Introduction
Mitochondria are semi-autonomous intracellular
organelles that play decisive roles in energy metabolism,
regulation of programmed cell death, and free radical production
(1). Mitochondrial dysfunction
occurs due to mitochondrial DNA (mtDNA) damage and mutation, and is
associated with aging. Recently, several studies have shown that
mitochondrial defects can play an important role in the progression
of cancer including hepatocellular carcinoma (HCC) (2–4).
While there are reports of these phenomena, the mechanisms
responsible for the initiation and evolution of mtDNA mutations, as
well as their roles in the development of cancer remain to be
elucidated.
HCC originates in hepatocytes or even in liver
progenitor cells (5). In
particular, epithelial hepatocytes are not proliferative under
physiological conditions and have highly diverse functions.
However, they have highly proliferative regeneration in response to
partial hepatectomies and chemical intoxication (6). Various complicated mechanisms have
been reported to cause abnormal proliferation and dedifferentiation
of hepatocytes in HCC pathogenesis, which leads to the subsequent
development of malignant tumors (7).
There is a critical point when epithelial cells
exhibit phenotypes with abnormal proliferation, invasion and
migration abilities, and drug resistance to malignant tumor
treatment. This point is called the epithelial-mesenchymal
transition (EMT). EMT represents the transition of epithelial cells
to a mesenchymal phenotype, and it features a loss of epithelial
cell markers (8). In adults, it
has been revealed that EMT plays an important role in tumor
formation and progression to meta-static carcinomas (9).
The mtDNA mutations leading to mitochondrial
dysfunction are expected to play a substantial role in the EMT
process. However, the details and molecular mechanisms underlying
this phenomenon are still unclear. In the present study,
Hep3B/ϱ0 cells, which are mtDNA-depleted cells, were
generated in order to investigate the role of mitochondrial
dysfunction and its regulatory mechanisms in the EMT phenomenon.
Using Hep3B/ϱ0 cells, EMT properties and an associated
signaling pathway in cells with mitochondrial dysfunction were
investigated.
Materials and methods
Materials and reagents
Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), antibiotics (penicillin and streptomycin), 10
Trypsin/EDTA solution, and phosphate-buffered saline (PBS) were
purchased from Welgen, Inc. (Daegu, Korea). Matrigel and 8-μm pored
Transwell filter chambers were purchased from Corning Inc.
(Tewksbury, MA, USA). All oligo primers were generated from Cosmo
Genetech Co., Ltd. (Seoul, Korea). The antibodies were purchased
from the following suppliers: Cell Signaling Technology Inc.
(Danvers, MA, USA), Santa Cruz Biotechnology Inc. (Dallas, TX,
USA), and R&D Systems Inc. (Minneapolis, MN, USA). SB431542 and
c-Jun peptide were purchased from Tocris Bioscience (Ellisville,
MO, USA). Alexa 488-conjugated secondary antibody and Fluo-4/AM
were obtained from Invitrogen (Carlsbad, CA, USA).
Cell culture
Human hepatocellular carcinoma cells (Hep3B) were
obtained from the Korean Cell Line Bank (Seoul, Korea). Hep3B cells
were grown in high-glucose-DMEM supplemented with 10% FBS, 100
units/ml of penicillin, and 100 μg/ml of streptomycin in a 37°C
incubator with a humidified atmosphere containing 5%
CO2.
Generation of Hep3B/ϱ0
cells
To develop Hep3B/ϱ0 cells, Hep3B was
chronically exposed to 50 ng/ml of EtBr in media supplemented with
sodium pyruvate (100 mM) and uridine (50 μg/ml). Subsequently, the
Hep3B/ϱ0 cell line was selected by treatment with media
consisting of the mitochondrial inhibitors rotenone (1 μg/ml) and
antimycin-A (1 μg/ml). After selection of the Hep3B/ϱ0
cell line, the cells were grown in high glucose DMEM supplemented
with 10% FBS, 50 μg/ml of uridine and 100 mM of sodium
pyruvate.
Reverse transcription-polymerase chain
reaction
Total RNA from the cells was isolated using TRIzol
reagent (Invitrogen Corp.). First-strand cDNA was synthesized by
M-MLV reverse transcriptase (Promega, Madison, WI, USA) with 1 μg
each of DNA-free total RNA sample and oligo-(dT)15 (Life
Technologies, Grand Island, NY, USA). Equal amounts of cDNA were
subsequently amplified by PCR in a 20-μl reaction mixture
containing 1X reaction buffer, dNTP mixture, i-Taq™ DNA polymerase
(iNtRON Biotechnology, Seongnam, Korea), and 10 μM of each specific
primer. Amplification products were electrophoresed on 1% agarose
gel and visualized by GelRed (Biotium Inc., Hayward, CA, USA)
staining under ultraviolet transillumination.
Electron microscopy
Cells grown on coverslips were fixed with a solution
of 2.5% glutaraldehyde and 2% formaldehyde in 100 mM cacodylate
buffer (pH 7.4) for 1.5 h at 4°C, washed twice with cacodylate
buffer, and were fixed with 2% osmium tetroxide in 50 mM cacodylate
buffer (pH 7.4). Specimens were washed twice with distilled-water
and stained overnight with aqueous 0.5% uranyl acetate at 4°C.
Cells were dehydrated and flat embedded in Epon 812. Ultra-thin
sections were analyzed with a Bio-TEM (Tecnai G2 Spirit; FEI Co.,
Hillsboro, OR, USA) at the Korea Basic Science Institute.
Assay for endogenous cellular oxygen
consumption
Exponentially growing cells (5×106) of
wild-type and Hep3B/ϱ0 cells were washed with TD
(Tris-based, Mg2+-, Ca2+-deficient) buffer
(0.137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and
25 mM Tris-HCl, pH 7.4) and collected after trypsinization. After
resuspending the cells (5×105) in 0.3 ml of a complete
medium without phenol red, the cells were transferred to a Mitocell
chamber equipped with a Clark oxygen electrode (782 Oxygen Meter;
Strathkelvin Instruments, Co., Motherwell, UK). Oxygen consumption
rates were measured after adding 30 μM 2,4-dinitrophenol (DNP) to
obtain a maximum respiration rate, and its specificity for
mitochondrial respiration was confirmed by adding 5 mM of potassium
cyanide (KCN). Maximum cellular respiration rates are expressed as
the ratio of DNP-uncoupled O2 consumption rate vs.
