Introduction
Acute myeloid leukemia (AML) is a genetically
heterogeneous disorder, characterized by uncontrolled clonal
proliferation of immature myeloid cells in the bone marrow and
blood with concurrent depletion of effective hematopoiesis
(1,2). The standard treatment of AML with
cytotoxic chemotherapy has remained mostly unchanged over the past
few decades, with dismal clinical outcome (3). One of the key issues of this poor
result is the development of resistance to chemotherapeutic agents,
which can lead to clinical relapse (4). Hence, novel anti-leukemia agents,
especially for relapsed leukemias resistant to existing
chemotherapeutic drugs are needed.
Multidrug resistance (MDR) is a major problem in the
treatment of various human hematological malignancies.
Overexpression of the drug transporter P-glycoprotein (P-gp) and/or
multidrug resistance-associated protein (MRP1) has generally been
reported to correlate with prognosis in AML (5,6).
MRP1, like P-gp belongs to the ATP binding cassette (ABC)
superfamily of membrane transport protein (6). The difference between them is that
the 190-kDa MRP1 can be localized on both the plasma and
intracytoplasmic membranes, which can cause intracellular or
cytoplasmic sequestration of the drug (5). MRP1 overexpression leads to the
failure of numerous chemotherapy protocols when there is without
P-gp overexpression, such as in HL60/A cells (7).
Sirtuins (SIRT1-7), the mammalian homologues of the
silent information regulator 2 (Sir2) in yeast, are members of
nicotinamide adenine dinucleotide (NAD+)-dependent class
III histone deacetylase family proteins (8–10).
Sirtuins have a critical role in genome stability, longevity,
metabolism and aging (9,10). Among them, SIRT1 is the direct
homologue of the yeast Sir2 and has a wide range of cellular
functions (11). Previous studies
have suggested that SIRT1 has oncogenic ability in hematological
malignancies, such as CLL, AML and even can be seen as a
therapeutic target for CML and AML treatment to overcome resistance
(12–14). Little is known about SIRT2, which
is the primary cytoplasmic Sirtuin but shuttles continuously
between the cytoplasmic and nuclear compartments during interphase
and thus, it has been reported to participate in cell cycle
progression, cell differentiation, and oxidative stress by modifing
other proteins and regulating gene expression (15–17,24,29).
SIRT2 has been suggested to participate in glioma, breast, lung
cancer, hepatocellular carcinoma, though, there are few studies in
leukemia (8,18–20).
In the present study, we found that SIRT2 was
overexpressed in the relapsed patients with AML compared to the
newly diagnosed, and both groups were increased from the normal
levels, and the MRP1-mediated multidrug resistance AML cell line
HL60/A could be reversed by inhibition of SIRT2.
Materials and methods
Patients and cell culture
Following informed consent, we collected bone marrow
samples of patients in the Hospital of Blood Diseases. The patients
included 25 children with acute lymphoblastic leukemia
(children-ALL), 30 acute lymphoblastic leukemia (ALL), 10 essential
thrombocythemia (ET), 11 chronic myeloid leukemia (CML), 8
polycythemia vera (PV), 11 myeloproliferative neoplasm (MPN), 25
acute myeloid leukemia (AML) and 6 healthy volunteers (Table I). All the patients were newly
diagnosed, except the 10 relapsed AML patients. Inclusion criteria
for the present study were based on the European Leukemia Net (ELN)
criteria. All the patient samples were treated in accordance with
the Helsinki Declaration.
 | Table ICharacteristics of the patients. |
Table I
Characteristics of the patients.
| Normal | Children-ALL | ALL | ET | CML | PV | MPN | AML |
|---|
| Number of
patients | 6 | 25 | 30 | 10 | 11 | 8 | 11 | 25 |
| Age (years) |
| Median | 28 | 6 | 34 | 40 | 39 | 40 | 32 | 37 |
| Range | 9–36 | 2–13 | 19–53 | 25–55 | 5–79 | 27–54 | 28–50 | 1–60 |
| Gender |
| Female | 2 | 11 | 9 | 3 | 4 | 4 | 5 | 11 |
| Male | 4 | 14 | 21 | 7 | 7 | 4 | 6 | 14 |
HL60 cells were preserved by our laboratory and its
MRP-mediated multidrug resistance HL60/A cells were provided by the
Pharmacology Laboratory at Institute of Hematology, Chinese Academy
of Medical Sciences. They were cultured in RPMI-1640 (Gibco-BRL
Life Technologies, Inc., Burlington, ON, Canada) containing 10%
fetal bovine serum (FBS; HyClone Laboratories, Logan, UT, USA), 100
U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humid
atmosphere with 5% CO2. HL60/A cells were grown in the
presence of 1 μg/ml doxorubicin (DOX) to maintain resistance
activity, which was removed 1 week before each assay.
Quantitative real-time PCR and western
blot analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen, Grand Island, NY, USA) and converted to cDNA by using
the SuperScript II RT (Invitrogen). Premier software 5 was used to
design the primers for real-time quantitative PCR. Human GAPDH
primers used as an internal control were 5′-GAAGGTGAAGGTCGGAGTC-3′
(forward) and 5′-GAA GATGGTGATGGGATTTC-3′ (reverse). Human SIRT2
primers were 5′-ACGCTGTCGCAGAGTCAT-3′ (forward) and 5′-CGCTCCAGG
GTATCTATGTT-3′ (reverse). Real-time quantitative PCR was performed
with SYBR-Green PCR kit (Takara, Shiga, Japan) on the ABI PRISM
7500 Sequence detection system. Thermal cycling conditions were
95°C for 30 sec, followed by 40 cycles of 5 sec at 95°C, and 34 sec
at 60°C. PCR reactions were performed in a total volume of 20 μl,
containing 2 μl of sample cDNA, 0.2 μM of each primer and the
SYBR-Green PCR kit following the manufacturer's instructions. The
expression level of SIRT2 was analyzed by the RQ value calculated
through 2−ΔΔCt method [ΔΔCt - (CtSIRT2
-CtGAPDH)sample - (CtSIRT2 -
CtGAPDH)calibrator]. Data are the means ± SD
of at least 3 independent experiments performed in triplicate.
Total protein was extracted using RIPA lysis buffer
with 1 mM PMSF (Sigma, St. Louis, MO, USA) and other protease
inhibitors. Protein concentration was determined by the BCA assay
(Beijing Solarbio Science and Technology Co., Ltd., Beijing, China)
according to the manufacturer's instructions. The total cell
lysates were heat-denatured at 100°C for 10 min before being run on
8–12% gradient SDS-PAGE. After SDS-PAGE, the proteins were
transferred onto polyvinylidene difluoride membranes (Millipore,
Bedford, MA, USA). Blots were incubated with the indicated
antibodies, incubated in secondary antibody and then developed by
using enhanced chemiluminescence detection reagent (GE Healthcare,
Buckinghamshire, UK) and determined by densitometric analysis with
a Lynx video densitometer (Biological Vision Inc., San Mateo, CA,
USA). For western blot analysis, we purchased antibodies against
GAPDH (sc-365062), SIRT2 (sc-20966), multidrug-resistance protein 1
(MRP1) (sc-7773), anti-phosphospecfic ERK1/2 (sc-101760) and
non-phosphorylated ERK1/2 (sc-135900) from Santa Cruz Biotechnology
(Santa Cruz, CA, USA); p53 (9282), BCL-2 (2872) and Apoptosis
Antibody Sampler kit (9915) from Cell Signaling Technology
(Danvers, MA, USA).
