Introduction
Nasopharyngeal carcinoma (NPC) is a squamous cell
carcinoma extremely common in southern regions of China and
Southeast Asia, characterized by a local invasion or early distant
metastasis at the time of diagnosis (1). Although it is radiosensitive
(2), a high number of patients
show local regional recurrence or metastatic spread (3). Therefore, it is of utmost importance
to understand the pathogenic mechanism of NPC for an early
diagnosis to apply effective therapeutic strategies.
TIGAR was first identified as a P53 target gene,
playing an important role in glycolysis and apoptosis in U2OS cells
(4). It represents a key gene in
the metabolism control mediated by P53 (5). Due to the enzymatic activity of the
encoded protein, TIGAR reduces fructose-2,6-bisphosphate (F-2,6-P2)
levels, leading to glycolysis inhibition and pentose phosphate
pathway (PPP) induction (4). In
addition, the PPP enhances the production of nicotinamide adenine
dinucleotide phosphate (NADPH), which scavenges intracellular
reactive oxygen species (ROS) and protects cells from oxidative
stress-induced apoptosis (4).
An increasing number of studies reported that TIGAR
modified expression is tightly correlated with cancer development.
A high expression level of TIGAR was observed in cancers such as
invasive breast cancer (6),
hepatocellular carcinoma (7),
intestinal cancer (8), and
glioblastoma (9,10). TIGAR protects cancer cells from
apoptosis in breast cancer and hepatocellular carcinoma (6,7). In
a mouse intestinal cancer model, transgenic mouse knockout for the
TIGAR gene showed a reduced tumor burden and an increased survival
(8). Knockdown of TIGAR in glioma
cells can enhance radiosensitivity by ROS accumulation, which
results in DNA damage and cellular senescence (11). These studies suggested that TIGAR
may act as an oncogene in some cancers to support cancer
progression.
However, the exact role of TIGAR in NPC has not been
yet reported. The present study aimed to investigate the role of
TIGAR in NPC tumorigenesis. Our results showed a high expression of
TIGAR in the tumor tissue of NPC patients compared with the
expression in the adjacent normal epithelium. Knockdown of TIGAR in
NPC cells reduced tumor growth and increased apoptosis via NF-κB
pathway. On the other hand, TIGAR overexpression promoted tumor
growth, although did not decrease apoptosis. These data strongly
suggested that TIGAR might represent an important oncogene in NPC
tumorigenesis.
Materials and methods
Clinical samples
A total of 96 NPC patients were selected at The
First Hospital of Sichuan Medical University, Luzhou, China.
Written informed consent was obtained from the patients, and this
program was approved by the Ethics Committee of the First Hospital
of Sichuan Medical University.
Immunohistochemical staining
The tissue specimens from the patients were
immunostained with a rabbit polyclonal TIGAR antibody (1:200, Santa
Cruz Biotechnology, Santa Cruz, CA, USA), according to the
manufacturer's instructions. Two pathologists independently scored
each slide. The percentage of positive tumor cells was evaluated
(0, 0%; 1, 1–25%; 2, 26–50%; 3, 51–75%; 4, 76–100%), as well as the
staining intensity (0, negative; 1, weak; 2, moderate; 3, strong;
4, very strong), as previously described (12). The intensity score × percentage
score value was used to obtain the final overall score for TIGAR
(0–16).
Cell culture
The human normal nasopharyngeal epithelial cell line
NP69-SV40T (Sun Yat-sen University Cancer Center, Guangdong,
China), was routinely maintained in keratinocyte serum-free medium
supplemented with human recombinant epidermal growth factor (EGF
1–53) and bovine pituitary extract (BPE) (Invitrogen, USA). The
human CNE-2 and 5–8F NPC cell lines were obtained from ATCC
(American Type Culture Collection), and routinely maintained in
RPMI-1640 medium supplemented with 10% fetal bovine serum (GE
Healthcare Life Sciences, Logan, UT, USA), 100 U/ml penicillin, and
100 μg/ml streptomycin (Beyotime Biotecnology, China).
Lentivirus-mediated small hairpin RNA
(Lenti-shRNA) against TIGAR
The Lenti-shRNA vector system against TIGAR was
constructed, packed, and purified by GeneChem (Shanghai, China).
The shRNA oligonucleotides were designed as TIGAR-shRNA
(GCCAGCTTTACTGGAGAACTT). A scramble sequence was synthesized as
control, and tagged as Scramble-shRNA (TTACCGAGACCGTACGTAT). Human
NPC cells CNE-2 and 5–8F were infected, and colonies expressing a
stable shRNA were selected using puromycin (Sigma-Aldrich, St.
Louis, MO, USA), according to the manufacturer's protocol.
Cell growth assay and colony formation
assay
For cell growth assay, stable cells were seeded at a
density of 5×104 per well. The cells were stained by
trypan blue and counted in the following 5 days. For colony
formation assay, stable cell lines transfected with TIGAR-shRNA and
Scramble-shRNA were seeded in 3.5-mm culture dishes at a density of
200. The colony formation was evaluated under the microscope after
10 days. Next, the cells were fixed with 4% PFA, and stained with
0.1% crystal violet. Data are expressed as the mean ± SD of five
independent experiments.
EdU assay
Stable cells were seeded on coverslips in 24-well
plate at a density of 2×104 per well. Twenty-four hours
later, cells were incubated with EdU (20 μM) for 1.5 h. Then, EdU
assay were continued using EdU DNA Proliferation In Vitro
Detection kit (RiboBio Co., Ltd., Guangzhou, China), according to
the manufacturer's instructions. In total, three fields per slice
were randomly selected and analyzed, and experiment was repeated
three times. The EdU incorporation rate was calculated as the ratio
of the EdU positive cell number to the total cell number in each
feld.