KCN-inhibited O2 consumption rate.
In vitro wounding migration assay
The cells were plated on 24-well plates (Nunc,
Rochester, NY, USA) at 90% confluence and left overnight. Cells
were scratched with a P200 pipette-tip. After wounding, the
cultures were further incubated in media supplemented with 1% FBS.
The cells were allowed to migrate for 16–24 h. Migration patterns
were observed under a phase contrast microscope and
photographed.
In vitro Transwell invasion assay
The invasion capacity of the cells was determined
using a 24-well Transwell system. The upper side of the Transwell
membrane was coated with 1 mg/ml Matrigel using 10 μl/well. The
cells were seeded at a density of 2×104 cells in 100 μl
of serum-free media to the upper compartment of the Transwell and
the full medium was added to the lower side. Cells were incubated
for 16–24 h at 37°C in 5% CO2. Cells that did not
penetrate the filter were wiped off with cotton swabs, and cells
that had invaded the lower surface of the filter were fixed with
methanol. The cells were then stained with hematoxylin/eosin,
observed under a phase contrast microscope and photographed.
Immunofluorescence
Cells were plated with submerged coverslips. The
primary antibodies used were mouse antiE-cadherin antibody and
mouse anti-vimentin monoclonal antibody. The Alexa 488-conjugated
secondary antibody was used for visualization of the primary
antibodies. The nuclei were counterstained by 300 mM of
4,6-diamidino-2-phenyl-indole (DAPI; Invitrogen). The coverslips
were mounted onto glass slides with Vectastain (Vector
Laboratories). The images were obtained with a fluorescence
microscope (Nikon, Tokyo, Japan) and laser-scanning confocal
microscope (LSM510; Carl Zeiss, Oberkochen, Germany) at ×200 or
×400 magnification.
Western blot analysis
Total cell lysates were prepared using PRO-PREP
protein extraction solution (iNtRON Biotechnology), including 1 mM
of sodium orthovanadate, which is an inhibitor of serine/threonine
protein phosphatase. Equal amounts (30 μg) of samples were resolved
by electrophoresis on 10% SDS-polyacrylamide gels, transferred to a
PVDF membrane, and sequentially probed with appropriate antibodies.
The primary antibodies were used at a dilution of 1:1,000 in 0.1%
TBS-T. This signal was developed with the enhanced
chemiluminescence (ECL) detection system (GE Healthcare, Uppsala,
Sweden).
RNA interference
The cells were transfected with siRNA specific for
Snail-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and control
siRNA according to manufacturer's instructions. After transfection,
the cells were incubated at 37°C for 48 h, and then used for
western blot analysis or in vitro wounding migration
assay.
Quantification of c-Jun/AP-1
activity
Activity of c-Jun/AP-1 was quantitatively measured
by enzyme-linked immunosorbent assay (ELISA) by using commercially
available TransAM kits (Active Motif, Carlsbad, CA, USA) following
the manufacturer's protocol. For the assay, 20 μg of nuclear