Lentiviral shRNA vectors and
transduction
Two different SIRT2-specific targeting sequences
were designed with software from Ambion. The hairpins were
synthesized and cloned into the lentiviral expression vector. The
two shRNA vectors and empty vector were transfected respectively
with packaging vectors into 293T producer cells using
Lipofectamine™ 2000 (Invitrogen) according to the protocol provided
by the company. The supernatants were harvested after culturing for
48 h and concentrated by ultracentrifugation. Packaged SIRT2-shRNAs
and the control vector were transfected into HL60/A cells
separately. The transfectants were selected with puromycin and
screened under a fluorescence microscope to observe GFP expression.
SIRT2 knockdown efficiency was measured by real-time PCR and
western blot analysis.
Lentiviral vector construction for SIRT2
overexpression and transfection
The complete coding region of SIRT2 gene was
amplified by PCR and cloned into the lentiviral expression vector
pCDH-CMV-MCS-EF1-GreenPuro-CD512B-1. The transfection protocol is
described above.
Immunofluorescence assay
Cells with indicated treatments were fixed with 4%
paraformaldehyde for 30 min and permeabilized with 0.5% Triton for
15 min. Cells were washed with ice-cold PBS, blocked with 0.5% BSA
in PBS for 30 min and then incubated with a rabbit polyclonal
antibody against SIRT2, followed by TRITC-conjugated goat
anti-rabbit IgG antibody and stained nuclei with DAPI. The images
were then visualized with Leica TCS SP2 confocal laser microscope
(Perkin-Elmer, Waltham, MA, USA).
Measurement of intracellular
doxorubicin
HL60/A, HL60 cells and their transfectants were
treated with DOX 10 μmol/l in serum-free RPMI-1640 for 1 h and
washed twice by PBS before imaging on Leica TCS SP2 confocal LAS
for 1 h before imaging on Leica TCS SP2 confocal laser microscope
(Perkin-Elmer). In another group, cells were incubated for 1.5 h in
serum-free RPMI-1640 containing doxorubicin 10 μmol/l and analyzed
by flow cytometry.
Cytotoxicity assay and analysis of
apoptosis
Cells were seeded into 96-well culture plates at a
density of 1×105 cells/ml and serial concentration of
daunorubicin (DNR) and arabinocytidine (Ara-C) were, respectively,
added in a final volume of 100 μl/well for 48 h. The cytotoxic
effects of drugs were determined by VersaMax microplate reader
(Molecular Devices, Sunnyvale, CA, USA) at 570 nm.
The apoptosis of the cells with or without DNR and
Ara-C were measured with Annexin V-APC/PI apoptosis analysis kit
(Tianjin Sungene Biotech Co., Ltd., Tianjin, China) as recommended
by the manufacturer and analyzed by flow cytometry.
Statistical analysis
Each experiment was repeated at least three times.
The data were summarized and presented as mean ± SD. The
statistical analyses were performed with Student's t-test using
GraphPad Prism software (GraphPad Prism, San Diego, CA, USA).
P<0.05 was considered as statistically significant.
Results
Expression of SIRT2 in leukemia
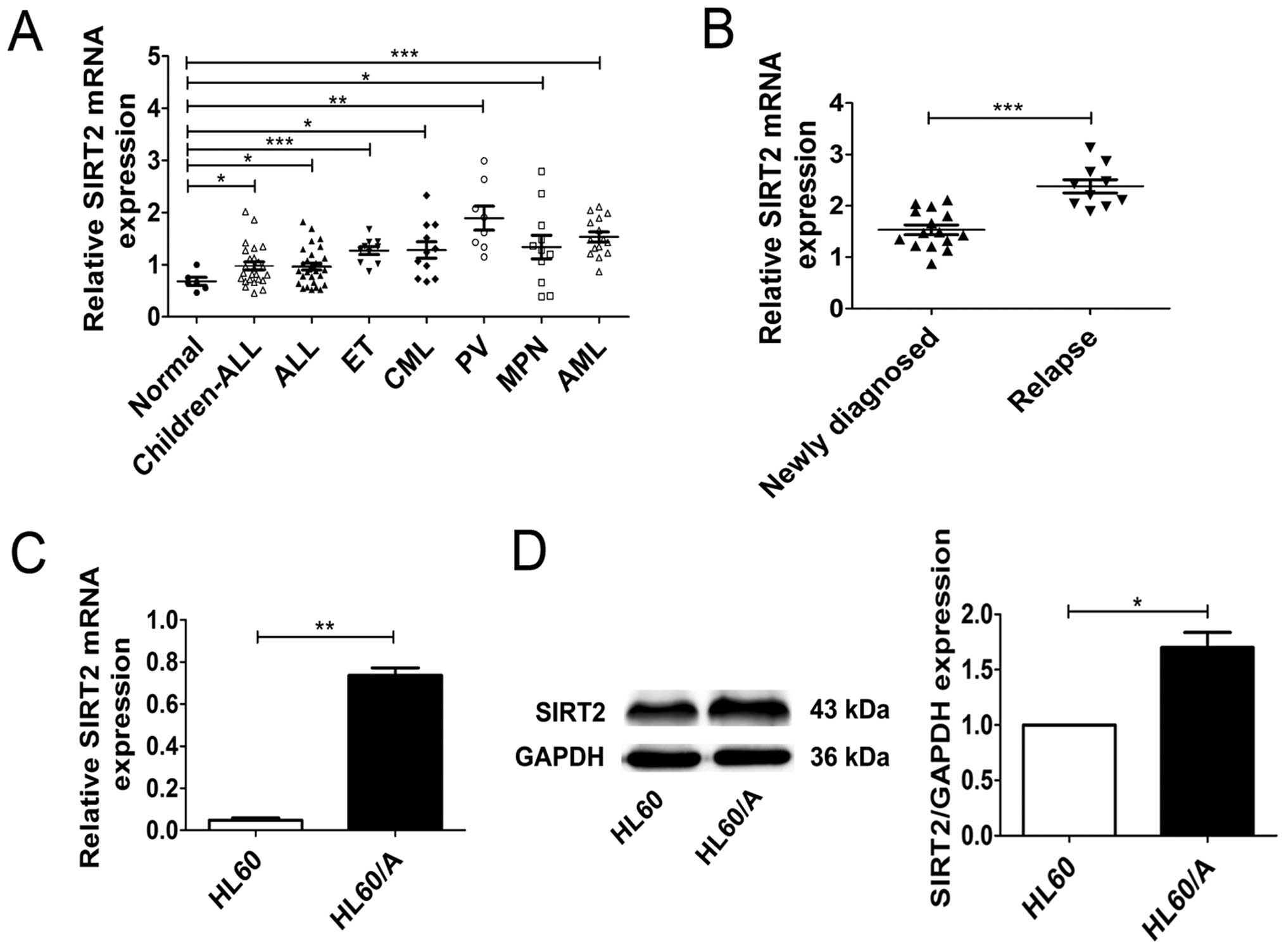
We first compared SIRT2 expression of patients with
different leukemias by real-time PCR. The real-time PCR analysis
revealed that the expressions of SIRT2 in leukemia patients was
higher than that in healthy subjects, although the degree of
discrepancy varied (Fig. 1A).
Then, we detected the expressions of SIRT2 in 10 relapsed AML
patients and 15 newly diagnosed AML patients and the result showed
that the expressions of SIRT2 in the relapsed AML patients were
higher than that in the newly diagnosed patients (Fig. 1B). In addition, we also detected
the expression of SIRT2 in HL60 acute myeloid leukemia cell lines
and its MRP-mediated multidrug resistance HL60/A cells through
real-time PCR and western blotting. Compared with SIRT2 expression
in HL60 cells, SIRT2 level in HL60/A was higher (Fig. 1C and D). Both on the level of mRNA
and protein the results showed that the expression of SIRT2 in HL60
cells and HL60/A cells were similar to our previous findings. These
data not only indicate that the expression of SIRT2 in leukemia
patients is higher than that in healthy volunteers, but also
suggest that the overexpression of SIRT2 may associate with
multidrug resistance in AML.
