Introduction
Pancreatic cancer is the fourth leading cause of
cancer-related deaths in men and women in the US (1). The most common type of pancreatic
cancer is pancreatic ductal adenocarcinoma (PDAC). PDAC is one of
the most lethal cancers with a low 5-year survival rate and limited
therapeutic options. Although the etiology of PDAC is still
unclear, chronic pancreatitis (CP) has been identified as one of
the potential precursors of PDAC (2,3). A
relationship between CP and PDAC has been elucidated in rodent
disease models (4). In both CP and
PDAC, a desmoplastic reaction occurs in the pancreas stroma,
characterized by a collagen-rich extracellular matrix (ECM) and
activated associated inflammatory cells (5–8). One
of the important reasons that chemotherapy does not work well in
PDAC patients is because drugs are poorly delivered to the tumor
due to this dense stroma abundant with collagens, glycoproteins and
low-density vasculature (9,10).
It has been demonstrated that activated pancreatic
stellate cells (PSCs) play a central role in the desmoplastic
reaction in the pancreas (11–13).
PSCs were first described by Watari et al in 1982 (14), and were successfully isolated by
Apte et al (15) and Bachem
et al in 1998 (16). Under
normal physiological conditions, PSCs are mainly located in
periacinar and interlobular areas of the pancreas and maintain a
quiescent phenotype characterized by abundant vitamin A-containing
lipid droplets in the cytoplasm (15). These quiescent PSCs keep the
balance between ECM synthesis and degradation. However, in diseases
such as CP and PDAC, PSCs transform or transdifferentiate into an
activated phenotype, which are devoted to stroma remodeling
(17). During this
transdifferentiation process, PSCs change their morphology and
function as follows: i) they change into a myofibroblastic shape
and lose their vitamin-A containing lipid droplets; ii) they
acquire migration and ECM modulation capacity; and iii) they
upregulate expression of α-SMA, vimentin, desmin and GFAP. While
scientists have previously tried to control or reverse this process
of PSC activation, the mechanisms involved in the activation
process are not fully clarified.
Activated PSCs not only act as positive regulators
of cell proliferation and migration, but inhibit tumor cell
apoptosis. In turn, cancer cells can facilitate the activation,
proliferation, migration and ECM production of PSCs. Such features
prompt local tumor progression as well as regional and distant
metastasis (18,19). Therefore, a better understanding of
the mechanisms of PSC transdifferentiation is crucial for research
on the pathogenesis and clinical treatment of CP and PDAC.
PSC activation shares many common morphological and
functional changes with the epithelial-mesenchymal transition (EMT)
process. EMT is the basis for wound healing in response to chronic
injury induced by toxic, viral, metabolic, or immunological factors
(20,21). The EMT process turns an epithelial
cell into a mesenchymal cell and has been widely studied in the
field of embryogenesis, tissue fibrosis and cancer metastasis
(22–24). Typically, EMT is characterized by
the expression of specific cell-surface proteins and cytoskeletal
proteins, as well as changes in ECM proteins production, followed
by activation of transcription factors. For example, EMT is
characterized by upregulated expression of vimentin and N-cadherin,
and downregulated expression of E-cadherin, accompanied by enhanced
expression of transcription factors, such as Snail, Slug, and Twist
(25,26). The EMT process is divided into
three different types (1–3). Type 2 EMT is characterized by
conversion from endothelial or epithelial cells into fibroblasts,
which are responsible for tissue inflammation and fibrosis, such as
that seen in PDAC (27,28). Morphological changes, with
redistribution of stress fibers and increased migratory capacity,
are other main features of the EMT process.
Due to these similarities between EMT and PSC
activation, we hypothesized that activation of PSCs may involve an
EMT-like process. Indeed, it has previously been reported that an
EMT-like process is involved in transdifferentiation of quiescent
hepatic stellate cells (HSCs), a homology of PSCs in the liver
(29). We tested our hypothesis by
examining the functional alterations and expression of EMT-related
genes during the activation process of PSCs in vitro. By
studying this process of PSC activation, we aim to uncover novel
therapeutic targets for treatment of CP and PDAC.
Materials and methods
Harvesting rat pancreas
All animal studies were reviewed and approved by the
Ethics Committee of Nanjing Medical University in accordance with
the established standards of the humane handling of research
animals. Male Sprague-Dawley rats (150–300 g) were obtained from
Vital River Laboratories (Beijing, China). Rats were kept in
standard laboratory conditions with light-dark cycles of 12–12 h
and free access to chow and water. Rats were sacrificed by
decapitation, before undergoing laparotomy. Rat pancreas was
dissected and placed into cold 0.9% NaCl solution.
Isolation of quiescent PSCs
Quiescent PSCs were isolated from rat pancreas with
enzyme digestion and Nycodenz gradient centrifugation as described
previously (15). Briefly, after
connective tissue and blood vessels were removed, pancreatic tissue
was slowly infused with 10 ml enzyme solution (mixture of
collagenase P, protease and DNase) until all lobules were swollen
and well infused. The pancreas tissue was pre-incubated, finely
minced and incubated again, in order to obtain a cell suspension.
After washing, PSCs were obtained by ladder centrifugation with
Nycodenz solution. Cells were cultured in DMEM/F12 (Wisent, Canada)
supplemented with 10% fetal bovine serum (FBS) (Wisent, Canada), 4
mM L-glutamine (Sigma-Aldrich, St. Louis, MO, USA) and 1%
penicillin-streptomycin (Thermo Scientific Hyclone, Waltham, MA,
USA) at 37°C with 5% CO2. Cells were subcultured at 90%
confluence in the following passages.
Characterization and identification of
isolated cells
Morphological appearances of isolated cells were
assessed under a contrast-phase microscope with blue-green
autofluorescence at 320 nm from lipid droplets within the
cytoplasm. PSCs were evaluated by
immunocytochemistry/immunofluorescence (ICC/IF) and
immunocytochemistry (ICC) using antibodies specific for PSC
markers, α-SMA, desmin, vimentin, and GFAP. Antibodies for
epithelium and macrophage markers (pan-CK, CK19, CD68) were used as
negative controls to rule out possible cell contamination. Cells
were seeded and stained in a 24-well plate (Corning Inc., NY, USA).
ICC/IF and ICC were performed with standard procedures. The primary
antibodies and their working dilutions were as follows: rabbit
anti-α-SMA 1:100 (Abcam, Cambridge, MA, USA); rabbit anti-desmin
1:200 (Abcam); rabbit anti-vimentin 1:100 (Cell Signaling
Technology, Beverly, MA, USA); mouse anti-GFAP 1:500
(Sigma-Aldrich); rabbit anti-CK19 1:300 (Abcam); mouse anti-CD68
1:100 (Abcam); and mouse anti-pan-CK 1:100 (Thermo Fisher
Scientific, USA). DAPI and hematoxylin were used for nuclear
counterstaining in ICC/IF and ICC, respectively. Cells were
observed under a fluorescence microscope (Nikon Ti, Japan).
