Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV), a
gamma-herpesvirus, is associated with Kaposi's sarcoma (KS) and
several lymphoproliferative diseases, including primary effusion
lymphoma (PEL) and multicentric Castleman's disease (MCD) (1). PEL, which was originally identified
in patients with advanced AIDS, belongs to a variant of
non-Hodgkin's B lymphoma showing serious lymphomatous effusion in
body cavities. Although treated with cytotoxic chemotherapeutic
agents, prognosis of patients is extremely poor upon diagnosis,
with a median survival times of 3–6 months (2). Similar to the other herpesviruses,
KSHV infection can be divided into latent and lytic stages and can
establish a life-long persistence in the host after primary
infection (3). During the latent
infection stage, only a limited set of genes are expressed,
including open reading frame (Orf) 71, Orf 72, Orf
73, viral-encoded microRNAs. After reactivating and entering
lytic stage, most viral genes are expressed in an orderly fashion
based on the time of expression, defined as immediate-early, early
and late genes, which facilitate genomic DNA replication and mature
virion production (4,5).
Currently, candidate drugs for management of
KSHV-related diseases can be divided in two groups, which include
compounds targeting viral DNA polymerase or not. DNA polymerase
inhibitors are mainly involved in nucleoside analogs, acyclic
nucleoside phosphonates, pyrophosphate analogs and non-nucleoside
analogs. Non-DNA polymerase inhibitors, which target viral or
cellular proteins, include compounds isolated from plants (6). For example, resveratrol, a
non-flavonoid polyphenol derived from Polygonum cuspidatum,
was demonstrated to decrease ERK1/2 activity and expression of
Egr-1 in KSHV-infected cells, resulting in inhibition of KSHV
reactivation from latency (7).
Additionally, DD2, a benzyl-substituted 4-(pyridine-2-amido)
benzoic acid, trapped an inactive monomeric conformation, resulting
in disruption of KSHV proteasomal dimerization (8). Despite the fact that various
antiviral agents are known to inhibit KSHV replication, studies on
these pre-drugs, mainly in vitro, are limited due to toxic
effect or incomplete efficiency. Moreover, these antiviral agents
can not eliminate persistent latent KSHV. To date, there are no
standard therapeutic guidelines for the treatment of KSHV-infected
diseases, and little progress has been made since the discovery of
KSHV 20 years ago.
Latency-associated nuclear antigen 1 (LANA1),
encoded by KSHV Orf 73, is a viral oncoprotein that
contributes to tumorigenesis (9).
LANA1 is a protein expressed in all KSHV-infected cells and is
required for viral genomic establishment and maintenance in the
nucleus (10). As a master
regulator of KSHV latency, LANA1 can also be considered as a
transcriptional modulator for host cell genes, resulting in
dysregulation of several pathways (11). For example, LANA1 can regulate the
Wnt pathway by nuclear trapping of glycogen synthase kinase 3β
(GSK3β), resulting in stabilizing of β-catenin (12). LANA1 is essential for KSHV-induced
angiogenesis and can activate phosphorylation of survivin, which
contributes to cell proliferation and viral latent replication
(13,14). LANA1 has also been shown to enhance
transactivation of human telomerase reverse transcriptase (hTERT)
promoter by interacting with transcription factor specificity
protein 1 (Sp1), therefore, contributing to immortalization of PEL
cells (15). Additionally, LANA1
targets degradation of the mitotic checkpoint kinase Bub1,
resulting in chromosomal instability, and facilitates establishment
of viral latency through interacting with Krüppel-associated box
domain associated protein-1 (KAP1) (16,17).
LANA1 also interacts with tumor suppressors, such as p53 and Rb
proteins, leading to disruption of their tumor-suppressive
functions (18,19). Moreover, LANA1 enhances
transcriptional activity of signal transducer and activator of
transcription 3 (STAT3) via physical and functional interaction
with STAT3 (20). Collectively,
the biological insights into LANA1 make it a promising rational
target for therapeutic strategies.
Triptolide, a diterpenoid triepoxide, is purified
from the roots of Chinese herb Tripterygium wilfordii
(21). Triptolide displays a
broad-spectrum bioactivity profile, including antitumor,
anti-inflammatory, immunosuppressive and anti-fertility effects. It
has been used for treatment of autoimmune diseases such as
rheumatoid arthritis (22). In
addition, triptolide has been reported to inhibit proliferation and
induce apoptosis against a variety of tumor cell lines in
vitro, including cholangiocarcinoma, melanoma, breast, gastric
cancer and non-small cell lung carcinoma (23). Triptolide can also induce
pancreatic cancer cell death via inhibition of Hsp70 and O-GlcNAc
modification of transcription factor Sp1 (24,25).
