Introduction
Liver fibrosis may be considered a wound healing
process induced in response to chronic liver injury resulting from
a variety of insults including viruses, autoimmunity, drugs and
metabolic diseases (1,2). As a consequence of fibrosis, there
may be distortion of the liver parenchyma because of nodule
formation, and altered blood flow, which lead to cirrhosis and
liver failure. Liver fibrosis is characterized by the accumulation
of extracellular matrix (ECM) and hepatic stellate cells (HSCs) are
responsible for most of the production and accumulation of collagen
in injured liver although, bone marrow-derived fibrocytes as well
as portal, and septal fibroblasts are also involved in this process
(3,4). Following liver injury, HSCs are
activated, and transition from quiescent cells into proliferative,
fibrogenic, and contractile myofibroblasts. This activation
increases the expression of α-smooth muscle actin (α-SMA), collagen
and tissue inhibitors of metalloproteinases (TIMPs), giving rise to
fibrotic liver (5). A variety of
antifibrotic therapeutic strategies designed to reverse the damage,
including suppression of HSC proliferation, or stimulation of
apoptosis, downregulation of collagen between production and
promotion of collagen degradation, as well as cell therapy using
mesenchymal stem cells, have been tried (6–11).
TGFβ signaling has long been believed to be a
central mediator of the fibrotic response (12). This signaling is mediated by
regulatory SMADs (SMAD2 or SMAD3) which are phosphorylated by the
activated TGFβ receptor complex and form heterodimers with SMAD4.
The dimerization induces translocation of the SMAD complex to the
nucleus where it exerts its regulatory function. As part of the
development of the fibrotic liver, an increased expression of TGFβ
recruits neutrophils, macrophages and fibroblasts, which exacerbate
the liver fibrosis (13,14). Recently, phosphorylation of SMAD2
[Ser245, Ser250, Ser255 and
Thr220 (15)], SMAD3
[Ser203, Ser207 and Thr178
(16)], by growth factor-mediated
ERK activation, has been shown to inhibit the TGFβ-mediated nuclear
translocation of the SMAD complex (17,18).
In addition, SMAD phosphorylation by Akt was shown to inhibit the
nuclear translocation of SMAD, leading to a reduced expression of
TGFβ target genes (19–21). Furthermore, it has been shown that
FGF inhibits the synthesis of type I collagen, the major ECM
component of the fibrotic tissue, at the level of transcription
(22,23).
AIMP1 is a cofactor of the multi-tRNA synthetase
complex (24) that is induced by a
variety of conditions including hypoxia, cytokines and
hypoglycemia, and has agonistic or antagonistic effects on
endothelial and immune cells (25–32).
Recently, we reported that the 6–46 aa domain of secreted AIMP1
activated ERK and Akt via FGFR2, leading to the increased
proliferation of mesenchymal stem cells (33). In the present study, we examined
whether the AIMP1 peptide could inhibit collagen synthesis via ERK
activation in TGFβ-stimulated HSCs and attenuate liver fibrosis in
a mouse model.
Materials and methods
Cell culture
LX2 cells, immortalized human HSCs, were kindly
provided by Professor S.L. Friedman (Liver Disease Research Center
of San Francisco General Hospital, San Francisco, CA, USA) and were
maintained in high-glucose Dulbecco's modified Eagle's medium
(DMEM) supplemented with 2% fetal bovine serum (FBS) and 1%
streptomycin/penicillin. For western blot analysis, LX2 cells were
seeded at ~70% confluence, on 60-mm dishes (2×105 cells)
or 100-mm dishes (7×105 cells) and cultured for 24 h.
After starvation for 12 h (reduction of FBS to 0.5%), LX2 cells
were treated with AIMP1 peptide as indicated. For the ERK
inhibition assay, LX2 cells, starved for 12 h as above, were
treated with AIMP1 peptide in the presence or absence of the
selective inhibitor of MAP kinases, U0126 (10 μM) for 30 min,
before TGFβ (2 ng/ml) was added.
Western blot analysis
For protein extraction, LX2 cells were washed with
cold 1X PBS, and lysed on ice for 30 min in cold RIPA buffer
(Sigma, 50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 10% Glycerol, 1%
Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, 12 mM
β-glycerophosphate, 10 mM NaF, 1 mM NaOV3, 1 mM PMSF, 2
mM β-mercaptoethanol) containing a protease inhibitor cocktail
(Cell Signaling Technology), followed by centrifugation at 15,000
rpm for 20 min. The cells were fractionated into nucleus and
cytoplasm using the anuclear/cytosol fractionation kit (BioVision)
according to the manufacturer's instructions, and confirmed by
western blot analysis using α-tubulin and lamin A/C for cytoplasm
and nucleus, respectively. Approximately 20 μg of total proteins
was loaded and separated by SDS-polyacrylamide gel electrophoresis
(PAGE). Proteins were transferred to polyvinylidene fluoride (PVDF)
membranes using the wet transfer kit (Bio-Rad Laboratories,
Hercules, CA, USA), followed by blocking for 30 min with 5% skim
milk in Tris-buffered saline (TBS) with 0.1% Tween-20 (TBS-T), and
then incubated overnight at 4°C in TBS containing 5% skim milk with
the indicated primary antibodies; anti-pSMAD2 (Ser465/467, 1:1,000;
Cell Signaling Technology), anti-pSMAD3 (Ser423/425, 1:1,000; Cell
Signaling Technology), anti-SMAD2/3 (1:1,000; Cell Signaling
Technology), anti-SMAD4 (1:1,000; Santa Cruz Biotechnology),
anti-collagen type I (1:1,000; Millipore), anti-Lamin A/C (1:1,000;
Abcam) anti-α-Tubulin (1:1,000; Sigma). The membranes were then
washed four times for 10 min in TBS-T solution and incubated with
horseradish peroxidase-conjugated secondary antibodies (0.1 μg/ml;
Santa Cruz Biotechnology). Immunoreactivity bands were detected
using the western blot detection kit (Abc-3001; AbClon), according
to the manufacturer's instructions.