Cell migration assay
Cell migration assays were carried out in 24-well
transwell chambers with polycarbonate filters (pore size, 8 μm;
Merck Millipore, Billerica, MA, USA). A total of 2×104
cultured cells, at 40 h and on day 10 after isolation, were plated
in serum-free medium in the upper chamber, and media containing 10%
FBS alone was added to the lower chamber. After 40 h of incubation,
residual medium in the upper chamber was removed carefully with a
pipette. Cells on the surface of the filter membrane were fixed and
stained with crystal violet staining solution (Beyotime, Shanghai,
China) according to the manufacturer's instructions. Penetrated
cells on the lower surface of the membrane were examined and
counted under the microscope, before being removed with a cotton
swab. Non-penetrated cells were assessed via the same method.
Migration rates were calculated with the following formula: number
of penetrated cells divided by the total number of both penetrated
and non-penetrated cells.
Live cell motility assay
For cell motility studies of PSC, a live cell
imaging system was used. There are many essential elements for this
system. Firstly, a camera was mounted on the pressurized chamber on
the stage of a Nikon TE 2000-E inverted optical microscope.
Secondly, the microscope employed a number of computer controlled
motorized systems in order to rapidly reconfigure the microscope
during automated image acquisition. Finally, a small cell incubator
on the microscope was used to control the cell environment and
maintain our samples in a humidified and CO2 enriched
atmosphere. The system was kept in a darkened room with ambient
temperature maintained close to 25°C. The entire process was
controlled by NIH-Elements AR software, and the following image
processing was performed using Image-Pro Plus 6.0. Cells were
cultured in 6-well plate and single cell movements was tracked with
automatic record of a series of continuous images at an interval of
2 min under 100 magnified vision over 72 h. Cell motility capacity
was assessed with the length of cell movement track over a fixed
time.
Total RNA isolation and quantitative
RT-PCR
Total RNA was extracted from quiescent and activated
PSCs cultured in 6-cm Petri dishes with Qiagen RNeasy Micro kit
(Qiagen, Mannheim, Germany) according to the manufacturer's
instructions. Agarose gel electrophoresis was used for quality
control of the isolated RNA. Reverse transcription was conducted
using PrimeScript RT Master Mix (Takara, Dalian, China), while
quantitative reverse transcription polymerase chain reaction
(qRT-PCR) was performed using FastStart Universal SYBR Green Master
(ROX) (Roche, Germany) on a StepOne Plus Real-Time-PCR System
(Applied Biosystems, Waltham, MA, USA) according to the
manufacturer's instructions. The PCR program consisted of an
initial enzyme activation step at 95°C for 10 min, followed by 45
cycles of amplification with 95°C for 15 sec, followed by
incubation at an appropriate temperature for 1 min. Finally, a
melting curve profile was set at 95°C (15 sec), 60°C (1 min), and
95°C (15 sec). β-actin was used as an endogenous control to which
each gene of interest was normalized. Relative quantification was
calculated by the ΔΔCT method and normalized based on the
designated control. Primer sequences used in qRT-PCR are shown in
Table I.
 | Table ISequences of primers used in qRT-PCR
for EMT related genes. |
Table I
Sequences of primers used in qRT-PCR
for EMT related genes.
| Gene | Accession no. | Primer sequence
(5′-3′) | Amplicon (bp) | Ta |
|---|
| rat S100A4 | NM_012618.2 | Forward:
TCCACCTTCCACACATACTCAG
Reverse: TTCATTGTCCCTGTTGCTGT | 168 | 55 |
| rat Vimentin | NM_031140.1 | Forward:
ATGTCCGCCAGCAGTATG
Reverse: CCTGTCTCCGGTATTCGTTT | 156 | 56 |
| rat
Collagen1α1 | NM_053304.1 | Forward:
CTGCCCAGAAGAATATGTATCAC
Reverse: GAAGCAAAGTTTCCTCCAAG | 198 | 58 |
| rat
Fibronectin1 | NM_019143.2 | Forward:
GCCCTTACAGTTCCAAGTTCC
Reverse: TTGTGCCTCCTCTGGTTGTG | 114 | 55 |
| rat N-cadherin | AB017695.1 | Forward:
CTGAATGGGATGCTGAGGT
Reverse: TTGAAAGGCCGTAAGTGGG | 193 | 55 |
| rat E-cadherin | NM_031334.1 | Forward:
ACAGGCCAGAGTTTATCCAGG
Reverse: TGAGGATGGTGTAGGCGATG | 144 | 55 |
| rat BMP7 | NM_001191856.1 | Forward:
AAAACAGCAGCAGTGACCAG
Reverse: TTCGTGTAGGAGTTCAGAGG | 157 | 54 |
| rat
Desmoplakin | BC098071.1 | Forward:
GGTCTGGTAGGCATTGAGTT
Reverse: AGTTCCTTGTTCATCGCTTG | 119 | 52 |
| rat Slug | U97061.1 | Forward:
GGAGCGTACAGCCCTATAACT
Reverse: CTAATGGGACTTTCTGAACCAC | 170 | 55 |
| rat Snail1 | NM_053805.1 | Forward:
AGTTGTCTACCGACCTTGCG
Reverse: TGCAGCTCGCTATAGTTGGG | 128 | 52 |
| rat HHIP | NM_001191817.1 | Forward:
AATGTGAGCCACCTTGTCGT
Reverse: TCACACTGAGGGCCGAGATA | 88 | 50 |
| rat Gli1 | NM_001191910.1 | Forward:
GGACTTTCTGGTCTGCCCTT
Reverse: AGATGGAAAGAGCCCGCTTC | 157 | 54 |
| rat Gli2 | NM_001107169.1 | Forward:
TAAGCGGAGCAAGGTCAAG
Reverse: GTGGCAGTTGGTCTCGTAGAT | 154 | 57 |
| rat Shh | NM_017221.1 | Forward:
CATCCCTTGGGAATGGCAGT
Reverse: TGCTTATCTGGCAGTCGCTT | 98 | 52 |
| rat ACTB | NM_031144.3 | Forward:
TGTGCTATGTTGCCTCAGACT
Reverse: CATTGCCGATAGTGATGACCT | 111 | 55 |
Western blotting
Total proteins were prepared with a protein
extraction kit (Beyotime) and quantified by the bicinchoninic acid
assay (BCA) method (Beyotime) according to the manufacturer's
instructions. Polyvinylidene fluoride (PVDF) membranes (Millipore)
were blocked in 5% non-fat dried milk in phosphate-buffered saline
with Tween-20 (PBST) and incubated overnight at 4°C with
appropriate primary antibodies [rabbit anti-α-SMA, dilution 1:200
(Abcam); rabbit anti-vimentin, dilution 1:1,000 (Cell Signaling
Technology); rabbit anti-S100A4, dilution 1:250 (Abcam); rabbit
anti-N-cadherin, dilution 1:200 (Santa Cruz Biotechnology, Dallas,
TX, USA); mouse anti-E-cadherin, dilution 1:1,000 (Abcam); rabbit
anti-BMP7, dilution 1:1,000 (Abcam); rabbit anti-Slug, dilution
1:700 (Abcam); and rabbit anti-Snail, dilution 1:1,000 (Cell
Signaling Technology)]. Membranes were then washed and incubated
for 2 h at room temperature with secondary horseradish
peroxidase-conjugated anti-mouse IgG or anti-rabbit IgG (Cell
Signaling Technology). All protein expression levels were
normalized to β-actin (Abcam).
