Introduction
Breast cancer is the most frequent malignancy in
women and the second leading cause of cancer death among women in
the United States (1,2). A family history of breast cancer is
one of the most important risk factors for the disease (3). In addition to the two major breast
cancer susceptibility genes, BRCA1 and BRCA2, several
other genes associated with breast cancer predisposition have been
identified, including ATM, CHEK2, PALB2, RAD51C and
BRIP1. Many of these genes are associated with BRCA1
and BRCA2 in the DNA damage response (DDR) pathway (4).
Germline mutations of BRCA1 predispose female
carriers to breast and ovarian cancers (5). Although germline mutations in
BRCA1 account for only 5% of breast cancer cases, silencing
of BRCA1 by promoter hypermethylation and other mechanisms
may contribute to ≤30% of sporadic breast cancers (6,7).
BRCA1-associated breast cancers usually contain p53
mutations and often exhibit a triple-negative phenotype (8,9).
BRCA1 and BRCA2 have roles in homologous
recombination (HR) for DNA repair (10,11).
When the remaining wild-type allele is lost in a tumor precursor
cell, this repair mechanism does not work, resulting in genomic
instability that is sufficient to enable tumor development
(12,13). Most cancers have defects in some
part of the DDR pathway. This provides an opportunity for
therapeutic intervention as genotoxic therapies cause significant
DNA damage, which is repairable in healthy cells but not in
DDR-defective cancer cells.
PARP family of proteins (PARP1 and PARP2), are
involved in a number of critical cellular processes, including DNA
damage repair and programmed cell death (14). When activated by DNA damage, these
proteins recruit other proteins that do the actual work of
repairing DNA. Inhibition of PARP is a recently developed strategy
for cancer therapy that exploits DDR defects in cancer cells
(14). PARP is responsible for the
sensing and repair of single-strand DNA breaks via base excision
repair (15). When a replication
fork encounters a single-strand break, the result is a
double-strand break. In wild-type cells, these double-strand breaks
are often repaired via homologous recombination (16). Cells deficient with BRCA1 and BRCA2
are unable to repair these double-strand breaks efficiently and
therefore undergo cell death (17,18).
Thus, PARP inhibitors exhibit efficacy in breast cancers with
inherited mutations in BRCA1 or BRCA2 (19). PARP inhibitors, Olaparib (AZD2281),
Veliparib (ABT-888), and Iniparib (BSI-201) have been shown to be
promising anti-cancer agents for breast and ovarian cancer and
being tested in clinical trials. Recently, the orally active PARP
inhibitor AZD2281 was evaluated as a single-agent therapy in humans
and showed clinical antitumor activity in BRCA-associated cancers
(19,20). However, the mechanism of action of
PARP inhibitors alone in cancer cells is not fully understood.
In this study, we investigated the effects of PARP
inhibitors in BRCA1 or BRCA2 mutant breast cancer
cell lines and in wild-type BRCA cell lines with and without
BRCA1 allelic loss. We provide evidence that the PARP inhibitor
AZD2281 inhibits the growth of breast cancer cells with BRCA1
allelic loss lacking mutation in BRCA1. These results might
lead the way to new approaches for treating a broad spectrum of
breast cancer subtypes. We also demonstrated that the PARP
inhibitor AZD2281 induces autophagy in BRCA mutated breast cancer
cells as well as breast cancer cells with BRCA1 allelic loss
lacking mutation in BRCA1. Our results also indicate
importance of selection of patients who would benefit from PARP
inhibitor therapy and molecular subclassifications of BRCA-related
breast cancers.
Materials and methods
Cell lines, culture conditions, and
reagents
We studied 14 human breast cancer cell lines: 3
BRCA1 mutant lines with BRCA1 allelic loss (HCC-1947,
MDA-MB-436, and SUM-149PT), 1 BRCA2 mutant line with
BRCA2 allelic loss (HCC-1428), 9 BRCA wild-type lines with
BRCA1 allelic loss (MCF-7, ZR75, MDA-MB-361, BT-474, SKBR3,
MDA-MB-231, BT-549, MDA-MB-468 and BT-20), and 1 BRCA wild-type
line without BRCA1 allelic loss (T47D). T47D, MCF-7, ZR75,
MDA-MB-361, BT-474, SKBR3, MDA-MB-231, BT-549, MDA-MB-468, and
BT-20 cells were cultured at 37°C in DMEM supplemented with 10% FBS
in a humid incubator with 5% CO2. SUM-149PT cells were
cultured in Ham’s F-12 supplemented with 5% FBS, insulin, and
hydrocortisone. The PARP inhibitors veliparib (ABT-888), olaparib
(AZD2281), and iniparib (BSI-201) were purchased from Selleck
Chemicals (Houston TX, USA).
WST-1 assay
Cell viability was assayed by applying the cell
proliferation reagent WST-1 (Roche Applied Science). First, a
suspension of 4,000 cells per 90 μl was seeded into each well of a
96-well plate and cultured overnight. Then, the necessary amount of
PARP inhibitor was added to the individual wells. After 3 days of
PARP inhibitor treatment, 10 μl of the ready-to-use WST-1 reagent
was added directly into the medium, the plates were incubated at
37°C for 30 min, and absorbance was measured on a plate reader at
450 nm. All experiments were done in triplicate. Cell viability was
calculated as the percentage of cells killed by the treatment as
measured by the difference in absorbance between treated and
untreated wells.
