma cells. Similarly, RNAi silencing
of TRPM2 in prostate cancer cells decrease their proliferation,
which suggests that TRPM2 has a role in facilitating prostate
cancer cell propagation and growth (17). In this same study, it was shown
that TRPM2, normally localized to the plasma membrane or lysosomal
membrane (7,12), was localized to the nucleus in
prostate cancer cells.
To date, no additional evidence has been reported
that shows a nuclear localization of TRPM2 in any other cancerous
or noncancerous cell line. Further, other TRPM subfamily members
have been shown to have novel roles in cancer. For example, TRPM8
was shown to be upregulated in pancreatic adenocarcinoma cells and
vital to their proliferation (2).
In summary, TRPM channels appear to have novel roles in various
types of human cancer. It is thus possible that the unique member,
TRPM2, may also have a novel role in human cancers as well.
In this study, our objective was to investigate
TRPM2 in two lines of human breast adenocarcinoma cells in order to
analyze its function in breast cancer cells and to provide a
preliminary evaluation of its potential as a pharmacologic target
in breast cancer. We show here that inhibition of TRPM2 function
causes decreased proliferation and increased levels of DNA damage
in breast adenocarcinoma cells, with minimal effects in normal
breast cells. Because these results have not been previously
reported in breast cancer cells, our studies present preliminary
evidence that TRPM2 is a potential therapeutic target in breast
cancer and its pharmacologic inhibition is expected to selectively
target breast cancer cells.
Materials and methods
Cell lines and cell culture reagents
HEK 293 (human embryonic kidney), MCF-10A (human
mammary epithelial), MCF-7 (human breast adenocarcinoma), and
MDA-MB-231 (human breast adenocarcinoma) cell lines were purchased
from American Type Culture Collection (ATCC; Manassas, VA, USA).
The HMEC (human mammary epithelial cell) line was purchased from
Lonza (Walkersville, MD, USA). Wild-type and poly(ADP-ribose)
glycohydrolase (PARG)-null embryonic trophoblast stem (TS) cells
were derived from E3.5 mouse blastocysts as previously described
(18). They were maintained in
growth medium containing fibroblast growth factor-4, heparin
sodium, murine embryonic feeder-conditioned medium, 15% FBS, and
0.5 mM benzamide (an inhibitor of poly(ADP-ribose) polymerase).
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from
Hyclone (Logan, UT, USA). Fetal bovine serum (FBS) was purchased
from Atlas Biologicals (Fort Collins, CO, USA). Mammary epithelial
growth medium (MEGM), which consists of mammary epithelial basal
medium (MEBM) plus growth supplements (see ‘Cell culture’ below),
was purchased from Lonza. Trypsin-EDTA (0.25%),
penicillin-streptomycin solution, and glutamine were purchased from
Invitrogen (Carlsbad, CA, USA).
Other reagents
OptiMEM reduced serum medium and Lipofectamine 2000
reagent were purchased from Gibco Life Technologies (Grand Island,
NY, USA). Protease inhibitor cocktail tablets (Complete Mini,
EDTA-free) were purchased from Roche (Mannheim, Germany). Primary
antibodies utilized were polyclonal rabbit anti-human TRPM2
antibody (Cat. #A300-414A, Bethyl Laboratories, Montgomery, TX,
USA), polyclonal rabbit anti-human β-actin (Cat. #600-401-886,
Rockland Immunochemicals, Limerick, PA, USA), polyclonal rabbit
anti-human manganese superoxide dismutase (MnSOD) (Cat. #06-984,
Millipore, Billerica, MA, USA), and monoclonal mouse anti-human
Lamin B2 clone LN43 (Cat. #MA1-06104, Thermo Fisher Pierce,
Pittsburgh, PA, USA). The secondary antibodies, horseradish
peroxidase (HRP)-conjugated goat anti-rabbit and HRP-conjugated
rabbit anti-mouse were purchased from Sigma (St. Louis, MO, USA).
2-Aminoethoxydiphenyl borate (2-APB), maintained as a 75 mM stock
solution in dimethyl sulfoxide (DMSO), and 30% hydrogen peroxide
solution were purchased from Sigma. The Fluo-4 NW Calcium Assay kit
was purchased from Life Technologies. Comet Assay kit, which
includes alkaline lysis solution, LMAgarose, 2-well CometSlides,
SYBR Green, and EDTA, was purchased from Trevigen (Gaithersburg,
MD, USA). CytoScan WST-1 cell proliferation assay was purchased
from VWR International (Radnor, PA, USA).
Cell culture
HEK 293, MDA-MB-231, and MCF-7 cells were grown and
maintained in DMEM supplemented with 10% FBS, 100 U/ml
penicillin/streptomycin, and 2 mM L-glutamine. Noncancerous human
mammary cells (MCF-10A and HMEC) were cultured in MEGM specialty
medium. MEGM consists of mammary epithelial cell basal medium
(MEBM) plus the following growth supplements: 5 μg/ml bovine
pituitary extract, 0.01 μg/ml human epidermal growth factor, 0.5
μg/ml hydrocortisone, GA-1000 (60 μg/ml gentamicin and 0.03 μg/mL
amphotericin B), and 5 μg/ml insulin. All cultures were incubated
at 37°C in 5% CO2 until treatments and analyses. Every
two days in culture, cells were washed once with phosphate-buffered
saline, pH 7.2 (PBS) and cultured in fresh growth medium.
RNA interference
The silencing of TRPM2 by RNAi was performed as
previously reported using siRNA specific to TRPM2
(5′-AUAGAUCAGGAACUCCGUCUC-3′) (17). This RNA oligo was purchased as
duplexed RNA from Integrated DNA Technologies (Coralville, IA,
USA). Each siRNA duplex was resuspended in RNase-free water at a
final concentration of 40 μM and stored at −20°C. Universal
scrambled control siRNA oligos were purchased as duplex RNA from
Sigma and were used for all negative controls (19).
For RNAi transfections, cells were plated in 0.5 ml
of medium per well without antibiotics in 24-well plates one day
before transfection. At the time of transfection, cells were ~50%
confluent. For each transfection, two mixtures were prepared: i)
duplex siRNA added to 50 μl OptiMEM medium, and ii) 1 μl
Lipofectamine 2000 added to 50 μl OptiMEM. After 5 min, the two
solutions were gently mixed and then incubated at room temperature
for 20 min. Final concentrations of siRNA were 100 nM for all cell
lines. The mixtures were added drop wise to each well and cells
were cultured for an additional 48–72 h.
Whole cell lysate extraction
Cells were grown on 6-well tissue culture plates,
harvested by trypsinization, washed once with 0.5 ml ice-cold PBS,
and resuspended in 0.5 ml lysis buffer containing 25 mM Tris-HCl
(pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, and protease
inhibitors (Complete Mini, EDTA-free tablets). Suspensions were
incubated for 30 min on ice and vortexed every 10 min. Cleared cell
lysates were obtained after centrifugation at 16,000 × g for 10
min. Sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) sample buffer (50 mM Tris-HCl pH 6.8, 1% SDS, 2.5%
glycerol, 0.005% bromophenol blue and 100 mM dithiothreitol) was
added to the supernatants. Samples were then heated for 2 min at
95°C in a digital dry bath incubator (Labnet International, Edison,
NJ, USA).