Currently, triptolide has entered clinical trials for treatment of
diseases including psoriasis vulgaris, diabetic nephropathy and
nephritic syndrome. Minnelide, a water-soluble pre-drug of
triptolide, has also entered clinical trials for treatment of
pancreatic cancer showing good patient toleration profile (26). However, little is known about
effect of triptolide on KSHV-associated PEL.
Previously, our laboratory found that Hsp90
inhibitors inhibit outgrowth of Epstein-Barr virus (EBV, another
gamma-herpesvirus closely related to KSHV)-infected malignant cells
via downregulation of EBV nuclear antigen 1 (EBNA1) (27). A small molecule pre-drug 17-DMAG
(an Hsp90 inhibitor) also decreases expression of conserved
herpesvirus protein kinases in EBV-infected malignant cells
(28). Recently, we found that
triptolide inhibited proliferation of latency III type infected
EBV-positive B lymphocytes through the virus latent membrane
protein 1 (LMP1)-dependent mechanism (29). In the present study, our results
demonstrate that triptolide can effectively inhibit cell
proliferation and induce cell apoptosis in the PEL cells.
Furthermore, triptolide specifically decreases LANA1 expression
without reducing the LANA1 transcript level. LANA1 accumulates
after treatment with proteasomal inhibitor MG-132 in the absence or
presence of triptolide. It is also indicated that triptolide
reduces the half-life of LANA1. In addition, the number of viral
DNA copies and the production of virions are reduced by triptolide.
Moreover, triptolide inhibited expression of the phosphorylated
STAT3 and secretion of IL-6. Finally, we also show that the
KSHV-induced lymphoproliferative diseases in non-obese
diabetic/severe combined immunodeficient (NOD-SCID) mice are
strongly inhibited by a non-toxic dose of triptolide.
Materials and methods
Cell lines and reagents
PEL cells (BCBL-1, JSC-1 and BC-3) and a Burkitt
lymphoma cell line, BJAB, were gifts kindly provided by Professor
K. Lan (Institute Pasteur of Shanghai, Chinese Academy of Sciences,
Shanghai, China). P3HR-1 cells (EBV-positive and KSHV-negative)
were kindly provided by Professor Y. Cao (Central South University,
Changsha, China). Peripheral blood mononuclear cells (PBMCs) were
isolated from healthy donors by using Ficoll-Paque Premium (GE
Healthcare, Uppsala, Sweden) according to the manufacturer's
instructions. Human renal embryonic 293T cells and HeLa cells,
which were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA), were obtained from Professor H. Li
(Wuhan University, Wuhan, China).
BJAB, P3HR-1, BCBL-1, BC-3, JSC-1 cells and PBMCs
were maintained in RPMI-1640 medium containing 10% fetal bovine
serum (FBS; Gibco-BRL, Gaithersburg, MD, USA). HeLa and 293T cells
were cultured in DMEM containing 10% FBS. The whole cell lines were
cultured at 37°C with 5% CO2 and 100% humidity.
Triptolide(Sigma, St.Louis, MO,
USA),12-O-Tetradecanoyl-phorbol-13-Acetate (TPA; Sigma), cidofovir
(CDV; Selleck Chemicals, Shanghai, China), cycloheximide (CHX;
Sigma), and MG-132 (Calbiochem, Billerica, MA, USA) were dissolved
in dimethylsulfoxide (DMSO). Sodium butyrate (NaB; Sigma) was
dissolved in PBS.
Plasmids
Plasmid DNA was purified through columns (Axygen
Scientific Inc., Union City, CA, USA) as described by the
manufacturer. The plasmid pSG5-LANA1 was constructed by inserting
the full LANA1 sequence into the MfeI/EcoRI of pSG5
vector (YouBio, Shanghai, China).
Cell viability assay
The viability of cells was determined by the Cell
Counting kit-8 assay (CCK-8; Dojindo Laboratories, Kumamoto,
Japan). In brief, cells were seeded in 96-well plates at a density
of 1×104/100 μl culture medium. All the cells were
treated with vehicle control (DMSO; 0.006%, vol/vol) or increasing
concentrations of triptolide for 24, 48 and 72 h. After different
treatments, 10 μl of tetrazolium substrate was added into each well
of the plate. The plates protected from light were incubated at
37°C for 1 h. The optical density (OD) was determined at an
absorbance of 450 nm using an ELx800 microimmunoanalyser (BioTek
Instruments, Inc., Winooski, VT, USA).