Wound healing assay and Transwell
migration assay
Cells at 80–90% confluence were scraped using a
sterile micro-pipette tip across the monolayer to obtain a
longitudinal scratch without cells, to perform the wound healing
assay. The empty area was monitored every 4 h. The Transwell assay
was performed using Transwell chambers with polycarbonate filters
(8-μm pore size, BD Biosciences, San Jose, CA, USA). The chambers
were coated with 50 μl Matrigel prior to cell seeding, and immersed
in a well containing 600 μl of complete medium, followed by the
addition of 4×104 cells in 100 μl serum-free medium to
the upper chamber. After an incubation of 24–48 h at 37°C in a 5%
CO2 incubator, cells were fixed with 4% PFA, and stained
with 0.1% crystal violet. The cells remained in the upper chamber
were removed with a cotton swab. The migrated cells were counted
under a microscope at ×400 magnification.
Apoptosis analysis
Stable cell lines were harvested at 48 h after
adriamycin (0.5 μg/ml) stimulation, and stained for Annexin V-FITC
and PI at room temperature for 15 min according to the
manufacturer's instructions (Invitrogen). Cells were analyzed by
flow cytometry (FACScan, Becton-Dickinson).
Xenograft tumor induction
Eight-week-old male nude mice (BALB/c-nu) were
bought from Beijing HFK Bioscience Co. Ltd., Beijing, China. The
cells expressing TIGAR-shRNA or Scramble-shRNA were collected and
resuspended in PBS at the density of 107/ml, and 100 μl
(106 cells) were subcutaneously inoculated in the flank
of each mouse. Tumor volumes were monitored every 3 days and the
length, width and height were measured to evaluate the tumor volume
through the following formula: Tumor volume (mm3) = 0.5
× length × width × height (13).
After the designated days, mice were sacrificed by carbon dioxide
asphyxiation, and the tumors were removed for analysis. The animal
experiments were approved by the Ethics Committee of Luzhou Medical
College, Luzhou, China.
Western blot analysis
Total cell proteins were extracted with RIPA lysis
buffer with cocktail of protease inhibitors (Beyotime
Biotecnology). The samples were resolved on a SDS-PAGE gel and then
transferred to a PVDF membrane (Amersham Biosciences, Fairfield,
CT, USA). The membranes were labeled with the following antibodies
of the proteins of interest: TIGAR (sc-166290; Santa Cruz
Biotechnology); Caspase-3 (19677-1-AP; Proteintech, Wuhan, China);
p65 (AN365, Beyotime Biotecnology); IkB-α (AI096, Beyotime
Biotecnology); Bcl-2 (12789-1-AP, Proteintech); MMP-2 (10373-2-AP,
Proteintech); MMP-9 (10375-2-AP, Proteintech); Oct-1 (10387-1-AP,
Proteintech); GAPDH (2118; Cell Signaling, Beverly, MA, USA).
Secondary antibodies were purchased from Proteintech. ECL was used
(Amersham Biosciences) for the detection of the target bands.
Statistical analysis
The experiments were performed at least in
triplicate. The results were expressed as the mean ± SD.
Statistical analysis was performed by SPSS software and GraphPad
Prism. P<0.05 was considered statistically significant.
Results
Enhanced expression of TIGAR in NPC
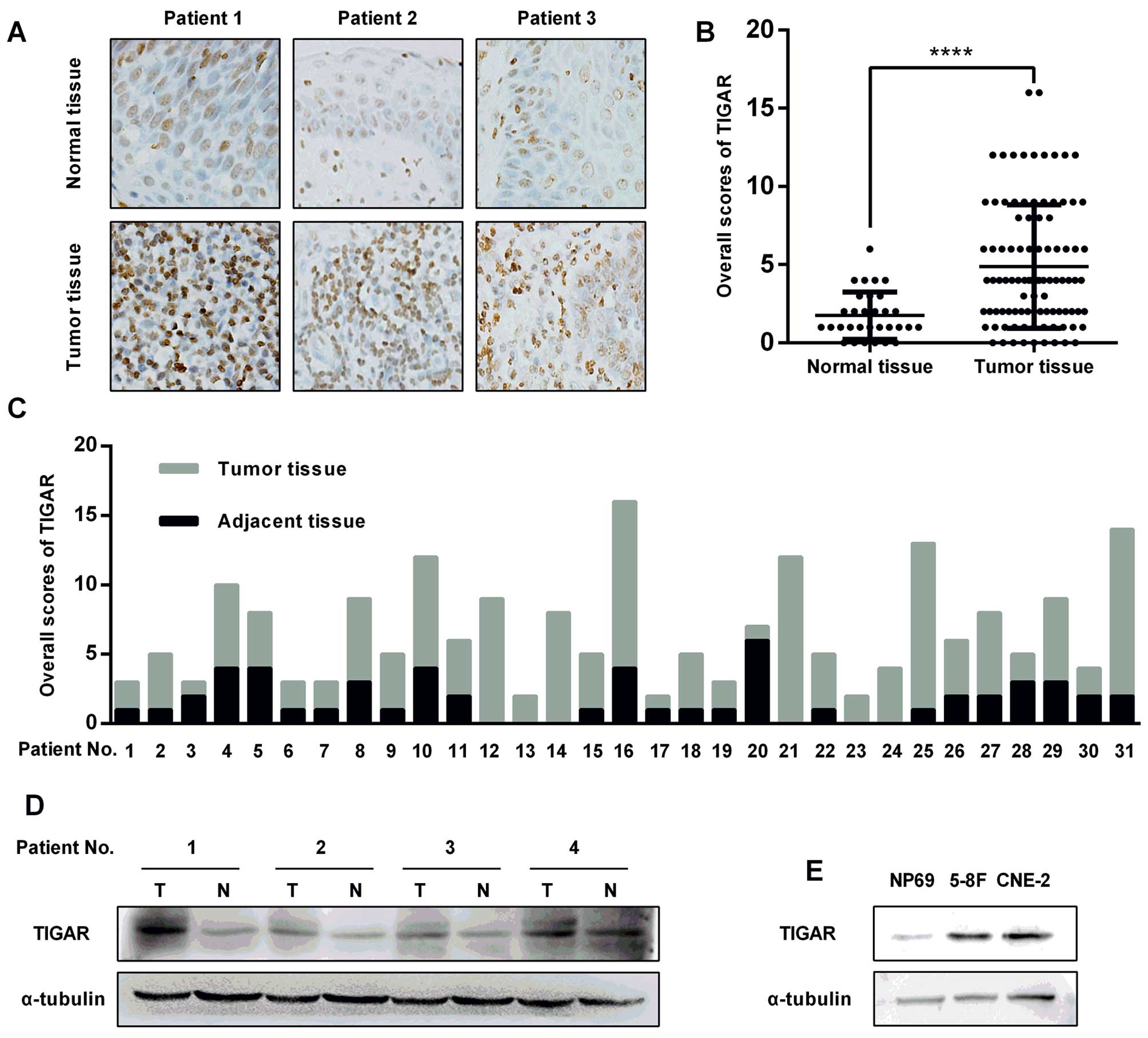
TIGAR expression was evaluated in NPC tissue
specimens by immunohistochemistry (IHC). TIGAR staining intensity
in NPC cells was significantly stronger compared to the staining in
the adjacent normal epithelial cells (Fig. 1A). Next, the immunostaining of the
96 slides from the NPC patients was scored, showing a higher
overall score in the tumor tissues compared with the normal
nasopharyngeal epithelium (Fig.
