Introduction
Lung cancer is the leading cause of cancer-related
mortality in the developed world, accounting for 26–29% of all
cancer deaths (1). Non-small cell
lung cancer (NSCLC) accounts for 80% of all lung cancer diagnosis
and the majority of these patients are diagnosed at an advanced
stage. Therefore, the prognosis of locally advanced NSCLC is poor
with a 5-year survival rate of <10% (2). Despite surgically complete resection,
lung cancer frequently relapses with metastasis (3). Understanding the mechanism of cancer
progression and metastasis could provide an opportunity to identify
novel targets for therapeutic intervention.
The metastatic cascade represents a multi-step
process that includes migration, invasion, adhesion, proliferation,
angiogenesis and lymphangiogenesis. Invasion and migration are
initial steps in the metastatic cascade (4). During cancer progression, some tumor
cells show changes in their plasticity by morphologic and
phenotypic conversion, as an expression of mesenchymal markers and
loss of epithelial markers, collectively referred to as
epithelial-mesenchymal transition (EMT), which is measured as
expression of mesenchymal markers and loss of epithelial markers
(5). EMT is increasingly
recognized as a critical phenomenon in lung cancer progression and
metastasis (6). EMT has been
considered as a general biological switch rendering NSCLC sensitive
or insensitive to EGFR inhibition. Increased expression of
E-cadherin has been associated with clinical activity of EGFR
inhibitors in NSCLC patients. Recent studies showed EMT was
observed in EGFR mutant lung cancers with acquired resistance to
EGFR TKIs (7).
We investigated the microRNA (miRNA) profiles of
mesenchymal-like lung cancer cell lines to evaluate the
relationship between EMT and miRNA in NSCLC. miR-146a is
downregulated in mesenchymal-like lung cancer cell lines. We
previously reported a significant association between the
pre-miR-146a rs2910164C>G single nucleotide polymorphism (SNP)
and lung cancer risk. The effect of this SNP on lung cancer risk
was more pronounced in never smokers (8). Insulin receptor substrate 2 (IRS2) is
a predicted target of miR-146a using a combined approach involving
computational prediction and a whole genome microarray experiment.
IRS adaptor proteins link signaling from upstream activators to
multiple downstream effectors which modulate normal growth,
metabolism, survival and differentiation (9).
In the present study, we determined the differential
expression of miR-146a in lung cancer tissues. We hypothesized that
miR-146a plays a role in EMT of lung cancer by directly targeting
IRS2. To test this hypothesis, we performed luciferase, cell
migration and invasion assays.
Materials and methods
Cell culture and growth conditions
Lung cancer cells (H226, H358, H460, HCC95, HCC827
and H1299) were maintained in RPMI-1640 medium (Gibco BRL,
Rockville, MD, USA) with 10% fetal bovine serum and antibiotics
(100 U/ml penicillin and 100 mg/ml streptomycin).
Patients and tissues samples
Tumor and corresponding normal lung tissue specimens
were obtained from 78 Korean patients with NSCLC who underwent
curative resection at the Kyungpook National University Hospital.
None of the patients had received chemotherapy or radiotherapy
prior to surgery. Appropriate Institutional Review Board permission
was obtained from the participating hospital, and written informed
consent was obtained from all subjects. All of the tumor and
macroscopically normal lung tissue samples were obtained at the
time of surgery, and were rapidly frozen in liquid nitrogen and
stored at −80°C until analysis. Tissue samples were histologically
confirmed by hematoxylin-eosin staining.
miRNA microarray and human whole genome
expression microarray
Agilent's Human miRNA (Rel 14.0) and Agilent Human
GE 4×44K (Agilent Technologies, Santa Clara, CA, USA) were used
according to the manufacturer's instructions.
TaqMan miRNA expression assay
qRT-PCR analysis for miRNAs was performed in
duplicate with MicroRNA assay kit (Applied Biosystems, Foster City,
CA, USA) according to the manufacturer's instructions, and RNU6B
was used for normalization.
miRNA mimic, miRNA inhibitor and siRNA
transfection
Cells were plated in 6-well plates at a density of
1.2×105 cells/well. The next day, cells were transfected
with 30 nM pre-miR miRNA mimic (Ambion, Austin, TX, USA), anti-miR
inhibitor (Ambion), premiR miRNA mimic-negative control#1 (Ambion)
and anti-miR negative control#1 inhibitor (Ambion) with
Lipofectamine™ RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to
the manufacturer's instructions. Also, cells were transfected with
specific Silencer® Select siRNA for IRS2 (s16487,
Ambion) and Silencer® Select Negative Control No. 1
siRNA (Ambion) with Lipofectamine RNAiMAX (Invitrogen).
qRT-PCR
Total RNA was isolated with TRIzol solution (Ambion)
according to the protocols of the manufacturer. The first strand of
cDNA was synthesized using the oligo(dT) primer system
(Super-Script III First-Strand Synthesis System; Invitrogen).
Aliquots of the reaction mixture were used for the qPCR
amplification with the CFX96 system (Bio-Rad Laboratories,
Hercules, CA, USA) using iQ SYBR Green Supermix (Bio-Rad
Laboratories). PCR was run for 40 cycles of denaturation at 95°C
for 1 sec, annealing at 56°C for 15 sec, and elongation at 72°C for
15 sec. Gene expression was quantified by the comparative CT
method, with normalization of CT values to the housekeeping gene
β-actin. After amplification, melting curve analysis was performed
to ensure the specificity of the products.
Western blot analysis
Cells were lysed in Pro-Prep protein extraction
solution (INtRON Biotechnology, Gyeonnggi-do, Korea) 72 h after
transfection. An equal amount of proteins were resolved on 8%
SDS-PAGE gels. The primary antibodies used for the analysis of
mouse anti-E-cadherin (1:1,000; BD, Franklin Lakes, NJ, USA),
vimentin (1:1,000; Cell Signaling Technology, Beverly, MA, USA),
IRS2 (1:1,000; Cell Signaling) antibody and β-actin antibodies
(1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Combined approach to identify the
potential target genes of miR-146a
Putative target genes of identified deregulated
miRNAs were detected using two different approaches. In the in
silico approach, we used the PicTar, TargetScan, miRanda and
RNAhybrid. To decrease the number of false-positive results, only
putative target genes predicted by at least two programs, were
accepted. In the experimental approach, we next examined the whole
genome expression microarray in mesenchymal lung cancer cell lines.