Cell transfection
For transfection, KSHV-negative HeLa cells were
transiently transfected using X-tremeGENE HP DNA Transfection
reagent (Roche, Basel, Switzerland) with pSG5 (the empty vector) or
pSG5-LANA1 according to the manufacturer's protocol. At 4 h
post-transfection, cells were treated with vehicle control (DMSO;
0.006%, vol/vol) or triptolide for further 44 h before cell
harvesting. In some experiments, cells were treated in the absence
or presence of MG-132 (10 μM) or CHX (25 μg/ml).
Immunoblot analysis
Briefly, cells treated in different conditions were
collected and washed with ice-cold PBS twice. Whole cell lysates
were prepared in RIPA lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) supplemented with 0.5% cocktail
protease inhibitor (Roche) and 0.5 mM phenylmethylsulfonyl fluoride
(PMSF). The cell lysates harvested were sonicated for 15 sec,
followed by centrifugation at 12,000 × g for 10 min. The
supernatants were collected and transferred into new tubes. Protein
concentrations were measured by the bicinchoninic acid method using
bovine serum albumin as a standard. Based on the measurement of
protein concentrations of all samples in the same experiment, the
loading volumes of samples were accordingly adjusted. Therefore,
equal amounts of proteins mixed with 5X loading buffer [250 nM
Tris-Hcl (pH 6.8), 10% SDS, 0.5% BPB, 50% glycerol, 5%
β-mercaptoethanol] were subjected to 10% SDS-PAGE gels. The gels
were run at 100 V, followed by transfer to PVDF membranes (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The membranes were blocked
with 5% skim milk in TBST, followed by incubation with primary
antibodies overnight at 4°C. After 3×5-min washes in TBST, the
membranes were incubated with appropriate
horseradish-peroxidase-conjugated secondary antibodies for 1 h. The
primary antibodies used were as follows: GAPDH (cat. no.
10494-1-AP, 1:5,000; Proteintech, Wuhan, China), β-actin (cat. no.
A5441, 1:5,000; Sigma), LANA-1 (cat. no. 13-210-100, 1:4,000;
Abionline, Cambridge, UK), vIL-6 (cat no. 13-214-050, 1:1,000;
Abionline), v-Cyclin (cat no. F105P, 1:1,000; Exalpha Biological,
Inc., Boston, MA, USA), STAT3 (cat no. 9139, 1:1,000; Cell
Signaling Technology, Danvers, MA, USA), phospho-STAT3 (cat. no.
4113, 1:1,000; Cell Signaling Technology) caspase-3 (cat. no. 9668,
1:1,000; Cell Signaling Technology), cleaved caspase-3 (cat. no.
9664, 1:1,000; Cell Signaling Technology), RTA (cat. no. 251345,
1:500; Abbiotec LLC, San Diego, CA, USA). A rabbit polyclonal
antibody against v-Flip was raised in our laboratory. The secondary
antibodies were horseradish-peroxidase-conjugated secondary
anti-mouse IgG (1:10,000; Wuhan Kerui Technology Co., Ltd., Wuhan,
China), anti-rabbit IgG (1:10,000; Wuhan Kerui Technology), or
anti-rat IgG (1:5,000; Wuhan Kerui Technology). Antibody-bound
proteins were detected using the ECL system (Bio-Rad Laboratories).
Image quantifications were performed using ImageJ software.