extract was used. Activated c-Jun/AP-1 levels were specifically
detected and quantified using a TransAM AP-1 kit. Active protein-1
(AP-1) dimers in the nuclear extracts bind to specific
oligonucleotides containing TPA-responsive elements (TREs)
immobilized onto a 96-well plate. Incubation with a c-Jun antibody
followed by addition of a secondary antibody conjugated to
horseradish peroxidase (HRP) provided sensitive colorimetric
readouts that were easily quantified by spectrophotometry
(A450nm).
Results
Hep3B/ϱ0 cells are
mtDNA-depleted cell model with EMT phenotypes
To investigate the effects of mitochondrial
dysfunction on the EMT process in liver cancer, ϱ0 cells
were first established using the Hep3B hepatocellular carcinoma
cell line. To construct ϱ0 cells, Hep3B cells were
chronically exposed to ethidium bromide (EtBr) in media. EtBr
preferentially intercalates into mtDNA and leads to depletion of
mitochondrial DNA (10,11). mtDNA depletion was first observed
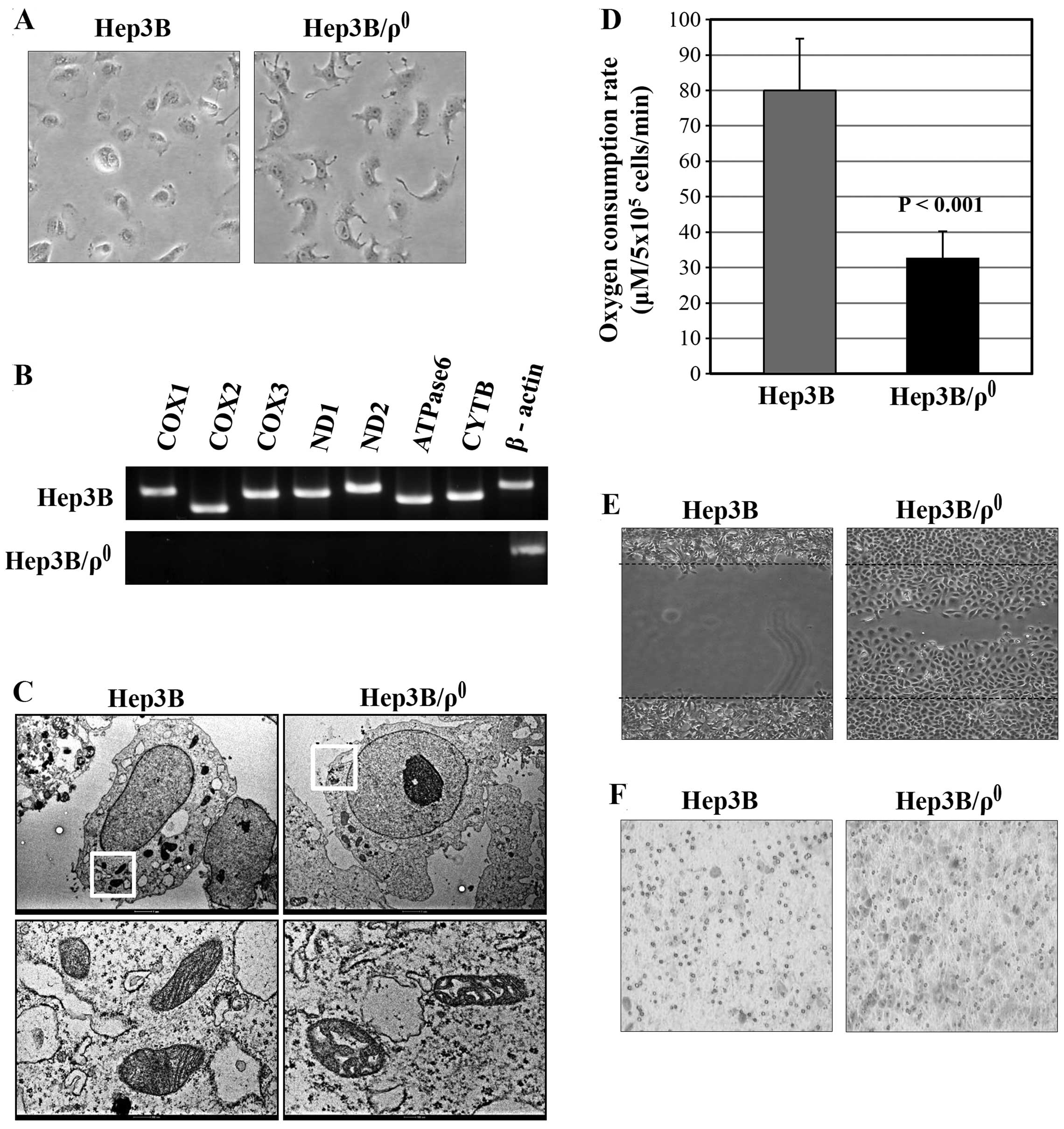
by morphological change (Fig. 1A)
and was monitored by RT-PCR for mitochondrial gene expression. The
mitochondrial gene expressions in Hep3B/ϱ0 cells were
substantially reduced, whereas those in parental Hep3B cells
amplified at the predicted size (Fig.
1B).
The morphological changes of the Hep3B/ϱ0
mitochondria were confirmed by ultra-structural analysis.
Cross-sections through the mitochondrial reticulum in the parental
Hep3B cells showed distinct outer and inner membranes with an
electron-dense matrix full of regularly arranged cristae structures
(Fig. 1C; Hep3B). In contrast, the
TEM images of the mtDNA-depleted cells showed an apparent
alteration of mitochondrial morphology (Fig. 1C; Hep3B/ϱ0). The network
was degraded into single mitochondrial units, often with a swollen
appearance.
Subsequently, the absence of respiratory chain
activity in Hep3B/ϱ0 cells was confirmed by measuring
oxygen consumption (Fig. 1D). The
basal oxygen consumption rate was low in Hep3B/ϱ0 cells
(32.49±7.72 μM/5×106 cells/min, n=7), with a mean that
is ~59.3% less than that of Hep3B cells (79.90±14.7
μM/5×106 cells/min, P<0.001). These data confirm that
mtDNA depletion in Hep3B/ϱ0 cells is associated with
depression of mitochondrial respiratory chain activity.
To examine the migration ability of
Hep3B/ϱ0 cells, wounding migration assay was performed.
The Hep3B/ϱ0 cells migrated substantially, whereas the
wild-type cells did not (Fig. 1E).
Next, the invasion capacity of Hep3B/ϱ0 cells was
examined using a Transwell based invasion assay. The
Hep3B/ϱ0 cells were more invasive than the wild-type
Hep3B cells (Fig. 1F). These
results indicate that mitochondrial dysfunction induces the EMT
phenotype by acquiring migration and invasion abilities in the
Hep3B/ϱ0 cells.
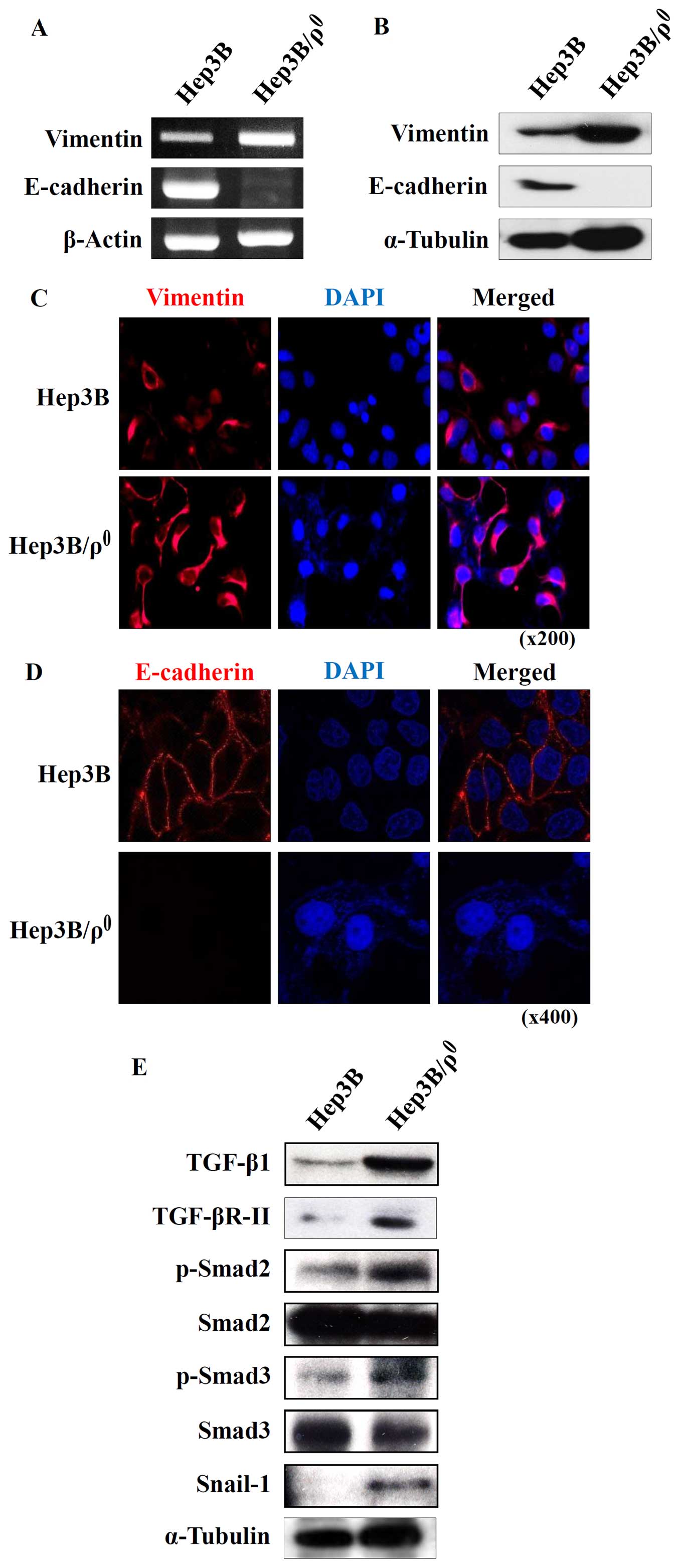
Hep3B/ϱ0 cells express
mesenchymal markers mediated by the TGF-β/Smad/Snail signaling
pathway
Invasive cell motility is a typical characteristic
of mesenchymal cells (12).