The overlap between the list of mRNA transcripts from the
microarray experiments and the putative targets of miR-146a miRNA
from in silico approach targets provided the potential
target genes regulated by miR-146a.
Luciferase reporter assays
To verify that miR-146a can regulate IRS2 gene
directly, we generated a Renilla luciferase reporter plasmid cloned
downstream to a segment of the IRS2 3′-UTR containing the putative
miR-146a binding sequences, which predicted two binding sites, at
position 5158–5186 bp of IRS2 3′-UTR, using the RNAhybrid 2.2
program. The constructs were then co-transfected into 293T cells
with miR-146a mimic, or mimic-negative control, and Renilla
luciferase activity was measured 48 h later.
Invasion and migration assays
The invasion assay was performed in triplicate using
48-well microchemotaxis chambers (Neuro. Probe, Inc., Gaithersburg,
MD, USA) with 8-μm pore membranes (Neuro. Probe) pre-coated with 10
μg/ml Matrigel (BD Bioscience). The cells (1×104) in 50
μl of serum-free medium were placed in the upper chamber, and the
lower chamber was filled with 26–27 μl of medium with 10% FBS.
After incubation for 24 h at 37°C, the cells that migrated to the
lower surface of the membranes were stained with a Diff-Quick kit
and then counted under a microscope. The migration assay was done
using the same procedure with membranes that were coated with 5
μg/ml collagen IV (Trevigen, Gaithersbug, MD, USA).
EGFR-tyrosine kinase inhibitor (EGFR-TKI)
treatment
The cells were treated with Gefitinib 0.01 M after
transfection. The media was changed every 24 h.
Statistical analyses
The statistical differences between groups were
analyzed using Student's t-test when two groups were compared. To
analyze the drug interaction for synergy, a one-way classification
ANOVA was used. All of the statistical analyses were performed with
SPSS 21.0 (SPSS, Chicago, IL, USA). The differences were considered
to be statistically significant at P<0.05.
Results
Identification of differentially
expressed microRNAs in mesenchymal lung cancer
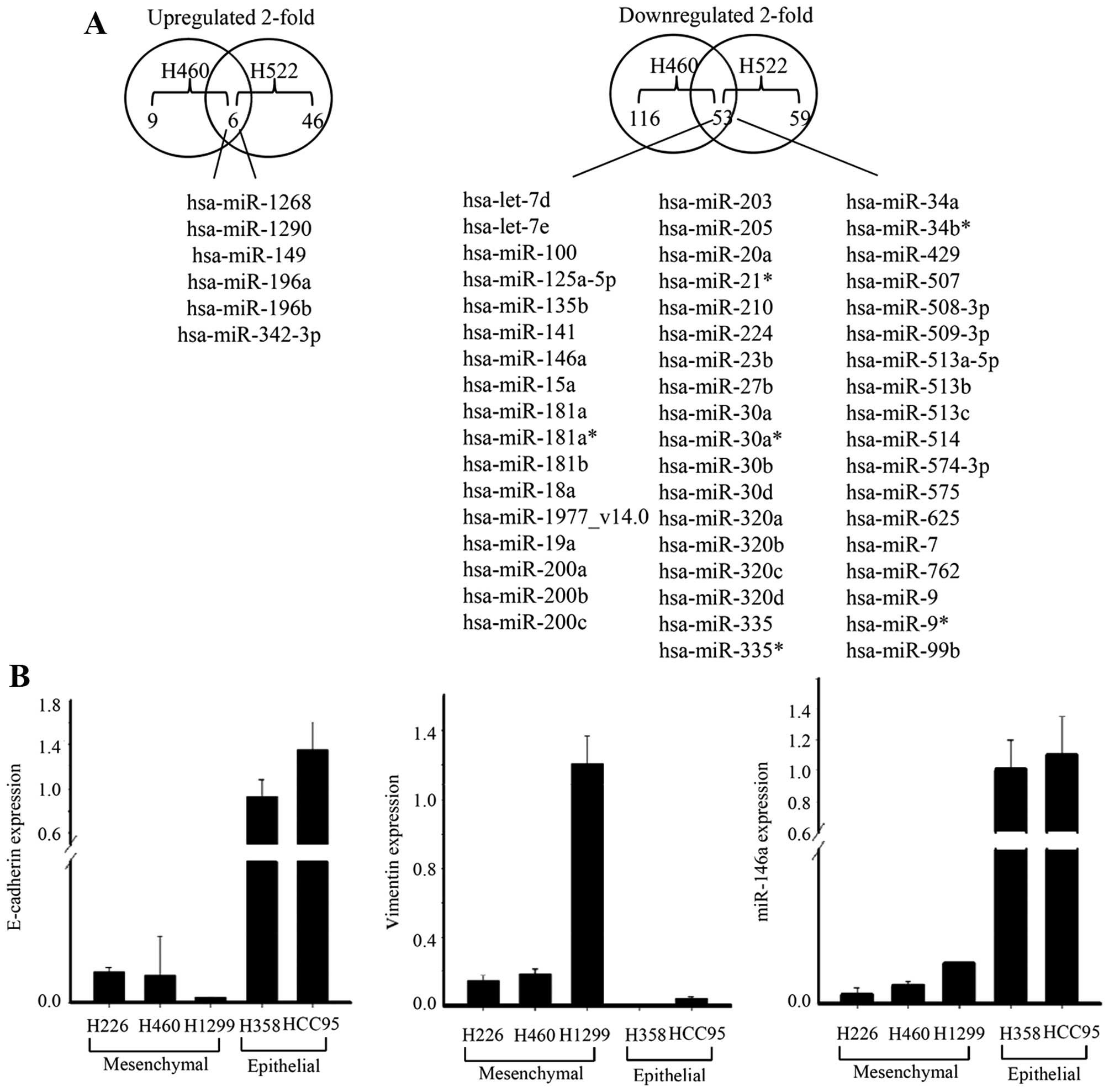
We first searched for the microRNAs that showed
differential expression between epithelial (H358) and
mesenchymal-like lung cancer cell lines (H460, H522) using miRNA
microarray (10). We identified 59
miRNAs that were differentially (≥2- to ≤0.5-fold) expressed in
both the mesenchymal-like lung cancer cell lines. Among the 59
differentially expressed miRNAs, six were downregulated and 53
miRNAs were underexpressed in mesenchymal-like lung cancer cell
lines. To validate the microarray results, the expression of
miR-146a was determined using TaqMan miRNA expression assay.