RNA extraction, cDNA preparation and
quantitative real-time PCR
PEL cells treated with vehicle control (DMSO;
0.006%, vol/vol) or triptolide for 24 and 48 h were harvested and
the total RNA was extracted using TRIzol reagent (Invitrogen)
according to the manufacturer's instructions. The concentration of
total RNA was determined by NanoDrop 2000 (Thermo Fisher
Scientific, Waltham, MA, USA), and all isolated RNA samples
(A260/A280 ratio >1.8) were stored at −80°C. Total RNA (1 μg)
was reversely transcribed using a PrimeScript RT reagent kit with
gDNA Eraser (cat. no. RR047A; Takara Bio, Tokyo, Japan). Genomic
DNA elimination reactions were performed in a volume of 10 μl
containing 2 μl of 5X gDNA Eraser buffer, 1 μl of gDNA Eraser, and
7 μl of RNase-free dH2O and the total RNA (1 μg),
followed by storage at room temperature for 30 min. The
reverse-transcription reaction mixture (20 μl) included 4 μl of 5X
PrimeScript buffer 2, 1 μl of PrimeScript RT Enzyme Mix I, 1 μl of
RT Primer Mixture containing oligo dT primer and random 6 mers, 4
μl of RNase-free dH2O, and 10 μl of genomic DNA
elimination reaction solution as mentioned above. The condition for
reverse-transcription reaction was 15 min at 37°C and 5 sec at
85°C. Expression of viral LANA1 mRNAs was quantified by
quantitative PCR using a SYBR-Green PCR kit (cat no. RR420A; Takara
Bio). Quantitative PCR reaction mixtures (20 μl) included 1X SYBR
Premix Ex Taq II (Tli RNaseH Plus), 0.4 μM each primer and 1 μl
cDNA reaction mixture (equivalent to 50 ng of input RNA). Reaction
was performed by a CFX96 Real-Time PCR detection system (Bio-Rad
Laboratories) and included an initial 30 sec denaturation step at
95°C, followed by 40 cycles of 10 sec at 95°C, 10 sec at 60°C and
15 sec at 72°C. Melting curve analysis was performed between 65 and
95°C (with 0.5°C increments) to verify amplicon specificity. The
average cycle threshold (CT) was determined by three independent
samples. Template-negative (quantitative PCR reaction mixtures
without cDNA) and RT-negative (RNA after genomic DNA elimination)
conditions were used as controls. The relative amounts of LANA1
transcript were normalized to the housekeeping gene GAPDH and
quantitative PCR data for the relative quantification were
calculated with the ΔΔCt method. The level of LANA1
transcript in cells treated with vehicle control (DMSO; 0.006%,
vol/vol) was set as 1. The primers were as follows: LANA1 forward,
5′-CGAGAGGAAGTTGTAGGAAACG-3′ and LANA1 reverse,
5′-CTTCCAGGTATAGGCAAGGTG-3′ (30);
GAPDH forward, 5′-ACATCGCTCAGACACCATG-3′ and GAPDH reverse,
5′-TGTAGTTGAGGTCAATGAAGGG-3′ (30). All experiments were repeated from
three independent cultures.
Flow cytometric analysis
PEL cells were seeded in 6-well plates and treated
with vehicle control (DMSO; 0.006%, vol/vol) or triptolide (50 or
100 nM) for 24 h, respectively. For apoptosis analysis, PEL cells
were evaluated with Annexin V-FITC/propidium iodide (PI) apoptosis
detection kit (Multisciences, Shanghai, China). Briefly,
2×105 cells were washed 3 times with PBS and resuspended
in 500 μl of 1X binding buffer, followed by adding 5 μl of Annexin
V-FIFC and 10 μl of PI. The whole cells were analyzed immediately
by a Beckman-Coulter system (EPICS Altra II; Beckman-Coulter,
Fullerton, CA, USA).
Progeny virus analysis
TPA (20 ng/ml) and NaB (1.2 mM) were used to induce
BCBL-1 cells entering lytic replication. At 3 h post-induction,
cell medium was replaced and cultured in the absence or presence of
triptolide for further 48 h. KSHV virion-associated DNA derived
from the supernatants was collected according to the method
previously described (31). Equal
amounts of the viral lysate were used for the quantitative
real-time PCR assay of the KSHV specific region, Orf K9. The
primer pair for Orf K9 was: Orf K9 forward,
5′-GTCTCTGCGCCATTCAAAAC-3′ and Orf K9 reverse,
5′-CCGGACACGACAACTAAGAA-3′. Intracellular viral DNA in BCBL-1 cells
was extracted with Phenol-Chloroform method according to the
manufacturer's instructions. The copy numbers of KSHV genome were
evaluated by a quantitative real-time PCR assay.
Enzyme-linked immunosorbent assay
(ELISA)
PEL cells were treated with or without 100 nM
triptolide for 24, 48, and 72 h, respectively. Cell supernatants
were collected and subjected to ELISA. IL-6 level of the cell
supernatants was measured using human IL-6 ELISA kit
(Multisciences) according to the manufacturer's instructions.
In vivo xenograft model
The animal protocol used in the present study was
approved by the Medical Ethics Committee of Wuhan University.
NOD/SCID mice were maintained under specific pathogen-free
conditions in the Animal Experiment Center of Wuhan University,
Animal Biosafety Level-II Laboratory.