1B). Among these slides, 31 specimens with both the NPC tissues
and the adjacent normal epithelium were selected, showing a similar
result as that shown in Fig. 1B
(Fig. 1C). The remarkably elevated
protein level in primary NPC tissues was confirmed using
immunoblotting (Fig. 1D). A
similar result was also observed in NPC cells (CNE-2, 5–8F) and
normal nasopharyngeal epithelial cells (NP69) (Fig. 1E).
Knockdown of TIGAR suppresses
proliferation, migration, invasion, and colony formation on NPC
cells
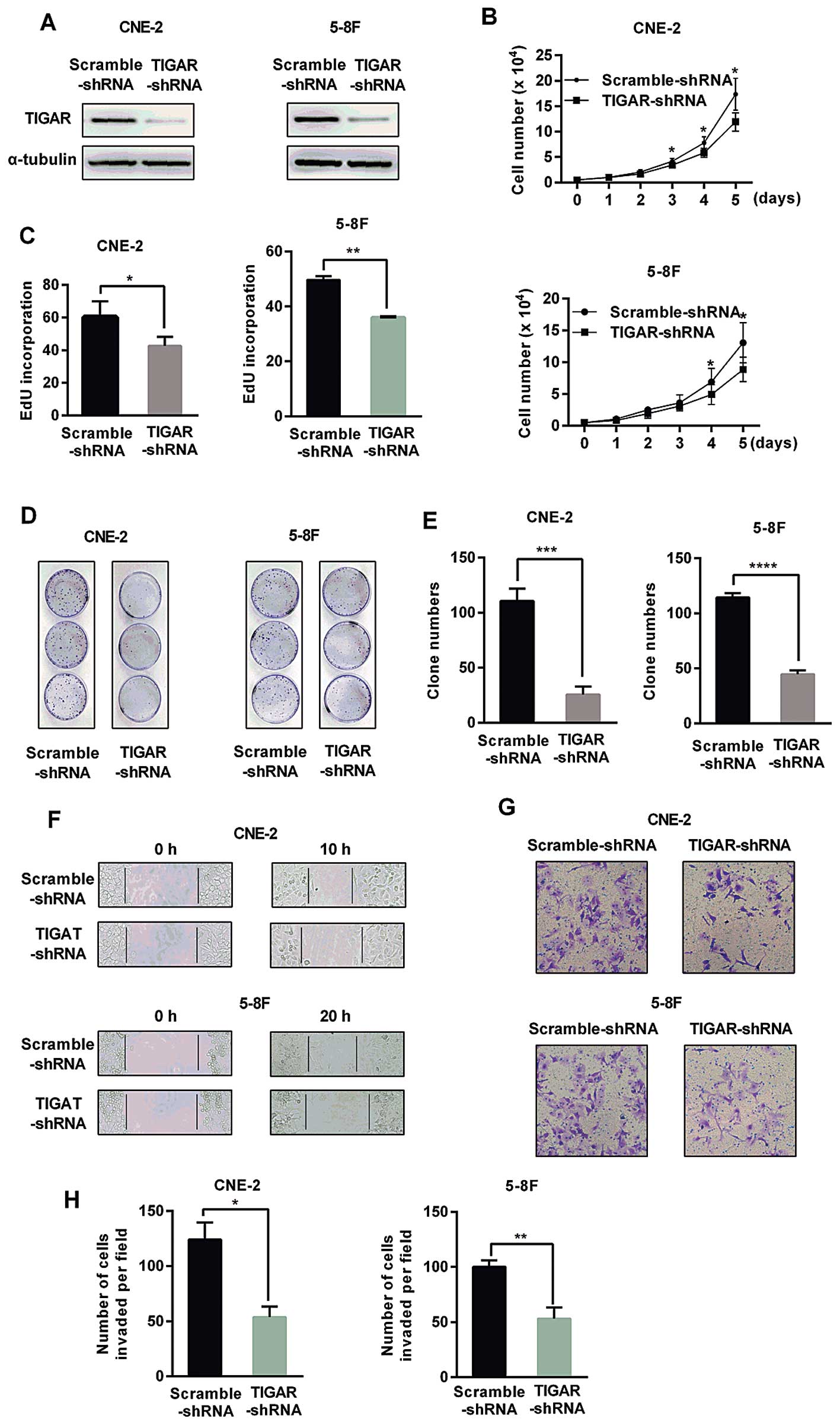
To explore the biological function of TIGAR in NPC
cells, lentivirus was used to introduce shRNA (Lenti-shRNA)
targeting TIGAR into CNE-2 and 5–8F cells (Fig. 2). The western blot analysis showed
a remarkable reduction of TIGAR level in TIGAR-shRNA cells compared
with TIGAR expression in Scramble-shRNA cells (Fig. 2A). ROS accumulation and GSH/GSSG
reduction also confirmed the high depletion efficiency of TIGAR
(data not shown). Cell growth assays revealed a reduced growth in
TIGAR-shRNA cells compared to the growth of the Scramble-shRNA
cells (Fig. 2B). EdU incorporation
confirmed the remarkable reduction of the proliferation in the
TIGAR-shRNA cells (Fig. 2C). The
number of colonies in TIGAR-shRNA cells was apparently lower than
the number in the Scramble-shRNA cells (Fig. 2D and E). In order to investigate
whether TIGAR was involved in migration, wound healing assay and
Transwell migration assay were performed, showing a significant
decreased migration ability in TIGAR-shRNA cells (Fig. 2F and G) as well as a reduced
invasion ability, as shown by Matrigel invasion assay (Fig. 2H).