Therefore, we speculate that Hep3B/ϱ0 cells might have
acquired mesenchymal phenotypes. During the EMT process, there is
diminished expression of E-cadherin, a marker of epithelial cells,
and increased expression of vimentin, a marker of mesenchymal cells
(13). As expected, the expression
changes of vimentin and E-cadherin showed mesenchymal features.
Vimentin in Hep3B/ϱ0 cells was highly expressed and
E-cadherin was eliminated in both mRNA and protein levels (Fig. 2A and B).
The expression changes in these molecules were then
confirmed by immunofluorescence. Parental Hep3B cells exhibited
epithelial properties; E-cadherin was expressed on the plasma
membrane and there was low vimentin expression (Fig. 2C and D). In contrast, the
Hep3B/ϱ0 cells had mesenchymal properties, including
increased vimentin in the cytosol and absence of E-cadherin on the
plasma membrane (Fig. 2C and D).
Because the Hep3B/ϱ0 cells originated from epithelial
cells, we are able to infer that the epithelial cells transitioned
to mesenchymal cells during the course of mitochondrial
dysfunction.
In malignant tumors, it is well known that EMT is
triggered by growth factors such as TGF-β (14). In particular, the EMT marker
changes are mainly regulated by TGF-β signaling (15). TGF-β elicits its cellular responses
via TGF-βRI and TGF-βRII (16).
Thus, we examined expression of TGF-β and its receptors in the
Hep3B/ϱ0 cells. The result in Fig. 2E shows increased expression of
TGF-β and TGF-βRII, which may indicate activation of TGF-β
signaling in the Hep3B/ϱ0 cells. Upon TGF-β-induced
heteromeric complex formation, TGF-βRII phosphorylates TGF-βRI. The
activated TGF-βRI then initiates its intracellular canonical
signaling pathway by phosphorylating receptor-regulated Smads such
as Smad2 and Smad3 (17,18).
To investigate the effect of increased TGF-β
signaling in the Hep3B/ϱ0 cells, the phosphorylation of
Smad2 and Smad3 was examined by western blot analysis. Both Smad2
and Smad3 were activated through phosphorylation in the
Hep3B/ϱ0 cells (Fig.
2E). These activated R-Smads then upregulated the expression of
their downstream gene, Snail-1, which is a positive regulator of
EMT and metastasis.
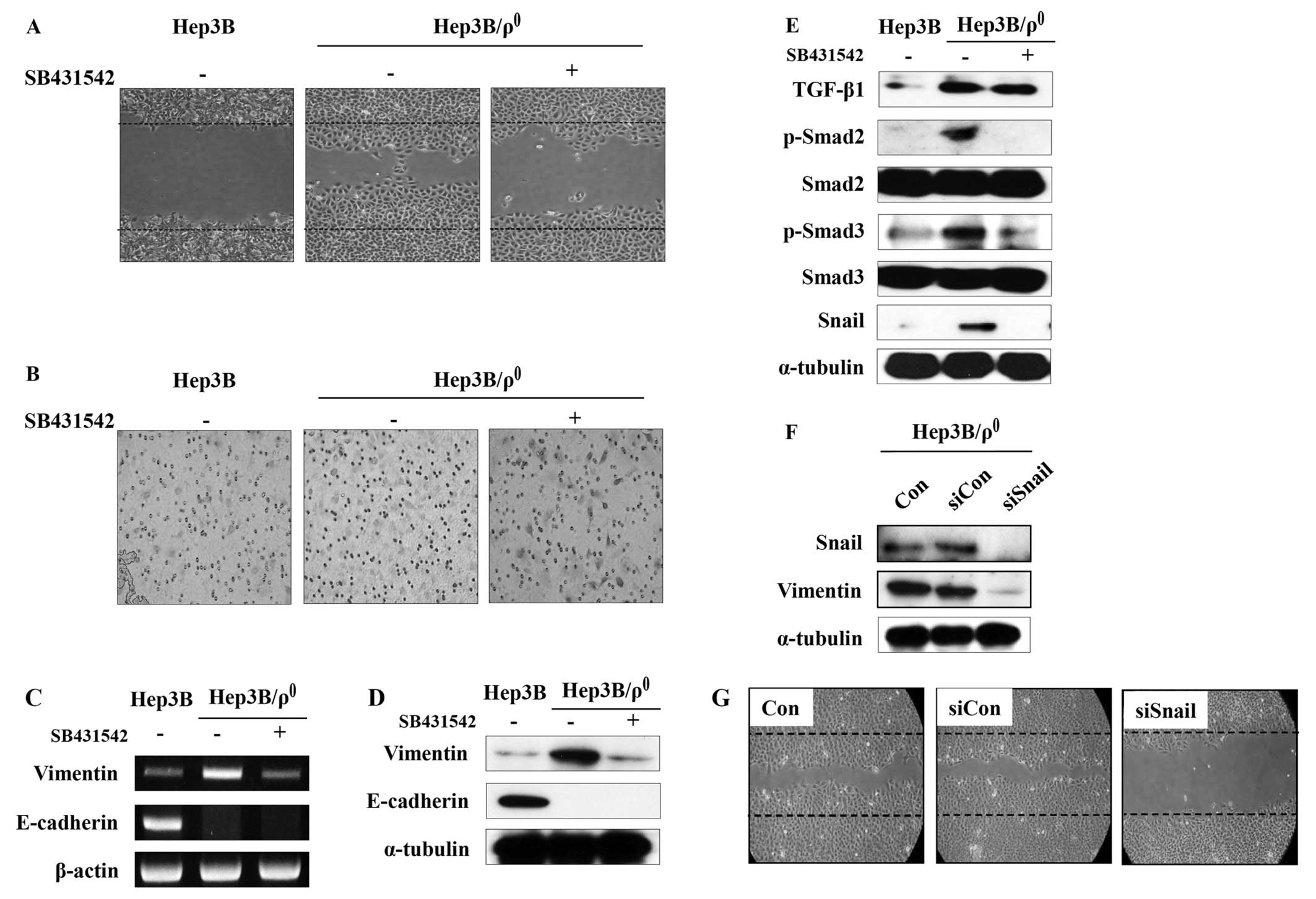
Blocking TGF-β signaling abolishes EMT
phenotypes in Hep3B/ϱ0 cells
We then determined whether the induced TGF-β
signaling plays a role on the migration and invasive abilities of
the Hep3B/ϱ0 cells. SB-431542, a novel small molecule
ATP-mimetic inhibitor of kinase activity, was used to inhibit TGF-β
signaling (19). As expected,
SB-431542 potently inhibited the motility and invasiveness of the
Hep3B/ϱ0 cells (Fig. 3A and
B).