Subcellular fractionations
Cells were grown in 60-mm tissue culture dishes
(approximately 2×106 cells/dish) and harvested by cell
scraping in ice-cold PBS. After mild centrifugation (200 × g for 5
min), pellets were then fractionated using the NE-PER Nuclear and
Cytoplasmic Extraction kit (Thermo Fisher Pierce, Rockfield, IL,
USA) according to the manufacturer’s protocol. Briefly, harvested
cells were washed with 1 ml ice-cold PBS and transferred to a 1.5
ml micro centrifuge tube. Cell pellets were obtained by
centrifugation at 500 × g for 3 min at 4°C. The supernatant was
removed and the pellet was resuspended in 0.2 ml ice-cold CER I
solution containing protease inhibitors. The suspension was
vortexed for 15 sec and placed on ice for 10 min. After addition of
11 μl of CER II solution, the suspension was vortexed for 5 sec,
placed on ice for 1 min, and vortexed again. The extract was
centrifuged at 16,000 × g for 5 min at 4°C and the supernatant,
which represents the cytoplasmic fraction, was removed. The pellet,
which contains the nuclei, was washed once with PBS as before and
then resuspended in 0.1 ml of ice-cold NER solution. The nuclei
were vortexed and placed on ice for 10 min. This was repeated 3
times for a total of 40 min. The extract was centrifuged as before
for 10 min and the supernatant, which represents the nuclear
fraction, was removed. Nuclear and cytoplasmic fractions were
prepared in SDS-PAGE sample buffer and heated in a standard manner,
as previously described.
Immunoblotting
The protein concentration for all samples was
obtained using the Pierce BCA Protein Assay kit (Thermo
Scientific). Approximately 20 μg of each lysate or subcellular
fraction sample was subjected to 7.5% SDS-PAGE. The proteins were
transferred to 0.45 μm nitrocellulose by semi-dry transfer at 25 V
for 1 h using a Trans-blot SD apparatus (Bio-Rad Laboratories,
Hercules, CA, USA). Membranes were blocked with PBS containing
0.05% Tween-20 (PBST) and 5% milk at room temperature for 1 h and
incubated with primary antibodies (1:1,000 anti-TRPM2, 1:3,000
anti-MnSOD, or 1:1,000 anti-Lamin B2) in PBST+5% milk overnight
(shaking) at 4°C. Membranes were then washed with PBST three times
and incubated with horseradish peroxidase (HRP)-conjugated goat
anti-rabbit or HRP-conjugated rabbit anti-mouse antibody (1:10,000)
for 1 h. The membranes were washed as described above, and
chemiluminescence was initiated using the SuperSignal West Pico
Chemiluminescent Substrate (Thermo Fisher Pierce). Immunoblots were
then developed on a ChemiDoc XRS gel imaging system (Bio-Rad
Laboratories). For quantification of protein levels for each blot,
immunoblots were examined by densitometry with the ChemiDoc imager
using Quantity One software. Relative densitometry ratios were then
calculated using β-actin as loading controls. The resulting ratio
of TRPM2/β-actin provided values that were then used to quantify
relative protein levels.
Proliferation assays
Cell proliferation assays using CytoScan WST-1
(Roche) were performed according to the manufacturer’s
specifications. This assay measures cellular dehydrogenase
activity, which is directly correlated to cell number. The
reduction of a tetrazolium salt by cellular dehydrogenases is
detected by spectrophotometer analysis at 425 nm. Briefly, cells
were seeded at 5,000 cells per well in triplicate in a 96-well
plate in growth medium appropriate to each cell type. Cells were
incubated overnight and then treated with 100 μM 2-APB, scrambled
siRNA or 100 nM TRPM2 siRNA the following day. At 0, 24, 48, 72 and
96 h time points, the media was removed, WST-1 reagent was added
(10 μl WST-1 premixed with 100 μl growth medium per well), and
cells were incubated for 1 h at 37°C. Analysis was performed using
a BioTek Synergy HT microplate reader using Gen5 software
(Winooski, VT, USA).
Intracellular calcium measurement
A Fluo-4 NW Calcium Assay kit (Invitrogen) was used
to measure intracellular calcium influx on a BioTek Synergy HT
microplate reader following the manufacturer’s protocols. Briefly,
approximately 5×104 cells per well were grown in a
96-well plate overnight. The next day, plates were washed twice in
Ca2+-free HBSS supplemented with HEPES buffer (pH 7.2),
and then the growth medium was replaced with 100 μl/well of the
Fluo-4 dye solution containing probenecid (to prevent extrusion of
the dye out of the cells). The plate was incubated at 37°C for 30
min and then at room temperature for an additional 30 min in the
dark. The loaded cells were then placed in the measurement position
in a BioTek Synergy fluorescence spectrophotometer. Changes in
fluorescence from the Fluo-4-NW dye quantify changes in
intracellular Ca2+ concentrations (excitation/emission
485/535 nm) after treatment with 5 mM hydrogen peroxide
(H2O2). Ca2+ influx was measured
up to 40 min.
Comet assays
Comet assays were performed using the CometAssay ES
system from Trevigen. The manufacturer’s instructions for alkaline
electrophoresis were followed. In brief, cells were seeded in
24-well tissue culture plates, incubated overnight in 0.5 ml growth
medium, then treated the following day with 100 μM 2-APB or
transfected with 100 nM TRPM2 siRNA. After collection by
trypsinization 24 h later, a cell concentration of 1×105
cells/ml was combined with molten low melting point agarose at a
ratio of 1:10 (v/v). Agarose/cell mix (50 μl) was immediately
pipetted onto a CometSlide and the slides were placed at 4°C in the
dark for 30 min to allow solidification of the gel. The slide was
then placed in lysis solution at 4°C overnight. The following day,
the lysis buffer was removed and cells were immersed in freshly
prepared alkaline unwinding solution for 20 min at room
temperature. Cold alkaline electrophoresis solution was added to
the CometAssay ES electrophoresis unit and slides were placed into
the electrophoresis chamber. Horizontal electrophoresis was
performed at 18 V for 30 min. When electrophoresis was complete,
the slides were immersed twice in water and once in 70% ethanol for
5 min each, and then dried at 37°C for 15 min to bring all the
cells into a single plane. The slides were then stored at room
temperature with a desiccant until ready for analysis.
To stain the CometSlides, 100 μl of SYBR Green
solution was added to each well and left for 30 min at room
temperature, and then allowed to dry completely at 37°C. The slides
were imaged using a Zeiss AxioObserver Z1 inverted fluorescence
microscope (Thornwood, NY) with Hamamatsu Orca-ER digital camera
and Axiovision software. Images were then analyzed using CometScore
software (Tritek Corp., Sumerduck, VA, USA). A minimum of 200 cells
for each treatment group were scored for quantification of ‘Percent
DNA in Tail’, a standard comet assay value that represents DNA
damage (20,21). This value was computed as the total
comet tail intensity divided by the total comet intensity,
multiplied by 100.
Statistical analyses
All error bars for proliferation, protein levels,
intracellular calcium influx, and comet assay quantifications
represented the standard error of the mean (SEM). Statistical
analyses were accomplished by one-way analysis of variance (ANOVA)
followed by Tukey’s test and unpaired Student’s t-test. Statistical
significance was defined as p<0.05.