SIRT2 knockdown decreased the MRP1 level,
enhanced drug accumulation and sensitivity to DNR and Ara-C
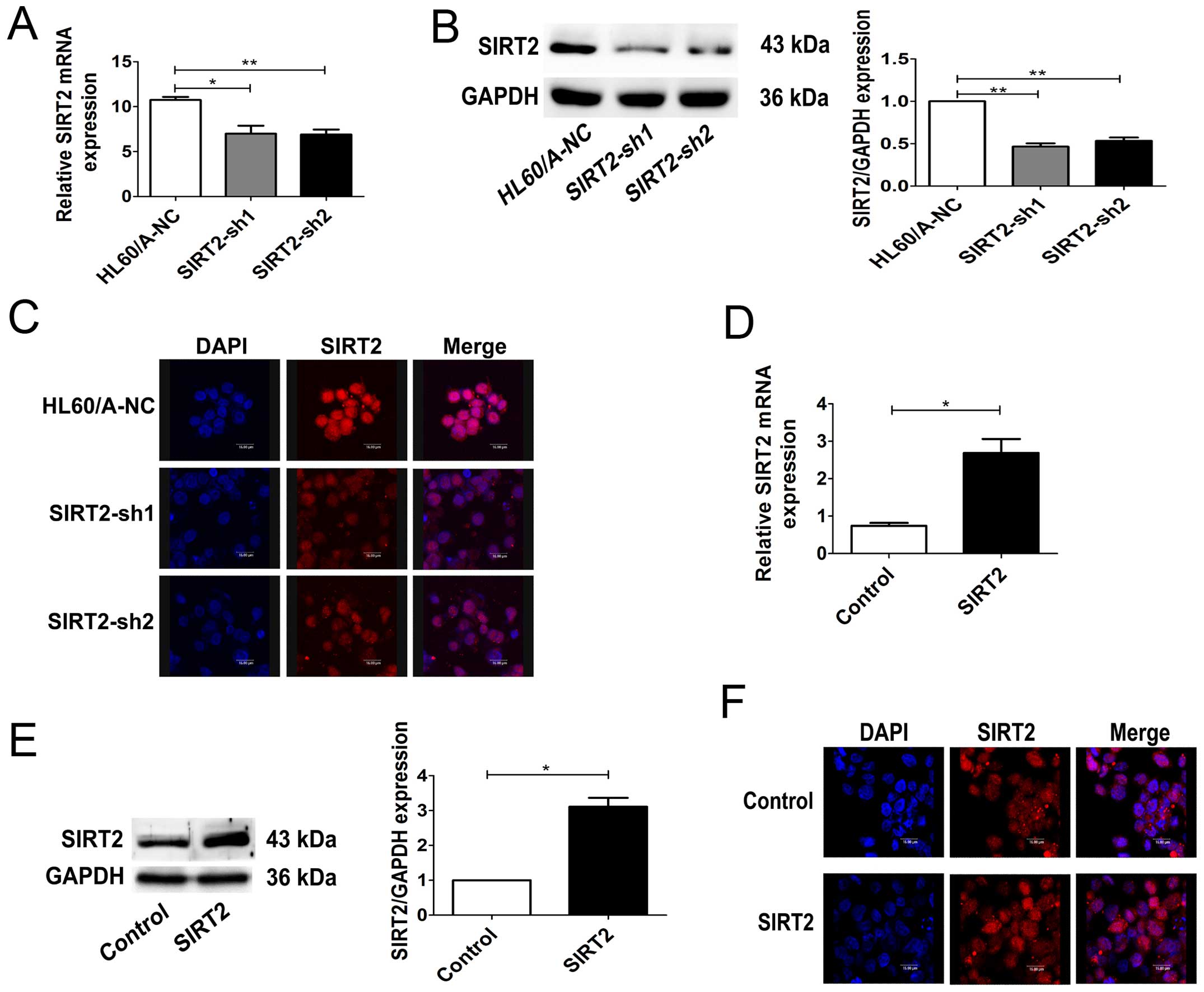
Two different SIRT2 specific shRNA plasmids were
constructed and transfected into HL60/A cells, which had a
relatively higher expression level of SIRT2. As expected, stable
transfection experiments showed both shRNA plasmids could
significantly inhibit SIRT2 expression in HL60/A cells in mRNA and
protein levels (Fig. 2A and B). As
both cytoplasmic and nuclear SIRT2 exist, the expression of SIRT2
in the HL60/A cells after transfection was detected by a confocal
laser microscope. The results showed that the SIRT2 expression was
significantly decreased, similarly to the results of western
blotting (Fig. 2C).
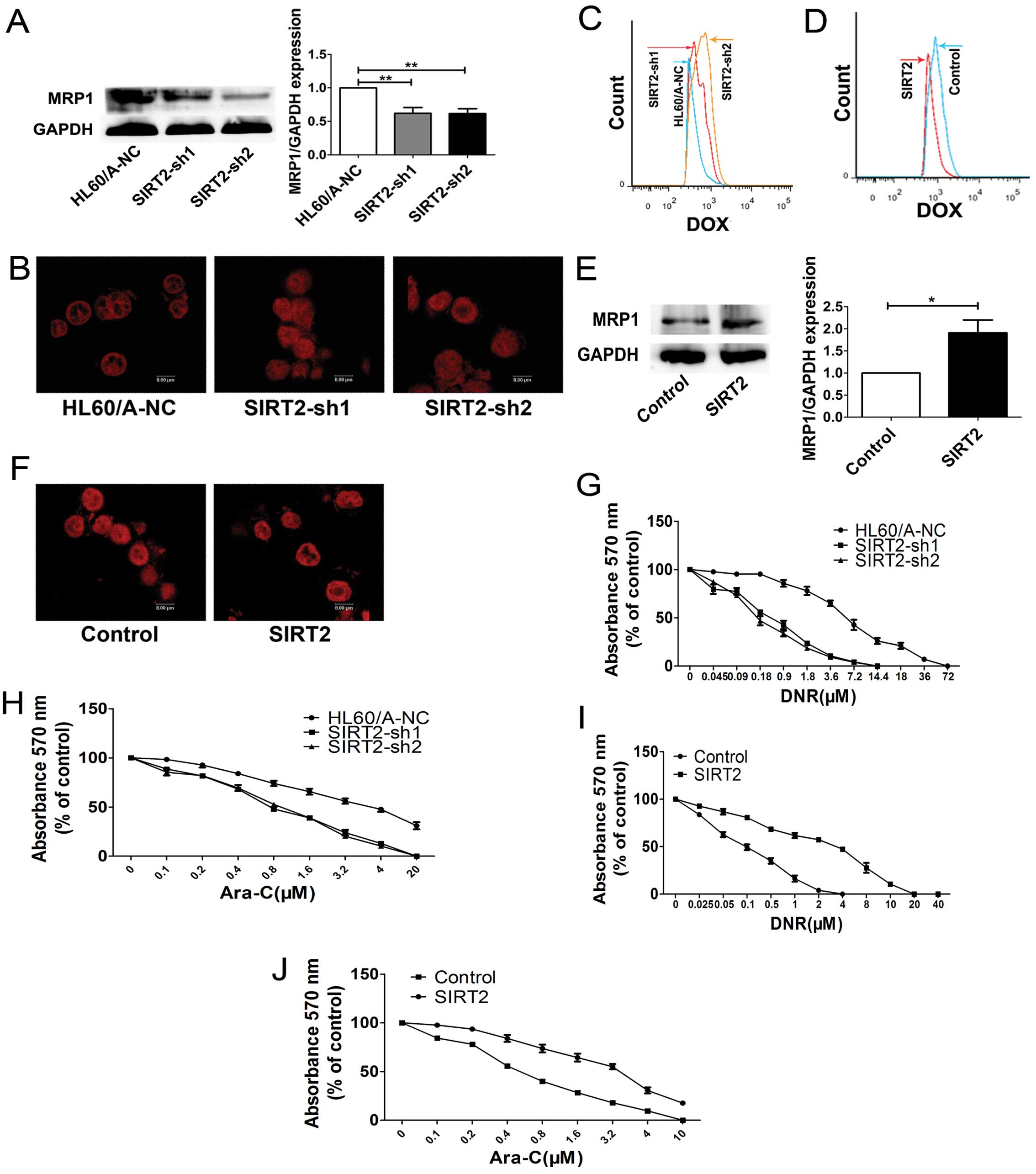
We detected the MRP1 protein, which is known as a
multidrug efflux pumps, and we found the MRP1 expression decreased
after inhibition of SIRT2 in HL60/A cells (Fig. 3A).