Statistical analysis
Statistical analysis was performed using SPSS,
version 13.0 (SPSS, Inc., Chicago, IL, USA). All experiments were
repeated in triplicate, and the most representative data are shown
in this report. Numeric data were expressed as mean ± standard
deviation (SD). Statistical significance between two groups was
determined by the Student's t-test, with P-values <0.05
considered statistically significant.
Results
Isolation and identification of rat
quiescent and activated PSCs
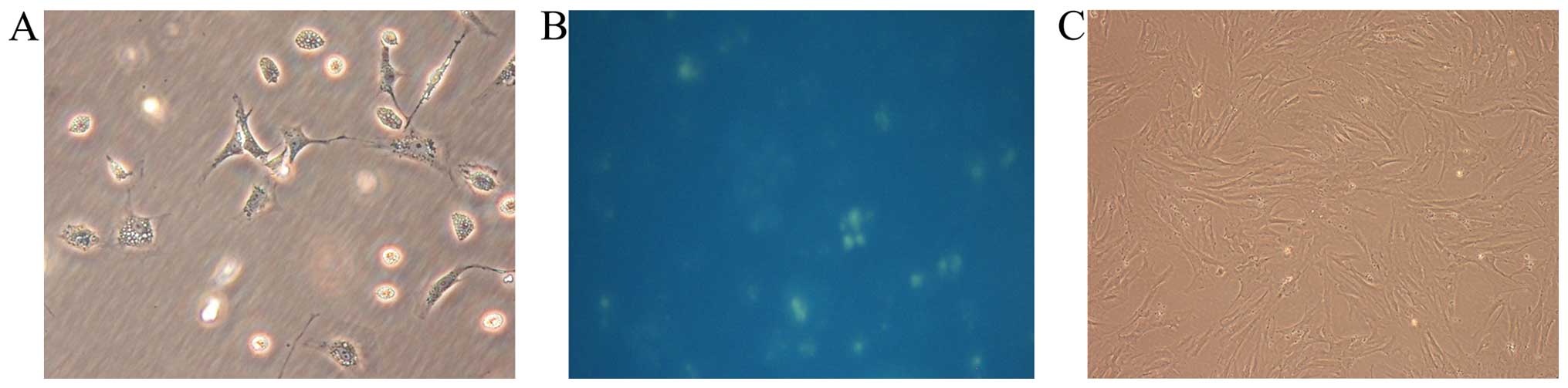
The yield of rat quiescent PSCs was ~4 to 7 million
cells per gram of pancreatic tissue. Freshly isolated PSCs remained
quiescent within the first 48 h of primary culture as indicated by
their typical round or polygon shape and autofluorescence from
lipid droplets in their cytoplasm (15). After 48 h, they began to transform
to an ‘activated’ form, acquiring fibroblastic characteristics and
losing their lipid droplets. Apte et al previously
demonstrated that freshly isolated PSCs cultured on plastic were
gradually activated after 48 h and fully activated after 7 days
in vitro (15). For this
reason, we chose cells cultured for 40 h after isolation as
representative of the quiescent state and cells cultured for 10
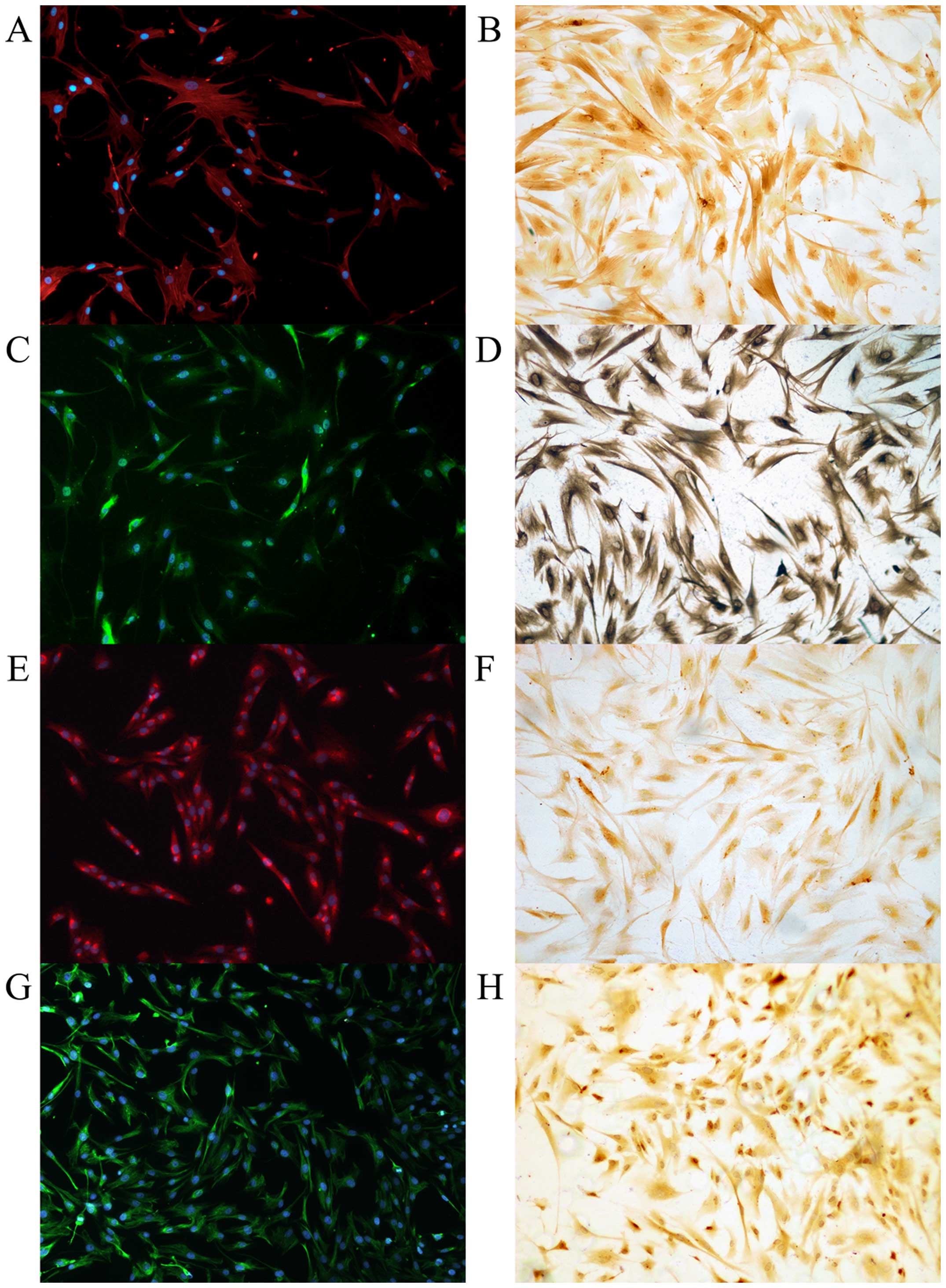
days as representing the fully activated state (Fig. 1). Primarily cultured cells were
positive for PSC marker (α-SMA, vimentin, GFAP and desmin) staining
in ICC and ICC/IF (Fig. 2). To
exclude possible contamination with macrophages and epithelial
cells, the purity of PSCs were confirmed with negative
immunostaining with CK19, CD68, and pan-CK (Fig. 3).