SB-431542 also reversed the expression of
mesenchymal markers. The increased expression of vimentin in the
Hep3B/ϱ0 cells was suppressed by SB-431542 in both mRNA
and protein levels. Unexpectedly, the expression of E-cadherin was
not restored by SB-431542 in either mRNA or protein levels
(Fig. 3C and D).
The expression of TGF-β and phosphorylation of Smad2
and Smad3 were blocked by SB-431542. Additionally, increased
expression of their target gene, Snail, was also reduced (Fig. 3E). These data indicate that EMT
phenotypes in the Hep3B/ϱ0 cells are mediated by the
TGF-β/Smad/Snail signaling pathway.
The above data indicate that TGF-β promotes
expression of Snail, which can regulate vimentin expression and
malignant phenotypes in the Hep3B/ϱ0 cells. To provide
additional evidence of the key role for Snail in the
Hep3B/ϱ0 cells, Snail was blocked with siRNA. The
Hep3B/ϱ0 cells were transfected with control or Snail
siRNA and incubated for 48 h. Transfection of Snail siRNA
significantly blocked Snail expression in the Hep3B/ϱ0
cells, whereas transfection of control siRNA had no effect
(Fig. 3F). Moreover, vimentin
expression was also reduced by blocking Snail expression.
Additionally, to examine the effect of Snail
downregulation on the Hep3B/ϱ0 cell migration,
Hep3B/ϱ0 cells were transfected with Snail siRNA. After
48 h of incubation, an in vitro wounding migration assay was
performed with the siRNA-transfected Hep3B/ϱ0 cells.
Although the control siRNA had no effect on migration, the
migration activity was reduced in the Hep3B/ϱ0 cells
transfected with Snail siRNA (Fig.
3G).
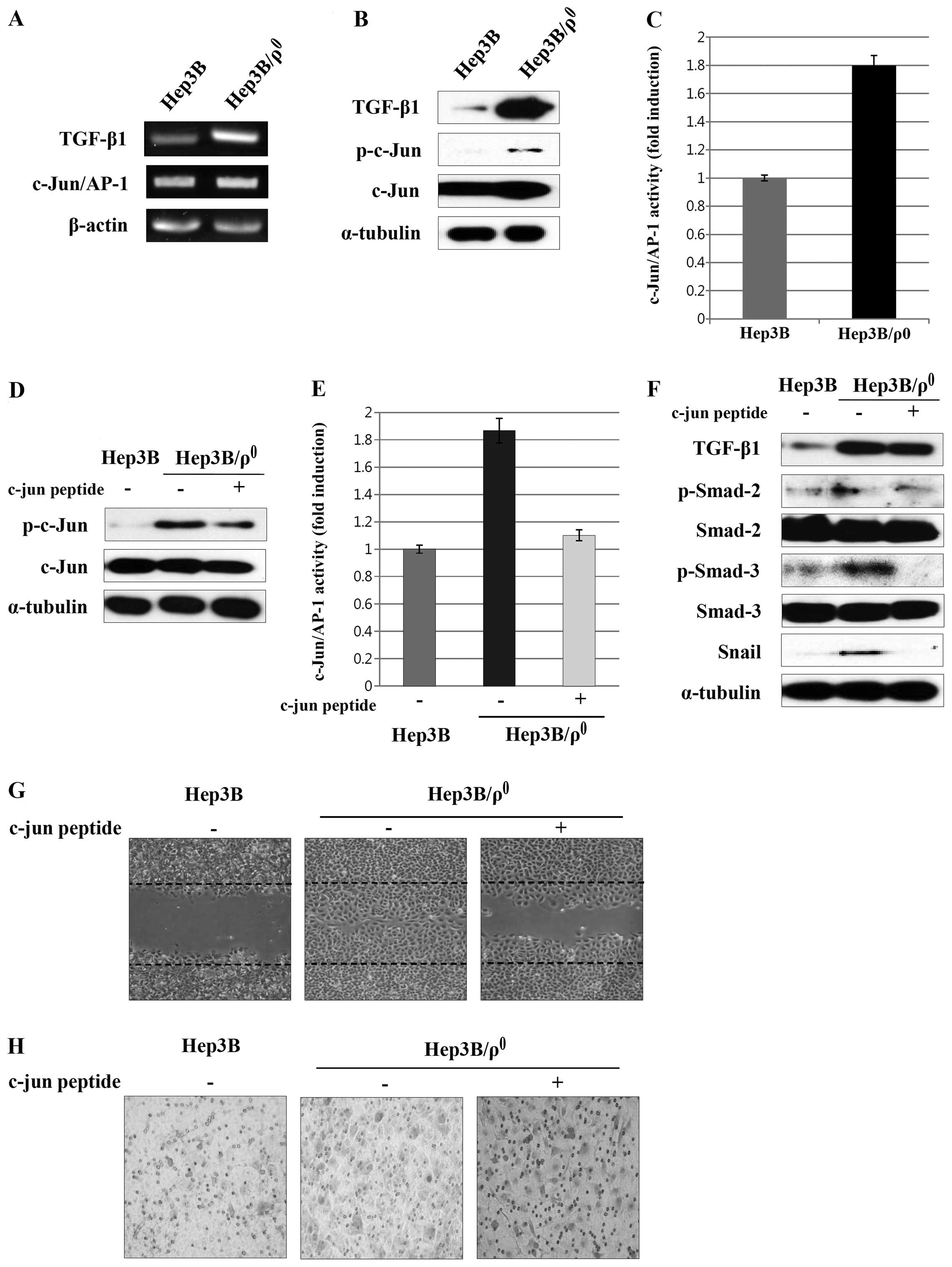
c-Jun/AP-1 activation triggers the
TGF-β/Smad/Snail signaling pathway and EMT phenotypes in the
Hep3B/ϱ0 cells
In previous experiments, we confirmed that the EMT
process induced by mitochondrial dysfunction occurs through
TGF-β/Smad/Snail signaling. Several studies have shown that TGF-β
can be increased by c-Jun/AP-1 activation (20). We examined the expression of
c-Jun/AP-1 in order to determine whether c-Jun/AP-1 is involved in
the increased TGF-β expression by mitochondrial dysfunction in the
Hep3B/ϱ0 cells. We observed that c-Jun/AP-1 expression
was upregulated and c-Jun phosphorylation was increased (Fig. 4A and B). In addition, when we
measured the activity of c-Jun/AP-1, we found that c-Jun/AP-1
activity in the Hep3B/ϱ0 cells was about two times
higher than that in parental cells (Fig. 4C).
We also examined the effect of c-Jun peptide on the
expression of c-Jun/AP-1 by adding c-Jun peptide directly to the
binding reaction. Phosphorylation of c-Jun was reduced by the c-Jun
peptide in the Hep3B/ϱ0 cells (Fig. 4D). To further verify these results,
we analyzed the effect of c-Jun peptide on c-Jun/AP-1 activation by
ELISA. As expected, the c-Jun/AP-1 activity in the
Hep3B/ϱ0 cells was disturbed by the c-Jun peptide
(Fig. 4E).
Then we examined the effect of blocking c-Jun/AP-1
activation by the c-Jun peptide on the TGF-β/Smad/Snail signaling
and EMT phenotypes in the Hep3B/ϱ0 cells. Expression of
the TGF-β was reduced by treatment with the c-Jun peptide.
Phosphorylation of Smad2 and Smad3 and expression of Snail was also
reduced (Fig. 4F). Additionally,
blocking of c-Jun/AP-1 activation reduced migration and
invasiveness (Fig. 4G and H).
Taken together, these results suggest that induction of EMT
phenotypes by the TGF-β/Smad/Snail signaling pathway is dependent