Consistent with the results of the microarray analysis, miR-146a
was downregulated in the mesenchymal-like lung cancer cell lines
(Fig. 1).
Quantitative analysis of miR-146a
expression in human lung cancer
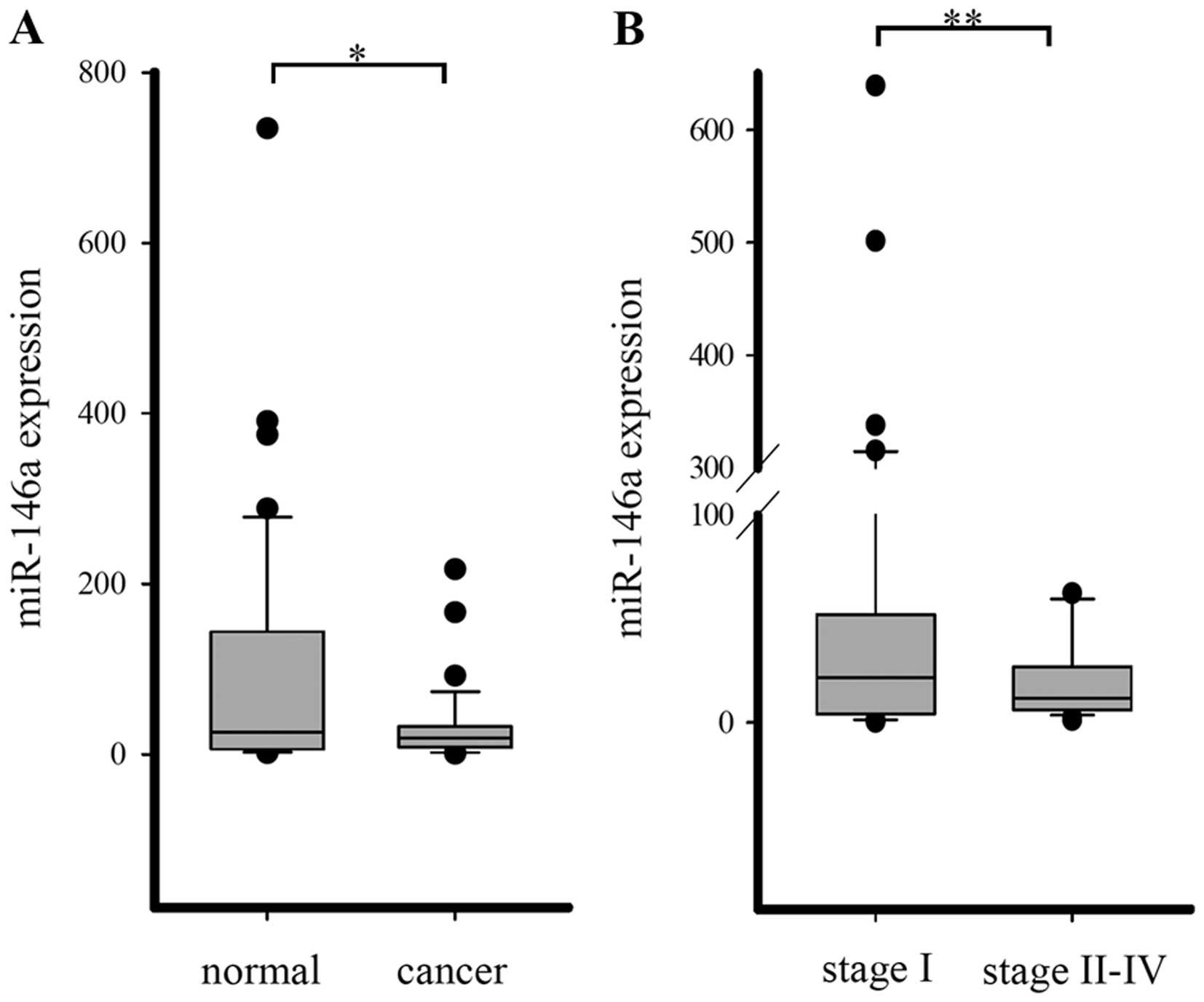
A TaqMan miRNA expression assay was applied to
detect the miRNA-146a expression in 78 pairs of lung cancer tissue
samples and matched normal lung tissue samples. miR-146a was
downregulated in lung cancer tissues compared with normal lung, but
there was no statistical significance. However, when patients were
categorized by lung cancer stages, the expression of miR-146a was
significantly down-regulated in advanced pathological stages
(Fig. 2).