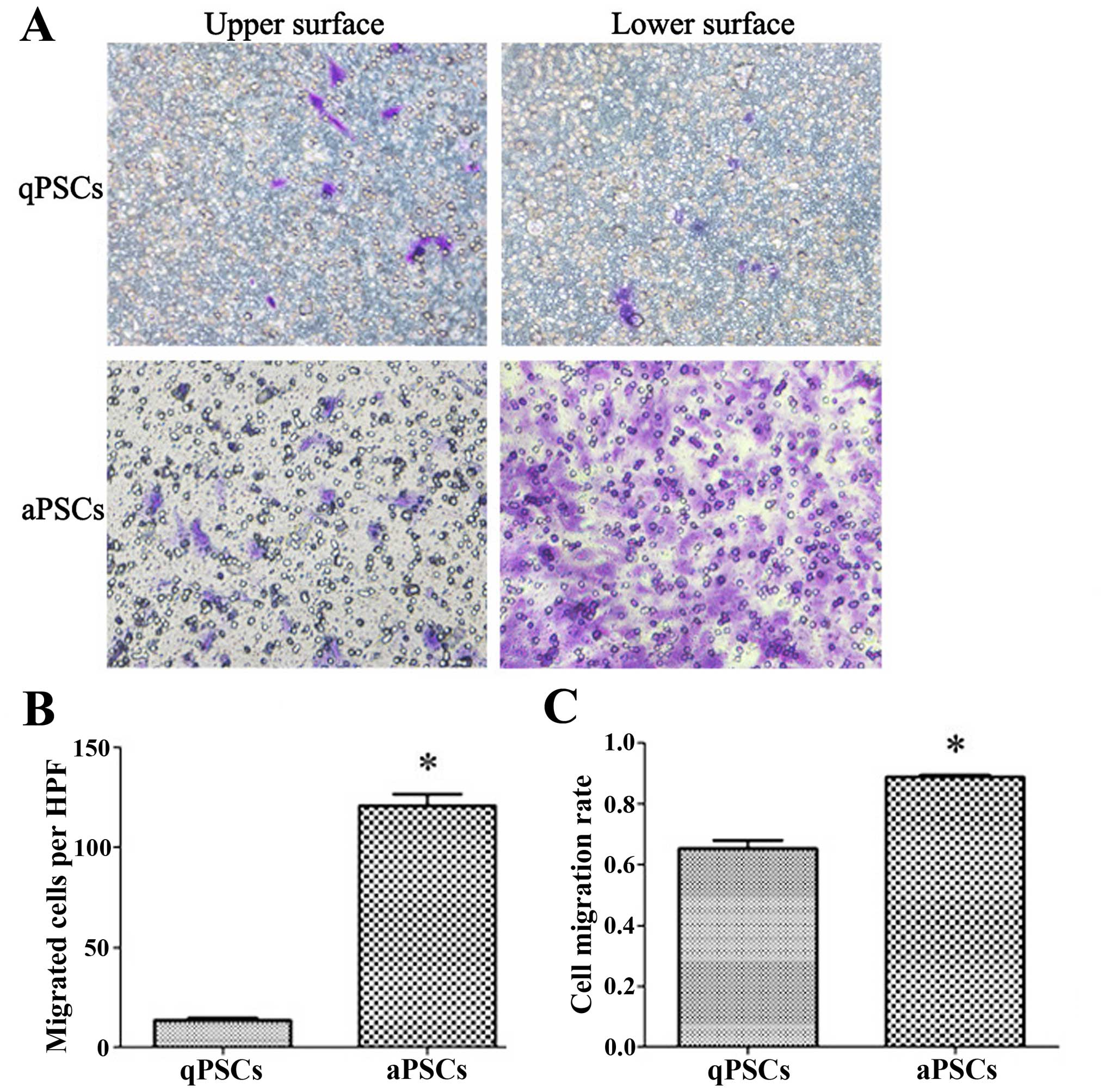
Cell migration ability is promoted after
PSC activation
PSC migration ability was assessed using transwell
chambers with 40-h incubation time. More cells migrated to the
lower surface of the membrane 10 days after isolation compared with
those isolated after only 40 h. In order to rule out possible
differences in cell adherence and proliferation ability after
activation, the cell migration rate was calculated. Increased cell
migration rate was observed after PSC activation (Fig. 4).