Immunofluorescence
LX2 cells (2×104 cells) were cultured for
24 h on round glass coverslips (VWR LabShop, Batavia, IL, USA) in
24-well plates. After 0.5% FBS starvation for 12 h, the cells were
treated with 5 μg/ml of AIMP1 peptide for 30 min, followed by 2
ng/ml of TGFβ for 1 h. The cells were fixed with 4% formaldehyde
freshly prepared from paraformaldehyde for 10 min and then
permeabilized with PBS supplemented with 0.1% Triton X-100 (PBST)
for 5 min. After washing with PBST three times for 5 min, fixed
cells were incubated for an additional 30 min in PBST containing 5%
BSA to prevent non-specific binding of the antibodies, followed by
an overnight incubation at 4°C with the indicated primary
antibodies. Cells were washed three times with PBST solution, and
then incubated with Alexa 488 and 594-conjugated goat anti-mouse or
anti-rabbit secondary antibodies (Molecular Probes) for 1 h at room
temperature. Nuclear DNA was counterstained with
4′,6′-diamidino-2-phenylindole (DAPI) and the fluorescence was
captured with the Leica confocal microscope TCS SP5 (Leica
Microsystems, Wetzlar, Germany).
Immunoprecipitation
After serum starvation for 4 h, LX2 cells were
treated with AIMP1 peptide (5 μg/ml) in the presence or absence of
U0126 (10 μM). Cells were harvested, lysed with RIPA buffer, and
incubated on ice for 30 min. Whole cell lysates were prepared by
centrifugation at 25,000 × g for 10 min at 4°C. The protein
extracts (300 μg) were incubated with anti-SMAD2/3 antibody (1.5
μg; Cell Signaling Technology) for 4 h at 4°C and then with protein
A agarose for 4 h, also at 4°C. The beads were washed three times
with RIPA buffer without β-mercaptoethanol, and resuspended in 1X
SDS sample buffer. The samples were boiled and loaded into 9%
SDS-PAGE. Phosphorylation of SMAD2/3 was confirmed by pSer antibody
(Abcam).
Luciferase activity assay
LX2 cells (5×104 cells) were seeded on a
12-well plate and cultured in DMEM supplemented with 2% fetal
bovine serum (FBS) and 1% streptomycin/penicillin. The cells were
then co-transfected with a SBE4-Luc vector containing the SMAD
binding element (SBE) and Renilla luciferase vector using
Lipofectamine 2000 (Invitrogen) for 12 h. After serum starvation
with 0.5% FBS for 4 h, LX2 cells were treated with AIMP1 peptide
for 10 min and then TGFβ was added for an additional 20 h.
Luciferase activity was determined using the Dual-Luciferase
Reporter Assay system (Promega) and quantified using GloMax
(Promega) according to the manufacturer's instructions. Luciferase
activity was normalized to Renilla luciferase activity.
RT-PCR
Total RNA was extracted using an RNA isolation kit
(iNtRON Biotechnology, Inc., Seoul, Korea) according to the
manufacturer's instructions. cDNA was prepared by reverse
transcription with 0.5 μg of total RNA. Type I collagen mRNA
(NM_000088.3) (forward, 5′-CCCCTGGAAAGAATGGAGATG-3′ and reverse,
5′-TCCAAACCACTGAAACCTCTG-3′); GAPDH (forward,
5′-CGAGATCCCTCCAAAATCAA-3′ and reverse, 5′-TGTGGTCATGAGTCCTTCCA-3′)
were amplified by real-time PCR, which was performed with the
AccuPower® GreenStar qPCR PreMix (SYBR-Green PreMix;
Bioneer Corp., Daejeon, Korea) and StepOne Real-Time PCR system
(Applied Biosystems). GAPDH was used as an endogenous control.
Animal study
Male BALB/c mice (6 weeks old, weighing 20–22 g)
were obtained from Orient Bio. Animal Inc. (Seoul, Republic of
Korea). Animal care and all experimental procedures were conducted
in accordance with the approval and guidelines of the Inha
Institutional Animal Care and Use Committee (Inha IACUC) of the
Medical School of Inha University (approval ID: 111024-1). The
animals were fed standard chow and tap water ad libitum, and
were maintained with a 12-h dark/light cycle at 21°C. Acute liver
damage was induced by intraperitoneal injection of 40%
CCl4 in corn oil at a single dose of (150 μl) twice a
week for one month. Control animals were treated with the same
volume of corn oil alone. Mice in the scrambled AIMP1 and AIMP1
peptide groups were treated twice-weekly with intraperitoneal
injections for 2 weeks during the CCl4 administration.
All mice were sacrificed by ether anesthesia after 4 weeks of
treatment. The livers were excised and weighed and the specimens
were immediately fixed in 10% neutral buffered formalin for
histochemical studies. Blood samples for biochemical analyses were
obtained by cardiac puncture.
Histopathology
Liver samples fixed in a 10% buffered formaldehyde
solution were processed using a paraffin slice technique. Sections
~4-μm thick were stained with hematoxylin and eosin (H&E) for
routine histological examination. The sections were first stained
with hematoxylin for 3 min, washed, and then stained with 0.5%
eosin for an additional 3 min. For staining of liver fibrosis, the
fixed liver tissue samples were stained with Masson's trichrome
according to standard protocol. After an additional wash with
water, the slides were sequentially dehydrated in 70, 95 and 100%
ethanol and cleared in xylene. The degree of liver damage was
examined in a blinded manner by a pathologist using a light
microscope (Olympus).
Assessment of biochemical parameters
Serum Total-Bilirubin, D-Bilirubin, aspartate
transaminase (AST), and alanine transaminase (ALT) levels were
measured at the Green Cross Reference Lab (Seoul, Republic of
Korea).
Immunohistochemistry
Immunohistochemical staining was performed using
formalin-fixed and deparaffinized tissue sections as previously
described (34,35). After blocking with normal goat
serum (Vector Laboratories, Burlingame, CA, USA) for 1 h, primary
antibodies specific for TGFβ, collagen I, and α-SMA (Sigma-Aldrich)
were used. After removing the unbound primary antibody, the
sections were incubated with secondary antibody in 1.5% horse
serum/PBS at room temperature for 1 h. The sections were visualized
by an avidin-biotin peroxidase complex solution using an ABC kit
(Vector Laboratories). The sections were washed in PBS, developed
with a diaminobenzidine tetrahydrochloride substrate for 15 min and
then the nucleus was counterstained with hematoxylin. Terminal
deoxynucleotidyl transferase-mediated nick end labelling (TUNEL)
was performed using the TUNEL kit (Millipore, Billerica, MA, USA)
according to the manufacturer's instructions.
Statistical analysis
Data are expressed as the mean ± SD, and analyzed
with an ANOVA and unpaired Student's t-test. A P-value ≤0.05 was
considered to indicate a statistically significant result.
Statistical calculations were performed using SPSS software for the
Windows operating system (version 10.0; SPSS, Inc., Chicago, IL,
USA).