miR-146a targets IRS2
To further analyze the targets of the differentially
expressed miRNAs in the mesenchymal-like lung cancer cell lines, we
generated mRNA microarray expression profiles in mesenchymal-like
lung cancer cell lines (data not shown). The genes changed by
2-fold matched with putative target genes in the in silico
approach were selected as candidates. Several miRNAs overlapped
between the list of mRNA transcripts from the microarray
experiments and the putative targets of miR-146a from in
silico approach. IRS2 was selected as a candidate target of
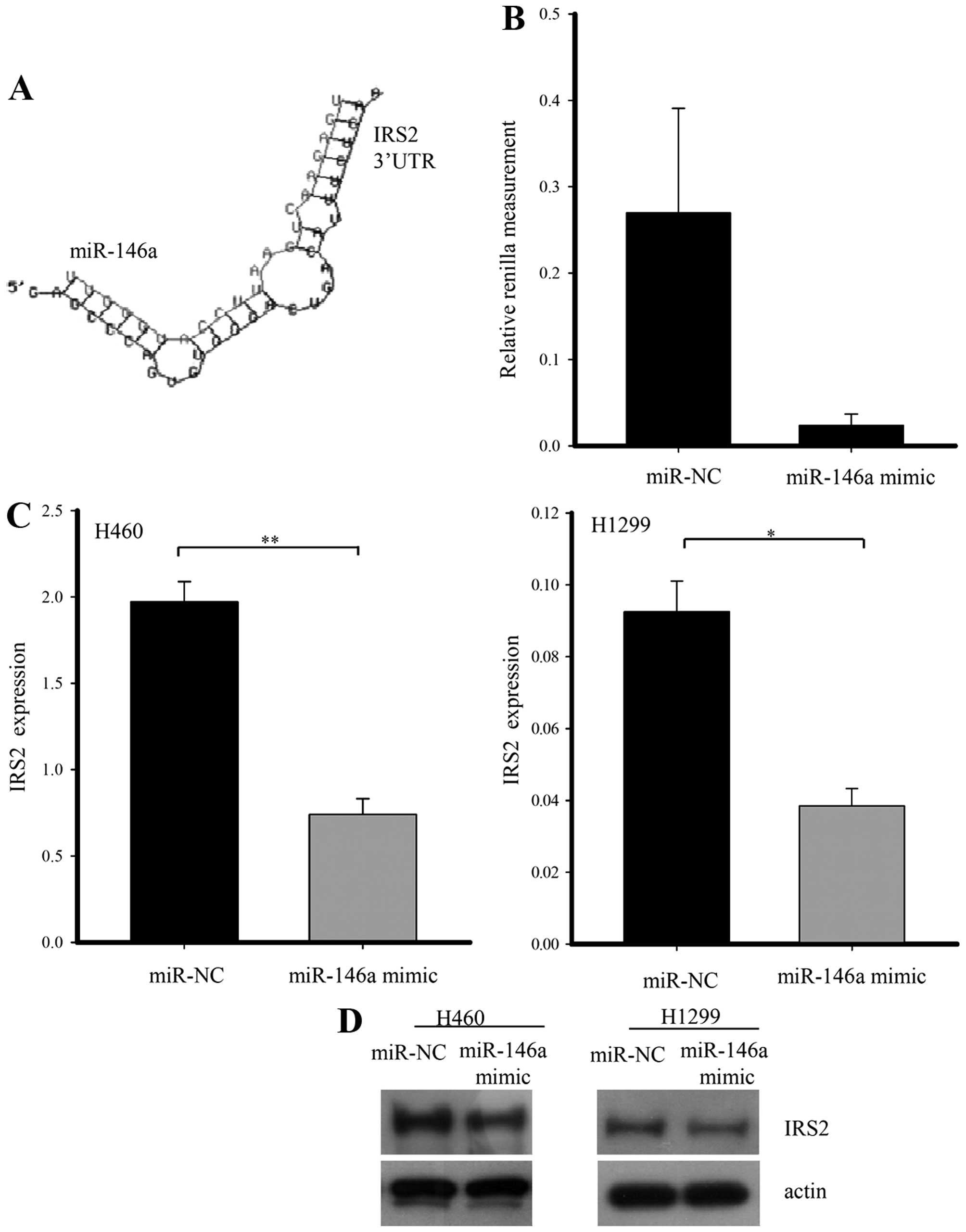
miR-146a through a literature search. To verify that miR-146a
regulates the IRS2 gene directly, we generated a Renilla luciferase
reporter plasmid cloned downstream to a segment of the IRS2 3-UTR
containing the putative miR-146a sequence. Renilla activity was
significantly lower by the miR-146a mimic as compared with negative
miRNA. To assess whether or not miR-146a has a functional role in
the downregulation of endogenous IRS2 expression, we performed
qRT-PCR and western blot analysis in H1299 and H460 cells after
transfection with negative control or miR-146a mimic. When miR-146a
was overexpressed, IRS2 mRNA and protein was diminished compared
with the control group (Fig.
3).
miR-146a inhibits lung cancer cell
migration and invasion in vitro
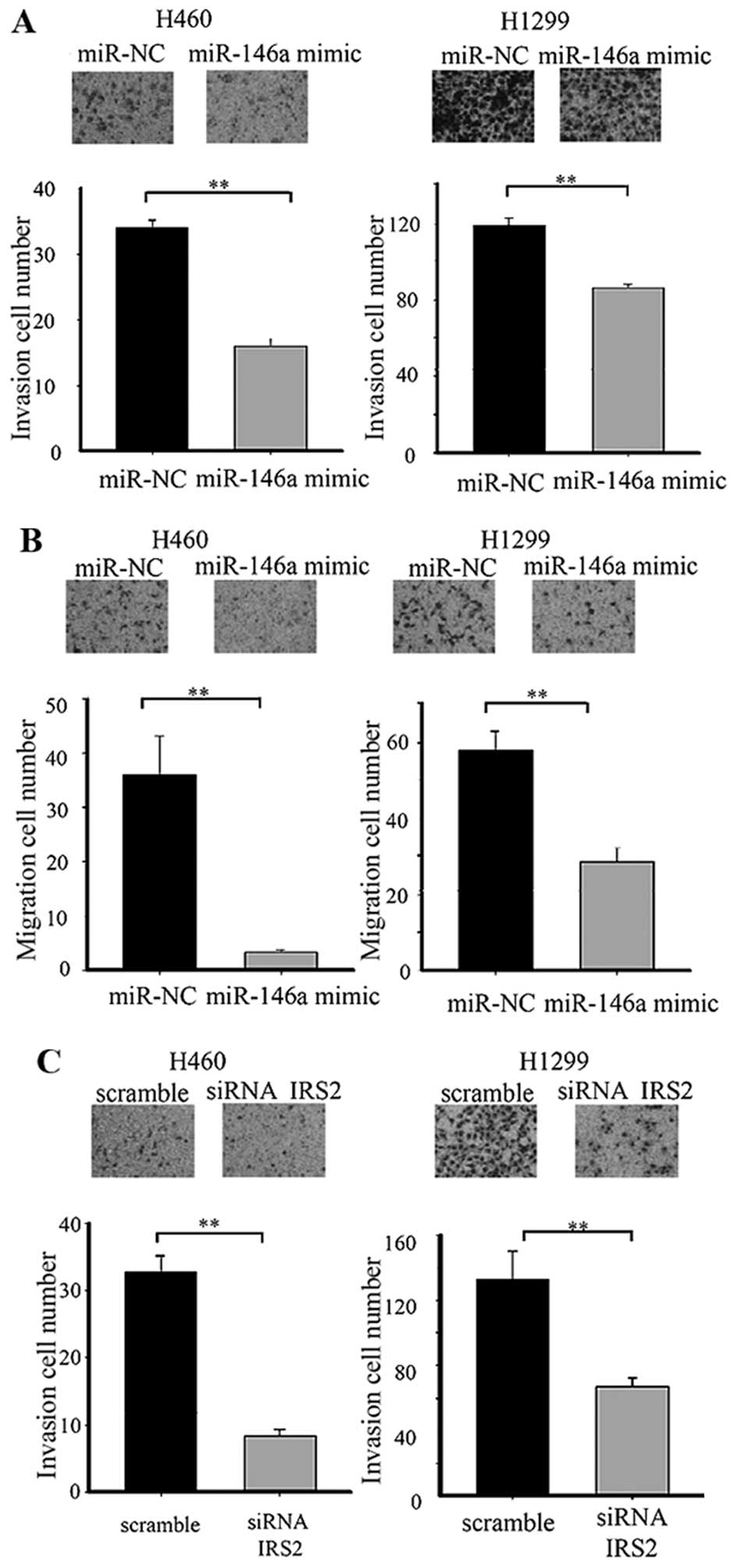
To determine if overexpression of miR-146a has an
effect on lung cancer progression, we performed invasion and
migration assays in a lung cancer cell line. Cells transfected with
miR-146a mimic displayed significant reduction of invasion and
migration to different degree in the H460 and H1299 cell lines. To
confirm that the effect of miR-146a in lung cancer cells occurred
through the IRS2 repression, IRS2 siRNA was used to transfect H460
and H1299 cells. IRS2 siRNA decreased lung cancer cell invasion
(Fig. 4).