on c-Jun/AP-1 activation in the Hep3B/ϱ0 cells.
Discussion
Cancer cells mainly depend on glycolysis for
production of energy they need. Otto Warburg defined the phenotype
as ‘aerobic glycolysis’ and proposed that the glycolytic phenotype
of cancer cells could be the result of mitochondrial defects.
Recent reports showed point mutations and deletions in mtDNA from a
wide range of tumor cells (21–24).
In particular, the frequency of mtDNA mutations markedly increased
in both non-cancerous and cancerous liver specimens from patients
with HCC compared to control liver tissues (2). It was, however, unclear whether these
mutations are simply the consequence of increased oxidative stress,
known to occur during tumor progression, or they have any direct
role in the progression of cancer.
EMT is a mechanism in which epithelial cells
differentiate into mesenchymal cells, and it plays an important
role in tumor formation and progression to metastatic carcinomas
(25,26). During EMT events, epithelial cells
lose cell polarity and show spindle-like morphology (27). The EMT process produces distinct
features, including disruption of epithelial cell markers, which
contain E-cadherin and β-catenin (28). Furthermore, EMT increases the
expression of various mesenchymal markers such as vimentin,
N-cadherin, and elevates cell invasion and metastasis (29).
Natio et al (30) reported that mitochondrial depletion
could induce EMT by c-Raf/MEK/Erk signaling in breast cancer cells.
Thus, we constructed a mitochondrial-depleted cell model with Hep3B
to determine the relationship between mitochondrial dysfunction and
EMT. In contrast to the parental cells, Hep3B/ϱ0 cells
are highly migratory and invasive (Fig. 1E and F). The expression pattern of
E-cadherin and vimentin also indicates EMT features of the
Hep3B/ϱ0 cells (Fig. 2A and
B).
The TGF-β signaling pathway is emerging as an
attractive target for cancer treatment (31). Blockade of TGF-β signaling may thus
be a promising target of therapeutic strategies for advanced
cancers. Many studies have demonstrated that blocking of TGF-β
action by SB-431542 inhibits tumor migration and metastasis
(32). The EMT features mediated
by TGF-β signaling in the Hep3B/ϱ0 cells were confirmed
with SB-431542 treatment. The migration and invasion were inhibited
by SB-431542 (Fig. 3A and B).
Additionally, increased vimentin expression was reversed by
treatment with SB-431542 in the Hep3B/ϱ0 cells. During
EMT, the expression of E-cadherin is known to be controlled by
Snail (33). Thus, we expected
that the expression of E-cadherin would be restored by SB-431542.
However, contrary to our expectation, the expression of E-cadherin
was not restored by SB-431542. As yet, we do not know why the
E-cadherin expression was not restored by SB-431542. Other factors
in addition to the TGF-β/Smad signaling may also be responsible for
the reduced expression of E-cadherin in the Hep3B/ϱ0
cells.
AP-1 has also been implicated in playing a pivotal
role during development and carcinogenesis (34,35).
The critical role of the c-Jun/AP-1 in the regulation of TGF-β
expression has been demonstrated in various cells (36). Expression of the c-Jun/AP-1 was
reported to be amplified by TGF-β during carcinogenesis (37). As shown in Fig. 4, the EMT induction by
TGF-β/Smad/Snail signaling in the Hep3B/ϱ0 cells depends
on c-Jun/AP-1 activation.