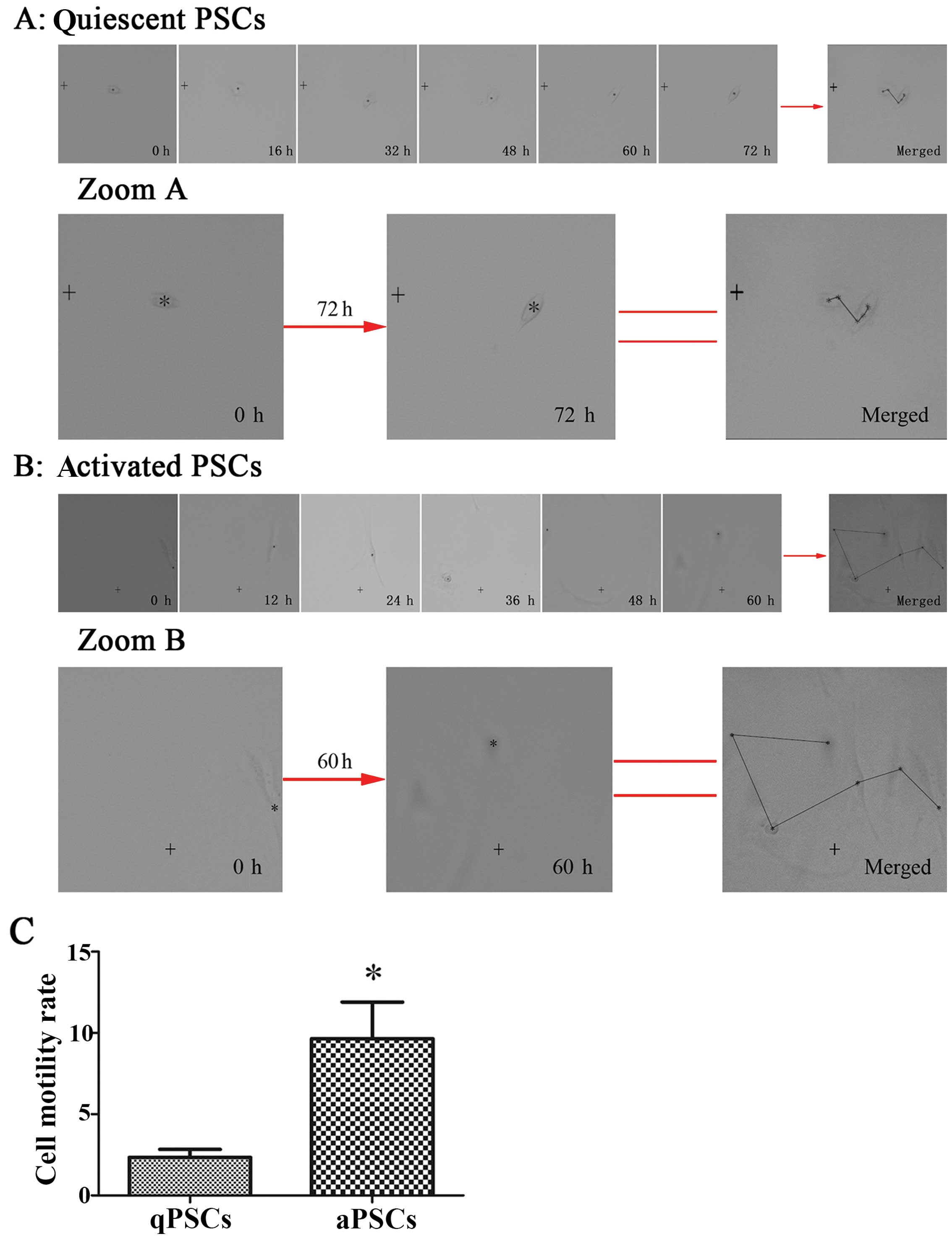
Live cell motility rate is increased
after PSC activation
Cell motility capacity was assessed by live cell
imaging system. PSC motility capacity was increased after cell
activation, as the activated PSCs moved a longer distance during a
fixed culture time. The real-time images of randomly chosen
monitored cells are showed in Fig.
5. At the same time, PSCs underwent characteristic
morphological changes in vitro, including loss of lipid
droplets, increased cell volume and transition to a myofibroblastic
phenotype, which is in accordance with our previous findings
(15,16).
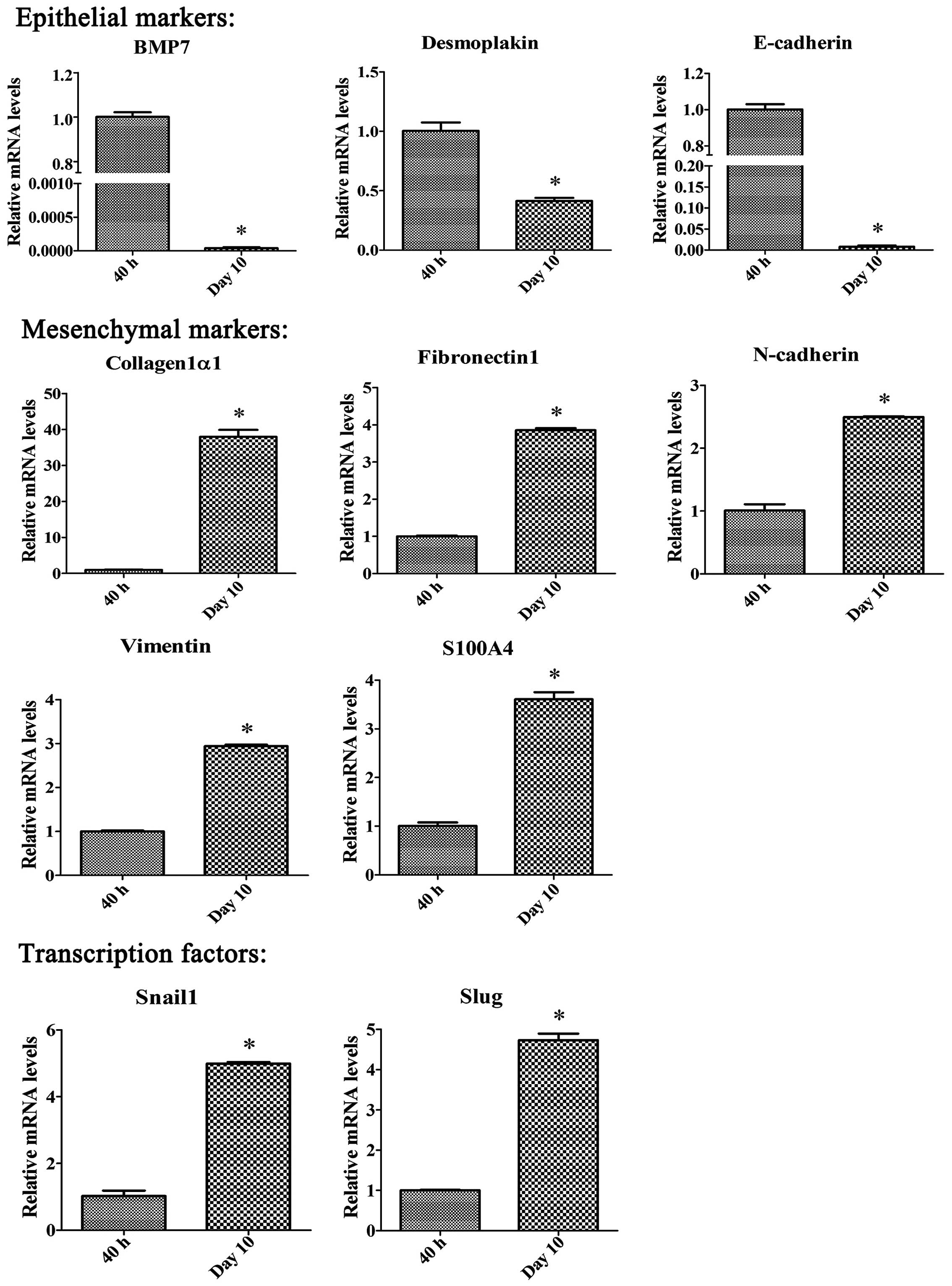
Changes in the expression of an
EMT-related gene panel after PSCs activation
To determine whether PSC activation involve an
EMT-like process, we analyzed expression of epithelial markers
E-cadherin, bone morphogenetic protein 7 (BMP7), and desmoplakin,
and the mesenchymal markers N-cadherin, fibroblast-specific protein
1 (S100A4), vimentin, fibronectin1, and collagen1α1, during the PSC
activation process by qRT-PCR. E-cadherin, BMP7 and desmoplakin
were significantly downregulated, while N-cadherin, S100A4,
vimentin, fibronectin1 and collagen1α1 were upregulated in
activated PSCs after 10 days of in vitro culture compared
with quiescent PSCs (Fig. 6).