BCBL-1 cells were inoculated intraperitoneally
(i.p.) into the flank of 4-week-old female NOD/SCID mice (purchased
from Beijing HFK Bioscience, Co., Ltd., Beijing, China). Twelve
mice were injected with BCBL-1 cells at 1×107 per mouse
on day 0. Three days post-injection, the mice were randomly divided
into vehicle and treatment groups (six mice per group), and daily
i.p. injection of triptolide (0.4 mg/kg) or PBS was performed.
After 21 days of treatment, all the mice were sacrificed by
cervical dislocation. Tumor burden was evaluated by measuring body
weight and volume of ascites. Tissue samples were collected and
fixed in 10% neutral buffered formalin, embedded and sectioned at 5
μm. Hematoxylin and eosin staining for histological analysis and
immunohistochemistry analysis were performed as previously
described (32).
Statistical analysis
Data were presented as the mean ± standard deviation
(mean ± SD) of three separate experiments. The statistical
significance of the difference was determined by the Student's
t-test, and P-values <0.05 were considered to indicate a
statistically significant result.
Results
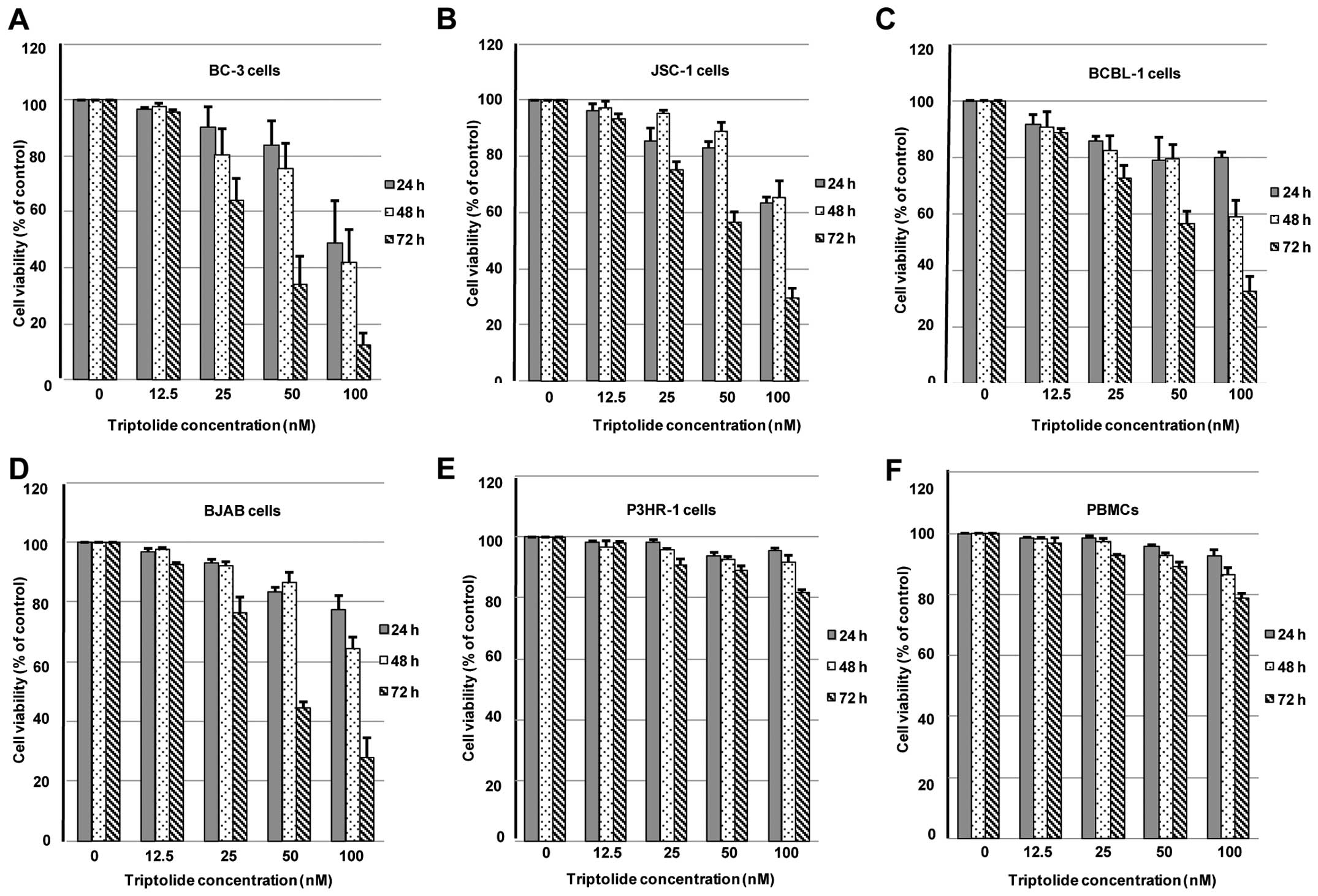
Triptolide inhibits proliferation of
KSHV-associated PEL cells
To determine whether triptolide specifically targets
cell viability in the KSHV-associated PEL cells, BC-3, BCBL-1 and
JSC-1 cells, together with KSHV-negative BJAB cells were treated
with vehicle control or a series of increasing concentrations of
triptolide for 24, 48 and 72 h, respectively. Cell viability was
determined by CCK-8 assays. Triptolide inhibited cell viability of
BC-3 cells in a dose- and time-dependent manner, with 50%
inhibitory concentration (IC50) values of 108.3, 89.5
and 46.4 nM by a 24-, 48- and 72-h trip-tolide treatment,
respectively (Fig. 1A). Similar
experiments were conducted in the other two PEL cells. The
IC50 values were calculated as 143.3, 128.3 and 67.9 nM