Results
TRPM2 levels in noncancerous and
cancerous breast cells
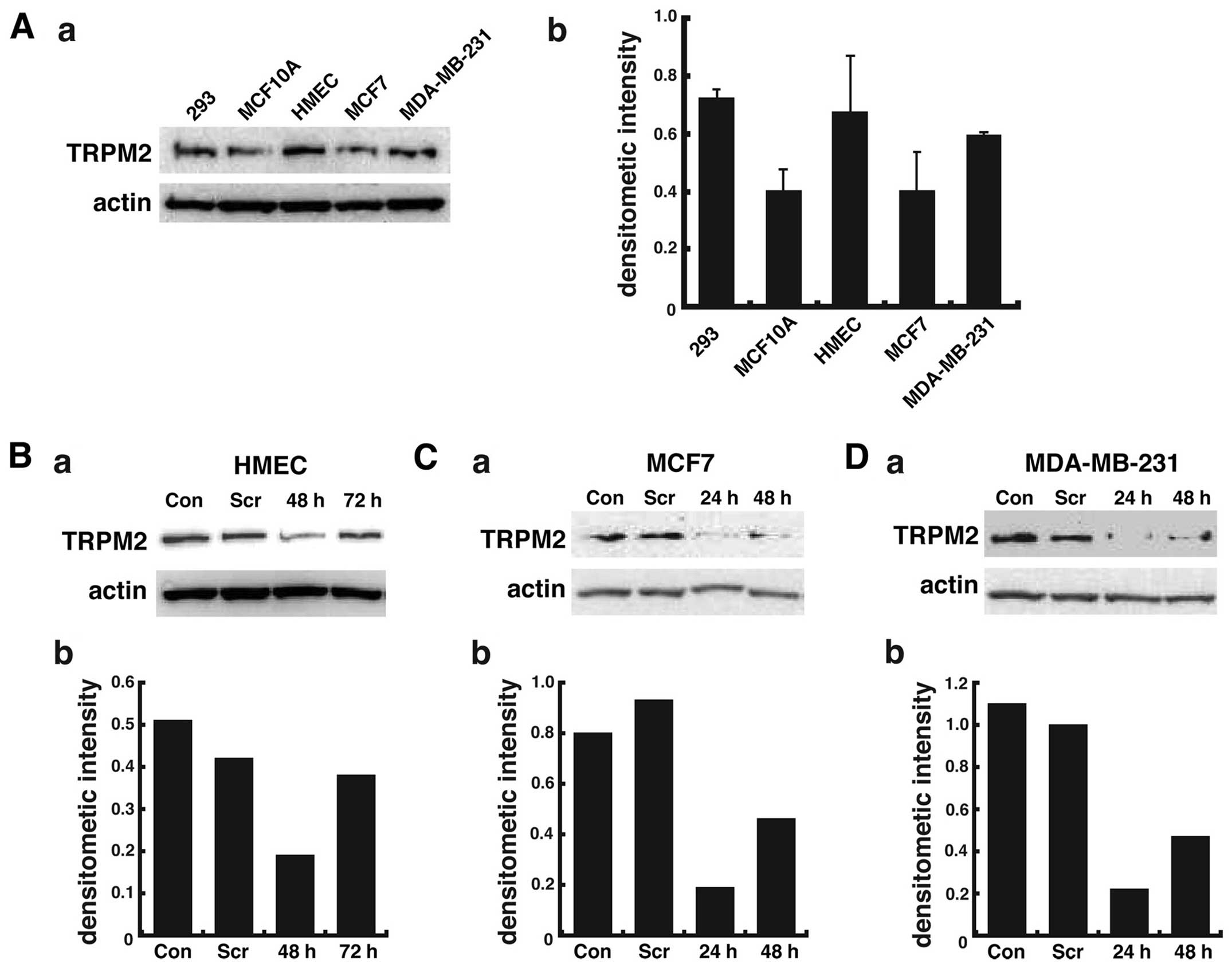
Although TRPM2 channels are nearly ubiquitously
expressed, we first analyzed the presence of TRPM2 channels in two
lines of noncancerous human mammary epithelial cells and two lines
of human breast adenocarcinoma cells by western blotting. The
positive control for TRPM2 levels was provided by human embryonic
kidney cells (HEK 293 cell line), as previous studies report the
presence of TRPM2 mRNA (22) and
endogenous protein expression (23) in these cells. Immunoblot analysis
demonstrated significant levels of TRPM2 in HEK 293 cells, as
expected (Fig. 1A). The results
also show that TRPM2 levels in the two noncancerous mammary
epithelial cells were variable, with greater levels in the HMEC
cell line as compared to the MCF-10A cell line (Fig. 1A-a). In two lines of metastatic
breast adenocarcinoma cells, greater levels of TRPM2 were observed
in the MDA-MB-231 cell line as compared to the MCF-7 cell line.
However, the increased levels of TRPM2 in HMEC cells (versus
MCF-10A cells) and MDA-MB-231 cells (versus MCF-7 cells) as
quantified by densitometry were not statistically significant
(Fig. 1A-b). However, these
results provide qualitative evidence that TRPM2 is present in each
breast cell line. In summary, these results demonstrate that TRPM2
is present in two lines of noncancerous and two lines of cancerous
breast cells.
Effects of TRPM2 pharmacologic inhibition
and TRPM2 RNAi silencing in breast adenocarcinoma cells
We utilized a TRPM2-specific siRNA sequence to knock
down TRPM2 expression in breast cells. The siRNA sequence we
utilized was previously shown to effectively knock down TRPM2
expression in both noncancerous prostate cells and prostate cancer
cells (17). Using this siRNA
sequence, we successfully decreased TRPM2 protein levels in
noncancerous HMEC cells, where levels were decreased >60% as
compared to control levels after 48 h (Fig. 1B). In breast adenocarcinoma cells
after 48 h of RNAi silencing, TRPM2 levels were decreased more than
75% in the MCF-7 cell line (Fig.
1C) and ~80% in the MDA-MB-231 cell line (Fig. 1D) as compared to control levels.
The results demonstrate the effective RNAi silencing of TRPM2 in
noncancerous and cancerous human breast cells.