Cell transfections
Lentiviral particles expressing BRCA1, BRCA2, ATG5,
or control shRNA were purchased from Sigma. MDA-MB-231, BT-20, and
HCC-1428 cells were transfected at a multiplicity of infection of
5. Five days after transfection, cells were treated with 5 μg/ml of
puromycin concentration to select cells stably expressing shRNA.
Lentiviral vector expressing mitochondrial yellow fluorescent
protein (mYFP) was purchased from Biogenova. HCC-1428 cells were
transfected at a multiplicity of infection of 5.
Western blot analysis
After treatment, the cells were trypsinized and
collected by centrifugation, and whole-cell lysates were obtained
by using a cell lysis buffer. Total protein concentration was
determined by using a detergent-compatible protein assay kit
(Bio-Rad Laboratories). Aliquots containing 30 μg of total protein
from each sample were subjected to SDS-PAGE with a 12% gradient and
electrotransferred to nitrocellulose membranes. The membranes were
blocked with 5% dry milk in TBS-Tween-20 and probed with primary
antibodies against BRCA1 and BRCA2 (Cell Signaling Technology) and
LC3 (Sigma). The antibodies were diluted in TBS-Tween-20 containing
2.5% dry milk and incubated at 4°C overnight. After the membranes
were washed with TBS-Tween-20, they were incubated with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody
(Amersham Life Sciences). Mouse anti-β-actin and donkey anti-mouse
secondary antibodies (Sigma) were used to monitor β-actin
expression to ensure equal loading of proteins. Chemiluminescence
was detected with ChemiGlow detection reagents (Alpha Innotech).
The blots were visualized with a FluorChem 8900 imager and
quantified with densitometer software (Alpha Innotech).
Evaluation of acidic vesicular
organelles
To detect and quantify acidic vesicular organelles,
cells were stained with acridine orange as described previously
(21). The number of acridine