After downregulation the expression of SIRT2, in
HL60/A cells showed increased uptake of DOX compared with the
control group HL60/A-NC cells (Fig.
3B). Flow cytometry was also used to measure the intracellular
fluorescent of DOX, and similar result was obtained (Fig. 3C).
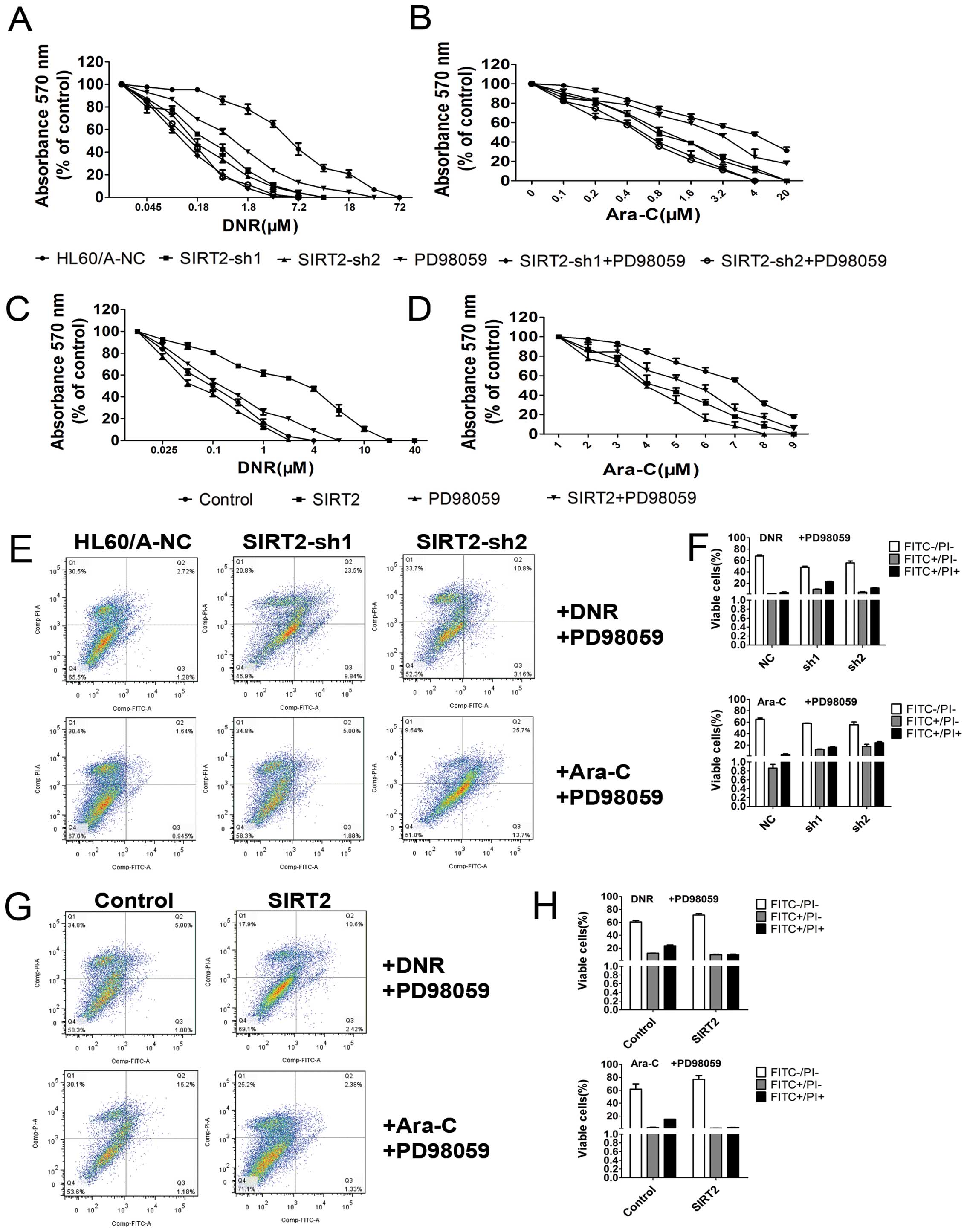
We evaluated the sensitivity of the HL60/A cells to
DNR and Ara-C after the SIRT2 was downregulated. The cells were
assessed for viability on exposure to DNR and Ara-C, respectively,
by using MTT assay and a clear enhancement of sensitivity to DNR
and Ara-C was observed in the HL60/A cells when the expression of
SIRT2 was downregulated (Fig. 3G and
H).
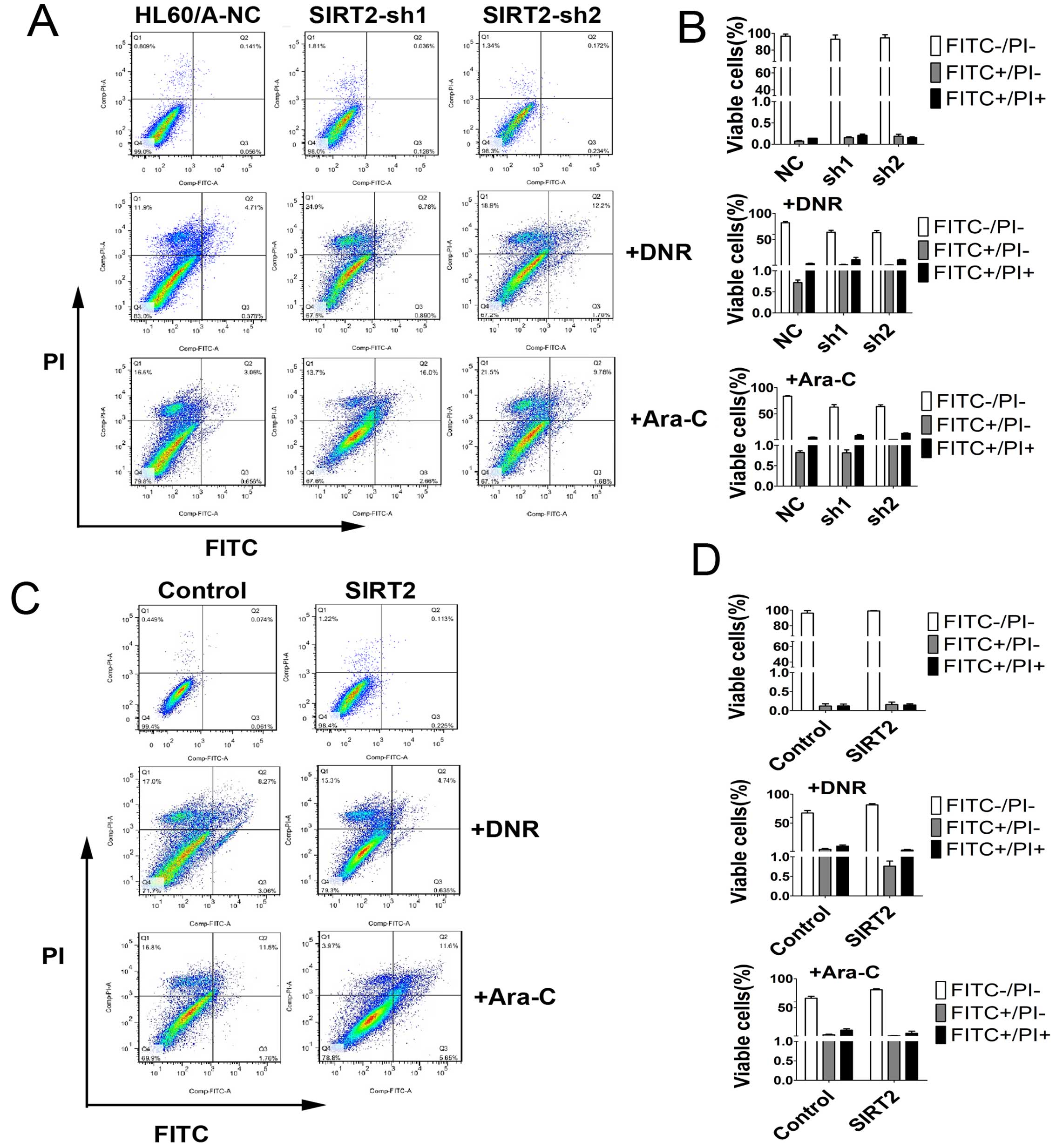
In order to detect and quantify apoptosis, we used
Annexin V-FITC/PI double staining to determine the effect of SIRT2
knockdown on apoptosis in HL60/A cells with or without DNR or Ara-C
treatment. The results showed that SIRT2 downregulation increased
slightly the percentage of apoptotic cells, but clearly only after
incubation with 0.1 μM DNR or 0.5 μM Ara-C for 48 h compared with
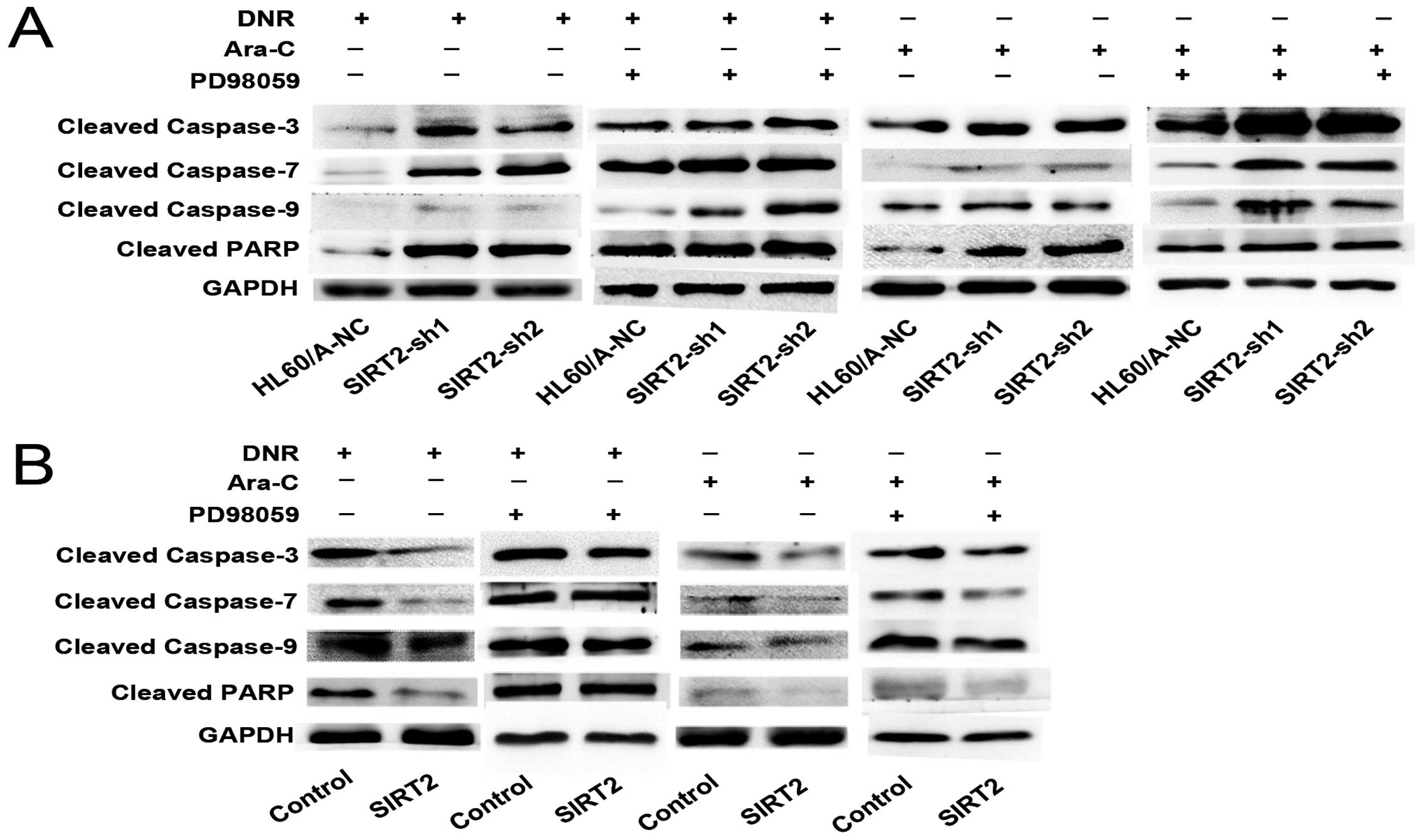
HL60/A-NC (Fig. 4A and B).
Caspase family proteins have been demonstrated as
key mediators of apoptosis, and their activation is taken as a
hallmark of apoptosis. We find that the expression of cleaved
caspase-3, -7 and -9 and PARP were higher in SIRT2-sh1 and
SIRT2-sh2 cells than that in HL60/A-NC after DNR or Ara-C treatment
(Fig. 5A). These data indicated
that knockdown of SIRT2 enhanced the sensitivity to DNR and Ara-C