Knockdown of TIGAR induces apoptosis in
human NPC cells
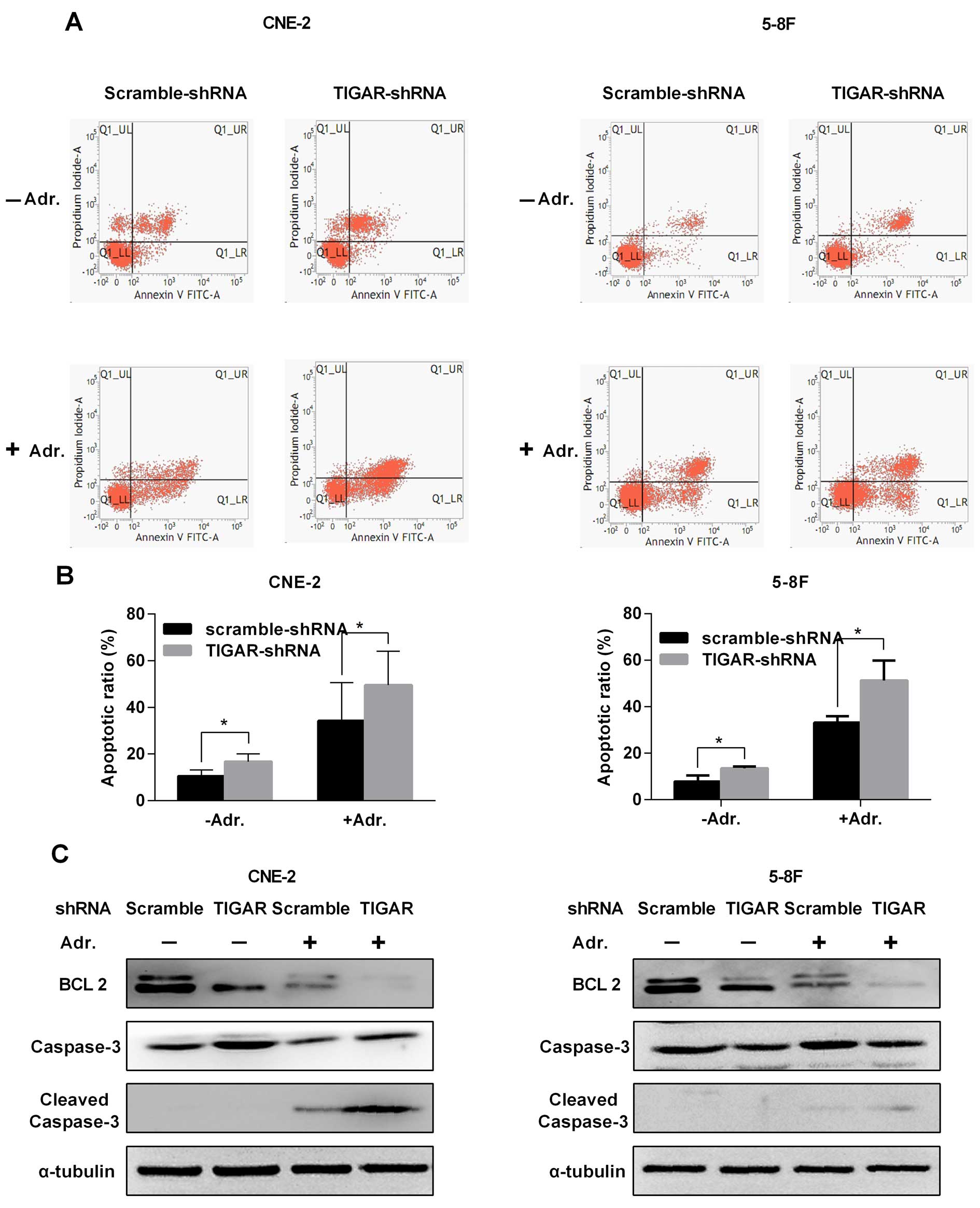
To evaluate whether TIGAR depletion affected
apoptosis in NPC cells, stable NPC cells were stained with Annexin
V/PI and analyzed by flow cytometry (Fig. 3). The results indicated that the
apoptotic rate was increased in TIGAR-shRNA cells compared to the
rate in the Scramble-shRNA cells (Fig.
3A and B). To test whether TIGAR depletion can improve the
adriamycin related apoptosis, cells were incubated with 0.5 μg/ml
adriamycin for 48 h, and analyzed by flow cytometry. As shown in
Fig. 3A and B, cell apoptosis was
remarkably induced in TIGAR-shRNA cells compared with apoptosis in
the Scramble-shRNA cells. We further examined the anti-apoptotic
protein BCL2, which was downregulated as we expected, while cleaved
caspase-3 was upregulated (Fig.
3C).