miR-146a overexpression or knockdown of
IRS2 regulates EMT-related genes
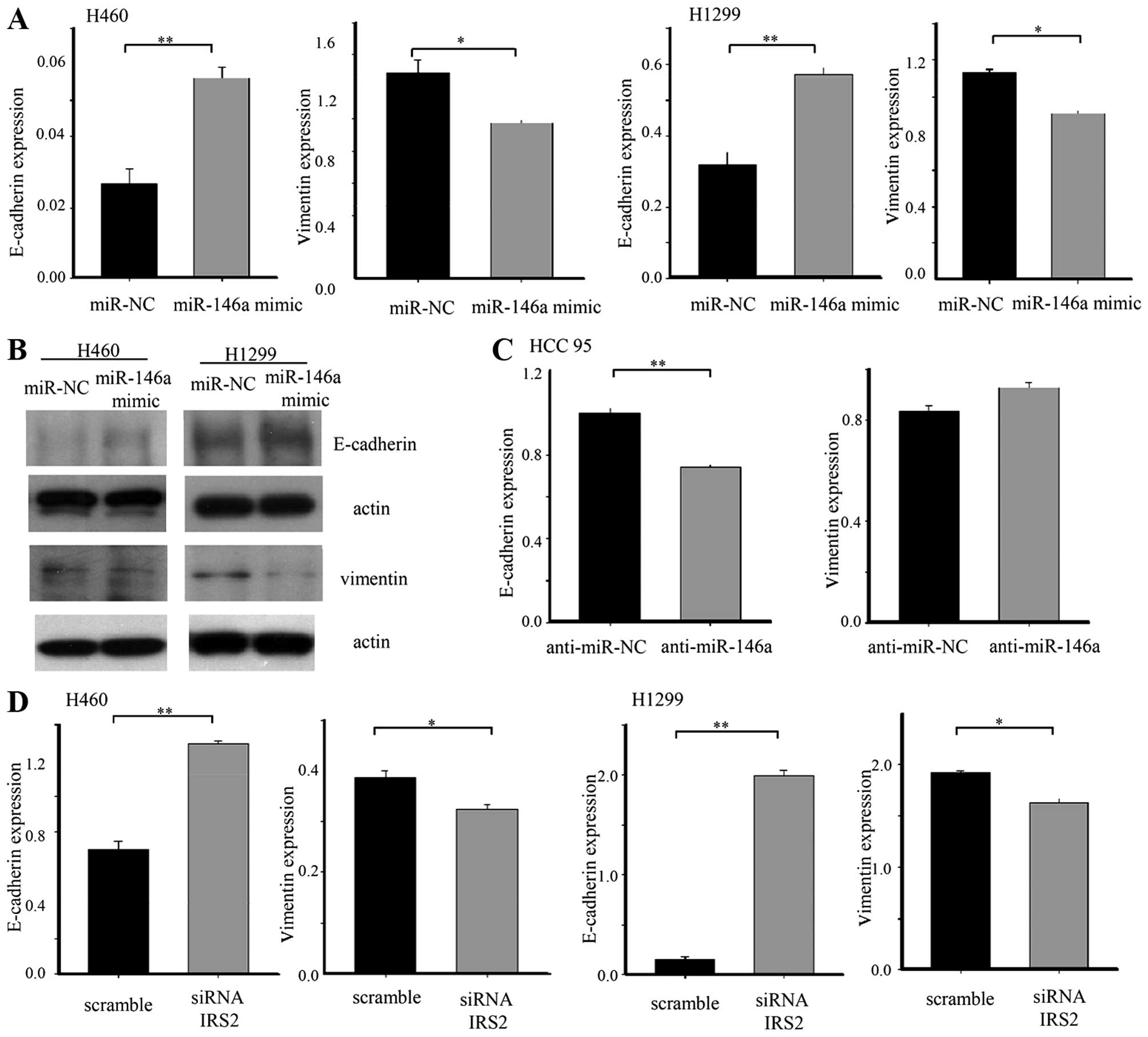
To elucidate the mechanism underlying impaired
invasion and migration capacity of miR-146a, we evaluated the
expression of an epithelial marker (E-cadherin) and mesenchymal
marker (vimentin). The E-cadherin was upregulated in lung cancer
cell lines after transfection with the 146a mimic. Ectopic
expression of miR-146a was sufficient to reduce expression of
vimentin. Conversely, anti-miR-146a induced downregulation of an
epithelial marker in HCC95 cells. On the other hand, anti-miR-146a
was not sufficient to increase the mesenchymal marker. To confirm
that inhibition of EMT by miR-146a in lung cancer occurred through
the IRS2 repression, we transfected IRS2 siRNA into lung cancer
cells. IRS2 siRNA changed the expression of the epithelial marker
and the mesenchymal marker, consistent with the effect of miR-146a
(Fig. 5).