orange-positive cells was determined by fluorescence-activated cell
sorting (FACS) analysis.
Transmission electron microscopy
Cells were grown on 6-well plates, treated with
AZD2281, ATG5 shRNA, or control shRNA, fixed for 2 h with 2.5%
glutaraldehyde in 0.1 mol/l cacodylate buffer (pH 7.4), and
postfixed in 1% OsO4 in the same buffer and then
subjected to the electron microscopic analysis as described
previously. Representative areas were chosen for ultrathin
sectioning and viewed with a Hitachi 7600 electron microscope
(Japan).
Flow cytometry analysis of apoptosis
Cells were collected and double-stained with Annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide using an
Annexin V-FITC apoptosis detection kit (BD Pharmingen) and
evaluated with a flow cytometer.
Results
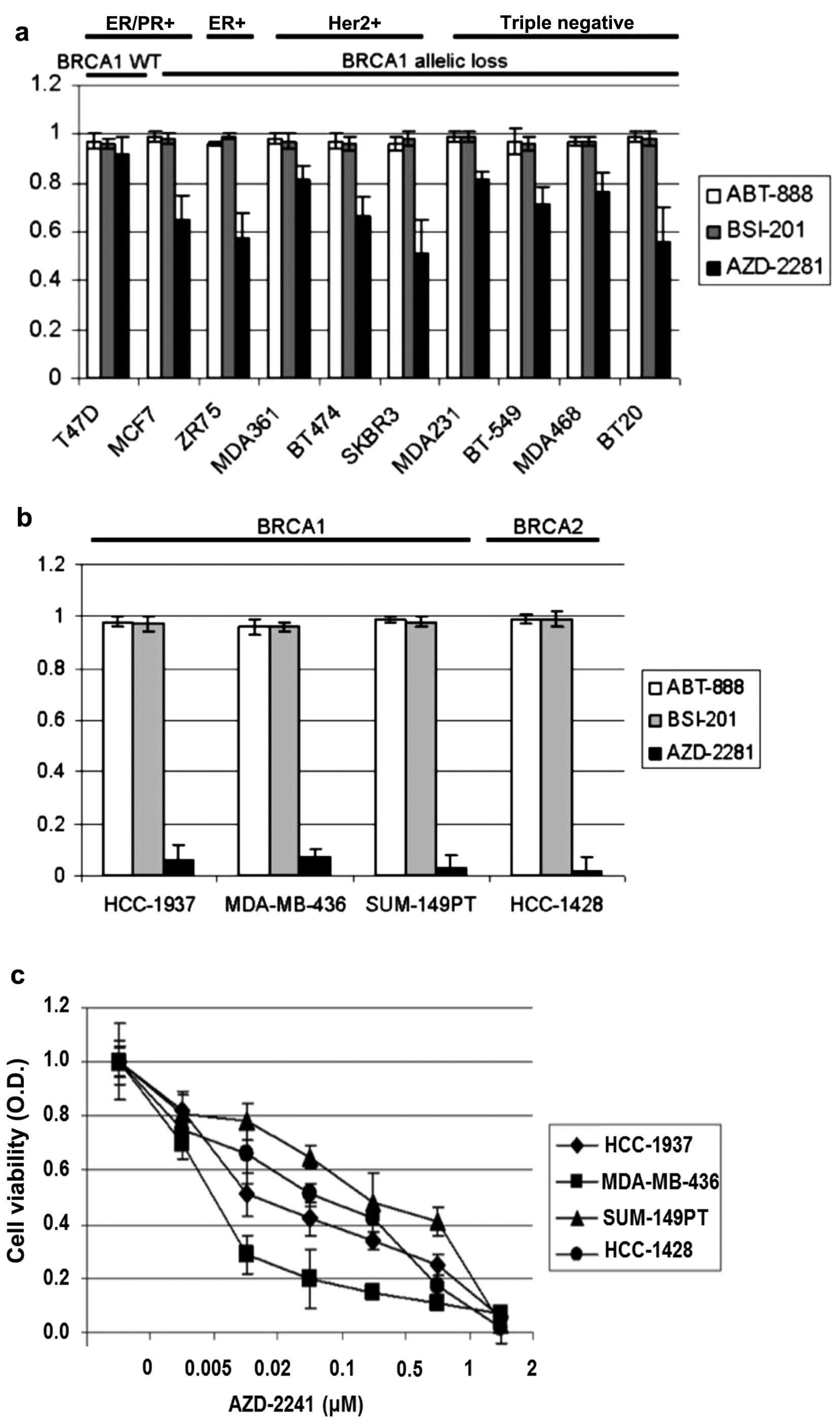
AZD2281 inhibits cell survival in BRCA1
or BRCA2 mutant breast cancer cell lines
According to the literature, 5 (12%) of 41 breast
cancer cell lines have BRCA mutations and 28 (68%) of the 41 cell
lines have BRCA1 allelic loss. To investigate the effects of
PARP inhibitors in BRCA wild-type breast cancer cell lines with
BRCA allelic loss we treated BRCA wild-type ER/PR+,
ER+, HER2+, and triple-negative cell lines
with 3 different PARP inhibitors, ABT-888, BSI-201, and AZD2281,
for 4 days. Growth rates were measured with the WST-1 assay.
Whereas AZD2281 induced an average growth inhibition of 33% in BRCA
wild-type cell lines at 2 μM, ABT-888 and BSI-201 did not induce
growth inhibition in the same cell lines at 2 μM concentration. The
growth inhibition effect of AZD2281 was significantly higher in the
BRCA wild-type cell lines with BRCA1 allelic loss than in
the BRCA wild-type cell line without BRCA1 allelic loss
(Fig. 1a). We also used the same
PARP inhibitors at the same concentration (2 μM) in the
BRCA1 mutant (HCC-1937, MDA-MB-436, and SUM-149PT) and
BRCA2 mutant (HCC-1428) cell lines. AZD2281 at 2 μM