in JSC-1 cells and 153.9, 119.6 and 69.1 nM in BCBL-1 cells by 24-,
48- and 72-h triptolide treatment, respectively (Fig. 1B and C). In addition, triptolide
inhibited cell proliferation to a similar degree in BJAB cells, and
the IC50 values were calculated as 146.6, 115.6 and 63.8
nM by 24-, 48- and 72-h triptolide treatment, respectively
(Fig. 1D).
To further evaluate the antiproliferative effects of
triptolide on KSHV-uninfected B cell line, P3HR-1 cells and PBMCs
isolated from healthy donors were treated with vehicle control or a
series of increasing concentrations of triptolide for 24, 48 and 72
h, respectively. Our previous results indicated that 100-nM
triptolide treatment for 4 days decreased cell viability by <20%
in P3HR-1 cells (28). As shown in
Fig. 1E, a series of increasing
concentrations of triptolide treatment for 24, 48 and 72 h showed
slight proliferation inhibition in P3HR-1 cells. Similar results
were also found in PBMCs treated by a series of increasing
concentrations of triptolide (Fig.
1F). These results suggested that triptolide efficiently
inhibited cell viability in PEL cells, but not significantly in
PBMCs and P3HR-1 cells.
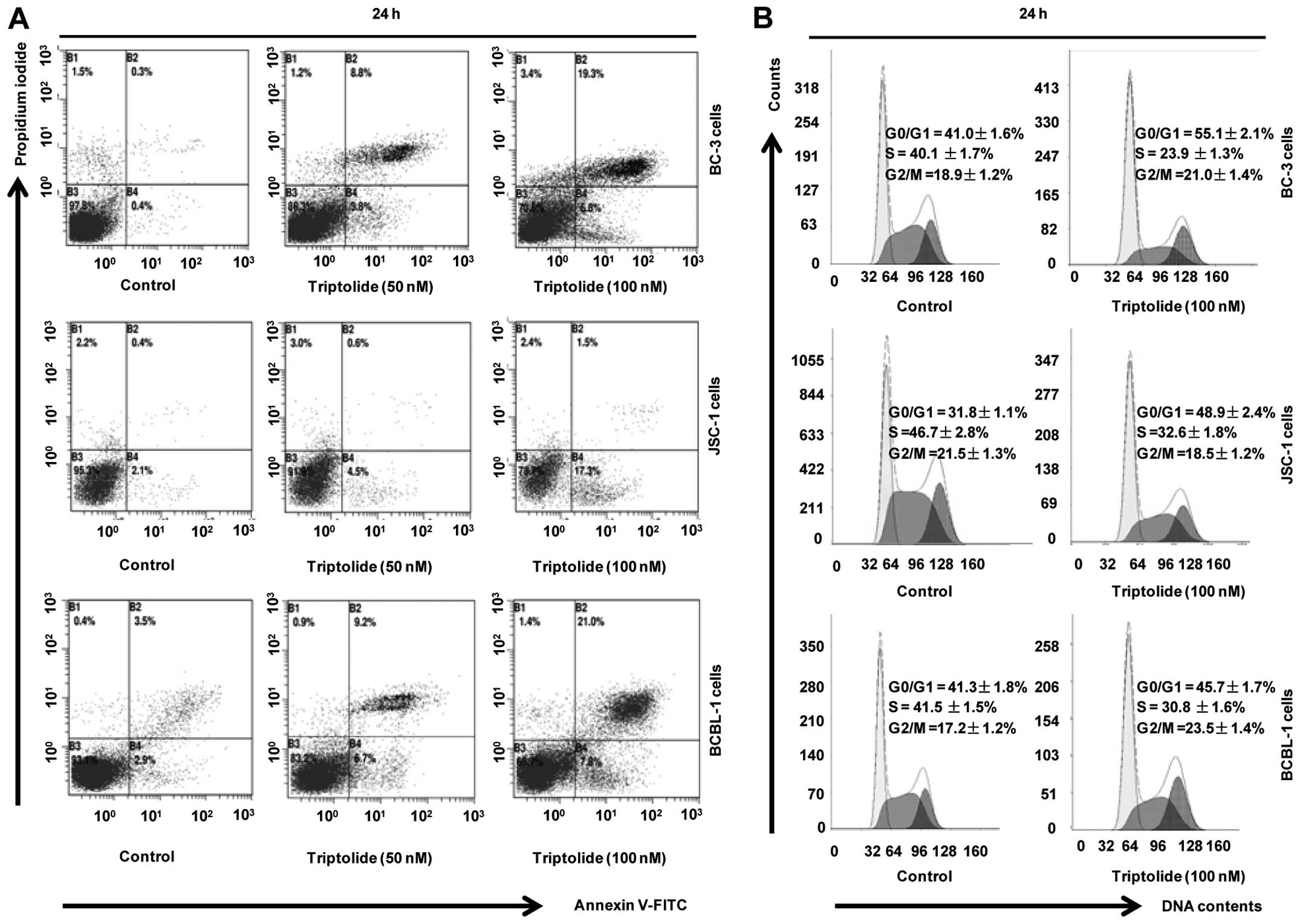
Triptolide induces cell cycle arrest and
apoptosis in the KSHV-associated PEL cells
To examine the effect of trip-tolide on induction of
cell cycle arrest and apoptosis, BC-3, JSC-1 and BCBL-1 cells were
treated with vehicle control or indicated triptolide for 24 h. Cell
cycle arrest and apoptosis were analyzed by flow cytometry. As
shown in Fig. 2A, treatment of PEL
cells with triptolide resulted in apoptosis in a dose-dependent
manner, which was confirmed by staining with Annexin V-FITC/PI. In
addition, treatment with 100 nM triptolide for 24 h could induce
cell apoptosis in BJAB cells, while not in P3HR-1 cells (data not
shown). For cell cycle analysis, treatment of BC-3 and JSC-1 cells
with triptolide resulted in G0/G1 arrest as observed by a marked