Results
The AIMP1 peptide regulates SMAD2
localization via ERK activation
We have previously reported that the AIMP1 peptide
activates MAPKs including ERK (33). In the present study, we used LX2
cells for human HSCs that were spontaneously immortalized in low
serum condition, which express α-smooth muscle actin, vimentin,
glial fibrillary acid protein and other HSC biomarkers upon
activation (36). We first
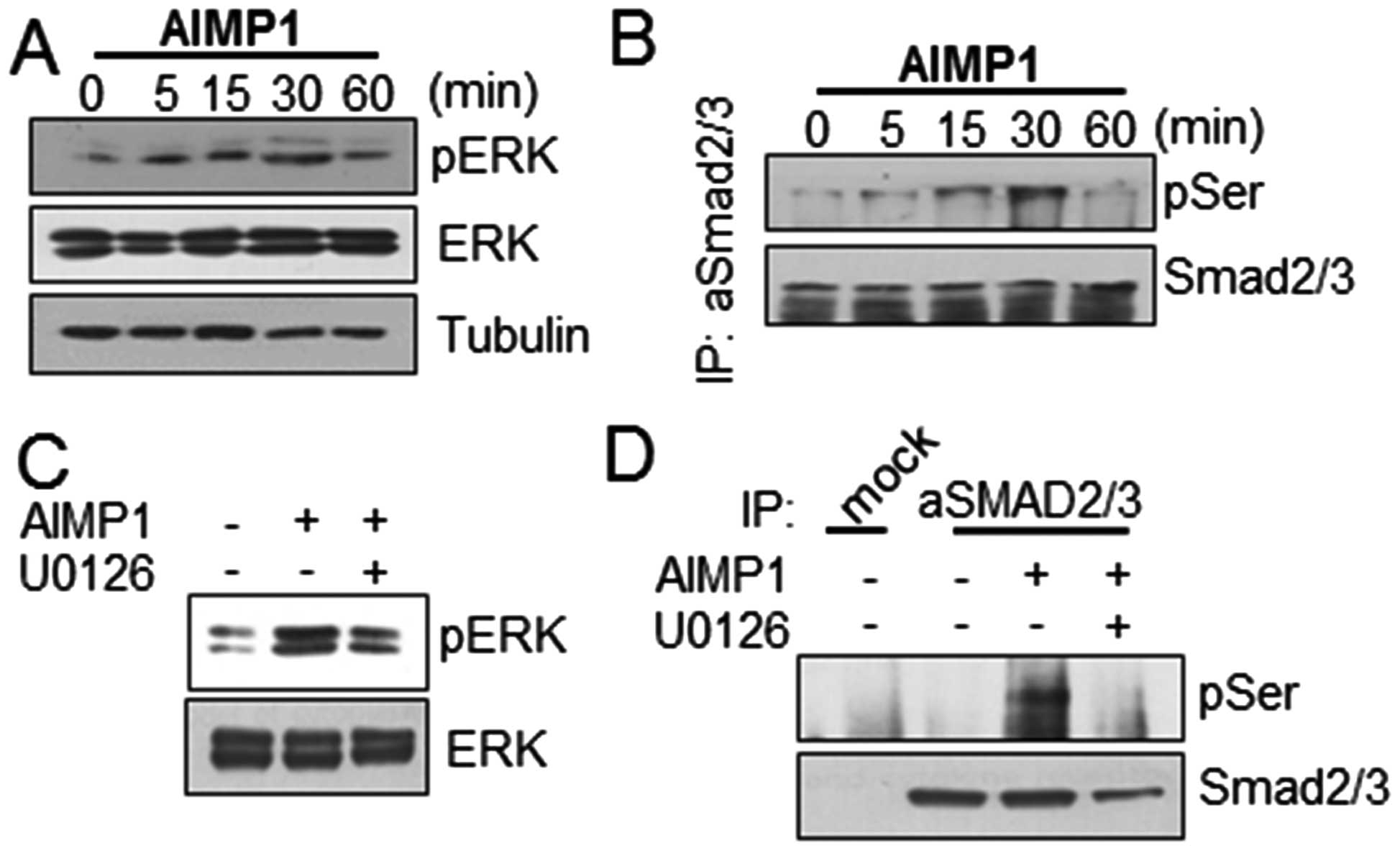
examined whether the AIMP1 peptide could induce ERK phosphorylation
in LX2 cells. AIMP1 peptide increased ERK phosphorylation in a
time-dependent manner (Fig. 1A).
Since the activation of ERK induces TGFβ-independent
phosphorylation of SMAD and inhibits nuclear translocation
(15,18,37),
we examined whether AIMP1-mediated ERK phosphorylation induced
SMAD2/3 phosphorylation. After treatment of LX2 cells with the
AIMP1 peptide as indicated, cell lysates were prepared and the
SMAD2/3 protein was immunoprecipitated using a specific antibody as
described in Materials and methods. The western blot analysis using
anti-phospho-serine antibody, showed that the AIMP1 peptide induced
phosphorylation of SMAD2 in a time-dependent manner (Fig. 1B). To verify whether the
phosphorylation at the serine residue of SMAD2/3 was induced by
AIMP1 peptide-mediated ERK phosphorylation, we treated the cells
with U0126, an ERK inhibitor, and then analyzed the SMAD2/3
phosphorylation. The suppression of ERK phosphorylation by U0126
indeed reduced the AIMP1 peptide-mediated SMAD2 phosphorylation
(Fig. 1C and D).
In order to transduce TGFβ signaling, the TGFβ
receptor, activated by the ligand binding, phosphorylates SMAD2/3.