TIGAR overexpression promotes
proliferation, colony formation, migration and invasion without
reducing apoptosis in human NPC cells
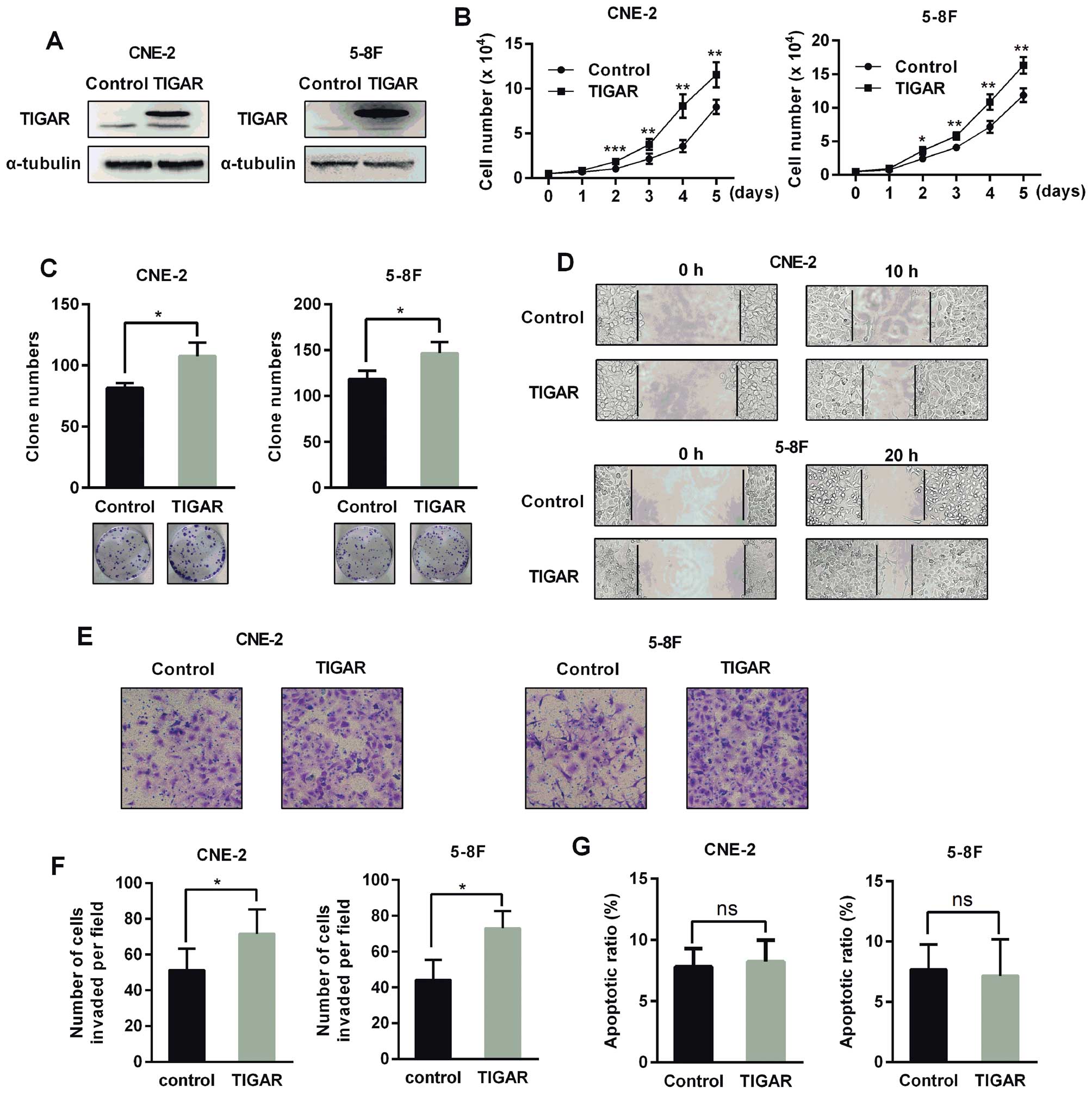
To evaluate the effect of TIGAR overexpression on
NPC cells, we constructed stable cells overexpressing TIGAR
(Fig. 4A). These cells exhibited
an increased growth rate, colony formation, and enhanced migration
and invasion (Fig. 4B–F). However,
TIGAR overexpression did not reduce apoptosis (Fig. 4G).
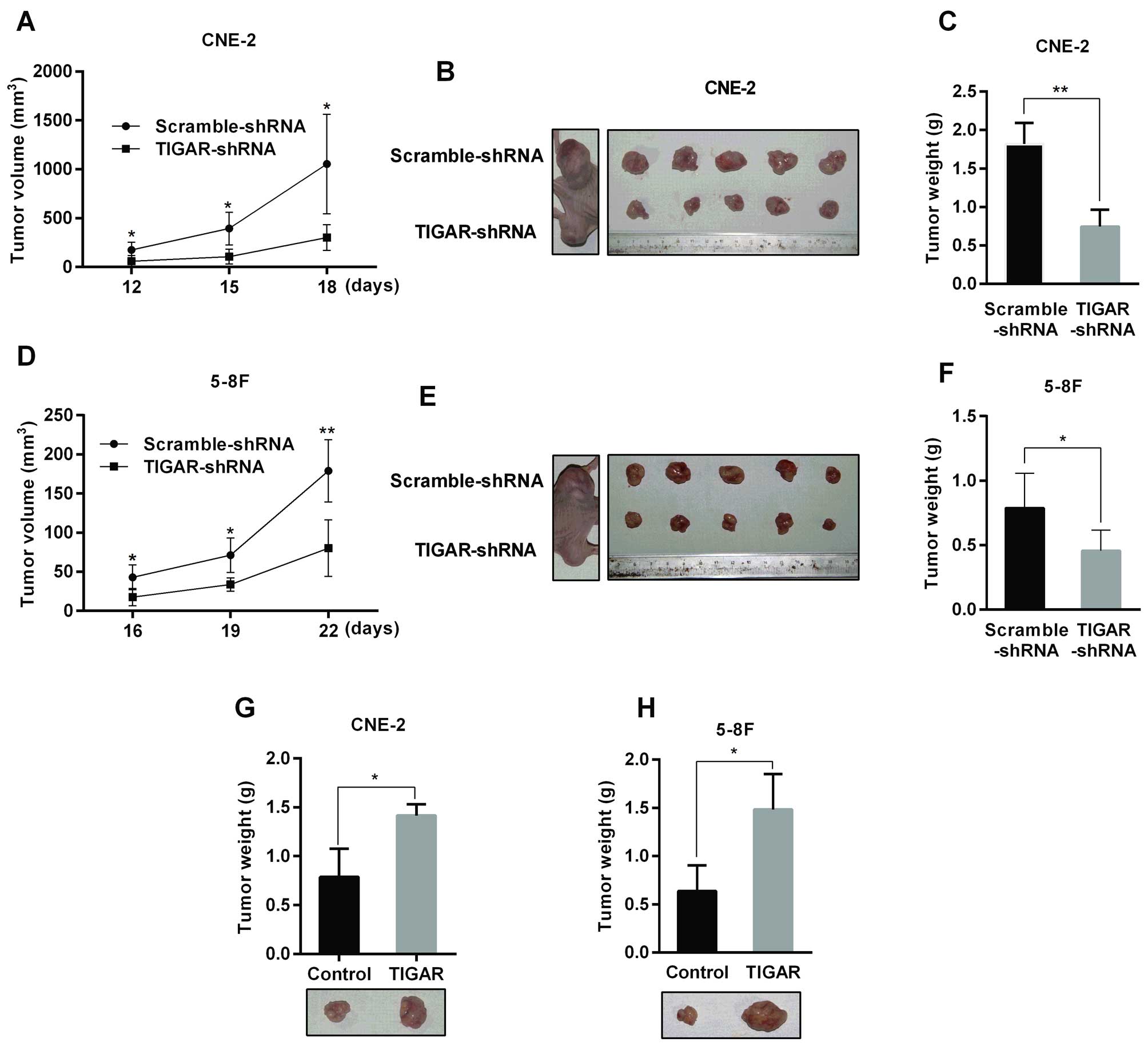
TIGAR increases NPC xenograft tumor
growth
To evaluate whether knockdown of TIGAR reduced tumor
growth in vivo, NPC stable cells were subcutaneously
inoculated into the flank of nude mice. As shown in Fig. 5, the tumor growth rate was
decreased in TIGAR-shRNA group compared with the corresponding
Scramble-shRNA group. In addition, the tumor weight in the
TIGAR-shRNA group was one-half of the tumor weight in the
Scramble-shRNA group (Fig. 5C and
F). On the contrary, TIGAR overexpression accelerated xenograft
tumor growth in nude mice (Fig. 5G and
H).