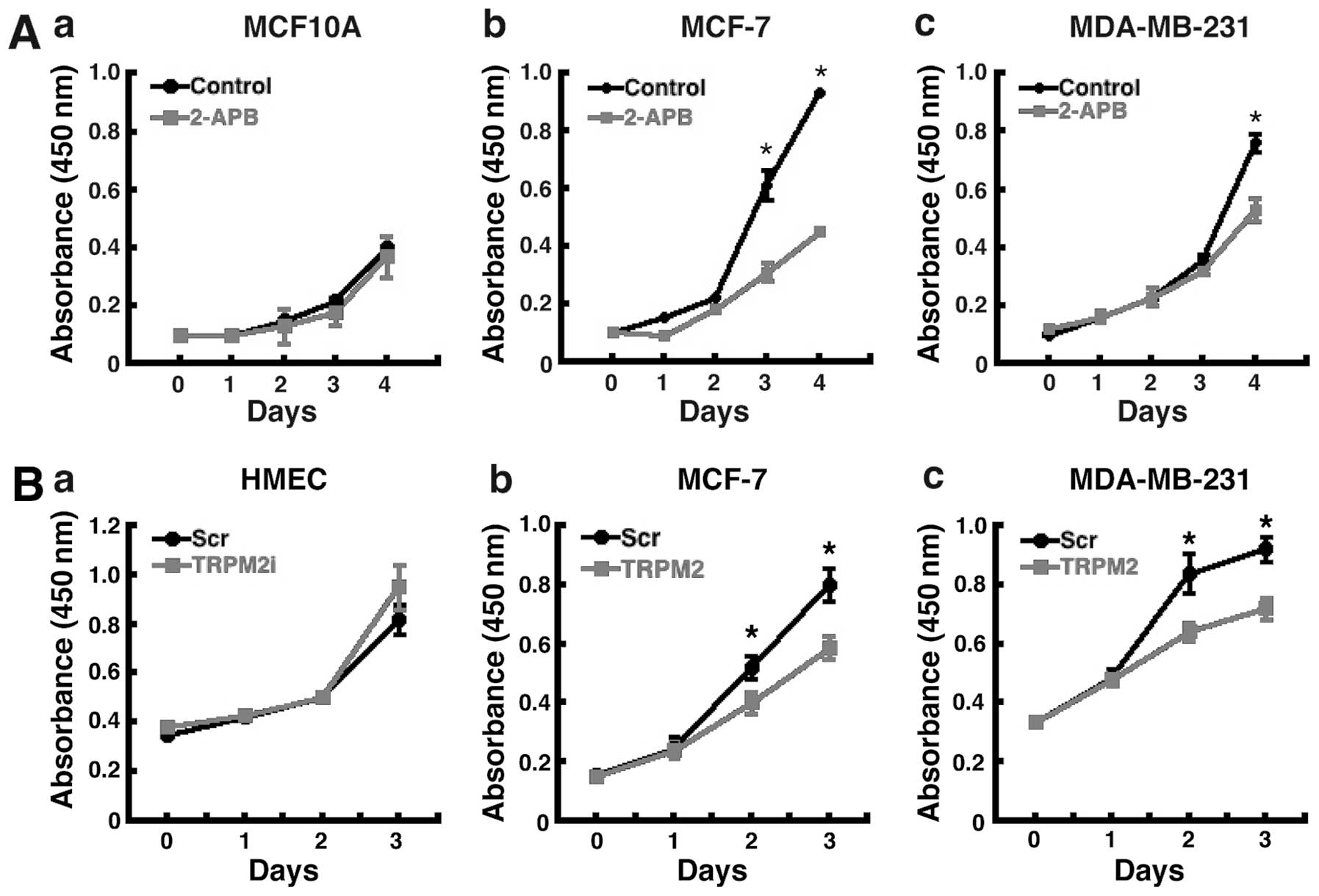
We next determined the effect of TRPM2 pharmacologic
inhibition and RNAi silencing on breast adenocarcinoma cell
proliferation. Treatment of the cells with 2-aminoethoxydiphenyl
borate (2-APB), a pharmacologic inhibitor of TRPM2 (24), led to decreased proliferation in
both lines of human breast adenocarcinoma cells (Fig. 2A-b and A-c). Treatment with 2-APB
did not significantly effect the proliferation of noncancerous
MCF-10A human breast epithelial cells (Fig. 2A-a). The effect was greater in
MCF-7 cells, where decreased proliferation was evident by
post-treatment day 3 (Fig. 2A-b).
After 4 days of 2-APB treatment, proliferation was decreased nearly
60% in MCF-7 cells. Further, an effect was also observed in
MDA-MB-231 breast adenocarcinoma cells, where proliferation was
decreased ~40% after four days of 2-APB treatment (Fig. 2A-c). These results show that
treatment with the TRPM2 inhibitor, 2-APB, leads to decreased
proliferation in human breast adenocarcinoma cells, but not in
noncancerous human mammary epithelial cells.
To verify that these effects were indeed due to the
inhibition of TRPM2 function, the effect of TRPM2 RNAi silencing on
cell proliferation was then analyzed. In both MCF-7 and MDA-MB-231
breast adenocarcinoma cells, decreased proliferation was observed 2
days after RNAi treatment (Fig. 2B-b
and B-c). After 3 days, RNAi silencing of TRPM2 led to a 30–40%
reduction in proliferation in these cells. No effect on
proliferation was observed after TRPM2 RNAi silencing in
noncancerous HMEC cells (Fig.
2B-a). These results thus demonstrate that the specific
knockdown of TRPM2 led to decreased proliferation in human breast
adenocarcinoma cells, but not in noncancerous human mammary
epithelial cells. Taken together, the results of the pharmacologic
inhibition of TRPM2 and RNAi silencing of TRPM2 indicate that TRPM2
has a role in facilitating the proliferation of human breast
adenocarcinoma cells.
Effect of TRPM2 inhibition on calcium
influx in breast adenocarcinoma cells
TRPM2 is recognized as a plasma membrane ionophore,
where it gates cations, including calcium, into the cell. Since
TRPM2-mediated calcium gating is not well studied in breast cells,
we again utilized 2-APB, as it was previously used to block the
influx of cations by the TRPM2 channel in several cell lines
(24). Thus, via the use of 2-APB,
we analyzed the ability of TRPM2 channels to promote calcium influx
in breast adenocarcinoma cells after oxidative stress. To analyze
TRPM2 function, we utilized the Fluo-4 NW calcium assay to quantify
calcium influx into breast adenocarcinoma cells after stimulation
by hydrogen peroxide, as this assay was previously utilized to