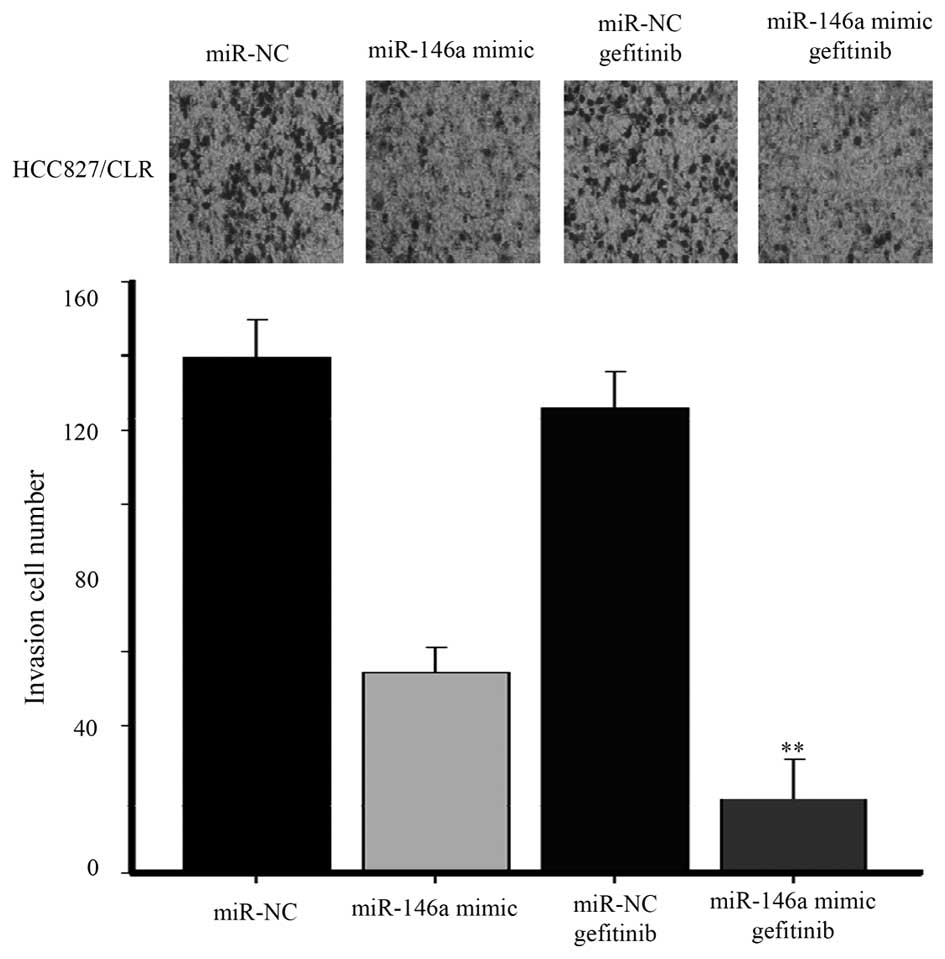
miR-146a mimic sensitizes
EFGR-TKI-resistant lung cancer line to gefitinib treatment
To investigate the role of miR-146a in the
chemoresistance induced by EMT, we performed invasion assays in
HCC827/CLR cells with acquired resistance to EFGR-TKIs induced by
EMT (11). miR-146a mimic markedly
decreased invasion of HCC827/CLR cells. Furthermore, combined
treatment with miR-146a mimic and gefitinib in HCC827/CLR cells
showed a significant reduction of invasion compared to
mono-treatment with EGFR-TKI (Fig.
6).
Discussion
Epithelial and mesenchymal cells are basic tissue
phenotypes. The epithelial cells that line the surface of a tissue
or organ are attached to the basement membrane, establishing an
apical-basal axis of polarity, and communicating with each other
through the gap junction, generally not exhibiting a regimented
structure or tight intracellular adhesion (12,13).
Mesenchymal cells form structures that are irregular in shape and
not uniform in composition or density. Adhesion between mesenchymal
cells is less strong than in their epithelial counterparts,
allowing for increased migratory capacity (14). EMT is a biologic process that
allows a polarized epithelial cell to convert into a mesenchymal
cell. EMT has an important role in the development of many tissues
during embryogenesis, but similar cell changes are recapitulated
during pathological processes, such as fibrosis and cancer
(15). EMT is a crucial mechanism
for cancer drug resistance, progression and metastasis in cancer
(16). Recent studies suggest that
EMT may occur during lung cancer development. The loss of
E-cadherin expression is a hallmark of EMT (17). In some studies, the loss of
E-cadherin function also correlates with poor patient prognosis in
lung cancers (18,19). The expression of HIF-1α, TWIST1 and
Snail, which are regulators of EMT, correlate with survival and
relapse in NSCLC (20). L1CAM and
SLUG are expressed in the tumor stroma of lung cancer specimens
while membranous E-cadherin is expressed in the central regions of
the tumor. These findings demonstrate that tumor cells at invasive
front have mesenchymal features in a pathologic section (6,21).
Serial biopsies demonstrate the transformation from epithelial to
mesenchymal cells from the same lung cancer patients. Chung et
al (11) reported the
morphology of the metastatic tumor was quite different from that of
the original lung cancer, which showed mixed acinar and
bronchioloalveolar features. Metastasis from lung cancer exhibited
spindle-shaped cells with the loss of epithelial polarity. EMT has
a role in determining sensitivity to EGFR-TKI or cytotoxic drugs
(22,23). Accumulating evidence suggests that
EMT is directly implicated in lung cancer.
miRNAs are non-coding, single-stranded RNAs that
repress gene expression by interacting with messenger RNA (mRNA) by
inhibiting mRNA translation or by inducing mRNA cleavage. Over
1,400 mature miRNAs have been identified in humans, mostly
clustered within the introns of protein coding genes, and targeting
roughly 70% of all genes (24).
The first evidence that miRNAs can act as regulators of EMT was
reported in 2008. The miR-200 family has a role in the control of
the epithelial/mesenchymal phenotype of cancer cells (25–27).