significantly inhibits cell survival in all 4 cell lines, whereas
ABT-888 and BSI-201 did not induce cell death at 2 μM (Fig. 1b). We also evaluated the effects of
AZD2281 at lower concentrations in the BRCA mutant breast cancer
cell lines, where it had a significant dose-dependent growth
inhibition effect (Fig. 1c).
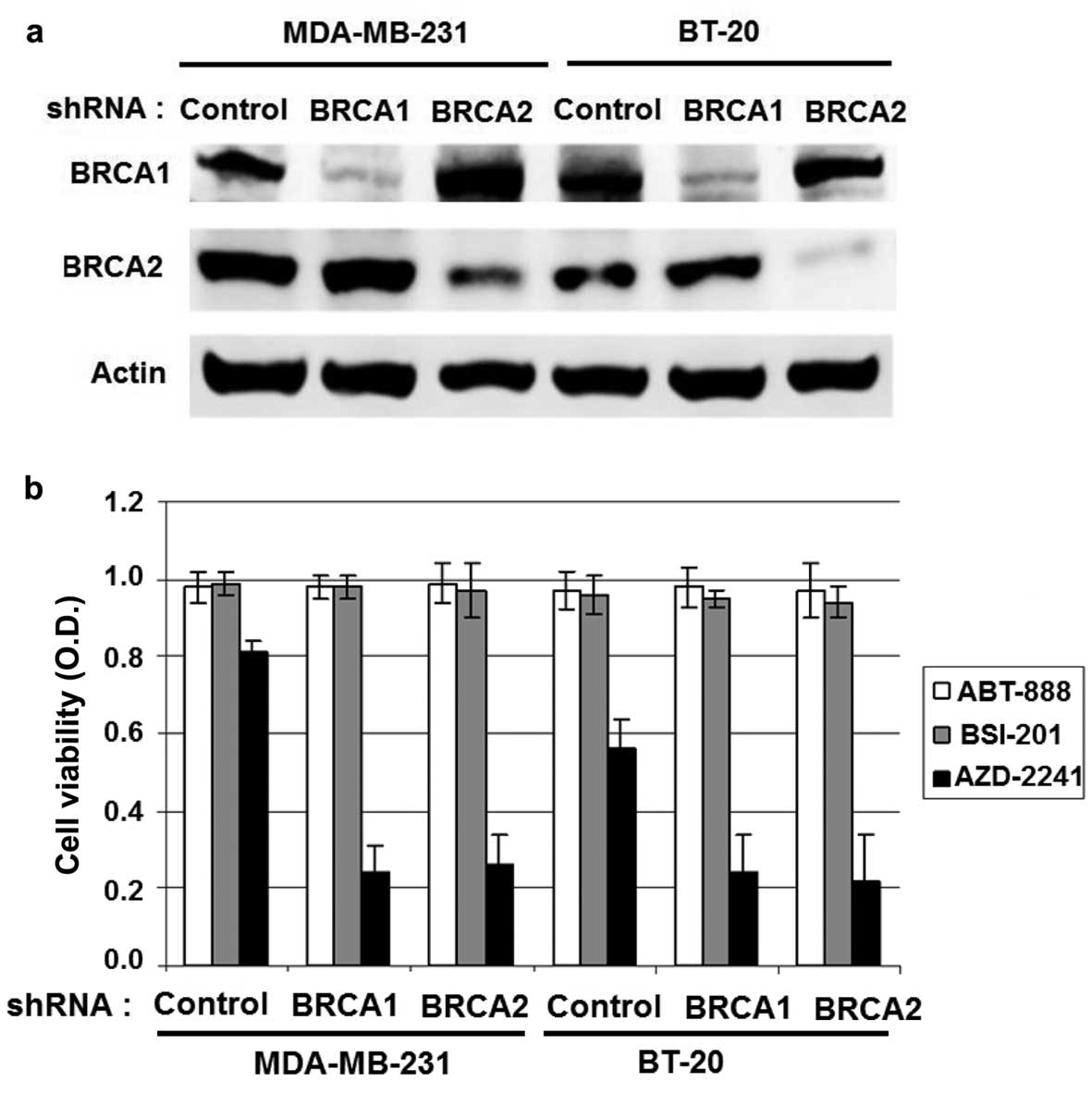
BRCA1 or BRCA2 downregulation in BRCA
wild-type breast cancer cell lines induces growth inhibition in
response to AZD2281 treatment
To determine the effect of BRCA1 or BRCA2 in
response to AZD2281 treatment, the BRCA wild-type MDA-MB-231 and
BT-20 cells were stably transfected with BRCA1, BRCA2, or control
lentiviral shRNA. BRCA1 or BRCA2 downregulation was demonstrated by
western blot analysis (Fig. 2a).
The 3 different PARP inhibitors used as single-agent treatments and
growth rates were measured with the WST-1 assay. AZD2281 induced
significantly superior growth inhibition compared with other PARP
inhibitors, such as ABT-888 and BSI-201 in BRCA1- or
BRCA2-knockdown cells than in control cells, indicating that the
growth inhibition effect of AZD2281 is dependent on BRCA deficiency
(Fig. 2b).
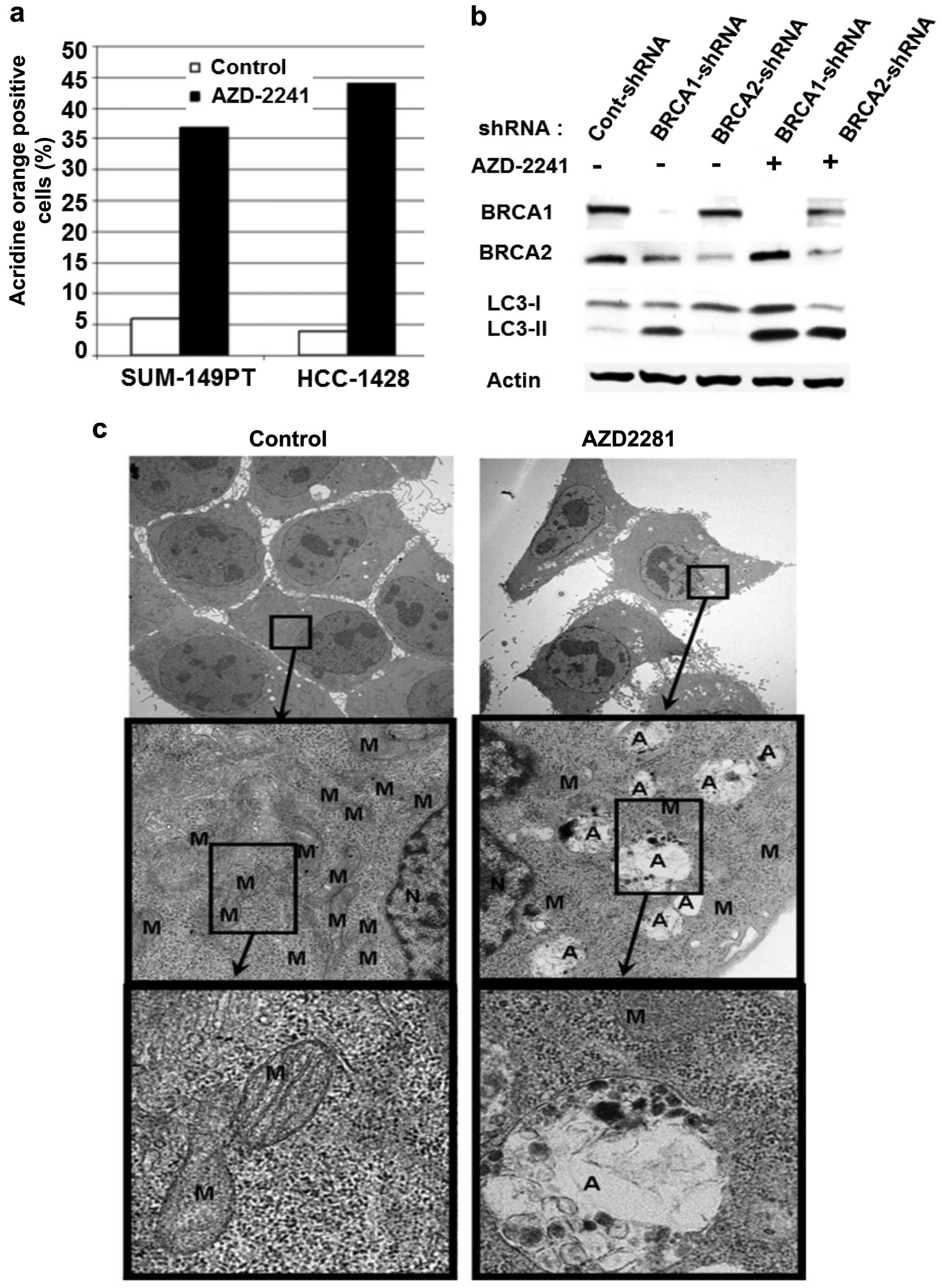
AZD2281 induces autophagy in BRCA1 or
BRCA2 mutant breast cancer cell lines
Autophagy is lysosomal degradation pathway
characterized by an increase in the number of autophagosomes that
surround organelles such as mitochondria, Golgi complexes,
polyribosomes, and the endoplasmic reticulum. Subsequently,
autophagosomes merge with lysosomes and digest damaged organelles
into amino acids to provide a new supply under stressful conditions
to protect the cells (22–24). Although activation of autophagy is
aimed at overcoming stressful situations, autophagy induction may
lead to cell death (25). To
determine effects of the most potent PARP inhibitor we investigated