Since the Snail transcription factor family, such as
Snail and Slug (also known as Snail1 and Snail2), is reported to
play a crucial role during the EMT process, we used qRT-PCR to
examine the expression of Snail1 and Slug during PSC activation.
Both Snail1 and Slug mRNA levels increased significantly in
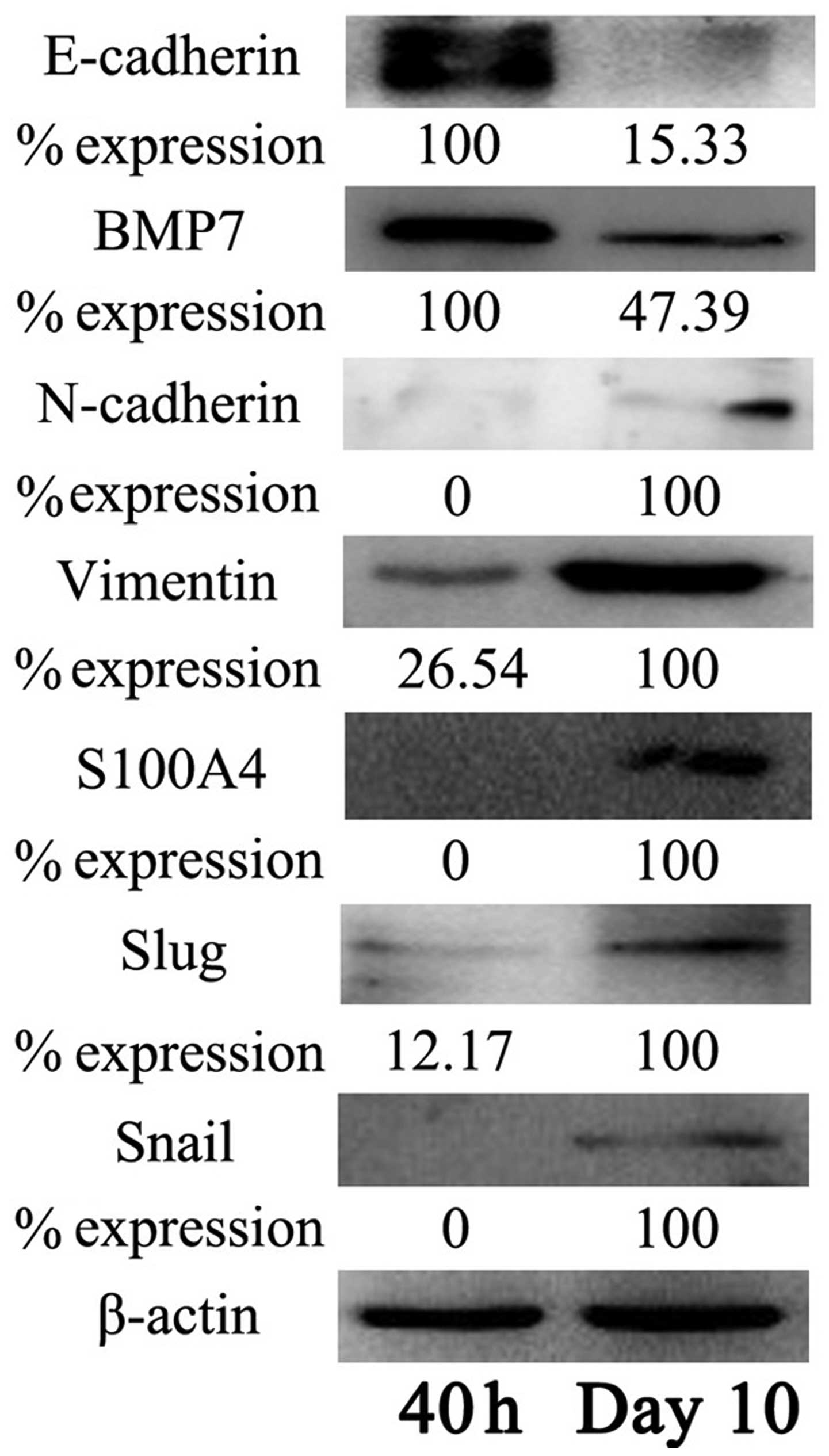
activated PSCs compared to quiescent PSCs. We also examined the
expression of EMT-related genes at the protein level by
immunoblotting. Western blot analysis confirmed that the decreased
protein expression of epithelial markers, including E-cadherin and
BMP-7, was accompanied by increased expression of mesenchymal
markers (i.e., N-cadherin, vimentin, S100A4) after PSCs were
activated (Fig. 7). Furthermore,
the two EMT-related transcription factors, Snail and Slug, also
showed increased expression at the protein level during PSC
activation.
Discussion
Previous studies proved that PSCs stimulated the EMT
process of cancer cells, and thereby elevated tumor migration and
invasion (30). The present study
provides novel evidence that PSC activation induced EMT-related
gene expression in vitro. We also observed significant
alterations in morphology and migration capacity when PSCs were
fully activated. We found that PSC activation was accompanied by
downregulation of E-cadherin and upregulation of N-cadherin,
vimentin, collagen1α1 and fibronectin1 gene expression.
Besides the classical molecular regulators of EMT
described above, we also examined expression of BMP7 and S100A4.
Bone morphogenetic proteins such as BMP7 belong to a large family
of cytokines, which regulate various biological processes including
cell proliferation, apoptosis, differentiation and morphogenesis
(31). A close relationship
between BMP signaling and EMT during embryogenesis, fibrosis and
cancer development has been elucidated by several studies (32–34).
BMP7, in particular is recognized as an antagonist of EMT induced
by TGF-β (35,36). In this study, BMP7 expression was
significantly decreased in activated PSCs.
S100A4 is a member of the calcium-binding S100
protein family, and has been associated with cell proliferation,
cellular adhesion, reconstruction of the ECM, angiogenesis and
cellular motility (37–39). Cells that express both α-SMA and
S100A4 are identified as fibroblasts (40) and S100A4 is expressed in
fibroblasts during type 2 EMT (41). In our study, when PSCs maintained a
fully activated state, S100A4 expression was notably increased.