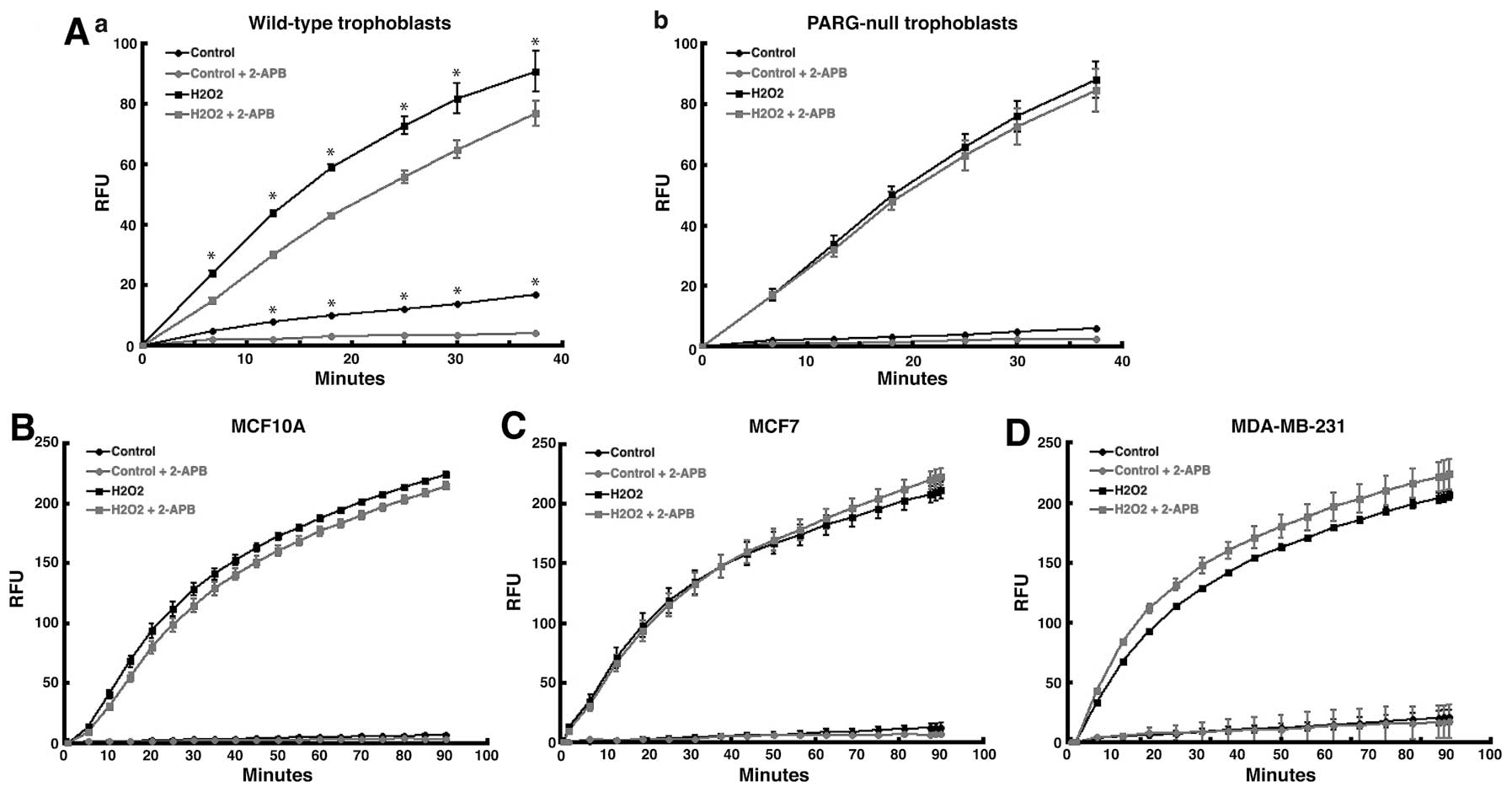
measure calcium influx due to TRPM2 channels (10). To validate the assay, we used the
assay to measure TRPM2-mediated calcium influx in wild-type and
poly(ADP-ribose) glycohydrolase (PARG)-null trophoblast stem (TS)
cells. We used these particular cell lines because the primary
molecule that activates TRPM2 channels is ADP-ribose. ADP-ribose, a
product produced by the hydrolysis of poly(ADP-ribose) (PAR)
polymers by PARG (25), binds
TRPM2, opens the channel, and causes the gating of cations into the
cell (6,26). Because of this, TRPM2-mediated
calcium influx should be minimal in PARG-null TS cells, which
contain no PARG enzymatic activity. In wild-type TS cells,
treatment with hydrogen peroxide led to significant levels of
calcium influx (Fig. 3A-a).
Pretreatment of these cells with 2-APB caused significant decrease
in calcium influx, which demonstrated that 2-APB blocked the influx
of calcium by TRPM2 channels in wild-type TS cells. However, in
PARG-null TS cells, pretreatment with 2-APB produced no significant
effect on calcium influx following H2O2
treatment (Fig. 3A-b). This
demonstrated that without PARG, ADP-ribose was not generated, the
TRPM2 channel was not activated, and calcium was thereby not gated
into the cell in PARG-null TS cells. Thus, the failure of 2-APB to
decrease calcium influx in PARG-null TS cells validated the Fluo-4
NW calcium assay.
Of note, pretreatment with 2-APB failed to decrease
the level of intracellular calcium stimulated by hydrogen peroxide
in either MCF-7 or MDA-MB-231 human breast adenocarcinoma cells
(Fig. 3C and D). Further,
intracellular calcium influx was greater in MDA-MB-231 cells
pretreated with 2-APB (Fig. 3D).
In noncancerous MCF-10A breast epithelial cells, pretreatment with
2-APB decreased calcium influx stimulated by
H2O2 at earlier time points (<15 min),
while no statistical difference was observed at later time points
(Fig. 3B). These results show that
the pharmacologic inhibition of TRPM2 channels in human breast
adenocarcinoma cells does not decrease calcium influx. This
suggests that TRPM2 may not primarily function as a calcium channel
in human breast adenocarcinoma cells, which further suggests that
the role for TRPM2 in breast cancer cells may be distinct from its
role in noncancerous cells.
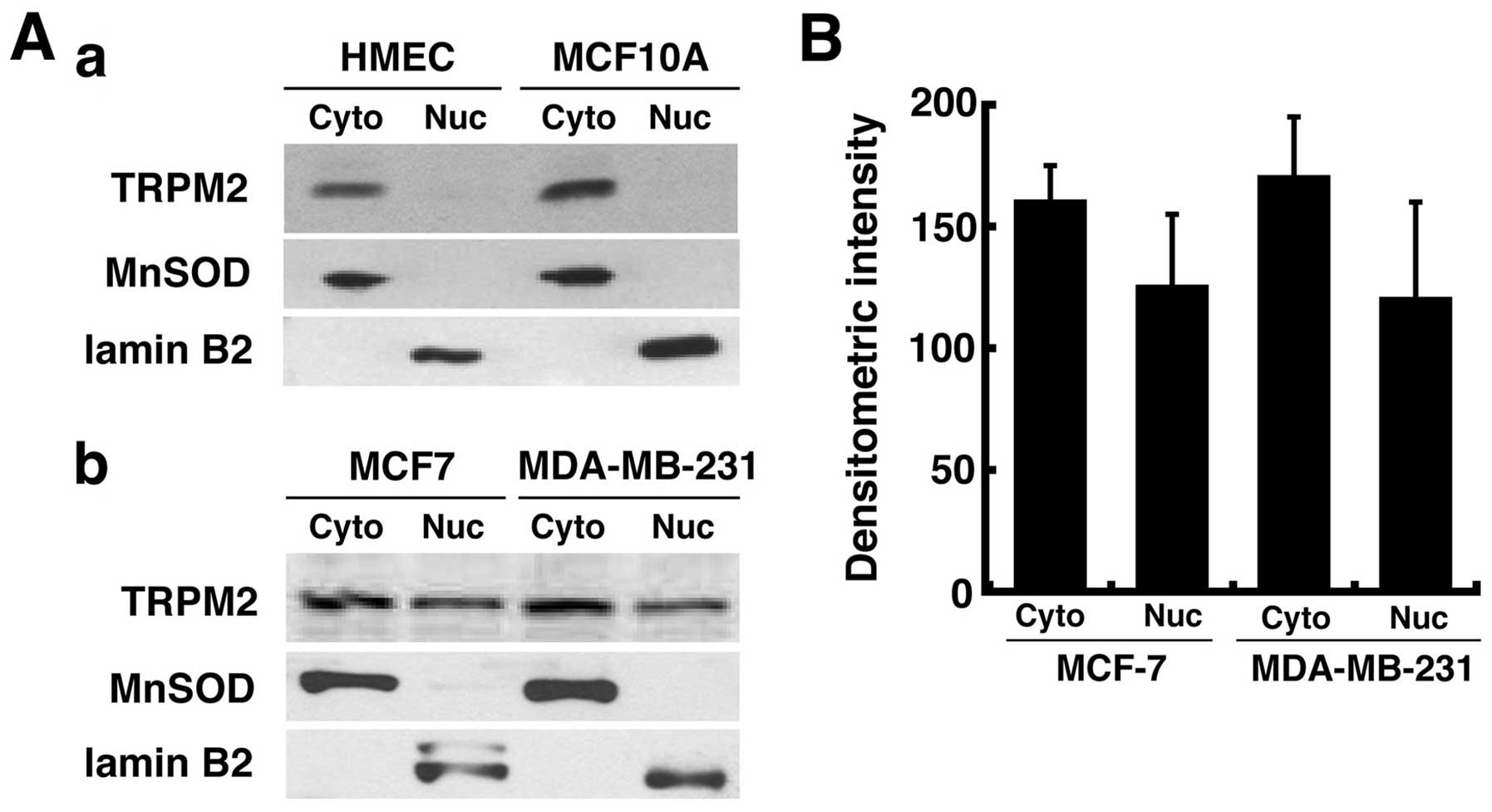
Nuclear localization of TRPM2 in human
breast adenocarcinoma cells
Because of the possibility that TRPM2 may have a
novel role in human breast adenocarcinoma cells, our next objective
was to determine its intracellular localization. In noncancerous
cells, TRPM2 is normally localized to the plasma membrane as a
non-specific cation channel (7).