whether AZD2281 induces autophagy in BRCA mutant breast cancer cell
lines. To this end we treated BRCA1 mutant (SUM-149PT) and
BRCA2 mutant (HCC-1428) breast cancer cells with 2 μM
AZD2281 for 1 day and stained them with acridine orange. Acridine
orange positive cells were counted using flow cytometry. AZD2281
induced significant autophagy (37 and 44%) in BRCA1 mutant
SUM-149PT and BRCA2 mutant HCC-1428 breast cancer breast
cancers, respectively, in 24 of treatment (Fig. 3a). We observed the same phenomenon
by AZD2281 in BRCA wild-type breast cancer cell line MDA-MB-231
with BRCA1 or BRCA2 downregulation. The knockdown of BRCA1 by
lenti-based stable shRNA in BRCA wild-type breast cancer cell line
MDA-MB-231 demostrated induction of autophagy as indicated by the
expression of LC3-II, an autophagy marker (Fig. 3b). AZD2281 treatment further
enhanced LC3-II expression in BRCA1- or BRCA2-knockdown cells
(Fig. 3b).
To further demonstrate the induction of autophagy we
also investigated ultrastructure by transmission electron
microscopy (TEM) before and after AZD2281 treatment. TEM images
clearly demonstrated that AZD2281 induces autophagy, which results
in mitochondrial degradation. AZD2281-treated cells had fewer
mitochondria and more autophagosomes compared with untreated cells
(Fig. 3c).
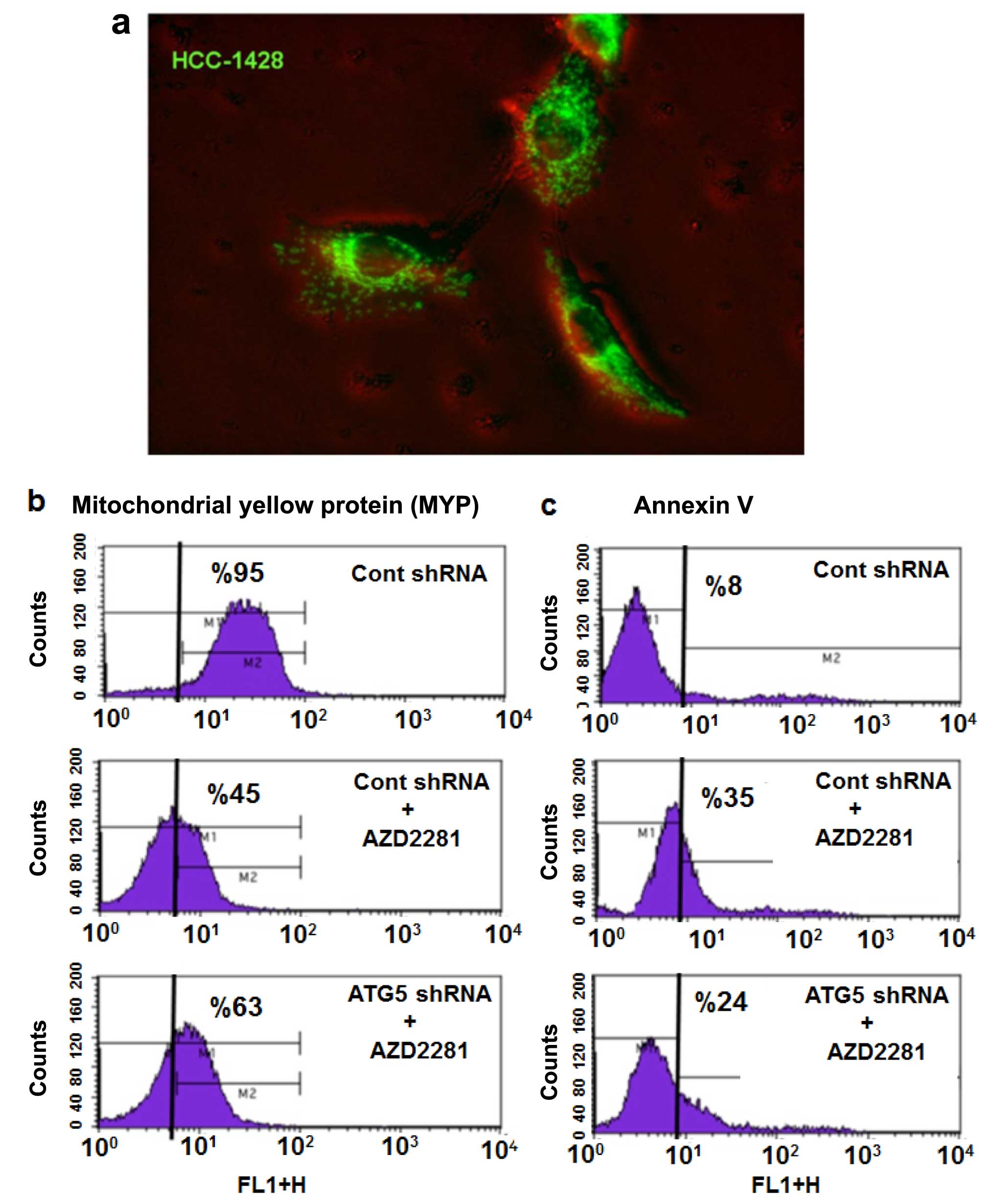
Inhibition of autophagy results in
partial inhibition of AZD2281-induced apoptosis
To investigate the roles of autophagy and
mitochondrial degradation under AZD2281 treatment, we stably
transfected HCC-1428 cells with mYFP using lentiviral vector.
Fluorescence microscope images clearly demonstrated the presence of
mYFP in the mitochondrial compartment of HCC-1428-mYFP cells
(Fig. 4a). HCC-1428-mYFP was
treated with AZD2281, and mitochondrial fluorescein was measured by
flow cytometry; untreated cells were used as a control. AZD2281
induced significant mitochondrial degradation (~45%) (Fig. 4b), which was also shown in the TEM