The pSMAD2/3 then forms a complex with SMAD4, resulting in
translocation to the nucleus, and regulation of a variety of TGFβ
target genes including type I collagen (38). To investigate whether the AIMP1
peptide-mediated ERK activation could inhibit the TGFβ-induced
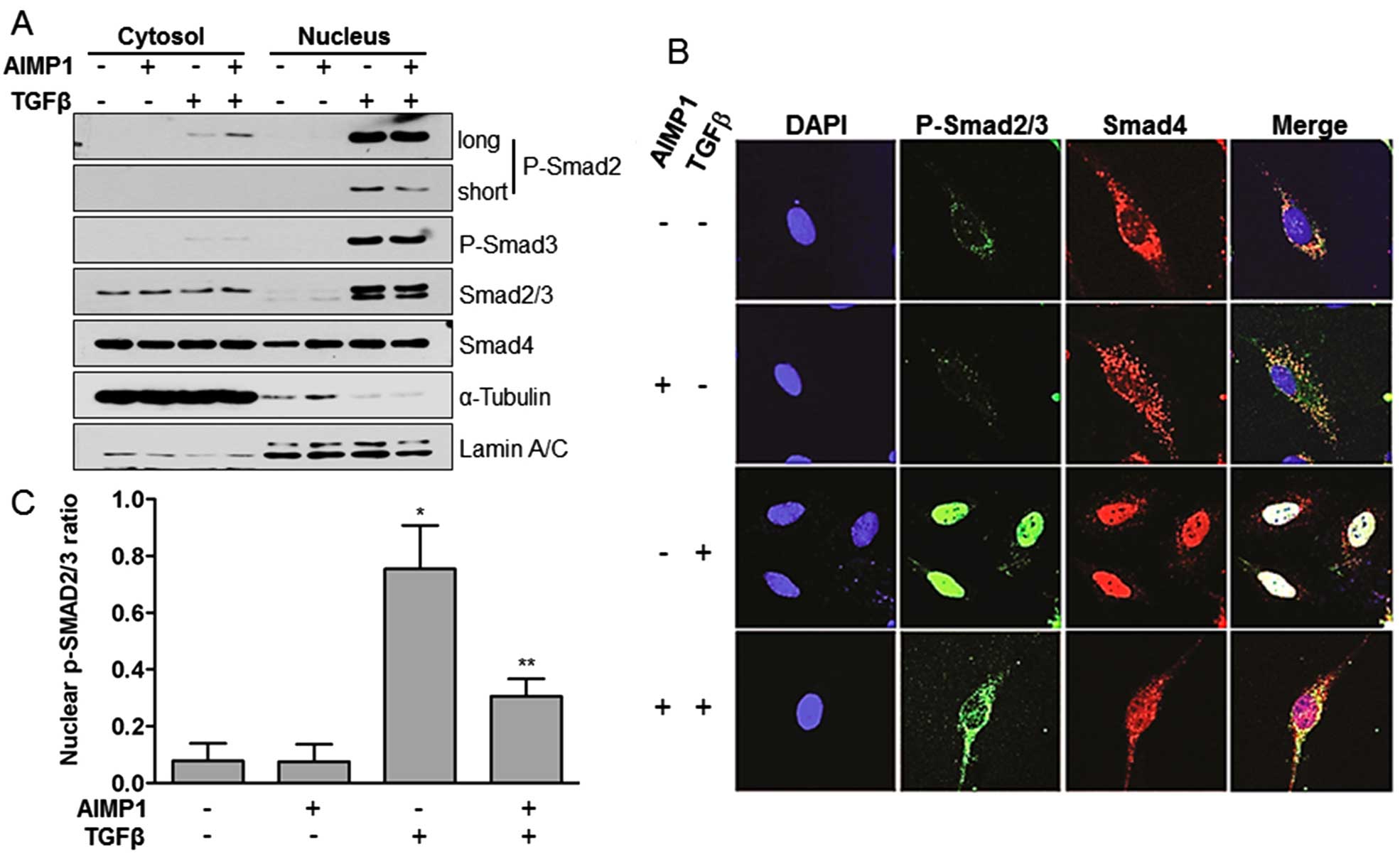
nuclear translocation of pSMAD2/3, we separated whole cell lysates
into the cytoplasmic and nuclear fractions after treatment with
AIMP1 peptide. Analysis of the separate fractions clearly showed
that pre-treatment of LX2 cells with AIMP1 peptide reduced the
nuclear translocation of pSMAD2, but not of pSMAD3 (Fig. 2A). We confirmed this result further
by immunofluorescence staining. TGFβ stimulation of LX2 cells
induced the nuclear translocation of pSMAD2/3 and SMAD4, whereas
pretreatment with AIMP1 peptide inhibited the nuclear translocation
of pSMAD2/3 (Fig. 2B and C). These
results suggest that the AIMP1 peptide inhibits the nuclear
translocation of pSMAD2/3 without affecting the phosphorylation of
SMAD2/3 by TGFβ.
The AIMP1 peptide inhibits the synthesis
of type I collagen
TGFβ has been reported to induce the deposition of
type I collagen at sites of injury in the liver, leading to liver
fibrosis (39–41). Thus, we examined whether the
reduction in the nuclear translocation of pSMAD2/3 seen after
treatment with the AIMP1 peptide could inhibit the TGFβ-mediated
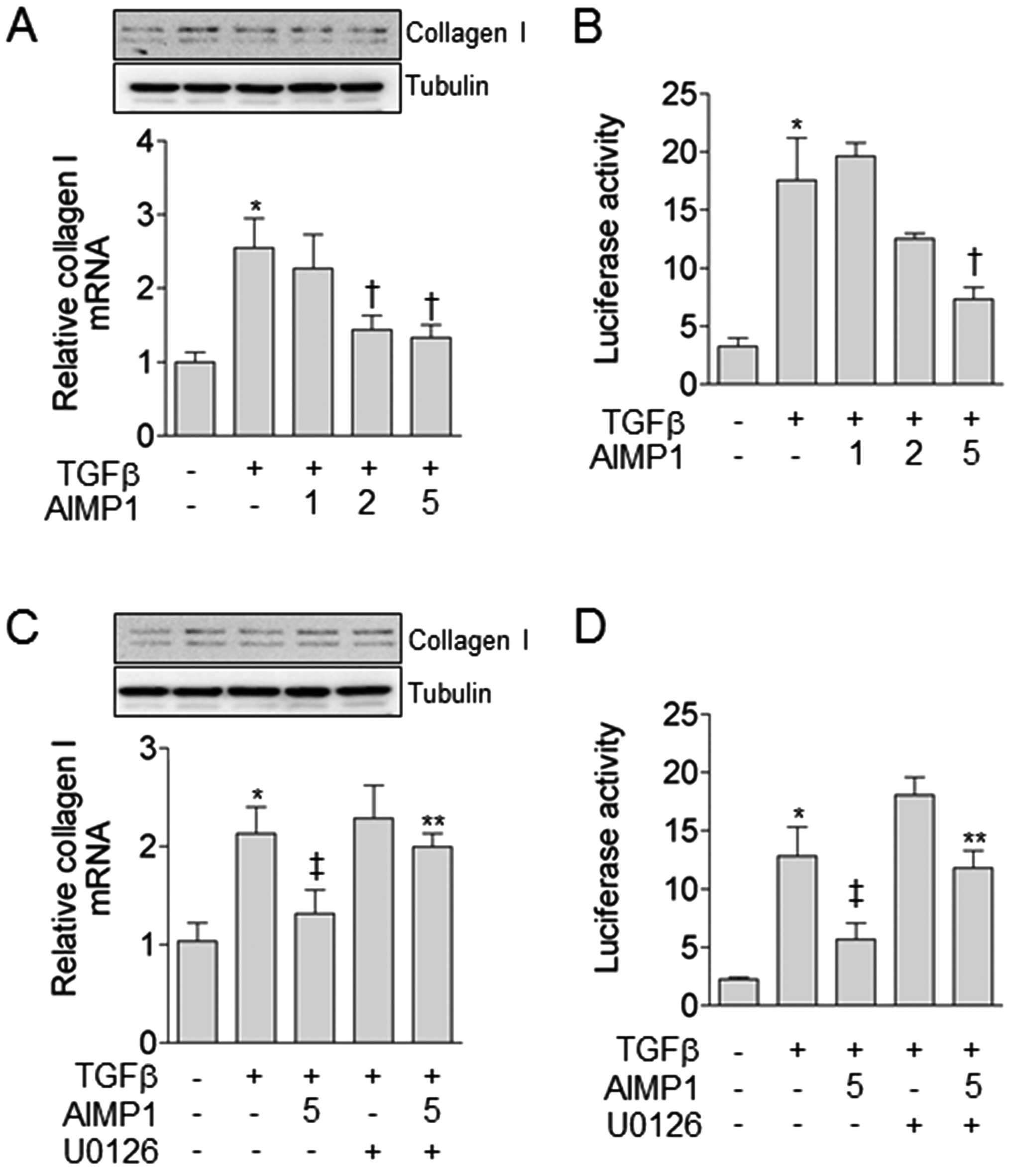
synthesis of type I collagen. Pretreatment with the AIMP1 peptide
decreased type I collagen synthesis in a dose-dependent manner at
the level of both mRNA and protein (Fig. 3A), suggesting that the decreased
expression level of type I collagen was due to the reduced
transcriptional activity of the SMAD complex in the nucleus. This
is a result of the fact that the AIMP1 peptide inhibited the
nuclear translocation of the pSMAD induced by TGFβ. To further
confirm the transcriptional activity of the SMAD complex, we
carried out a luciferase activity assay using the SBE4-Luc vector
containing SMAD binding element (SBE). The AIMP1 peptide attenuated
the TGFβ-mediated increase of luciferase activity in a
dose-dependent manner (Fig. 3B).
In addition, we investigated whether the AIMP1 peptide-mediated
decrease of type I collagen expression was dependent on ERK
activation. As shown in Fig. 3C,
pre-treatment with U0126 abolished the AIMP1 peptide-mediated
reduction of type I collagen expression. The luciferase activity