Recently it was reported that mitochondrial
retrograde signaling induces EMT in breast cancer cells (38). It was shown that reduction of mtDNA
activates the Ca2+/calcineurin/Akt-dependent
mitochondrial retrograde signaling pathway. Since Akt
phosphorylation in the Hep3B/ϱ0 cells was rather
downregulated (data not shown), there may be more than one
mechanism for a mitochondrial dysfunctioned cell to acquire EMT
phenotypes.
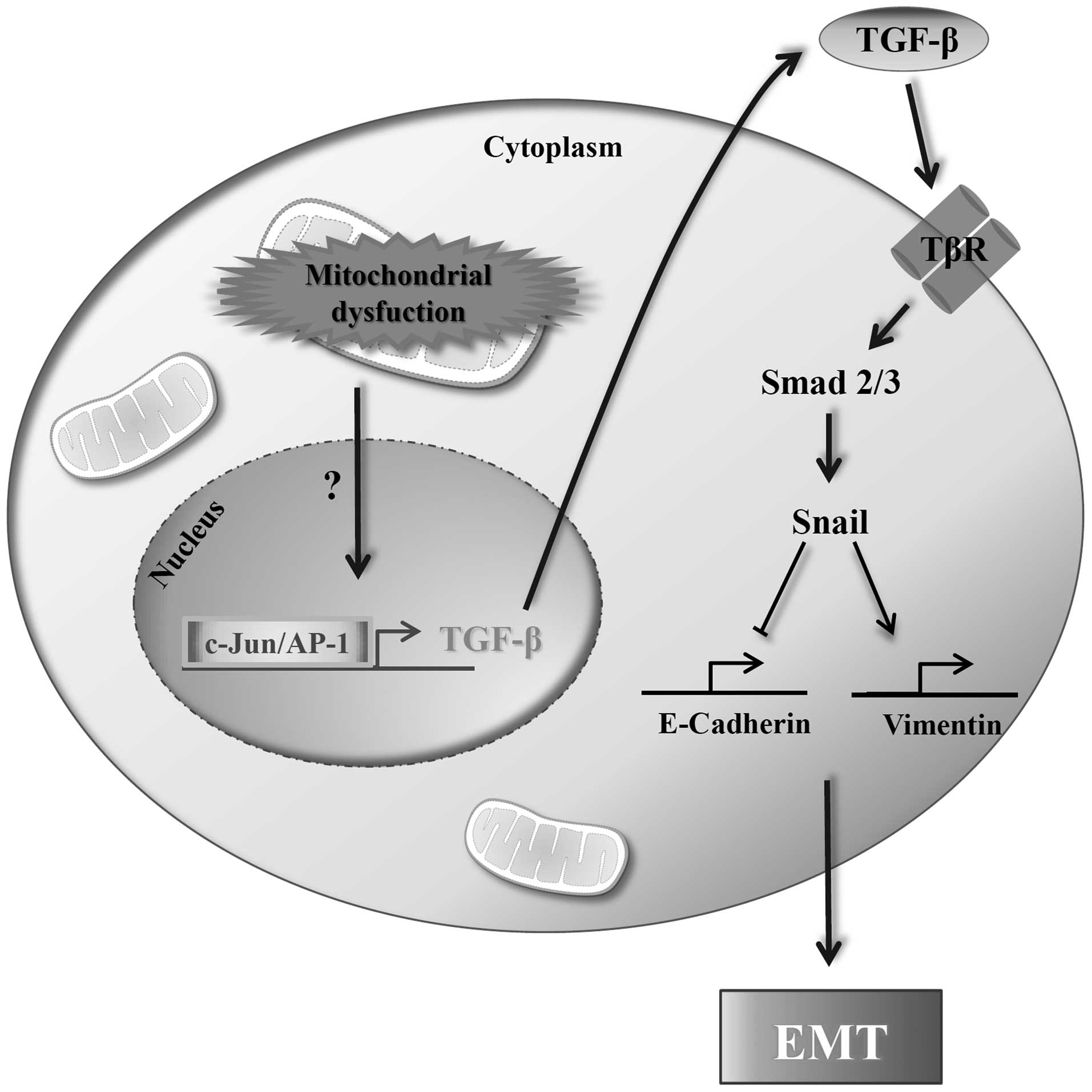
In conclusion, we demonstrated that the
mitochondrial dysfunctioned cell line Hep3B/ϱ0, acquired
EMT features by activation of the TGF-β/Smad/Snail signaling
pathway (Fig. 5). Collectively,
these results indicate that inhibition of TGF-β/Smad/Snail
signaling under conditions of mitochondrial dysfunction may thus be
a potential strategy for therapy of malignant cancers.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea (NRF) Grant funded by the Korean
Government (MOE) (no. 2010-0024330) and by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (NRF-2013R1A1A2062885)
and in part by the Research Fund Program of Genetic Engineering
Institute, Pusan National University, Korea, 2015.
References
|
1
|
Brandon M, Baldi P and Wallace DC:
Mitochondrial mutations in cancer. Oncogene. 25:4647–4662. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nishikawa M, Nishiguchi S, Shiomi S,
Tamori A, Koh N, Takeda T, Kubo S, Hirohashi K, Kinoshita H, Sato
E, et al: Somatic mutation of mitochondrial DNA in cancerous and
noncancerous liver tissue in individuals with hepatocellular
carcinoma. Cancer Res. 61:1843–1845. 2001.PubMed/NCBI
|
|
3
|
Nomoto S, Sanchez-Cespedes M and Sidransky
D: Identification of mtDNA mutations in human cancer. Methods Mol
Biol. 197:107–117. 2002.PubMed/NCBI
|
|
4
|
Wheelhouse NM, Lai PB, Wigmore SJ, Ross JA
and Harrison DJ: Mitochondrial D-loop mutations and deletion
profiles of cancerous and noncancerous liver tissue in hepatitis B
virus-infected liver. Br J Cancer. 92:1268–1272. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Zijl F, Zulehner G, Petz M, Schneller
D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H and Mikulits
W: Epithelial-mesenchymal transition in hepatocellular carcinoma.