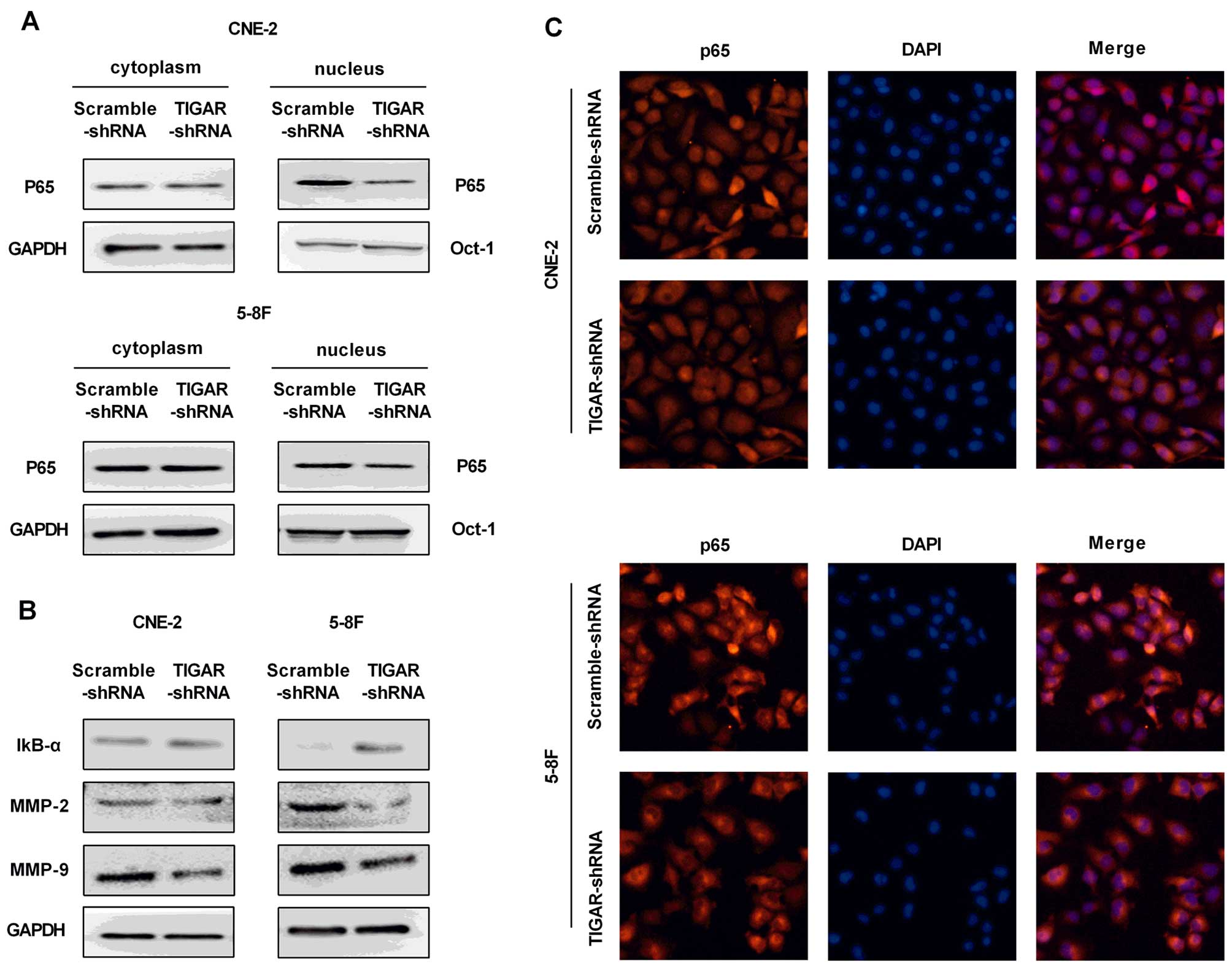
Knockdown of TIGAR inhibits the NF-κB
signaling pathway
The IκB-α and p65 were analyzed to evaluate whether
NF-κB pathway was involved in TIGAR-regulated apoptosis in NPC
cells. Our results showed increased expression of IκB-α, and
inhibited translocation of p65 into the nucleus in both CEN-2 and
5–8F TIGAR-shRNA cells, indicating an inhibited NF-κB pathway
(Fig. 6A and B).
Immunofluorescence staining of p65 confirmed the reduced level of
p65 in the nucleus (Fig. 6C).