in HL60/A cells.
SIRT2 overexpression increases MRP1 level
attenuated drug accumulation and sensitivity to DNR and Ara-C
In order to further study the effect of SIRT2
expression on multidrug resistance, we transfected HL60 cells with
SIRT2-overexpression-plasmid and observed an obviously increased
expression of SIRT2 in mRNA and protein levels (Fig. 2D and E). The expression of SIRT2 in
the HL60 cells after transfection was also detected by a confocal
laser microscope (Fig. 2F). The
MRP1 expression increased after overexpression of SIRT2 (Fig. 3E).
Confocal laser microscope was used to detect the
accumulation of the drug in the cells after SIRT2 overexpression.
As expected, after upregulation the expression of SIRT2, in HL60
cells showed decreased uptake of DOX compared with the HL60-control
cells (Fig. 3F). We also measured
the intracellular fluorescence of DOX by using flow cytometry and
obtained similar results (Fig.
3D). The cytotoxicity assay result showed that the sensitivity
to DNR and Ara-C was attenuated in the HL60 cells when the
expression of SIRT2 was upregulated (Fig. 3I and J). The overexpression of
SIRT2, decreased the percentage of apoptotic cells with incubation
of DNR and Ara-C (Fig. 4C and D),
less cleaved caspase-3, -7 and -9 and PARP protein expression was
observed in contrast to HL60-control (Fig. 5B). These results indicated that
SIRT2 overexpression enhanced the resistance to DNR and Ara-C in
HL60 cells, which had a relatively lower expression level of SIRT2,
compared to the multidrug resistance HL60/A cells.