images. Next, we inhibited autophagy by knocking down the key
autophagosome structural protein ATG5 using lentiviral shRNA vector
in HCC-1428-mYFP cells. Mitochondrial degradation was markedly
rescued in ATG5-knockdown HCC-1428.mYFP-shATG5 cells compared with
HCC-1428-mYFP-sh-control cells under AZD2281 treatment (Fig. 4b). Inhibition of autophagy by
knocking down ATG5 also partially inhibited AZD2281-induced
apoptosis (Fig. 4c), suggesting
that autophagy contributes to AZD2281-induced cell death in BRCA
mutated breast cancer cells.
Discussion
In this study, we show for the first time that a
PARP inhibitor as a single agent induces significant
autophagy/mitophagy in BRCA mutant cell lines. In addition,
we demonstrated that AZD2281 induces growth inhibition in BRCA
wild-type breast cancer cell lines with BRCA1 allelic loss,
indicating that breast cancer patients with BRCA1 allelic
loss may benefit from PARP inhibitors.
Previously, AZD2281 was evaluated in a genetically
engineered mouse model of BRCA1 breast cancer (26). Treatment of tumor-bearing mice with
AZD2281 inhibits tumor growth and prolonged survival. Combination
treatment with AZD2281 plus cisplatin or carboplatin increased
recurrence-free survival and overall survival (26). AZD2281 has also been used as a
single agent in clinical trials in breast and ovarian cancer
patients with BRCA mutations (19,20).
In this study, we evaluated the effects of 3 different PARP
inhibitors, ABT-888, BSI-201 and AZD228, in BRCA mutant breast
cancer cell lines as single agents without DNA damaging agents;
such a study has not been performed previously. BRCA mutations in
breast cancer cell lines were not well described until 2006, when
Elstrodt et al, reported a detailed BRCA1 mutation
analysis of 41 breast cancer cell lines (5). Before the report was published, only
one of the 41 cell lines was known to have BRCA1 mutation.