Next, NF-κB target genes matrix metalloproteinase- 2 (MMP-2) and
MMP-9 were examed for a significant reduction in TIGAR-shRNA cells
(Fig. 6B).
Discussion
Our results showed that the expression of TIGAR was
upregulated in NPC tumor tissues compared with the adjacent normal
epithelium. Knockdown of TIGAR in NPC cells CNE-2 or 5–8F and
xenograft tumor models indicated a reduced tumor progression and
enhanced apoptosis. These results suggest that TIGAR may act as an
oncogene in NPC tumorigenesis. Recently, Cheung et al
reported an increased TIGAR expression in primary human colon
cancer, and showed that TIGAR was required for intestinal
tumorigenesis (8). Our current
results are consistent with these already published results.
As a glycolysis regulator, TIGAR degrades
intracellular fructose-2,6-bisphosphate (F-2,6-P2), which resulted
in a shift from glycolysis to the pentose phosphate pathway (PPP).
On the other hand, knockdown of TIGAR results in increased F-2,6-P2
levels and glycolytic flux (4,11,14).
As PPP plays an important role in NAPDH production, knockdown of
TIGAR results in decreased levels of NAPDH (15–17)
and reduced glutathione (4,9,17),
contributing to the accumulation of ROS (18). Through knockdown of TIGAR, our
results confirmed the elevated ratio of GSH/GSSG and accumulation
of ROS in NPC cells.
We further investigated the underlying mechanisms
involved in NPC progress and we discovered a correlation between
TIGAR and NF-κB pathway in NPC. NF-κB is a transcription factor
composed of five subunits, including RelA (p65), RelB, cRel, NFKB1
(p50/p105) and NFKB2 (p52/p100) (19). In unstimulated cells, NF-κB binds
to a class of inhibitory proteins called IκB (inhibitor of κB),
which mask the nuclear localization signals (NLS) of NF-κB proteins
and keeps them sequestered in an inactive state in the cytoplasm
(20). Upon stimulation, IκB is
phosphorylated by IκB kinase (IKK) and subsequently degraded,
resulting in a rapid NF-κB translocation into the nucleus (21). Then, specific genes with NF-κB
binding sites are activated, such as matrix metalloproteinase
(MMPs) that degrade the extracellular matrix to facilitate cell
invasion (22).
Numerous studies reported that NF-κB was
constitutively active in many different types of human tumors, and
exerted pro-tumorigenic functions (23). The oncogenic role of NF-κB in NPC
was widely investigated in the past decade, revealing its function
as a regulator of genes that control cell proliferation and cell
survival, and protect the cell from apoptosis (24–28).
Herein, we discovered a correlation between TIGAR and NF-κB pathway
in NPC. Our results showed that knockdown of TIGAR contributed to
NF-κB pathway inactivation in NPC cells, with an increased IκB-α
expression, and an inhibited translocation of p65 into the nucleus,
indicating an inhibited NF-κB pathway, thus supporting the role of
TIGAR as a tumor promoter.
In the present study, we evaluated the effect of
chemotherapeutics in combination with TIGAR depletion on apoptosis
in vitro. Our results showed that TIGAR depletion can
significantly promote the adriamycin-induced apoptosis. However, it
is still unknown whether this effect can be obtained in
vivo. Radiation destroys genomic DNA to induce apoptosis
(29). As a scavenger of
intracellular ROS, TIGAR is capable of maintaining genomic DNA
stability. Therefore, we assume that depletion of TIGAR may enhance
the radiosensitivity of tumors. However, this aspect needs further
clarification.
In conclusion, this study reported a higher
expression of TIGAR in NPC tissues, compared with the adjacent
normal epithelium. Knockdown of TIGAR by lentivirus-shRNA in NPC
cells CNE-2 or 5–8F contributed to a reduction in cell growth and
increased apoptotic rate. In addition, xenograft tumor models
revealed the tumor promotor role of TIGAR. Furthermore, to our
knowledge, this is the first report introducing the involvement of
the NF-κB pathway in the TIGAR-inducing NPC tumorigenesis. The
present study highlighted the oncogenic role of TIGAR in NPC
tumorigenesis, underlining a potential role of TIGAR as a
therapeutic target for cancer treatment.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation (grant no. 81201784), Scientific
Research Foundation of the Education Department of Sichuan Province
(15ZA0163), the Union Project of Luzhou City and Sichuan Medical
University (2013LZLY-J40), and The First Hospital of Sichuan
Medical University Foundation (grant no. 201519).
Abbreviations:
|
TIGAR
|
TP53-induced glycolysis and apoptosis
regulator
|
|
NPC
|
nasopharyngeal carcinoma
|
|
NF-κB
|
nuclear factor κB
|
|
Lenti-shRNA
|
lentivirus-mediated small hairpin
RNA
|
|
MMP-2
|
matrix metalloproteinase-2
|
|
MMP-9
|
matrix metalloproteinase-9
|
References
|
1
|
Li XJ, Ong CK, Cao Y, Xiang YQ, Shao JY,
Ooi A, Peng LX, Lu WH, Zhang Z, Petillo D, et al: Serglycin is a
theranostic target in nasopharyngeal carcinoma that promotes
metastasis. Cancer Res. 71:3162–3172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiao WW, Huang SM, Han F, Wu SX, Lu LX,
Lin CG, Deng XW, Lu TX, Cui NJ and Zhao C: Local control, survival,
and late toxicities of locally advanced nasopharyngeal carcinoma
treated by simultaneous modulated accelerated radiotherapy combined
with cisplatin concurrent chemotherapy: Long-term results of a
phase 2 study. Cancer. 117:1874–1883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee AW, Lin JC and Ng WT: Current
management of nasopharyngeal cancer. Semin Radiat Oncol.
22:233–244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bensaad K, Tsuruta A, Selak MA, Vidal MN,
Nakano K, Bartrons R, Gottlieb E and Vousden KH: TIGAR, a
p53-inducible regulator of glycolysis and apoptosis. Cell.
126:107–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vousden KH and Ryan KM: p53 and
metabolism. Nat Rev Cancer. 9:691–700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Won KY, Lim SJ, Kim GY, Kim YW, Han SA,
Song JY and Lee DK: Regulatory role of p53 in cancer metabolism via
SCO2 and TIGAR in human breast cancer. Hum Pathol. 43:221–228.