The ERK1/2 signaling pathway is involved
in SIRT2-mediated multidrug resistance in AML cell lines
We next analyzed the mechanism changing the
multidrug resistance effects of the SIRT2 expression in the HL60/A
and HL60 cell lines. In our previous study we demonstrated that the
MAPKs signaling pathways participate in mediation of the imatinib
resistance in CML (21). In the
present study, we detected the expression level of ERK1/2, an
upstream regulator of p53 as well as BCL-2, an anti-apoptotic
protein expressed in most cases of AML and may contribute to drug
resistance in AML (22,23). It has been observed that
downregulation of both SIRT1 and SIRT2 can enhance the sensitivity
of chemotherapy in mammalian cells via p53-independent mechanism
(24), thus, we also evaluated
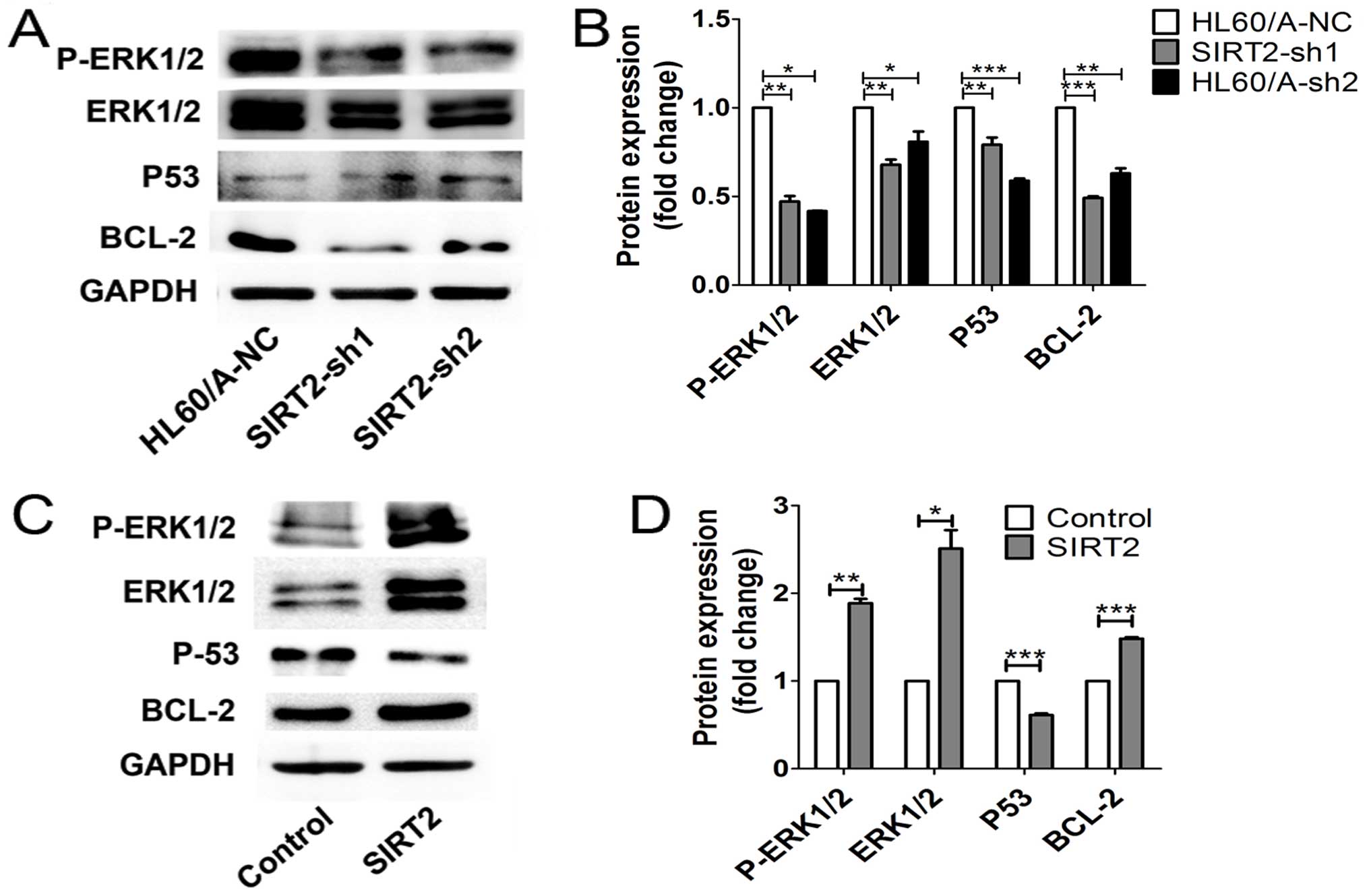
p53. We found that the expression of ERK1/2 and its phosphorylation
as well as BCL-2 were downregulated, while p53 was upregulated in
the SIRT2-depleted HL60/A cells (Fig.
6A and B). In contrast, overexpression of the SIRT2 in HL60
cells upregulated ERK1/2 and its phosphorylation and BCL-2,
decreased the p53 level (Fig. 6C and
D). Thus, these results implied the activation of ERK1/2
signaling might be important to this process.
To further measure the potential influence of
manipulating ERK1/2 signaling pathway to this change of multidrug
resistance induction exerted by the level of SIRT2, we took
advantage of PD98059, a specific MEK1 pharmacological inhibitor, to
block its activation and then measured the changes affected by
SIRT2 depletion or overexpression. The inhibitor added to the
HL60/A cells with SIRT2 depletion did not alone influence the
viability of cells on exposure to DNR (P<0.05) and Ara-C
(P<0.05). Pre-incubation with PD98059 additionally increased the
sensitivity of HL60/A (Fig. 7A and
B; P<0.05). The apotosis results show that pre-incubation
with PD98059 additionally increased the percentage of apoptotic
cells under incubation with DNR and Ara-C (Fig. 7E and F) as well as the caspase
family proteins (Fig. 5A).