Loss of cellular miR-200 results in a more mesenchymal-like, highly
motile and aggressive phenotype of lung cancer cells (28). Decreased miR-193a-3p/5p expression
was significantly associated with lymph node metastasis in lung
cancer. The overexpression of miR-193a-3p/5p can inhibit NSCLC cell
migration, invasion, and EMT in vitro and lung metastasis
formation in vivo. ERBB4 and S6K2 are the direct targets of
miR-193a-3p and that PIK3R3 and mTOR were the direct targets of
miR-193a-5p in NSCLC (29). Many
secreted polypeptides are implicated in the EMT process and their
corresponding intracellular transduction pathways form highly
interconnected networks. Transforming growth factor-β, Wnt, Notch,
and growth factors acting through tyrosine kinase receptors induce
EMT (30). Along with the
identification of more signatures of EMT, miRNAs will be have a
role as regulators of EMT in lung cancer.
Aberrant expression of miR-146a is involved in
progression of various types of cancers (31–33).
Chen et al (34) reported
the relative miR-146a expression was significantly lower in NSCLC
tissues than in the normal lung tissues. The expression of miR-146a
was significantly lower in cases with advanced clinical stages and
in the cases with metastasis. Overall survival of patients with
high miR-146a expression was longer than that of patients with low
expression. In their experiments, miR-146a suppressed cell growth
and migratory capacity though inhibited EGFR and NF-κB signaling.
In this study, we discovered that IRS2 was a novel direct target of
miRNA-146a. IRS2 is a member of insulin receptor substrate (IRS)
proteins, an adaptor molecule that integrate extracellular signals
from insulin and other ligands to intracellular effectors such as
phosphoinositide 3-kinase and mitogen activated protein kinase.
IRS2 is expressed in almost all mammalian tissues and cells.
E-cadherin is a key regulator of epithelial cell structure and
function, and a loss of E-cadherin is recognized as one of the
first steps in the sequence of events in EMT. siRNA-mediated
reduction in IRS2 expression significantly increases basal levels
of E-cadherin mRNA and protein (35). Wnt signaling plays a pivotal role
in cell proliferation, tissue homeostasis, and tumorigenesis.
Dishevelled (Dvl) is a central node of Wnt signaling. IRS1/2
promotes Wnt/β-catenin signaling by stabilizing Dvl2.
Overexpression of IRS1/2 increases the protein level of Dvl2 and
promotes canonical Wnt signaling, whereas depletion of IRS1/2
reduces the level of Dvl2 and attenuates Wnt/β-catenin signaling.
IRS1/2 promote the induction of EMT and cell proliferation in
response to Wnt stimulation, whereas depletion of Dvl2 impaired the
IRS1/2-mediated EMT and cell growth (36). EGFR signaling activated by hypoxia
also induces EMT (37).
Interestingly, EGF increased IRS-2 promoter activity, while
repression of the induction of IRS-2 levels abolishes the EGF
enhancement of cell motility (38). Overexpression of IRS1 or IRS2 in
the mammary gland showed progressive mammary hyperplasia,
tumorigenesis, and metastasis (9,39,40).
IRS1 plays a central role in cancer cell proliferation, while IRS2
is associated with cancer cell motility and metastasis. The
elimination of IRS1/2 results in long-term inhibition of IGF-IR
signaling and powerful inhibition of tumor cell growth (41). This evidence suggested that IRS2 is
a major adaptor molecule that links extracellular signals and their
corresponding intracellular transduction pathways in EMT.
In this study, we observed substantial IRS2
suppression by miR-146a at the mRNA and protein levels in lung
cancer. IRS2 was directly regulated by miR-146a through binding of
the 3′-UTR. miR-146a was downregulated in advanced pathological
stages and suppressed lung cancer invasion and migration by
repressing IRS2 expression. Ectopic expression of miR-146a caused
upregulation of E-cadherin in lung cancer cell lines and reduced
their motility. Conversely, inhibition of miR-146a reduced
E-cadherin expression. miR-146a enhanced the sensitivity of
HCC827/CLR to gefitinib. Our data identify miR-146a as a powerful
marker for determining the epithelial phenotype of lung cancer
cells.
Acknowledgements
This study was supported by the National Research
Foundation of Korea Grant funded by the Korean Government
(NRF-2012R1A1A4A01005638) and by Konyang University Myunggok
Research Fund, 2013.
References
|
1
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang P: Epidemiology of lung cancer
prognosis: Quantity and quality of life. Cancer Epidemiology. Verma
M: Humana Press; pp. 469–486. 2009, View Article : Google Scholar
|
|
3
|
Detterbeck FC: Lobectomy versus limited
resection in T1N0 lung cancer. Ann Thorac Surg. 96:742–744. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Zijl F, Krupitza G and Mikulits W:
Initial steps of metastasis: Cell invasion and endothelial
transmigration. Mutat Res. 728:23–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shih J-Y and Yang P-C: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin L and Bivona TG: Mechanisms of
resistance to epidermal growth factor receptor inhibitors and novel
therapeutic strategies to overcome resistance in NSCLC patients.
Chemother Res Pract. 2012:8172972012.PubMed/NCBI
|
|
8
|
Jeon H-S, Lee YH, Lee SY, Jang JA, Choi
YY, Yoo SS, Lee WK, Choi JE, Son JW, Kang YM, et al: A common
polymorphism in pre-microRNA-146a is associated with lung cancer
risk in a Korean population. Gene. 534:66–71. 2014. View Article : Google Scholar
|
|
9
|
Dearth RK, Cui X, Kim H-J, Hadsell DL and
Lee AV: Oncogenic transformation by the signaling adaptor proteins
insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle.
6:705–713. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomson S, Petti F, Sujka-Kwok I, Epstein
D and Haley JD: Kinase switching in mesenchymal-like non-small cell
lung cancer lines contributes to EGFR inhibitor resistance through
pathway redundancy. Clin Exp Metastasis. 25:843–854. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chung J-H, Rho JK, Xu X, Lee JS, Yoon HI,
Lee CT, Choi YJ, Kim HR, Kim CH and Lee JC: Clinical and molecular
evidences of epithelial to mesenchymal transition in acquired
resistance to EGFR-TKIs. Lung Cancer. 73:176–182. 2011. View Article : Google Scholar
|
|
12
|
Chen J, Han Q and Pei D: EMT and MET as
paradigms for cell fate switching. J Mol Cell Biol. 4:66–69. 2012.
View Article : Google Scholar
|
|
13
|
Hay ED: The mesenchymal cell, its role in
the embryo, and the remarkable signaling mechanisms that create it.