However, it has also been found to be localized to the lysosomal
membrane (12). Thus, present
studies show that TRPM2 has an extra-nuclear localization in normal
cells. In agreement with these studies, our TRPM2 localization
analyses demonstrated that TRPM2 has an extra-nuclear localization
in noncancerous HMEC and MCF-10A breast cells (Fig. 4A-a), as TRPM2 protein was observed
in the cytoplasmic fractions of these cells after subcellular
fractionations. However, in MCF-7 and MDA-MB-231 human breast
adenocarcinoma cells, TRPM2 was present in the nuclear fractions of
these cells (Fig. 4A-b). This
localization was not exclusive, as TRPM2 was also observed in the
cytoplasmic fractions in these cells. Quantification of protein
levels by densitometry indicated that ~40–45% of the TRPM2 protein
present in human breast adenocarcinoma cells was localized to the
nucleus (Fig. 4B). As these
results are consistent with the nuclear TRPM2 localization in
prostate cancer cells shown previously (17), our data thus demonstrate that TRPM2
is present in the nuclei of MCF-7 and MDA-MB-231 human breast
adenocarcinoma cells. This suggests that TRPM2 may have a novel
role in the nuclei of these cells.
Inhibition or RNAi silencing of TRPM2
causes increased DNA damage in breast adenocarcinoma cells
To investigate a possible nuclear role of TRPM2 in
human breast adenocarcinoma cells, we determined the effect of
TRPM2 pharmacologic inhibition and TRPM2 RNAi silencing on the
levels of DNA damage in MCF-7 and MDA-MB-231 cells. This was
performed by utilizing the single cell gel electrophoresis (comet)
assay. The comet assay is based on the alkaline lysis of labile DNA
at sites of damage (27). The
unwound, relaxed DNA is able to migrate out of the cell during
electrophoresis and can be visualized using SYBR green nucleic acid
stain. Cells that have accumulated DNA damage appear as fluorescent
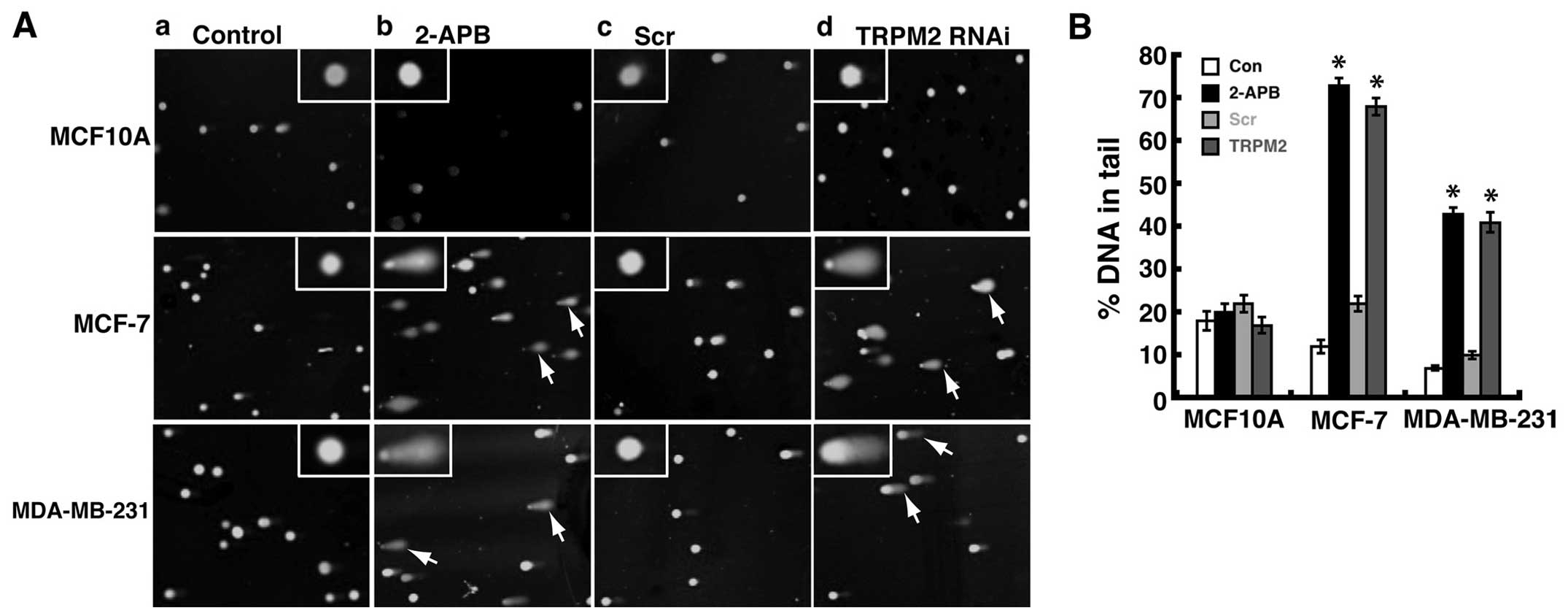
comets with tails that represent DNA fragmentation. In noncancerous
human MCF-10A breast epithelial cells, pretreatment with 2-APB did
not cause the formation of significant levels of comets (Fig. 5A-b), which indicates minimal levels
of DNA damage. However, in MCF-7 human breast adenocarcinoma cells,
significant levels of comets were observed after 2-APB treatment
(Fig. 5A-b) as compared to
untreated cells (Fig. 5A-a).
Similar results were observed in MDA-MB-231 human breast
adenocarcinoma cells, where 2-APB treatment led to significant
amounts of comets as compared to untreated cells (Fig. 5A-a and A-b). These results
demonstrated that treatment with the TRPM2 inhibitor, 2-APB, led to
increased levels of DNA damage in human breast adenocarcinoma
cells.