Elstrodt et al, identified BRCA1 mutations in three
cell lines that had not been described as BRCA1 mutant
before. They also found that 28 (68%) of the 41 cell lines had
BRCA1 allelic loss (5). On
the basis of these results, we evaluated PARP inhibitors as
single-agent therapy in 14 breast cancer cell lines: 4 BRCA mutant
lines with BRCA1 allelic loss, 9 BRCA wild-type lines with
BRCA1 allelic loss, and 1 BRCA wild-type line without
BRCA1 allelic loss. Our data clearly demonstrated that BRCA
mutant breast cancer cell lines with BRCA allelic loss were highly
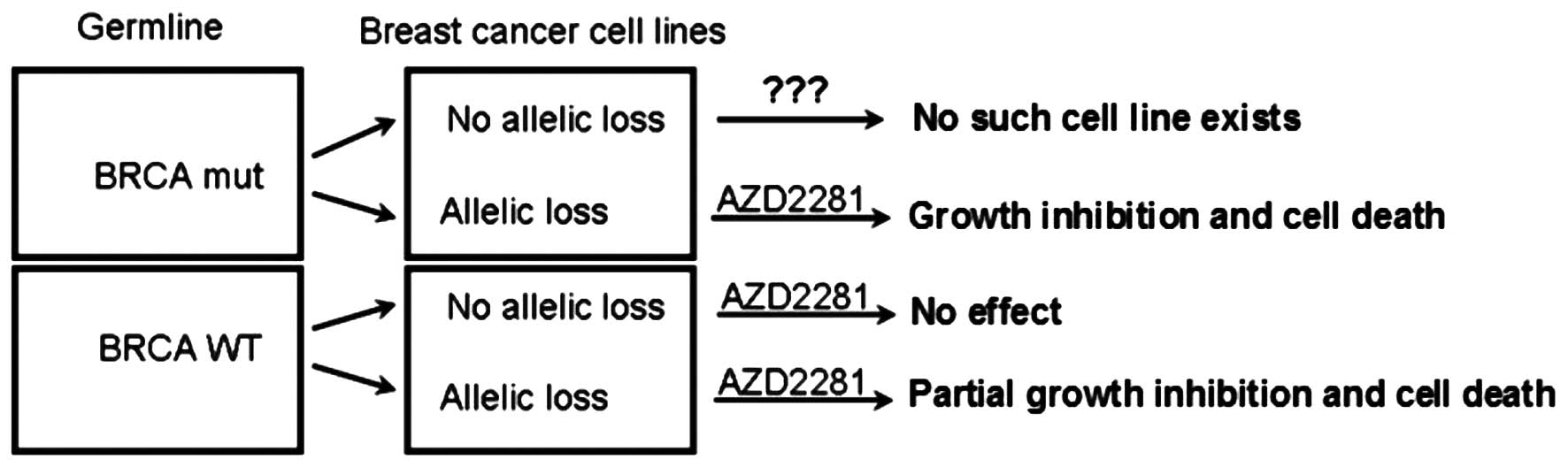
sensitive to AZD2281 as monotherapy (Fig. 5). Unfortunately, no cell line
exists with BRCA mutation and without BRCA allelic loss; such cells
may be resistant to PARP inhibitors because of a functional BRCA
allele. When we investigated whether BRCA allelic loss results in
sensitivity to PARP inhibitors in BRCA wild-type cell lines, we
found significant growth inhibition, but not cell death, such as
that seen in BRCA mutant cell lines.
Autophagy is lysosomal degradation pathway that is
induced as a protective and prosurvival pathway against nuclear DNA
damage and metabolic and therapeutic stress, if excessive this
process can also lead to cell death in breast and other cancers
(22–25,29,30).
To the best of our knowledge, our study is the first to show that
AZD2281 induces complete cell death (95–99%) and autophagy, which
targets mitochondria. Our findings indicate that autophagy is
involved in cell death mechanism as AZD2281-induced apoptosis was
reversed by genetic inhibition of autophagy. Here, we speculate
that AZD2281 not only induces nuclear DNA damage but may also
induce elimination of mitochondria by autophagy by a process called
mitophagy and may contribute to the cell death process (28). Although the clinical implications
of this finding are not yet known, we speculate that autophagy
could serve as a predictive marker for PARP inhibition therapy.
Furthermore, our study points out that BRCA wild-type cells with
BRCA allelic loss may be more sensitive to PARP inhibitors than are
those without BRCA allelic loss. This observation may potentially
explain why differential response rates are being observed in
clinical trials, even in homogeneous cohorts of germline BRCA
mutation carriers. For example, the reported response rate is ~40%
for AZD2281 and ~37.5% for ABT-888 (in combination with
temozolamide), indicating that almost half of the patients with
germline BRCA mutations are not responsive to these agents
(20,27). Therefore, the results of our
current study might shed further light on the molecular
subclassifications of BRCA-related breast cancers and ultimately
lead to a better characterization of the molecular tumor type that
would benefit from PARP inhibitors.
References
|
1
|
Baynes RD, Dansey RD, Klein JL, Hamm C,
Campbell M, Abella E and Peters WP: High-dose chemotherapy and
hematopoietic stem cell transplantation for breast cancer: Past or
future? Semin Oncol. 28:377–388. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Collaborative Group on Hormonal Factors in
Breast Cancer. Familial breast cancer: Collaborative reanalysis of
individual data from 52 epidemiological studies including 58,209
women with breast cancer and 101,986 women without the disease.
Lancet. 358:1389–1399. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ripperger T, Gadzicki D, Meindl A and
Schlegelberger B: Breast cancer susceptibility: Current knowledge
and implications for genetic counselling. Eur J Hum Genet.