increase in the number of cells in the G1 phase (Fig. 2B). In contrast, treatment of BCBL-1
cells with triptolide resulted in cell cycle arrest in G2/M phase
as observed by an increase in the percentage of cells in the G2/M
phase (Fig. 2B). These results
suggest that triptolide induces cell cycle arrest and apoptosis in
the KSHV-associated PEL cells.
Triptolide decreases LANA1 expression
without reducing LANA1 transcript level in KSHV-associated PEL
cells
To determine whether triptolide alters expression of
LANA1, three different latently infected KSHV-associated PEL cells
were treated with vehicle control or triptolide for 48 h.
Whole-cell extracts were prepared and subjected to western blot
analysis. In addition to inhibition of LANA1 expression, two other
KSHV-encoded latent proteins, v-Cyclin and v-Flip, were reduced
after triptolide treatment in the three KSHV-associated PEL cells
(Fig. 3A). When compared to the
vehicle control, triptolide reduced the LANA1 expression level to
9.8, 16.8 and 23.4% in BC-3, BCBL-1 and JSC-1 cells, respectively.
When compared to the vehicle control, triptolide reduced the
v-Cyclin expression level to 3.4, 17.4 and 19.6%, the v-Flip
expression level to 14.2, 22.3 and 24.6% in BC-3, BCBL-1 and JSC-1
cells, respectively. Furthermore, consistent with the results of
cell apoptosis assays, triptolide resulted in increased caspase-3
cleavage in the three KSHV-associated PEL cells. In addition,
expression of STAT3, which is one of transcription factors,
remained unchanged with triptolide treatment, suggesting that the
effect of triptolide on LANA1 was a selective event. To further
determine if triptolide specifically decreases LANA1 expression,
KSHV-negative HeLa cells were transiently transfected with
pSG5-LANA1, followed by treatments with or without triptolide
beginning at 4 h after transfection. As shown in Fig. 3B, triptolide treatment
significantly decreased the expression of transfected pSG5-LANA1 in
HeLa cells.