To verify that these effects were due to the
inhibition of TRPM2, we performed TRPM2 RNAi silencing in these
cells. RNAi silencing of TRPM2 in noncancerous MCF-10A cells did
not lead to significant numbers of cells with comets (Fig. 5A-d). However, in both MCF-7 and
MDA-MB-231 breast adenocarcinoma cells, RNAi silencing of TRPM2 led
to increased amounts of cells with comets as compared to cells
transfected with negative control scrambled siRNA oligos (Fig. 5A-c and A-d). These results verify
that the increased level of DNA damage in human breast
adenocarcinoma cells is mediated via the inhibition or knockdown of
TRPM2. Since DNA in the tail of the comets represents DNA damage, a
common quantification of comet assay results is the ‘percent DNA in
tail’ (20). Thus, using
CometScore software and analyzing a minimum of 200 cells per
treatment group, we calculated this value for the human breast cell
lines treated with 2-APB or RNAi silencing of TRPM2. The resulting
values demonstrated that increased levels of DNA damage were
quantified in MCF-7 and MDA-MB-231 cells as compared to
noncancerous control cells (Fig.
5B). Further, there appears to be even greater levels of DNA
damage in MCF-7 cells as compared to MDA-MB-231 cells. This is seen
through the ‘percent DNA in tail’ values for 2-APB treated or TRPM2
RNAi-silenced MCF-7 cells, which were ~70% each, as compared to
~40% in MDA-MB-231 cells (Fig.
5B). No significant increases in percent DNA in tail values for
2-APB treated or TRPM2 RNAi-silenced MCF-10A cells were observed,
which demonstrates minimal effects of TRPM2 inhibition or knockdown
on DNA damage levels in noncancerous human mammary epithelial
cells. Taken together, the results show that the pharmacologic
inhibition or RNAi silencing of TRPM2 led to increased levels DNA
damage in human breast adenocarcinoma cells. This indicates that
TRPM2 has a protective role in these lines of human breast
adenocarcinoma cells, where it somehow minimizes damage to genomic
DNA.
Discussion
This study provides insight into the role of TRPM2
in breast cancer cells that potentially provides a foundation for
investigating TRPM2 inhibition to selectively target the DNA of
breast adenocarcinoma cells in the future. The discovery of a
unique role for TRPM2 in breast cancer cells, along with the
ability of TRPM2 inhibition or RNAi silencing to increase DNA
damage and decrease cell proliferation specifically in breast
cancer cells, suggests that the pharmacologic inhibition of TRPM2
may specifically target breast tumors. Further, current studies
that show protective effects in noncancerous cells due to TRPM2
inhibition (7–9), along with our studies here that show
the absence of harmful effects in noncancerous breast cells after
TRPM2 inhibition or RNAi silencing, suggest that pharmacologic
agents that inhibit TRPM2 are expected to produce deleterious
effects only in breast cancer cells. Thus, our study provides the
preliminary results necessary to further study the ability of TRPM2
pharmacologic inhibition to prevent the survival, proliferation,
and/or metastasis of breast adenocarcinoma cells.
Our results are in agreement with a previous study
that utilized prostate cancer cell lines (17). This previous study demonstrated a
nuclear localization of TRPM2 in prostate cancer cells. RNAi
knockdown of TRPM2 decreased the proliferation of these cells.
Further, a nuclear localization and decreased proliferation were
not observed in noncancerous prostate cells. While the authors of
this study presented these effects in prostate cancer cells, we
provide here the first study that demonstrates such effects in
breast adenocarcinoma cells. Further, we expand upon these findings
by identifying a novel protective effect of TRPM2 in breast
adenocarcinoma cells. Loss of TRPM2 function causes significant
increases in DNA damage. This function is distinct from its
function in non cancerous cells. Because this role involves genomic
DNA, it thus appears that the nuclear localization of TRPM2
facilitates its ability to somehow provide protective effects
toward genomic DNA. Future studies will be required to determine
exactly how TRPM2 accomplishes this function. Possibilities include
the facilitation of DNA repair by nuclear TRPM2 or the stimulation
of Ca2+-mediated functions in the nucleus by promoting
nuclear calcium influx.
A recent study demonstrated a protective role for
TRPM2 in cardiac myocytes in response to reperfusion injury
(28). This is in contrast to the
role of TRPM2 in noncancerous cells, where the activation of cation
gating by TRPM2 exacerbates injury in response to oxidative stress.
Further, it is potentially consistent with our studies that show a
protective role for TRPM2 in breast cancer cells. However, this
study demonstrated the ability of TRPM2 to facilitate mitochondrial
bioenergetics and electron transport in cardiac cells. Also, no
nuclear localization of TRPM2 was shown in this study. It is
possible that the effects of TRPM2 inhibition on genomic DNA damage
in breast cancer cells are partially due to effects on the
mitochondria. However, the lack of deleterious effects in
noncancerous breast cells provides evidence against the ability of
TRPM2 inhibition to disrupt mitochondrial function and promote
reactive oxygen species generation in breast cells. It is thus
possible that TRPM2 may have different roles in cardiac cells and
breast cancer cells.
We show here minimal effects of TRPM2 inhibition on
calcium influx in breast cancer cells. This suggests that TRPM2 may
not exclusively function as a cation channel in breast cancer
cells. However, it is possible that TRPM2 may have a lesser role as
an ionophore or ion channel, as TRPM2 was shown to also be
localized to the cytoplasmic fraction in breast cancer cells.
Further, the abundance of TRPM2 protein levels in breast cancer
cells may be significantly decreased as compare to their levels in
neurons and developmental cells, where TRPM2-mediated calcium
influx is robust. Moreover, it is possible that TRPM2 may control
the influx of calcium in the nucleus. Further studies will be
necessary to determine if TRPM2 mediates significant levels of
calcium influx into the cytoplasm or nuclei of breast
adenocarcinoma cells.
Future studies may also include the investigation of
the ADP-ribose pyrophosphatase enzymatic activity of TRPM2 channels
in breast cancer cells. Since this ability is a unique feature of
TRPM2 channels, it may prove to be significant in maintaining the
survival, proliferation, or limiting DNA damage in breast
adenocarcinoma cells. For example, the hydrolysis of the
nucleotide, ADP-ribose, may facilitate prolonged survival in breast
cancer cells via a nucleotide signaling pathway. These studies,
along with the aforementioned calcium influx studies, are
important, because they will provide the necessary information to
determine if preventing TRPM2 cation gating (i.e. inhibiting the
ion channel) or antagonizing the enzymatic activity of TRPM2 is the
key determinant for successfully targeting the DNA of breast
adenocarcinoma cells. Thus, this present study, along with these
future investigations, may provide the foundation necessary for the
initial rational drug design of TRPM2 inhibitors to be used in the
treatment of breast cancer.
In summary, we discovered a novel role for TRPM2 in
breast adenocarcinoma cells. This role appears to be essential for
these cells to survive and proliferate. Thus, this study fits with
a new paradigm for cancer drug development that identifies a
vulnerability that can lead to agents with a much higher
therapeutic index because of greatly reduced general toxicity.
Indeed, our data showing that reducing TRPM2 activity is not toxic
in normal cells, but is toxic in breast cancer cells, supports the
new paradigm leading to new cancer drugs. As there is growing
evidence that specific types of cancer contain specific
vulnerabilities, the presence of nuclear TRPM2 may therefore
represent one such vulnerability.
Acknowledgements
This study was supported in part by NIH/NIAAA grant
K05AA017149 to Dr Gary G. Meadows at Washington State University
(WSU), the James and Diann Robbers Student Research Fund at WSU to
X.F., the Summer Undergraduate Research Fellowship at WSU supported
by ASPET to L.P.P., and NIH/NIGMS Training Grant T32GM08336 for the
NIH Protein Biotechnology Program at Washington State University to
M.M.H. We would like to thank Daniel P. Powell, Steven D. Blake,
and Joy L. Hoffman, all at Ohio Northern University, for their
critical readings of this manuscript.