17:722–731. 2009. View Article : Google Scholar
|
|
5
|
Elstrodt F, Hollestelle A, Nagel JH, Gorin
M, Wasielewski M, van den Ouweland A, Merajver SD, Ethier SP and
Schutte M: BRCA1 mutation analysis of 41 human breast cancer cell
lines reveals three new deleterious mutants. Cancer Res. 66:41–45.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Birgisdottir V, Stefansson OA,
Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG and Eyfjord JE:
Epigenetic silencing and deletion of the BRCA1 gene in sporadic
breast cancer. Breast Cancer Res. 8:R382006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson CA, Ramos L, Villaseñor MR, Anders
KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D,
et al: Localization of human BRCA1 and its loss in high-grade,
non-inherited breast carcinomas. Nat Genet. 21:236–240. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lakhani SR, Van De Vijver MJ, Jacquemier
J, Anderson TJ, Osin PP, McGuffog L and Easton DF: The pathology of
familial breast cancer: Predictive value of immunohistochemical
markers estrogen receptor, progesterone receptor, HER-2, and p53 in
patients with mutations in BRCA1 and BRCA2. J Clin Oncol.
20:2310–2318. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Palacios J, Honrado E, Osorio A, Cazorla
A, Sarrió D, Barroso A, Rodríguez S, Cigudosa JC, Diez O, Alonso C,
et al: Phenotypic characterization of BRCA1 and BRCA2 tumors based
in a tissue microarray study with 37 immunohistochemical markers.
Breast Cancer Res Treat. 90:5–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moynahan ME, Chiu JW, Koller BH and Jasin
M: Brca1 controls homology-directed DNA repair. Mol Cell.
4:511–518. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tutt A, Bertwistle D, Valentine J, Gabriel
A, Swift S, Ross G, Griffin C, Thacker J and Ashworth A: Mutation
in Brca2 stimulates error-prone homology-directed repair of DNA
double-strand breaks occurring between repeated sequences. EMBO J.
20:4704–4716. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tutt A and Ashworth A: The relationship
between the roles of BRCA genes in DNA repair and cancer
predisposition. Trends Mol Med. 8:571–576. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huen MS and Chen J: Assembly of checkpoint
and repair machineries at DNA damage sites. Trends Biochem Sci.
35:101–108. 2010. View Article : Google Scholar
|
|
14
|
Ashworth A: A synthetic lethal therapeutic
approach: Poly(ADP) ribose polymerase inhibitors for the treatment
of cancers deficient in DNA double-strand break repair. J Clin
Oncol. 26:3785–3790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herceg Z and Wang ZQ: Functions of
poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity
and cell death. Mutat Res. 477:97–110. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wiltshire TD, Lovejoy CA, Wang T, Xia F,
O’Connor MJ and Cortez D: Sensitivity to poly(ADP-ribose)
polymerase (PARP) inhibition identifies ubiquitin-specific
peptidase 11 (USP11) as a regulator of DNA double-strand break
repair. J Biol Chem. 285:14565–14571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bryant HE, Schultz N, Thomas HD, Parker
KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ and Helleday T:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farmer H, McCabe N, Lord CJ, Tutt AN,
Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I,
Knights C, et al: Targeting the DNA repair defect in BRCA mutant
cells as a therapeutic strategy. Nature. 434:917–921. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fong PC, Boss DS, Yap TA, Tutt A, Wu P,
Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et
al: Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA
mutation carriers. N Engl J Med. 361:123–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tutt A, Robson M, Garber JE, Domchek SM,
Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler
RK, et al: Oral poly(ADP-ribose) polymerase inhibitor olaparib in
patients with BRCA1 or BRCA2 mutations and advanced breast cancer:
A proof-of-concept trial. Lancet. 376:235–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Murai J, Huang SY, Das BB, Renaud A, Zhang
Y, Doroshow JH, Ji J, Takeda S and Pommier Y: Differential trapping
of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res.
72:5588–5599. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
23
|
Dalby KN, Tekedereli I, Lopez-Berestein G
and Ozpolat B: Targeting the prodeath and prosurvival functions of
autophagy as novel therapeutic strategies in cancer. Autophagy.
6:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Akar U, Ozpolat B, Mehta K, Fok J, Kondo Y
and Lopez-Berestein G: Tissue transglutaminase inhibits autophagy
in pancreatic cancer cells. Mol Cancer Res. 5:241–249. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rottenberg S, Jaspers JE, Kersbergen A,
van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M,
Zevenhoven J, Lau A, et al: High sensitivity of BRCA1-deficient
mammary tumors to the PARP inhibitor AZD2281 alone and in
combination with platinum drugs. Proc Natl Acad Sci USA.
105:17079–17084. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: Disease entity or title of
convenience? Nat Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ashrafi G and Schwarz TL: The pathways of
mitophagy for quality control and clearance of mitochondria. Cell
Death Differ. 20:31–42. 2013. View Article : Google Scholar
|
|
29
|
Akar U, Chaves-Reyez A, Barria M, Tari A,
Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G and Ozpolat
B: Silencing of Bcl-2 expression by small interfering RNA induces
autophagic cell death in MCF-7 breast cancer cells. Autophagy.
4:669–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tekedereli I, Alpay SN, Akar U, Yuca E,
Ayugo-Rodriguez C, Han HD, Sood AK, Lopez-Berestein G and Ozpolat
B: Therapeutic silencing of Bcl-2 by systemically
administered-siRNA nanotherapeutics inhibits tumor growth by
autophagy and apoptosis and enhances the efficacy of chemotherapy
in orthotopic xenograft models of ER (−) and ER (+) breast cancer.
Mol Ther Nucleic Acids. 2:e1212013. View Article : Google Scholar
|