2012. View Article : Google Scholar
|
|
7
|
Ye L, Zhao X, Lu J, Qian G, Zheng JC and
Ge S: Knockdown of TIGAR by RNA interference induces apoptosis and
autophagy in HepG2 hepatocellular carcinoma cells. Biochem Biophys
Res Commun. 437:300–306. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheung EC, Athineos D, Lee P, Ridgway RA,
Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ and Vousden KH:
TIGAR is required for efficient intestinal regeneration and
tumorigenesis. Dev Cell. 25:463–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wanka C, Steinbach JP and Rieger J:
Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects
glioma cells from starvation-induced cell death by up-regulating
respiration and improving cellular redox homeostasis. J Biol Chem.
287:33436–33446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sinha S, Ghildiyal R, Mehta VS and Sen E:
ATM-NFκB axis-driven TIGAR regulates sensitivity of glioma cells to
radio-mimetics in the presence of TNFα. Cell Death Dis. 4:e6152013.
View Article : Google Scholar
|
|
11
|
Pena-Rico MA, Calvo-Vidal MN,
Villalonga-Planells R, Martinez-Soler F, Gimenez-Bonafe P,
Navarro-Sabate A, Tortosa A, Bartrons R and Manzano A: TP53 induced
glycolysis and apoptosis regulator (TIGAR) knockdown results in
radio-sensitization of glioma cells. Radiother Oncol. 101:132–139.
2011. View Article : Google Scholar
|
|
12
|
Wang L, Wei D, Huang S, Peng Z, Le X, Wu
TT, et al: Transcription factor Sp1 expression is a significant
predictor of survival in human gastric cancer. Clin Cancer Res.
9:6371–6380. 2003.PubMed/NCBI
|
|
13
|
Somasundaram K and El-Deiry WS: Inhibition
of p53-mediated transactivation and cell cycle arrest by E1A
through its p300/CBP-interacting region. Oncogene. 14:1047–1057.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kimata M, Matoba S, Iwai-Kanai E, Nakamura
H, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Mita Y, Okigaki M, et
al: p53 and TIGAR regulate cardiac myocyte energy homeostasis under
hypoxic stress. Am J Physiol Heart Circ Physiol. 299:H1908–H1916.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lui VW, Lau CP, Cheung CS, Ho K, Ng MH,
Cheng SH, Hong B, Tsao SW, Tsang CM, Lei KI, et al: An RNA-directed
nucleoside anti-metabolite, 1-
(3-C-ethynyl-beta-d-ribo-pentofuranosyl) cytosine (ECyd), elicits
antitumor effect via TP53-induced Glycolysis and Apoptosis
Regulator (TIGAR) downregulation. Biochem Pharmacol. 79:1772–1780.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lui VW, Wong EY, Ho K, Ng PK, Lau CP, Tsui
SK, Tsang CM, Tsao SW, Cheng SH, Ng MH, et al: Inhibition of c-Met
down-regulates TIGAR expression and reduces NADPH production
leading to cell death. Oncogene. 30:1127–1134. 2011. View Article : Google Scholar
|
|
17
|
Yin L, Kosugi M and Kufe D: Inhibition of
the MUC1-C oncoprotein induces multiple myeloma cell death by
down-regulating TIGAR expression and depleting NADPH. Blood.
119:810–816. 2012. View Article : Google Scholar :
|
|
18
|
Bensaad K, Cheung EC and Vousden KH:
Modulation of intra-cellular ROS levels by TIGAR controls
autophagy. EMBO J. 28:3015–3026. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Napetschnig J and Wu H: Molecular basis of
NF-κB signaling. Annu Rev Biophys. 42:443–468. 2013. View Article : Google Scholar :
|
|
20
|
Jacobs MD and Harrison SC: Structure of an
IkappaBalpha/NF-kappaB complex. Cell. 95:749–758. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perkins ND and Gilmore TD: Good cop, bad
cop: The different faces of NF-kappaB. Cell Death Differ.
13:759–772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li F, Zhang J, Arfuso F, Chinnathambi A,
Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G: NF-κB in cancer
therapy. Arch Toxicol. 89:711–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ren Q, Sato H, Murono S, Furukawa M and
Yoshizaki T: Epstein-Barr virus (EBV) latent membrane protein 1
induces interleukin-8 through the nuclear factor-kappa B signaling
pathway in EBV-infected nasopharyngeal carcinoma cell line.
Laryngoscope. 114:855–859. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo AK, Lo KW, Tsao SW, Wong HL, Hui JW, To
KF, Hayward DS, Chui YL, Lau YL, Takada K, et al: Epstein-Barr
virus infection alters cellular signal cascades in human
nasopharyngeal epithelial cells. Neoplasia. 8:173–180. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheung AK, Ko JM, Lung HL, Chan KW,
Stanbridge EJ, Zabarovsky E, Tokino T, Kashima L, Suzuki T, Kwong
DL, et al: Cysteine-rich intestinal protein 2 (CRIP2) acts as a
repressor of NF-kappaB-mediated proangiogenic cytokine
transcription to suppress tumorigenesis and angiogenesis. Proc Natl
Acad Sci USA. 108:8390–8395. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun W, Guo MM, Han P, Lin JZ, Liang FY,
Tan GM, Li HB, Zeng M and Huang XM: Id-1 and the p65 subunit of
NF-κB promote migration of nasopharyngeal carcinoma cells and are
correlated with poor prognosis. Carcinogenesis. 33:810–817. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kan R, Shuen WH, Lung HL, Cheung AK, Dai
W, Kwong DL, Ng WT, Lee AW, Yau CC, Ngan RK, et al: NF-κB p65
subunit is modulated by latent transforming growth factor-β binding
protein 2 (LTBP2) in nasopharyngeal carcinoma HONE1 and HK1 cells.
PLoS One. 10:e01272392015. View Article : Google Scholar
|
|
29
|
Pawlik TM and Keyomarsi K: Role of cell
cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol
Biol Phys. 59:928–942. 2004. View Article : Google Scholar : PubMed/NCBI
|