Abbreviations:
|
2-APB
|
2-aminoethoxydiphenyl borate
|
|
ADP-ribose
|
adenosine diphosphoribose
|
|
MEBM
|
mammary epithelial basal medium
|
|
MEGM
|
mammary epithelial growth medium
|
|
MnSOD
|
manganese superoxide dismutase
|
|
PAR
|
poly(ADP-ribose)
|
|
PARG
|
poly(ADP-ribose) glycohydrolase
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
TRP
|
transient receptor potential
superfamily of cation channels
|
|
TRPM
|
transient receptor potential,
melastatin subfamily
|
|
TS cell
|
trophoblast stem cell
|
References
|
1
|
Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng
T, Zou Q, Shen F and Wang Y: Critical role of TRPC6 channels in
VEGF-mediated angiogenesis. Cancer Lett. 283:43–51. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yee NS, Zhou W and Lee M: Transient
receptor potential channel TRPM8 is over-expressed and required for
cellular proliferation in pancreatic adenocarcinoma. Cancer Lett.
297:49–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu G, Wang X, Yang Z, Cao H, Meng Z, Wang
Y and Chen D: Effects of TRPM8 on the proliferation and
angiogenesis of prostate cancer PC-3 cells in vivo. Oncol Lett.
2:1213–1217. 2011.
|
|
4
|
Bessac BF and Fleig A: TRPM7 channel is
sensitive to osmotic gradients in human kidney cells. J Physiol.
582:1073–1086. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Knowlton WM, Daniels RL, Palkar R, McCoy
DD and McKemy DD: Pharmacological blockade of TRPM8 ion channels
alters cold and cold pain responses in mice. PLoS One.
6:e258942011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perraud AL, Fleig A, Dunn CA, et al:
ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed
by Nudix motif homology. Nature. 411:595–599. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nadler MJ, Hermosura MC, Inabe K, et al:
LTRPC7 is a Mg.ATP-regulated divalent cation channel required for
cell viability. Nature. 411:590–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hara Y, Wakamori M, Ishii M, et al: LTRPC2
Ca2+-permeable channel activated by changes in redox
status confers susceptibility to cell death. Mol Cell. 9:163–173.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang W, Hirschler-Laszkiewicz I, Tong Q,
et al: TRPM2 is an ion channel that modulates hematopoietic cell
death through activation of caspases and PARP cleavage. Am J
Physiol Cell Physiol. 290:C1146–C1159. 2006. View Article : Google Scholar
|
|
10
|
Blenn C, Wyrsch P, Bader J, Bollhalder M
and Althaus FR: Poly(ADP-ribose)glycohydrolase is an upstream
regulator of Ca2+ fluxes in oxidative cell death. Cell
Mol Life Sci. 68:1455–1466. 2011. View Article : Google Scholar :
|
|
11
|
Cook NL, Vink R, Helps SC, Manavis J and
van den Heuvel C: Transient receptor potential melastatin 2
expression is increased following experimental traumatic brain
injury in rats. J Mol Neurosci. 42:192–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lange I, Yamamoto S, Partida-Sanchez S,
Mori Y, Fleig A and Penner R: TRPM2 functions as a lysosomal
Ca2+-release channel in beta cells. Sci Signal.
2:ra232009.
|
|
13
|
Ishii M, Hagiwara T, Mori Y and Shimizu S:
Involvement of TRPM2 and L-type Ca2+ channels in
Ca2+ entry and cell death induced by hydrogen peroxide
in rat β-cell line RIN-5F. J Toxicol Sci. 39:199–209. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kheradpezhouh E, Ma L, Morphett A, Barritt
GJ and Rychkov GY: TRPM2 channels mediate acetaminophen-induced
liver damage. Proc Natl Acad Sci USA. 111:3176–3181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Cotrim A, Teos L, Zheng C, Swaim W,
Mitchell J, Mori Y and Ambudkar I: Loss of TRPM2 function protects
against irradiation-induced salivary gland dysfunction. Nat Commun.
4:15152013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Orfanelli U, Wenke AK, Doglioni C, Russo
V, Bosserhoff AK and Lavorgna G: Identification of novel sense and
antisense transcription at the TRPM2 locus in cancer. Cell Res.
18:1128–1140. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zeng X, Sikka SC, Huang L, Sun C, Xu C,
Jia D, Abdel-Mageed AB, Pottle JE, Taylor JT and Li M: Novel role
for the transient receptor potential channel TRPM2 in prostate
cancer cell proliferation. Prostate Cancer Prostatic Dis.
13:195–201. 2010. View Article : Google Scholar :
|
|
18
|
Koh DW, Lawler AM, Poitras MF, Sasaki M,
Wattler S, Nehls MC, Stöger T, Poirier GG, Dawson VL and Dawson TM:
Failure to degrade poly(ADP-ribose) causes increased sensitivity to
cytotoxicity and early embryonic lethality. Proc Natl Acad Sci USA.
101:17699–17704. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu T, Biddle D, Hanks AN, Brouha B, Yan
H, Lee RM, Leachman SA and Grossman D: Activation of dual apoptotic
pathways in human melanocytes and protection by survivin. J Invest
Dermatol. 126:2247–2256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hartmann A, Agurell E, Beevers C, et al:
4th International Comet Assay Workshop: Recommendations for
conducting the in vivo alkaline Comet assay. 4th International
Comet Assay Workshop. Mutagenesis. 18:45–51. 2003. View Article : Google Scholar
|
|
21
|
Zhou Y, Feng X and Koh DW: Enhanced DNA
accessibility and increased DNA damage induced by the absence of
poly(ADP-ribose) hydrolysis. Biochemistry. 49:7360–7366. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang W, Chu X, Tong Q, Cheung JY, Conrad
K, Masker K and Miller BA: A novel TRPM2 isoform inhibits calcium
influx and susceptibility to cell death. J Biol Chem.
278:16222–16229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun L, Yau HY, Wong WY, Li RA, Huang Y and
Yao X: Role of TRPM2 in H(2)O(2)-induced cell apoptosis in
endothelial cells. PLoS One. 7:e431862012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Togashi K, Inada H and Tominaga M:
Inhibition of the transient receptor potential cation channel TRPM2
by 2-aminoethoxy-diphenyl borate (2-APB). Br J Pharmacol.
153:1324–1330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koh DW, Patel CN, Ramsinghani S, Slama JT,
Oliveira MA and Jacobson MK: Identification of an inhibitor binding
site of poly(ADP-ribose) glycohydrolase. Biochemistry.
42:4855–4863. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Buelow B, Song Y and Scharenberg AM: The
Poly(ADP-ribose) polymerase PARP-1 is required for oxidative
stress-induced TRPM2 activation in lymphocytes. J Biol Chem.
283:24571–24583. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Singh NP, McCoy MT, Tice RR and Schneider
EL: A simple technique for quantitation of low levels of DNA damage
in individual cells. Exp Cell Res. 175:184–191. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller BA, Hoffman NE, Merali S, et al:
TRPM2 channels protect against cardiac ischemia-reperfusion injury:
Role of mitochondria. J Biol Chem. 289:7615–7629. 2014. View Article : Google Scholar : PubMed/NCBI
|