Moreover, the inhibitor was added to the HL60 cells with SIRT2
overexpression, we found that pre-incubation with PD98059 abolished
the resistance changes to DNR and Ara-C of HL60 cells (Fig. 7C and D; P<0.05), reversed the
apoptosis changes under treatment with DNR and Ara-C (Fig. 7G and H) and the changes of caspase
family proteins, which was caused by SIRT2 overexpressing HL60
cells after treatment with DNR or Ara-C (Fig. 5B). To further elucidate the role of
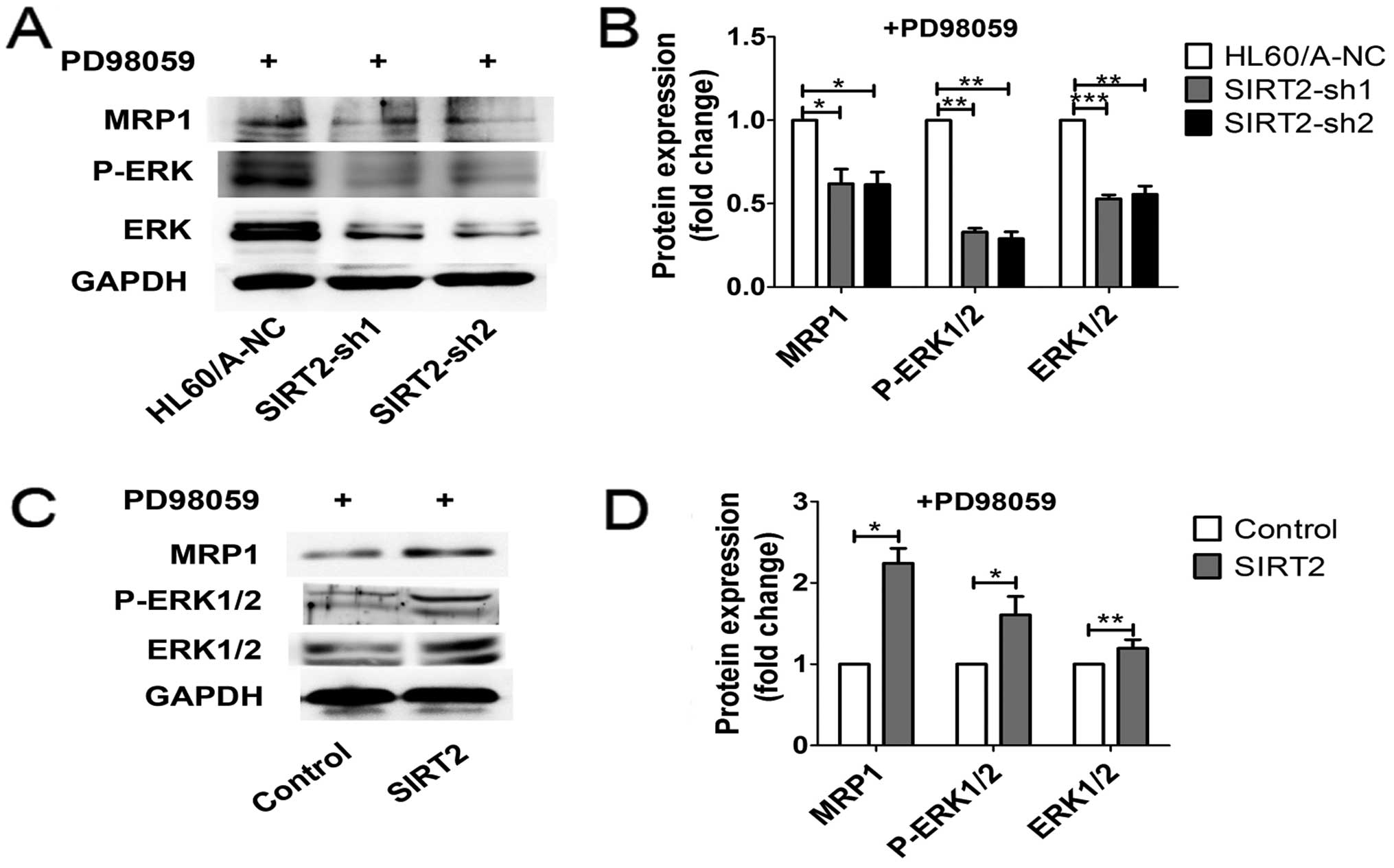
ERK1/2 MAPK signaling in the regulation of MRP1 expression in AML
cell lines, we analyzed MRP1 levels in the presence of PD98059 (50
μM). MRP1 expression was reduced in HL60/A cells and HL60 cells,
additionally decreased in HL60/A cells with SIRT2 depletion and
reversed in HL60 cells with SIRT2 overexpression (Fig. 8). According to these results, we
suggest that ERK1/2 signaling pathway is involved in these changes
of multidrug resistance induction exerted by the level of
SIRT2.
Discussion
Sirtuins, a complex family of proteins (SIRT1-7),
are enzymes that require nicotinamide adenine dinucleotide
(NAD+) to catalyze their reactions are present in
organisms ranging from bacteria to plants to animals (8–10).
Impressive recent progress has been shown with SIRT1, one of the
most extensively studied sirtuins. It has been reported to
deacetylate the histones H1, H3 and H4 and regulate transcription
factors such as tumor suppressor p53, NF-κB and Foxo3a (12–14,24–27).
Unlike SIRT1, SIRT2 is mostly found in the cytoplasm and
deacetylates a number of cytoplasmic proteins, such as α-tubulin
(28). In addition to this, SIRT2
has also been observed in the nucleus and target histones, NF-κB,
p53 and FOXO transcription factors (9,16,17).
A recent study showed that SIRT2 expression is significantly
upregulated in leukemic cells from AML patients and higher in the
patients with high-risk AML, which demonstrates the involvement of
SIRT2 in the resistance against chemotherapy (29).
In the present study, we not only demonstrated that
SIRT2 expression was higher in the AML patients but also higher in
other leukemia such as ALL, ET, CML, PV and MPN patients compared
to healthy volunteers. As expected, we demonstrated that SIRT2 was
higher expressed in the relapsed patients suffering from AML than
the newly diagnosed AML patients. The results on the expression of
SIRT2 in the AML were consistent with the aforementioned report and
further demonstrated that SIRT2 participates in the resistance
against chemotherapy. To further understand this phenomenon, we
used the AML cell line HL60 and its multidrug resistance cell line
HL60/A as the cell model to develop the present study. First, we
detected the expression of SIRT2 in HL60 and HL60/A cells and the
result was consistent with in the patients. Based on this, we
constructed two kinds of lentiviral vectors, one for knockdown of
SIRT2 in the HL60/A cells and the other for overexpression of SIRT2
in the HL60 cells. We demonstrated that SIRT2-silencing HL60/A
cells show more sensitivity to DNR and Ara-C. We observed that more
DOX accumulated in SIRT2-silenced HL60/A cells than HL60/A-NC
cells. The results we observed in the SIRT2 overexpressing HL60
cells were consistent with the SIRT2-silenced HL60/A cells, the
SIRT2 overexpression enhanced the drug resistance ability in HL60
cells and more DOX was discharged. All of the data showed that
SIRT2 enhanced to multidrug resistance in AML.
To gain more insights into how SIRT2 mediated AML
cell multidrug resistance, we measured the activities of multiple
signaling pathways after changing the SIRT2 level. We found that
SIRT2 knockdown significantly downregulated the phosphorylation of
ERK1/2. Overexpression of SIRT2 led to upregulation of the
phosphorylation of ERK1/2. To further investigate whether the
multidrug resistance of AML cells was mediated by SIRT2 though
ERK1/2 pathway, the MEK1 specific inhibitor PD98059 was used. As
shown in the results, inhibition of ERK1/2 by chemical inhibitor
indeed assisted the SIRT2 effects. These results implied that SIRT2
mediated the multidrug resistance of AML cells though ERK1/2
signaling pathway.
Furthermore, the expression of p53 and BCL-2
proteins were evaluated. The classical function of p53 is
regulating apoptosis of both the cytosol and mitochondria. These
functions involve a direct interaction of p53 with members of the
BCL2 family and allowing p53 to function as a so-called
BCL2-homology domain-3 (BH3)-only protein (30,31).
BCL2 family proteins have a central role in the intrinsic apoptotic
pathway. BAX and BAK promote apoptosis by regulating mitochondrial
membrane potential, BCL2 and BCL-Xl negatively regulate them,
whereas, the BCL2-homology domain-3 (BH3)-only proteins including
p53 control these survival proteins (32). Based on the above, a number of p53
inhibitors have been considered as a patron to protect the normal
tissues from many of the deleterious side effects of chemotherapies
(33). Another report have shown
that the balance of Mdm2/p53 ratio is involved in the sensitization
in human breast-, endometrial- and hepatocellular-carcinoma cell
lines via EGFR-Raf-MEK-ERK1/2-Mdm2 signaling pathway (34). We hypothesized that
ERK1/2-Mdm2-p53-BCL2 may be a pathway related to the multidrug
resistance in AML cell lines. However, this hypothesis needs
further study.
In conclusion, our results demonstrated that lower
expression of SIRT2 declines the multidrug resistance in
chemoresistant AML cells. SIRT2 affects the multidrug resistance of
AML cell lines via ERK1/2 signaling pathway. These findings shed
light on the effect of SIRT2 on the multidrug resistance of AML
cells and implied that targeting SIRT2 may yield therapeutic
efficacy in the treatment of leukemia diseases.
Acknowledgements
The present study was supported by grants from the
Natural Science Key Program Foundation of Tianjin Technology
Commission of China (grant no. 14JCZDJC34900), the National Natural
Science General Program Foundation of China (no. 81170510) and the
Major Program Foundation of China (no. 81090410).