Future Oncol. 5:1169–1179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Taub R: Liver regeneration: From myth to
mechanism. Nat Rev Mol Cell Biol. 5:836–847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyazono K, Ehata S and Koinuma D:
Tumor-promoting functions of transforming growth factor-β in
progression of cancer. Ups J Med Sci. 117:143–152. 2012. View Article : Google Scholar :
|
|
10
|
Chandel NS, Maltepe E, Goldwasser E,
Mathieu CE, Simon MC and Schumacker PT: Mitochondrial reactive
oxygen species trigger hypoxia-induced transcription. Proc Natl
Acad Sci USA. 95:11715–11720. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pelicano H, Feng L, Zhou Y, Carew JS,
Hileman EO, Plunkett W, Keating MJ and Huang P: Inhibition of
mitochondrial respiration: A novel strategy to enhance drug-induced
apoptosis in human leukemia cells by a reactive oxygen
species-mediated mechanism. J Biol Chem. 278:37832–37839. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sahai E: Mechanisms of cancer cell
invasion. Curr Opin Genet Dev. 15:87–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wiercinska E, Naber HP, Pardali E, van der
Pluijm G, van Dam H and ten Dijke P: The TGF-β/Smad pathway induces
breast cancer cell invasion through the up-regulation of matrix
metalloproteinase 2 and 9 in a spheroid invasion model system.
Breast Cancer Res Treat. 128:657–666. 2011. View Article : Google Scholar
|
|
17
|
Moustakas A and Heldin CH: The regulation
of TGFbeta signal transduction. Development. 136:3699–3714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
ten Dijke P and Hill CS: New insights into
TGF-beta-Smad signalling. Trends Biochem Sci. 29:265–273. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hjelmeland MD, Hjelmeland AB,
Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS,
Bigner DD, Wang XF and Rich JN: SB-431542, a small molecule
transforming growth factor-beta-receptor antagonist, inhibits human
glioma cell line proliferation and motility. Mol Cancer Ther.
3:737–745. 2004.PubMed/NCBI
|
|
20
|
Sullivan BP, Kassel KM, Manley S, Baker AK
and Luyendyk JP: Regulation of transforming growth
factor-β1-dependent integrin β6 expression by p38 mitogen-activated
protein kinase in bile duct epithelial cells. J Pharmacol Exp Ther.
337:471–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han JS, Choi BS, Yang CW and Kim YS:
Aldosterone-induced TGF-β1 expression is regulated by
mitogen-activated protein kinases and activator protein-1 in
mesangial cells. J Korean Med Sci. 24(Suppl): S195–S203. 2009.
View Article : Google Scholar
|
|
23
|
Warburg O, Wind F and Negelein E: The
metabolism of tumours. J Gen Physiol. 8:519–530. 1927. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chatterjee A, Mambo E and Sidransky D:
Mitochondrial DNA mutations in human cancer. Oncogene.
25:4663–4674. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kalluri R: EMT: When epithelial cells
decide to become mesenchymal-like cells. J Clin Invest.
119:1417–1419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cannito S, Novo E, di Bonzo LV, Busletta
C, Colombatto S and Parola M: Epithelial-mesenchymal transition:
From molecular mechanisms, redox regulation to implications in
human health and disease. Antioxid Redox Signal. 12:1383–1430.
2010. View Article : Google Scholar
|
|
27
|
Schock F and Perrimon N: Molecular
mechanisms of epithelial morphogenesis. Annu Rev Cell Dev Biol.
18:463–493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Acloque H, Adams MS, Fishwick K,
Bronner-Fraser M and Nieto MA: Epithelial-mesenchymal transitions:
The importance of changing cell state in development and disease. J
Clin Invest. 119:1438–1449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naito A, Cook CC, Mizumachi T, Wang M, Xie
CH, Evans TT, Kelly T and Higuchi M: Progressive tumor features
accompany epithelial-mesenchymal transition induced in
mitochondrial DNA-depleted cells. Cancer Sci. 99:1584–1588. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cicchini C, Laudadio I, Citarella F,
Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L and
Tripodi M: TGFbeta-induced EMT requires focal adhesion kinase (FAK)
signaling. Exp Cell Res. 314:143–152. 2008. View Article : Google Scholar
|
|
32
|
Davies M, Robinson M, Smith E, Huntley S,
Prime S and Paterson I: Induction of an epithelial to mesenchymal
transition in human immortal and malignant keratinocytes by
TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell
Biochem. 95:918–931. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SJ, Angel P, Lafyatis R, Hattori K,
Kim KY, Sporn MB, Karin M and Roberts AB: Autoinduction of
transforming growth factor beta 1 is mediated by the AP-1 complex.
Mol Cell Biol. 10:1492–1497. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holzberg D, Knight CG, Dittrich-Breiholz
O, Schneider H, Dörrie A, Hoffmann E, Resch K and Kracht M:
Disruption of the c-JUN-JNK complex by a cell-permeable peptide
containing the c-JUN delta domain induces apoptosis and affects a
distinct set of interleukin-1-induced inflammatory genes. J Biol
Chem. 278:40213–40223. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin WN, Luo SF, Lin CC, Hsiao LD and Yang
CM: Differential involvement of PKC-dependent MAPKs activation in
lipopoly-saccharide-induced AP-1 expression in human tracheal
smooth muscle cells. Cell Signal. 21:1385–1395. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Y, Peng F, Gao B, Ingram AJ and
Krepinsky JC: High glucose-induced RhoA activation requires
caveolae and PKCβ1-mediated ROS generation. Am J Physiol Renal
Physiol. 302:F159–F172. 2012. View Article : Google Scholar
|
|
37
|
Lv ZM, Wang Q, Wan Q, Lin JG, Hu MS, Liu
YX and Wang R: The role of the p38 MAPK signaling pathway in high
glucose-induced epithelial-mesenchymal transition of cultured human
renal tubular epithelial cells. PLoS One. 6:e228062011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guha M, Srinivasan S, Ruthel G, Kashina
AK, Carstens RP, Mendoza A, Khanna C, Van Winkle T and Avadhani NG:
Mitochondrial retrograde signaling induces epithelial-mesenchymal
transition and generates breast cancer stem cells. Oncogene.
33:5238–5250. 2014. View Article : Google Scholar
|