assay further confirmed that the AIMP1 peptide-mediated decrease of
type I collagen expression is ERK-dependent (Fig. 3D).
The AIMP1 peptide decreases the severity
of liver fibrosis in CCl4-induced mouse model
We next assessed whether the AIMP1 peptide could
have a therapeutic effect in the CCl4-induced mouse
model of liver, by inhibiting the expression of type I collagen
induced by TGFβ. The AIMP1 peptide was administered
intraperitoneally twice weekly as described in Materials and
methods. Histological analysis showed that treatment with the AIMP1
peptide decreased the CCl4-induced liver damage in a
dose-dependent manner (Fig. 4A).
In addition, Masson-Trichrome staining further clearly showed that
collagen deposition was reduced by AIMP1 peptide treatment
(Fig. 4A). The level of liver
toxicity was examined by analysis of blood chemistry parameters.
Treatment with the AIMP1 peptide significantly decreased the levels
of total (T) and indirect (D) bilirubin, which were increased by
CCl4. In addition, the AIMP1 peptide significantly
decreased the levels of alanine aminotransferase (ALT) and
aspartate aminotransferase (AST), suggesting that it could have a
therapeutic effect on liver fibrosis in this model (Fig. 4B and C). To further confirm whether
the therapeutic effect of the AIMP1 peptide is specific, we used
scrambled AIMP1 peptide (sAIMP1) as a negative control, as
previously described for a defective mutant Y24T (42). H&E and Masson-Trichrome
staining showed that the wild-type AIMP1 peptide significantly
attenuated CCl4-induced liver fibrosis, whereas the
sAIMP1 peptide did not, thereby confirming the specificity of the
therapeutic effect (Fig. 4D and
E). In addition, immunohistochemical staining clearly showed
that the AIMP1, but not the sAIMP1, peptide reduced the levels of
expression of α-SMA, TGFβ and collagen (Fig. 4D and E). Furthermore, the AIMP1
peptide decreased hepatic apoptosis induced by CCl4 as
measured by TUNEL (Fig. 4D and E).
Serum markers for liver fibrosis including AST, ALT, albumin and
BUN showed that the AIMP1 peptide significantly improved liver
fibrosis, while the sAIMP1 peptide had no effect (Table I). In conclusion, these results
suggest that the AIMP1 peptide attenuates liver fibrosis by
inhibiting apoptosis induced by CCl4, which, in turn,
leads to decreased collagen deposition, α-SMA activation and TGFβ
accumulation.