Furthermore, the present study proves that the Snail
and Slug transcription factors were related to the activation of
PSCs. The vertebrate Snail genes mediating EMT are divided into two
subtypes: Snail1 and Snail2 (also known as Slug) (42,43).
Snail1 and Snail2 are known as repressors of E-cadherin and can
upregulate mesenchymal markers such as vimentin (44,45).
Previous research showed that Snail expression was associated with
tissue fibrosis (42). In
addition, Scarpa et al reported that Snail1 plays a pivotal
role in HSC activation (46).
Similarly, in this study, Snail1 and Slug were upregulated in PSC
activation.
Based on the results of this study and previous
studies, it is clear that PSCs complete an EMT-like process that
promotes the development of a migratory, mesenchymal phenotype from
a quiescent phenotype. Therefore, the EMT process is a key event in
PSC activation. It follows that reversing the EMT process could be
a potential therapeutic strategy for chronic pancreatitis and
pancreatic cancer. However, little is known about the reverse
process of EMT, which is called mesenchymal-epithelial transition
(MET). The process of MET is well known in kidney formation, and
multiple genes are involved, including BMP7 (47–49).
Similarly, BMP7 has been reported to act as a positive regulator of
MET in mice (35). BMP-7
antagonizes TGF-β1-induced fibrotic effects in vitro and
reverses fibrosis in various organs such as the kidney, heart and
colon (50). Therefore, reducing
the expression of BMP7 is likely to restore quiescence in activated
PSCs. However, further work is required to clarify the molecular
mechanisms underlying the signaling pathways involved in EMT and
MET processes.
One pathway known to be involved in EMT is the
Hedgehog pathway. This pathway promotes formation of mesenchymal
cells and has been shown to participate in HSC transdifferentiation
from quiescent HSCs to myofibroblastic HSCs in vitro
(51–53). Similarly, in vivo studies
have indicated that Hedgehog pathway activation is associated with
EMT, myofibroblast proliferation and liver fibrosis while
inhibition of this pathway attenuates EMT, myofibroblast
accumulation and fibrosis in mouse models (29,54,55).
For this reason, genes related to the Hedgehog pathway were
examined by qRT-PCR in the present study (data not shown). We found
the in the activated PSCs, Gli1, Gli2 (i.e., Hedgehog target genes)
and sonic hedgehog (i.e., the Hedgehog ligand) were all
upregulated, and HHIP (i.e., a Hedgehog ligand antagonist) was
downregulated. However, additional research is needed to confirm
whether the Hedgehog signaling pathway modulates the process of PSC
transdifferentiation.
In conclusion, the present study supports that the
transition of quiescent PSCs to activated myofibroblastic PSCs
involves an EMT-like process in vitro. This knowledge
improves our understanding of the pathogenesis of pancreatic
fibrogenesis, and offers a potential theoretical basis for future
research on the treatment of PDAC and chronic pancreatitis.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation of China (81272239, 81170336, 81272382,
81300351), the Natural Science Foundation of Jiangsu Province
(BK2012881), the Program for Development of Innovative Research
Team in the First Affiliated Hospital of NJMU, the Priority
Academic Program Development of Jiangsu Higher Education
Institutions (PAPD, JX10231801), and the Translational Research of
Early Diagnosis and Comprehensive Treatment in Pancreatic Cancer
(The research Special Fund For Public Welfare Industry of Health,
201202007).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pandol S, Gukovskaya A, Edderkaoui M,
Dawson D, Eibl G and Lugea A: Epidemiology, risk factors, and the
promotion of pancreatic cancer: Role of the stellate cell. J
Gastroenterol Hepatol. 27(Suppl 2): 127–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chu GC, Kimmelman AC, Hezel AF and DePinho
RA: Stromal biology of pancreatic cancer. J Cell Biochem.
101:887–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Apte MV, Park S, Phillips PA, Santucci N,
Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA,
et al: Desmoplastic reaction in pancreatic cancer: Role of
pancreatic stellate cells. Pancreas. 29:179–187. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamaguchi K: How to define patients at
high risk for pancreatic cancer. Pancreatology. 11(Suppl 2): 3–6.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang W, Liao Z, Li G, Li ZS, Chen J, Zhan
XB, Wang LW, Liu F, Hu LH, Guo Y, et al: Incidence of pancreatic
cancer in Chinese patients with chronic pancreatitis.
Pancreatology. 11:16–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kudo Y, Kamisawa T, Anjiki H, Takuma K and
Egawa N: Incidence of and risk factors for developing pancreatic
cancer in patients with chronic pancreatitis.
Hepatogastroenterology. 58:609–611. 2011.PubMed/NCBI
|
|
8
|
Pezzilli R, Vecchiarelli S, Di Marco MC,
Serra C, Santini D, Calculli L, Fabbri D, Rojas Mena B and Imbrogno
A: Pancreatic ductal adenocarcinoma associated with autoimmune
pancreatitis. Case Rep Gastroenterol. 5:378–385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liotta LA and Kohn EC: The
microenvironment of the tumour-host interface. Nature. 411:375–379.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Wever O and Mareel M: Role of tissue
stroma in cancer cell invasion. J Pathol. 200:429–447. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guerra C, Schuhmacher AJ, Cañamero M,
Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP and
Barbacid M: Chronic pancreatitis is essential for induction of
pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice.
Cancer Cell. 11:291–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Erkan M, Adler G, Apte MV, Bachem MG,
Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch
HJ, et al: StellaTUM: Current consensus and discussion on
pancreatic stellate cell research. Gut. 61:172–178. 2012.
View Article : Google Scholar
|
|
13
|
Jaster R: Molecular regulation of
pancreatic stellate cell function. Mol Cancer. 3:262004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Watari N, Hotta Y and Mabuchi Y:
Morphological studies on a vitamin A-storing cell and its complex
with macrophage observed in mouse pancreatic tissues following
excess vitamin A administration. Okajimas Folia Anat Jpn.
58:837–858. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Apte MV, Haber PS, Applegate TL, Norton
ID, McCaughan GW, Korsten MA, Pirola RC and Wilson JS: Periacinar
stellate shaped cells in rat pancreas: Identification, isolation,
and culture. Gut. 43:128–133. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bachem MG, Schneider E, Gross H,
Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grünert A and
Adler G: Identification, culture, and characterization of
pancreatic stellate cells in rats and humans. Gastroenterology.