Dev Dyn. 233:706–720. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Denlinger CE, Ikonomidis JS, Reed CE and
Spinale FG: Epithelial to mesenchymal transition: The doorway to
metastasis in human lung cancers. J Thorac Cardiovasc Surg.
140:505–513. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nakata S, Sugio K, Uramoto H, Oyama T,
Hanagiri T, Morita M and Yasumoto K: The methylation status and
protein expression of CDH1, p16(INK4A), and fragile histidine triad
in nonsmall cell lung carcinoma: Epigenetic silencing, clinical
features, and prognostic significance. Cancer. 106:2190–2199. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu D, Huang C, Kameyama K, Hayashi E,
Yamauchi A, Kobayashi S and Yokomise H: E-cadherin expression
associated with differentiation and prognosis in patients with
non-small cell lung cancer. Ann Thorac Surg. 71:949–954; discussion
954–945. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hung J-J, Yang M-H, Hsu H-S, Hsu W-H, Liu
J-S and Wu K-J: Prognostic significance of hypoxia-inducible
factor-1α, TWIST1 and Snail expression in resectable non-small cell
lung cancer. Thorax. 64:1082–1089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tischler V, Pfeifer M, Hausladen S,
Schirmer U, Bonde AK, Kristiansen G, Sos ML, Weder W, Moch H,
Altevogt P, et al: L1CAM protein expression is associated with poor
prognosis in non-small cell lung cancer. Mol Cancer. 10:1272011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhuo WL, Wang Y, Zhuo XL, Zhang YS and
Chen ZT: Short interfering RNA directed against TWIST, a novel zinc
finger transcription factor, increases A549 cell sensitivity to
cisplatin via MAPK/mitochondrial pathway. Biochem Biophys Res
Commun. 369:1098–1102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Kim CH and Lee JC: Epithelial to mesenchymal transition
derived from repeated exposure to gefitinib determines the
sensitivity to EGFR inhibitors in A549, a non-small cell lung
cancer cell line. Lung Cancer. 63:219–226. 2009. View Article : Google Scholar
|
|
24
|
Friedman RC, Farh KK-H, Burge CB and
Bartel DP: Most mammalian mRNAs are conserved targets of microRNAs.
Genome Res. 19:92–105. 2009. View Article : Google Scholar :
|
|
25
|
Ceppi P and Peter ME: MicroRNAs regulate
both epithelial-to-mesenchymal transition and cancer stem cells.
Oncogene. 33:269–278. 2014. View Article : Google Scholar
|
|
26
|
Park S-M, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ceppi P, Mudduluru G, Kumarswamy R, Rapa
I, Scagliotti GV, Papotti M and Allgayer H: Loss of miR-200c
expression induces an aggressive, invasive, and chemoresistant
phenotype in non-small cell lung cancer. Mol Cancer Res.
8:1207–1216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu T, Li J, Yan M, Liu L, Lin H, Zhao F,
Sun L, Zhang Y, Cui Y, Zhang F, et al: MicroRNA-193a-3p and -5p
suppress the metastasis of human non-small-cell lung cancer by
downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway.
Oncogene. 34:413–423. 2015. View Article : Google Scholar
|
|
30
|
Moustakas A and Heldin C-H: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou L, Zhao X, Han Y, Lu Y, Shang Y, Liu
C, Li T, Jin Z, Fan D and Wu K: Regulation of UHRF1 by miR-146a/b
modulates gastric cancer invasion and metastasis. FASEB J.
27:4929–4939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Karakatsanis A, Papaconstantinou I,
Gazouli M, Lyberopoulou A, Polymeneas G and Voros D: Expression of
microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c,
miR-221, miR-222, and miR-223 in patients with hepatocellular
carcinoma or intrahepatic cholangiocarcinoma and its prognostic
significance. Mol Carcinog. 52:297–303. 2013. View Article : Google Scholar
|
|
33
|
Xu B, Wang N, Wang X, Tong N, Shao N, Tao
J, Li P, Niu X, Feng N, Zhang L, et al: MiR-146a suppresses tumor
growth and progression by targeting EGFR pathway and in a
p-ERK-dependent manner in castration-resistant prostate cancer.
Prostate. 72:1171–1178. 2012. View Article : Google Scholar
|
|
34
|
Chen G, Umelo IA, Lv S, Teugels E, Fostier
K, Kronenberger P, Dewaele A, Sadones J, Geers C and De Grève J:
miR-146a inhibits cell growth, cell migration and induces apoptosis
in non-small cell lung cancer cells. PLoS One. 8:e603172013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carew RM, Browne MB, Hickey FB and Brazil
DP: Insulin receptor substrate 2 and FoxO3a signalling are involved
in E-cadherin expression and transforming growth factor-β1-induced
repression in kidney epithelial cells. FEBS J. 278:3370–3380. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Geng Y, Ju Y, Ren F, Qiu Y, Tomita Y,
Tomoeda M, Kishida M, Wang Y, Jin L, Su F, et al: Insulin receptor
substrate 1/2 (IRS1/2) regulates Wnt/β-catenin signaling through
blocking autophagic degradation of dishevelled2. J Biol Chem.
289:11230–11241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Misra A, Pandey C, Sze SK and Thanabalu T:
Hypoxia activated EGFR signaling induces epithelial to mesenchymal
transition (EMT). PLoS One. 7:e497662012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cui X, Kim H-J, Kuiatse I, Kim H, Brown PH
and Lee AV: Epidermal growth factor induces insulin receptor
substrate-2 in breast cancer cells via c-Jun NH(2)-terminal
kinase/activator protein-1 signaling to regulate cell migration.
Cancer Res. 66:5304–5313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dearth RK, Cui X, Kim H-J, Kuiatse I,
Lawrence NA, Zhang X, Divisova J, Britton OL, Mohsin S, Allred DC,
et al: Mammary tumorigenesis and metastasis caused by
overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2.
Mol Cell Biol. 26:9302–9314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chan BT and Lee AV: Insulin receptor
substrates (IRSs) and breast tumorigenesis. J Mammary Gland Biol
Neoplasia. 13:415–422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reuveni H, Flashner-Abramson E, Steiner L,
Makedonski K, Song R, Shir A, Herlyn M, Bar-Eli M and Levitzki A:
Therapeutic destruction of insulin receptor substrates for cancer
treatment. Cancer Res. 73:4383–4394. 2013. View Article : Google Scholar : PubMed/NCBI
|