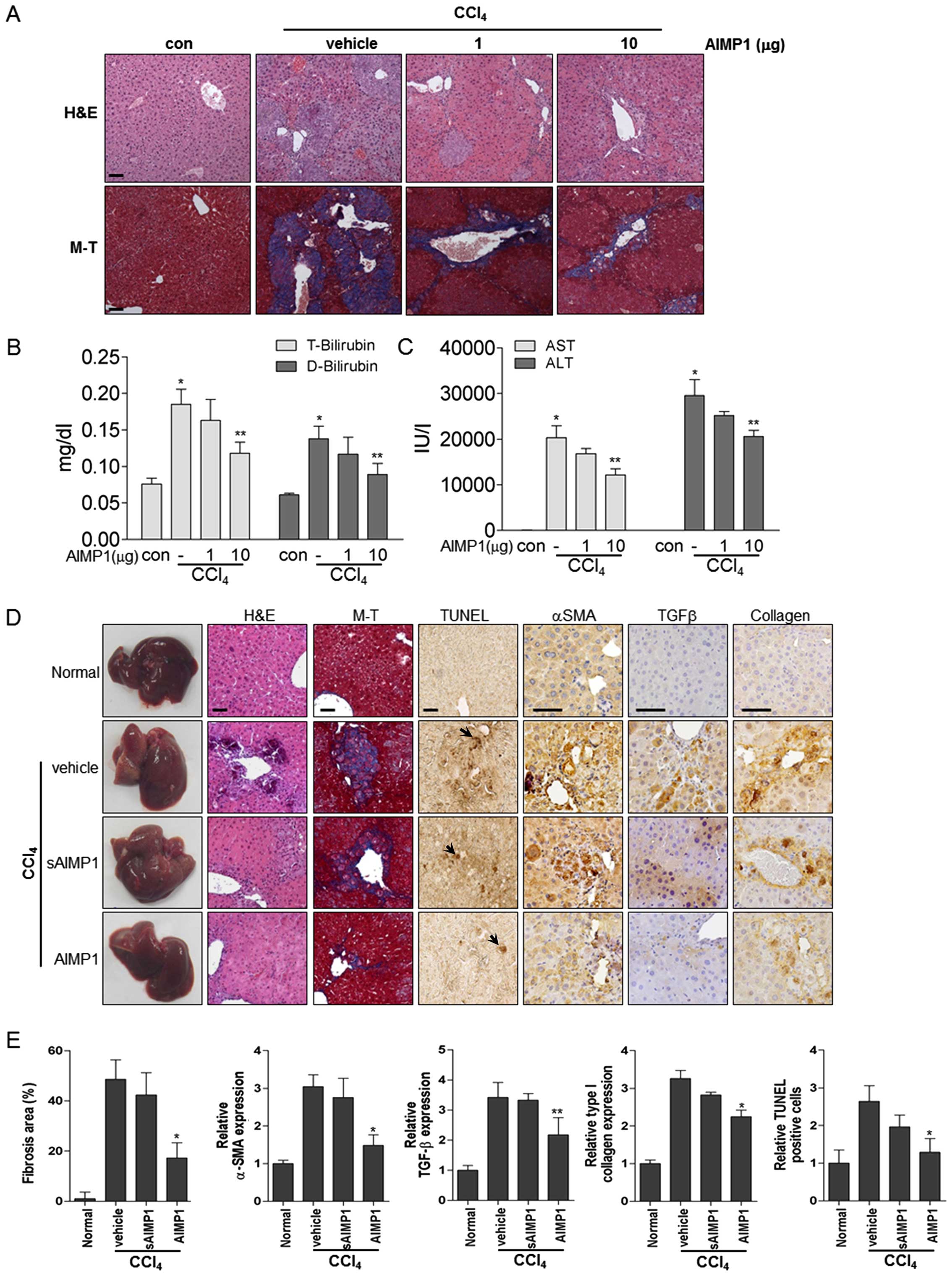 | Figure 4The AIMP1 peptide attenuates
CCl4- induced liver fibrosis. The AIMP1 peptide was
administered intraperitoneally twice weekly to mice with
CCl4-induced liver fibrosis (each group, n=5). (A)
Histological analysis was by H&E and Masson-Trichrome staining
(magnification, ×200). (B and C) In addition, T-Bilirubin,
D-Bilirubin, AST and ALT levels were quantified using blood.
*P<0.01 vs. con; **P<0.05, vs.
AIMP1−/CCl4. (D and E) Vehicle, sAIMP1
peptide (10 μg) or AIMP1 peptide (10 μg) was administered as
described above. Histological analyses [H&E and
Masson-Trichrome staining (magnification, ×200)] and
immunohistochemical analysis [α-SMA, TGFβ, collagen and TUNEL
(magnification, ×400)] were carried out and evaluated. Apoptotic
cells are indicated by an arrow. Scale bar, 50 μm. |
 | Table IEffect of AIMP1 on serum parameters
in liver fibrosis. |
Table I
Effect of AIMP1 on serum parameters
in liver fibrosis.
| |
CCl4 |
|---|
| |
|
|---|
| Control | Vehicle | sAIMP1 | AIMP1 |
|---|
| AST | 155.50±23.46 |
232.17±21.60a | 195.20±17.68 |
169.33±26.91c |
| ALT | 57.83±5.81 |
135.00±12.07b | 123.60±19.17 | 102.00±9.16c |
| Albumin | 2.34±0.33 | 1.66±0.03a | 2.06±0.26 | 2.60±0.26d |
| BUN | 38.00±4.82 | 56.53±10.95a | 56.90±4.47 | 28.82±11.5c |
Discussion
Cytokines from a number of sources, including
inflammatory cells, injured hepatocytes, and Kupffer cells
(43), can activate quiescent HSCs
and transform them into critical players in the exacerbation of
liver fibrosis. In addition, HSCs, together with Kupffer cells and
platelets represent the major reservoir of TGFβ, a factor, which
also plays an important role in the induction of liver fibrosis and
cirrhosis (44–46) by inducing the excessive deposition
of ECM including type I collagen. Furthermore, phosphorylation of
SMAD by activated ERK inhibits SMAD activity by suppression of
nuclear translocation (15,18,37).
In the present study, we examined whether the AIMP1 peptide, an
activator of ERK, could inhibit TGFβ signaling by suppression of
the nuclear translocation of SMADs and thereby inhibit the
synthesis of type I collagen.
The AIMP1 peptide induced the phosphorylation of ERK
in a time-dependent manner, and this lead to phosphorylation of the
serine residue of SMAD2/3. Notably, western blot analysis after
immunoprecipitation with SMAD2/3 antibody showed only the
phosphor-serine of SMAD2, which suggests that only the SMAD2
molecule in the SMAD2/3 is phosphorylated by activated ERK
(Fig. 1). Separate analysis of
nuclear and cytoplasmic fractions clearly showed that the AIMP1
peptide inhibits the TGFβ-mediated nuclear translocation of SMAD2,
but not of SMAD3 (Fig. 2A).
Further investigation will be needed to determine the reason for
the selective phosphorylation of SMAD2 alone. The observation that
SMAD2 phosphorylation by activated ERK inhibits nuclear
translocation is in accordance with previous reports (15,18,37).
We also investigated whether the SMAD phosphorylation by the AIMP1
peptide-mediated ERK activation could affect the SMAD
phosphorylation (SMAD2, Ser465/467; SMAD3,
Ser423/425) by TGFβ. However, the results showed no
effect on the phosphorylation of SMAD2/3 by TGFβ (data not shown),
which suggests that the AIMP1 peptide decreases the nuclear
translocation by acting downstream of the TGFβ-mediated
phosphorylation of SMAD.
The entrance of the phosphorylated SMAD complex into
the nucleus results in an increase in the levels of expression of
type I collagen. Since the nuclear translocation of SMAD complex is
decreased by the AIMP1 peptide-mediated ERK activation we examined
whether treatment with the AIMP1 peptide could also decrease the
type I collagen synthesis in HSCs. Western blot analysis showed
that the AIMP1 peptide decreased the level of type I collagen
expression in a dose-dependent manner. In addition, qRT-PCR and the
luciferase activity assay confirmed that this decrease is regulated
at the level of transcription level (Fig. 3).