References
|
1
|
Löwenberg B, Downing JR and Burnett A:
Acute myeloid leukemia. N Engl J Med. 341:1051–1062. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Estey E and Döhner H: Acute myeloid
leukaemia. Lancet. 368:1894–1907. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DiNardo CD and Cortes JE: New treatment
for acute myelogenous leukemia. Expert Opin Pharmacother.
16:95–106. 2015. View Article : Google Scholar
|
|
4
|
Vyas P, Appelbaum FR and Craddock C:
Allogeneic hematopoietic cell transplantation for acute myeloid
leukemia. Biol Blood Marrow Transplant. 21:8–15. 2015. View Article : Google Scholar
|
|
5
|
Ross DD: Novel mechanisms of drug
resistance in leukemia. Leukemia. 14:467–473. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Legrand O, Zittoun R and Marie JP: Role of
MRP1 in multidrug resistance in acute myeloid leukemia. Leukemia.
13:578–584. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Niewiarowski W, Gendaszewska E, Rebowski
G, Wójcik M, Mikołajczyk B, Goss W, Soszyński M and Bartosz G:
Multidrug resistance-associated protein - reduction of expression
in human leukaemia cells by antisense phosphorothioate
olignucleotides. Acta Biochim Pol. 47:1183–1188. 2000.
|
|
8
|
Li Z, Xie QR, Chen Z, Lu S and Xia W:
Regulation of SIRT2 levels for human non-small cell lung cancer
therapy. Lung Cancer. 82:9–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harting K and Knöll B: SIRT2-mediated
protein deacetylation: An emerging key regulator in brain
physiology and pathology. Eur J Cell Biol. 89:262–269. 2010.
View Article : Google Scholar
|
|
10
|
Guarente L: Frankin H. Epstein Lecture:
Sirtuins, aging, and medicine. N Engl J Med. 364:2235–2244. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peck B, Chen CY, Ho KK, Di Fruscia P,
Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD and Lam EW: SIRT
inhibitors induce cell death and p53 acetylation through targeting
both SIRT1 and SIRT2. Mol Cancer Ther. 9:844–855. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Audrito V, Vaisitti T, Rossi D, Gottardi
D, D'Arena G, Laurenti L, Gaidano G, Malavasi F and Deaglio S:
Nicotinamide blocks proliferation and induces apoptosis of chronic
lymphocytic leukemia cells through activation of the
p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res.
71:4473–4483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bradbury CA, Khanim FL, Hayden R, Bunce
CM, White DA, Drayson MT, Craddock C and Turner BM: Histone
deacetylases in acute myeloid leukaemia show a distinctive pattern
of expression that changes selectively in response to deacetylase
inhibitors. Leukemia. 19:1751–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Wang L, Li L, Wang Z, Ho Y, McDonald
T, Holyoake TL, Chen W and Bhatia R: Activation of p53 by SIRT1
inhibition enhances elimination of CML leukemia stem cells in
combination with imatinib. Cancer Cell. 21:266–281. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
North BJ and Verdin E: Interphase
nucleo-cytoplasmic shuttling and localization of SIRT2 during
mitosis. PLoS One. 2:e7842007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sunami Y, Araki M, Hironaka Y, Morishita
S, Kobayashi M, Liew EL, Edahiro Y, Tsutsui M, Ohsaka A and Komatsu
N: Inhibition of the NAD-dependent protein deacetylase SIRT2
induces granulocytic differentiation in human leukemia cells. PLoS
One. 8:e576332013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang F, Nguyen M, Qin FX and Tong Q: SIRT2
deacetylates FOXO3a in response to oxidative stress and caloric
restriction. Aging Cell. 6:505–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hiratsuka M, Inoue T, Toda T, Kimura N,
Shirayoshi Y, Kamitani H, Watanabe T, Ohama E, Tahimic CG, Kurimasa
A, et al: Proteomics-based identification of differentially
expressed genes in human gliomas: Down-regulation of SIRT2 gene.
Biochem Biophys Res Commun. 309:558–566. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McGlynn LM, Zino S, MacDonald AI, Curle J,
Reilly JE, Mohammed ZM, McMillan DC, Mallon E, Payne AP, Edwards J,
et al: SIRT2: Tumour suppressor or tumour promoter in operable
breast cancer? Eur J Cancer. 50:290–301. 2014. View Article : Google Scholar
|
|
20
|
Chen J, Chan AW, To KF, Chen W, Zhang Z,
Ren J, Song C, Cheung YS, Lai PB, Cheng SH, et al: SIRT2
overexpression in hepatocellular carcinoma mediates epithelial to
mesenchymal transition by protein kinase B/glycogen synthase
kinase-3β/β-catenin signaling. Hepatology. 57:2287–2298. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jin W, Li Q, Lin Y, Lu Y, Li H, Wang L, Hu
R, Ma L, Wang J and Pang T: Reversal of Imatinib resistance in
BCR-ABL-positive leukemia after inhibition of the
Na+/H+ exchanger. Cancer Lett. 308:81–90.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kang MA, Kim MS, Kim JY, Shin YJ, Song JY
and Jeong JH: A novel pyrido-thieno-pyrimidine derivative activates
p53 through induction of phosphorylation and acetylation in
colorectal cancer cells. Int J Oncol. 46:342–350. 2015.
|
|
23
|
Konopleva M, Tari AM, Estrov Z, Harris D,
Xie Z, Zhao S, López-Berestein G and Andreeff M: Liposomal Bcl-2
antisense oligonucleotides enhance proliferation, sensitize acute
myeloid leukemia to cytosine-arabinoside, and induce apoptosis
independent of other antiapoptotic proteins. Blood. 95:3929–3938.
2000.PubMed/NCBI
|
|
24
|
Matsushita N, Takami Y, Kimura M, Tachiiri
S, Ishiai M, Nakayama T and Takata M: Role of NAD-dependent
deacetylases SIRT1 and SIRT2 in radiation and cisplatin-induced
cell death in vertebrate cells. Genes Cells. 10:321–332. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vaquero A, Scher M, Lee D,
Erdjument-Bromage H, Tempst P and Reinberg D: Human SirT1 interacts
with histone H1 and promotes formation of facultative
heterochromatin. Mol Cell. 16:93–105. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yeung F, Hoberg JE, Ramsey CS, Keller MD,
Jones DR, Frye RA and Mayo MW: Modulation of NF-kappaB-dependent
transcription and cell survival by the SIRT1 deacetylase. EMBO J.
23:2369–2380. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakagawa T and Guarente L: Sirtuins at a
glance. J Cell Sci. 124:833–838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
North BJ and Verdin E: Mitotic regulation
of SIRT2 by cyclin-dependent kinase 1-dependent phosphorylation. J
Biol Chem. 282:19546–19555. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dan L, Klimenkova O, Klimiankou M, Klusman
JH, van den Heuvel-Eibrink MM, Reinhardt D, Welte K and Skokowa J:
The role of sirtuin 2 activation by nicotinamide
phosphoribosyl-transferase in the aberrant proliferation and
survival of myeloid leukemia cells. Haematologica. 97:551–559.
2012. View Article : Google Scholar :
|
|
30
|
Vousden KH and Lane DP: p53 in health and
disease. Nat Rev Mol Cell Biol. 8:275–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moll UM, Wolff S, Speidel D and Deppert W:
Transcription-independent pro-apoptotic functions of p53. Curr Opin
Cell Biol. 17:631–636. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cory S and Adams JM: The Bcl2 family:
Regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Komarov PG, Komarova EA, Kondratov RV,
Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV: A chemical
inhibitor of p53 that protects mice from the side effects of cancer
therapy. Science. 285:1733–1737. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ali I, Damdimopoulou P, Stenius U and
Halldin K: Cadmium at nanomolar concentrations activates
Raf-MEK-ERK1/2 MAPKs signaling via EGFR in human cancer cell lines.
Chem Biol Interact. 231:44–52. 2015. View Article : Google Scholar : PubMed/NCBI
|