115:421–432. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Apte M, Pirola R and Wilson J: The
fibrosis of chronic pancreatitis: new insights into the role of
pancreatic stellate cells. Antioxid Redox Signal. 15:2711–2722.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Apte MV, Pirola RC and Wilson JS:
Pancreatic stellate cells: A starring role in normal and diseased
pancreas. Front Physiol. 3:3442012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Apte MV, Wilson JS, Lugea A and Pandol SJ:
A starring role for stellate cells in the pancreatic cancer
microenvironment. Gastroenterology. 144:1210–1219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boyer B, Vallés AM and Edme N: Induction
and regulation of epithelial-mesenchymal transitions. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scheel C and Weinberg RA: Phenotypic
plasticity and epithelial-mesenchymal transitions in cancer and
normal stem cells? Int J Cancer. 129:2310–2314. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kalluri R and Neilson EG:
Epithelial-mesenchymal transition and its implications for
fibrosis. J Clin Invest. 112:1776–1784. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neilson EG: Plasticity, nuclear diapause,
and a requiem for the terminal differentiation of epithelia. J Am
Soc Nephrol. 18:1995–1998. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guarino M: Epithelial-mesenchymal
transition and tumour invasion. Int J Biochem Cell Biol.
39:2153–2160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Moustakas A: Integrins open the way to
epithelial-mesenchymal transitions. Cell Cycle. 9:16822010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi SS, Omenetti A, Witek RP, Moylan CA,
Syn WK, Jung Y, Yang L, Sudan DL, Sicklick JK, Michelotti GA, et
al: Hedgehog pathway activation and epithelial-to-mesenchymal
transitions during myofibroblastic transformation of rat hepatic
cells in culture and cirrhosis. Am J Physiol Gastrointest Liver
Physiol. 297:G1093–G1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kikuta K, Masamune A, Watanabe T, Ariga H,
Itoh H, Hamada S, Satoh K, Egawa S, Unno M and Shimosegawa T:
Pancreatic stellate cells promote epithelial-mesenchymal transition
in pancreatic cancer cells. Biochem Biophys Res Commun.
403:380–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hogan BL: Bone morphogenetic proteins:
Multifunctional regulators of vertebrate development. Genes Dev.
10:1580–1594. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang MH, Kim JS, Seo JE, Oh SC and Yoo YA:
BMP2 accelerates the motility and invasiveness of gastric cancer
cells via activation of the phosphatidylinositol 3-kinase
(PI3K)/Akt pathway. Exp Cell Res. 316:24–37. 2010. View Article : Google Scholar
|
|
33
|
Klahr S: The bone morphogenetic proteins
(BMPs). Their role in renal fibrosis and renal function. J Nephrol.
16:179–185. 2003.PubMed/NCBI
|
|
34
|
Ohta S, Schoenwolf GC and Yamada G: The
cessation of gastrulation: BMP signaling and EMT during and at the
end of gastrulation. Cell Adhes Migr. 4:440–446. 2010. View Article : Google Scholar
|
|
35
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zeisberg M, Shah AA and Kalluri R: Bone
morphogenic protein-7 induces mesenchymal to epithelial transition
in adult renal fibroblasts and facilitates regeneration of injured
kidney. J Biol Chem. 280:8094–8100. 2005. View Article : Google Scholar
|
|
37
|
Tarabykina S, Griffiths TR, Tulchinsky E,
Mellon JK, Bronstein IB and Kriajevska M: Metastasis-associated
protein S100A4: Spotlight on its role in cell migration. Curr
Cancer Drug Targets. 7:217–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mazzucchelli L: Protein S100A4: Too long
overlooked by pathologists? Am J Pathol. 160:7–13. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li ZH and Bresnick AR: The S100A4
metastasis factor regulates cellular motility via a direct
interaction with myosin-IIA. Cancer Res. 66:5173–5180. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zeisberg EM, Potenta S, Xie L, Zeisberg M
and Kalluri R: Discovery of endothelial to mesenchymal transition
as a source for carcinoma-associated fibroblasts. Cancer Res.
67:10123–10128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Strutz F, Okada H, Lo CW, Danoff T, Carone
RL, Tomaszewski JE and Neilson EG: Identification and
characterization of a fibroblast marker: FSP1. J Cell Biol.
130:393–405. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nieto MA: The snail superfamily of
zinc-finger transcription factors. Nat Rev Mol Cell Biol.
3:155–166. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Scarpa M, Grillo AR, Brun P, Macchi V,
Stefani A, Signori S, Buda A, Fabris P, Giordani MT, De Caro R, et
al: Snail1 transcription factor is a critical mediator of hepatic
stellate cell activation following hepatic injury. Am J Physiol
Gastrointest Liver Physiol. 300:G316–G326. 2011. View Article : Google Scholar
|
|
47
|
Lipschutz JH: Molecular development of the
kidney: A review of the results of gene disruption studies. Am J
Kidney Dis. 31:383–397. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hogan BL and Kolodziej PA: Organogenesis:
Molecular mechanisms of tubulogenesis. Nat Rev Genet. 3:513–523.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rothenpieler UW and Dressler GR: Pax-2 is
required for mesenchyme-to-epithelium conversion during kidney
development. Development. 119:711–720. 1993.PubMed/NCBI
|
|
50
|
Mizuiri S, Hemmi H, Arita M, Tai R,
Hattori Y, Muto A, Suzuki Y, Ohashi Y, Sakai K and Aikawa A:
Effluent markers related to epithelial mesenchymal transition with
adjusted values for effluent cancer antigen 125 in peritoneal
dialysis patients. Int J Nephrol. 2011:2610402011.PubMed/NCBI
|
|
51
|
Bailey JM, Singh PK and Hollingsworth MA:
Cancer metastasis facilitated by developmental pathways: Sonic
hedgehog, Notch, and bone morphogenic proteins. J Cell Biochem.
102:829–839. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Katoh Y and Katoh M: Hedgehog signaling,
epithelial-to-mesenchymal transition and miRNA (review). Int J Mol
Med. 22:271–275. 2008.PubMed/NCBI
|
|
53
|
Choi SS, Syn WK, Karaca GF, Omenetti A,
Moylan CA, Witek RP, Agboola KM, Jung Y, Michelotti GA and Diehl
AM: Leptin promotes the myofibroblastic phenotype in hepatic
stellate cells by activating the hedgehog pathway. J Biol Chem.
285:36551–36560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Omenetti A, Porrello A, Jung Y, Yang L,
Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, et
al: Hedgehog signaling regulates epithelial-mesenchymal transition
during biliary fibrosis in rodents and humans. J Clin Invest.
118:3331–3342. 2008.PubMed/NCBI
|
|
55
|
Syn WK, Jung Y, Omenetti A, Abdelmalek M,
Guy CD, Yang L, Wang J, Witek RP, Fearing CM, Pereira TA, et al:
Hedgehog-mediated epithelial-to-mesenchymal transition and
fibrogenic repair in nonalcoholic fatty liver disease.
Gastroenterology. 137:1478–1488. e14782009. View Article : Google Scholar : PubMed/NCBI
|