The fact that treatment with the AIMP1 peptide
reduced the level of expression of type I collagen in HSCs,
suggests that this peptide may be applicable to treat liver
fibrosis in vivo. We first examined whether the AIMP1
peptide was cytotoxic for hepatocytes. A cytotoxicity assay using
HepG2 cells in vitro did not show any evidence for
cytotoxicity of the AIMP1 peptide at concentrations up to 100 μg/ml
(data not shown). To assess the in vivo efficacy of the
AIMP1 peptide, we generated a liver fibrosis model using
CCl4 and found that the AIMP1 peptide significantly
attenuates both histopathological and biomarker parameters for
liver fibrosis in the mice (Fig.
4A–C). To assess the specificity of the AIMP1 peptide effect,
we used an sAIMP1 peptide as a negative control since the Y24T
mutation of the AIMP1 peptide was previously shown to lack
biological activity on fibroblasts (42). H&E and MT staining showed that
the AIMP1 peptide attenuated liver fibrosis compared to the vehicle
and the sAIMP1 peptide (Fig. 4D and
E). Immunohistochemical staining of α-SMA, TGFβ, and collagen
and fibrogenic markers also showed that the AIMP1 peptide clearly
attenuated CCl4-induced liver fibrosis. In the fibrotic
liver, damaged tissues undergo apoptosis and release metabolites
into the blood. An assay for apoptosis using TUNEL
immunohistochemical staining showed that the AIMP1 peptide
decreased apoptosis in the liver tissue. In addition, analysis of
liver fibrosis biomarkers in the serum clearly showed that
treatment with the AIMP1 peptide improved the levels of the
relevant parameters (Table I).
In conclusion, the AIMP1 peptide inhibited the
nuclear translocation of the SMAD complex, resulting in a reduced
level of expression of type I collagen, and thereby efficiently
inhibited the development of liver fibrosis in a
CCl4-induced mouse model. We therefore suggest that the
AIMP1 peptide might be useful to treat liver fibrosis as an
inhibitor of TGFβ signaling and should be investigated further.
Acknowledgements
The present study was supported by the Bio and
Medical Technology Development Program [NRF-2012M3A9C6049719 (to
S.G.P.)], the Bio-Synergy Research Project (NRF-2015M3A9C4075818),
and the Medical Research Center (2014009392, MSIP) through the
National Research Foundation, Korea (to S.S.H).
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li JT, Liao ZX, Ping J, Xu D and Wang H:
Molecular mechanism of hepatic stellate cell activation and
antifibrotic therapeutic strategies. J Gastroenterol. 43:419–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Russo FP, Alison MR, Bigger BW, Amofah E,
Florou A, Amin F, Bou-Gharios G, Jeffery R, Iredale JP and Forbes
SJ: The bone marrow functionally contributes to liver fibrosis.
Gastroenterology. 130:1807–1821. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pinzani M and Rombouts K: Liver fibrosis:
From the bench to clinical targets. Dig Liver Dis. 36:231–242.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iredale JP, Benyon RC, Arthur MJ, Ferris
WF, Alcolado R, Winwood PJ, Clark N and Murphy G: Tissue inhibitor
of metal-loproteinase-1 messenger RNA expression is enhanced
relative to interstitial collagenase messenger RNA in experimental
liver injury and fibrosis. Hepatology. 24:176–184. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng JH, She H, Han YP, Wang J, Xiong S,
Asahina K and Tsukamoto H: Wnt antagonism inhibits hepatic stellate
cell activation and liver fibrosis. Am J Physiol Gastrointest Liver
Physiol. 294:G39–G49. 2008. View Article : Google Scholar
|
|
7
|
Yoshiji H, Noguchi R, Kuriyama S, Ikenaka
Y, Yoshii J, Yanase K, Namisaki T, Kitade M, Masaki T and Fukui H:
Imatinib mesylate (STI-571) attenuates liver fibrosis development
in rats. Am J Physiol Gastrointest Liver Physiol. 288:G907–G913.
2005. View Article : Google Scholar
|
|
8
|
Son MK, Ryu YL, Jung KH, Lee H, Lee HS,
Yan HH, Park HJ, Ryu JK, Suh JK, Hong S, et al: HS-173, a novel
PI3K inhibitor, attenuates the activation of hepatic stellate cells
in liver fibrosis. Sci Rep. 3:34702013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bueno M, Salgado S, Beas-Zárate C and
Armendariz-Borunda J: Urokinase-type plasminogen activator gene
therapy in liver cirrhosis is mediated by collagens gene expression
down-regulation and up-regulation of MMPs, HGF and VEGF. J Gene
Med. 8:1291–1299. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Anselmi K, Stolz DB, Nalesnik M, Watkins
SC, Kamath R and Gandhi CR: Gliotoxin causes apoptosis and necrosis
of rat Kupffer cells in vitro and in vivo in the absence of
oxidative stress: Exacerbation by caspase and serine protease
inhibition. J Hepatol. 47:103–113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shackel N and Rockey D: In pursuit of the
‘Holy Grail’ - stem cells, hepatic injury, fibrogenesis and repair.
Hepatology. 41:16–18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
LeRoy EC, Trojanowska MI and Smith EA:
Cytokines and human fibrosis. Eur Cytokine Netw. 1:215–219.
1990.PubMed/NCBI
|
|
13
|
Kane CJ, Hebda PA, Mansbridge JN and
Hanawalt PC: Direct evidence for spatial and temporal regulation of
transforming growth factor beta 1 expression during cutaneous wound
healing. J Cell Physiol. 148:157–173. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wahl SM, Hunt DA, Wakefield LM,
McCartney-Francis N, Wahl LM, Roberts AB and Sporn MB: Transforming
growth factor type beta induces monocyte chemotaxis and growth
factor production. Proc Natl Acad Sci USA. 84:5788–5792. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Funaba M, Zimmerman CM and Mathews LS:
Modulation of Smad2-mediated signaling by extracellular
signal-regulated kinase. J Biol Chem. 277:41361–41368. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuura I, Wang G, He D and Liu F:
Identification and characterization of ERK MAP kinase
phosphorylation sites in Smad3. Biochemistry. 44:12546–12553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mulder KM: Role of Ras and Mapks in
TGFbeta signaling. Cytokine Growth Factor Rev. 11:23–35. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kretzschmar M, Doody J, Timokhina I and
Massagué J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Conery AR, Cao Y, Thompson EA, Townsend CM
Jr, Ko TC and Luo K: Akt interacts directly with Smad3 to regulate
the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol.
6:366–372. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song K, Wang H, Krebs TL and Danielpour D:
Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated
Smad3 activation. EMBO J. 25:58–69. 2006. View Article : Google Scholar
|
|
21
|
Remy I, Montmarquette A and Michnick SW:
PKB/Akt modulates TGF-beta signalling through a direct interaction
with Smad3. Nat Cell Biol. 6:358–365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takayama S, Murakami S, Miki Y, Ikezawa K,
Tasaka S, Terashima A, Asano T and Okada H: Effects of basic
fibroblast growth factor on human periodontal ligament cells. J
Periodontal Res. 32:667–675. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Silverio-Ruiz KG, Martinez AE, Garlet GP,
Barbosa CF, Silva JS, Cicarelli RM, Valentini SR, Abi-Rached RS and
Junior CR: Opposite effects of bFGF and TGF-beta on collagen
metabolism by human periodontal ligament fibroblasts. Cytokine.
39:130–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Quevillon S, Agou F, Robinson JC and
Mirande M: The p43 component of the mammalian multi-synthetase
complex is likely to be the precursor of the endothelial
monocyte-activating polypeptide II cytokine. J Biol Chem.
272:32573–32579. 1997. View Article : Google Scholar
|
|
25
|
Matschurat S, Knies UE, Person V, Fink L,
Stoelcker B, Ebenebe C, Behrensdorf HA, Schaper J and Clauss M:
Regulation of EMAP II by hypoxia. Am J Pathol. 162:93–103. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park SG, Shin H, Shin YK, Lee Y, Choi EC,
Park BJ and Kim S: The novel cytokine p43 stimulates dermal
fibroblast proliferation and wound repair. Am J Pathol.
166:387–398. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park SG, Kang YS, Kim JY, Lee CS, Ko YG,
Lee WJ, Lee KU, Yeom YI and Kim S: Hormonal activity of AIMP1/p43
for glucose homeostasis. Proc Natl Acad Sci USA. 103:14913–14918.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ko YG, Park H, Kim T, Lee JW, Park SG,
Seol W, Kim JE, Lee WH, Kim SH, Park JE, et al: A cofactor of tRNA
synthetase, p43, is secreted to up-regulate proinflammatory genes.
J Biol Chem. 276:23028–23033. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park SG, Kang YS, Ahn YH, Lee SH, Kim KR,
Kim KW, Koh GY, Ko YG and Kim S: Dose-dependent biphasic activity
of tRNA synthetase-associating factor, p43, in angiogenesis. J Biol
Chem. 277:45243–45248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park H, Park SG, Lee JW, Kim T, Kim G, Ko
YG and Kim S: Monocyte cell adhesion induced by a human
aminoacyl-tRNA synthetase-associated factor, p43: Identification of
the related adhesion molecules and signal pathways. J Leukoc Biol.
71:223–230. 2002.PubMed/NCBI
|
|
31
|
Kim E, Kim SH, Kim S and Kim TS: The novel
cytokine p43 induces IL-12 production in macrophages via NF-kappaB
activation, leading to enhanced IFN-gamma production in
CD4+ T cells. J Immunol. 176:256–264. 2006. View Article : Google Scholar
|
|
32
|
Kim E, Kim SH, Kim S, Cho D and Kim TS:
AIMP1/p43 protein induces the maturation of bone marrow-derived
dendritic cells with T helper type 1-polarizing ability. J Immunol.
180:2894–2902. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SY, Son WS, Park MC, Kim CM, Cha BH,
Yoon KJ, Lee SH and Park SG: ARS-interacting multi-functional
protein 1 induces proliferation of human bone marrow-derived
mesenchymal stem cells by accumulation of β-catenin via fibroblast
growth factor receptor 2-mediated activation of Akt. Stem Cells
Dev. 22:2630–2640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee JH, Lee H, Joung YK, Jung KH, Choi JH,
Lee DH, Park KD and Hong SS: The use of low molecular weight
heparin-pluronic nanogels to impede liver fibrosis by inhibition
the TGF-β/Smad signaling pathway. Biomaterials. 32:1438–1445. 2011.
View Article : Google Scholar
|
|
35
|
Coffer PJ, Jin J and Woodgett JR: Protein
kinase B (c-Akt): A multifunctional mediator of
phosphatidylinositol 3-kinase activation. Biochem J. 335:1–13.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu L, Hui AY, Albanis E, Arthur MJ,
O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL and Eng FJ: Human
hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis
of hepatic fibrosis. Gut. 54:142–151. 2005. View Article : Google Scholar
|
|
37
|
Kretzschmar M, Doody J and Massagué J:
Opposing BMP and EGF signalling pathways converge on the TGF-beta
family mediator Smad1. Nature. 389:618–622. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen SJ, Yuan W, Mori Y, Levenson A,
Trojanowska M and Varga J: Stimulation of type I collagen
transcription in human skin fibroblasts by TGF-beta: Involvement of
Smad 3. J Invest Dermatol. 112:49–57. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qi Z, Atsuchi N, Ooshima A, Takeshita A
and Ueno H: Blockade of type beta transforming growth factor
signaling prevents liver fibrosis and dysfunction in the rat. Proc
Natl Acad Sci USA. 96:2345–2349. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wynn TA: Common and unique mechanisms
regulate fibrosis in various fibroproliferative diseases. J Clin
Invest. 117:524–529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Han JM, Park SG, Lee Y and Kim S:
Structural separation of different extracellular activities in
aminoacyl-tRNA synthetase-interacting multi-functional protein,
p43/AIMP1. Biochem Biophys Res Commun. 342:113–118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Friedman SL: Molecular regulation of
hepatic fibrosis, an integrated cellular response to tissue injury.
J Biol Chem. 275:2247–2250. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gressner AM: Cytokines and cellular
crosstalk involved in the activation of fat-storing cells. J
Hepatol. 22(Suppl): 28–36. 1995.PubMed/NCBI
|
|
45
|
Bissell DM, Wang SS, Jarnagin WR and Roll
FJ: Cell-specific expression of transforming growth factor-beta in
rat liver. Evidence for autocrine regulation of hepatocyte
proliferation. J Clin Invest. 96:447–455. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen A and Davis BH: The DNA binding
protein BTEB mediates acetaldehyde-induced, jun N-terminal
kinase-dependent alphaI(I) collagen gene expression in rat hepatic
stellate cells. Mol Cell Biol. 20:2818–2826. 2000. View Article : Google Scholar : PubMed/NCBI
|