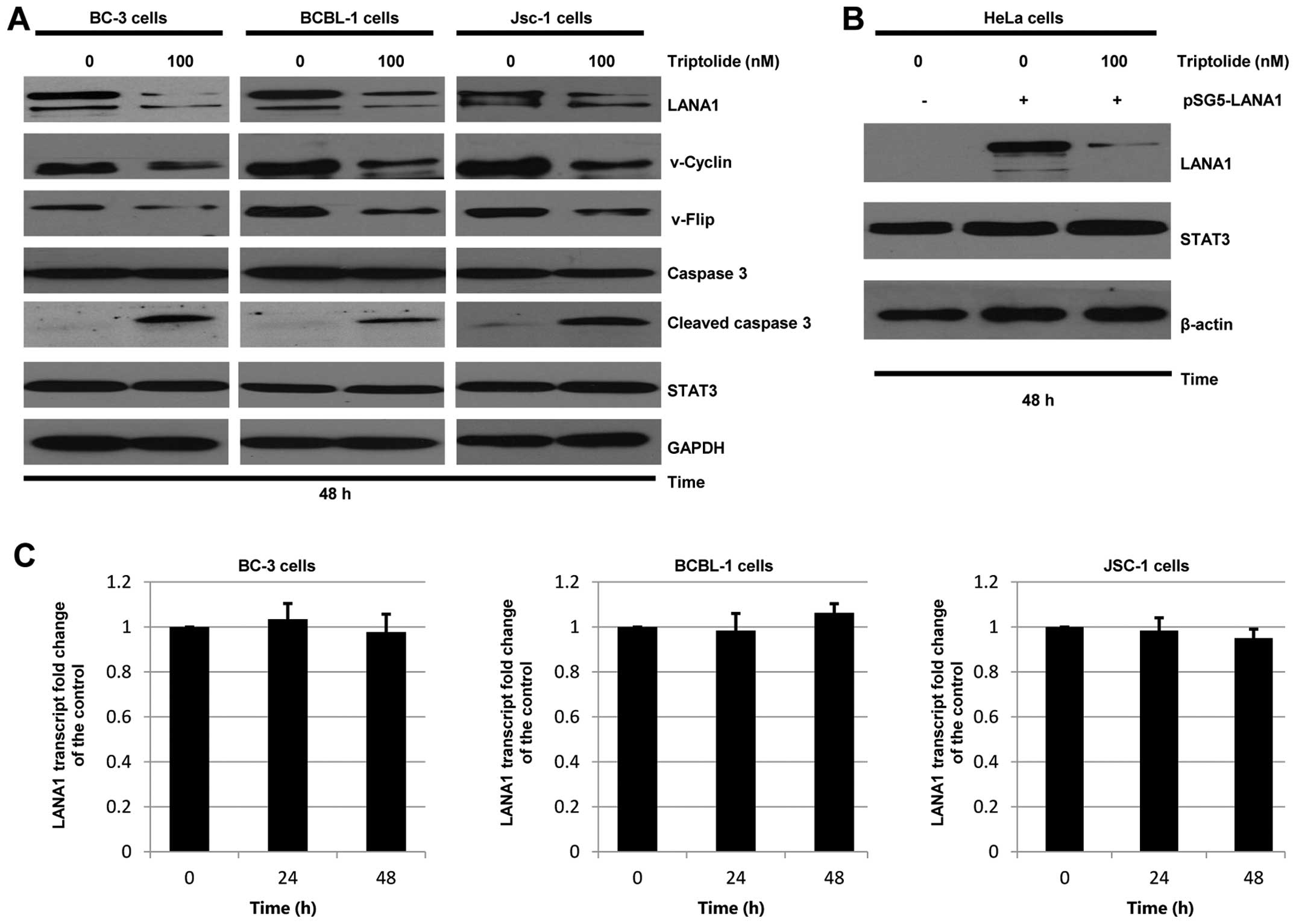 | Figure 3Triptolide downregulates LANA1
expression. (A and B) LANA1 degradation induced by triptolide. (A)
BC-3, BCBL-1 and JSC-1 cells were treated with vehicle control or
triptolide (100 nM) for 48 h, respectively. (B) HeLa cells were
transfected with empty vector (pSG5) or pSG5-LANA1 as indicated,
followed by a 44-h treatment with 100 nM triptolide beginning at 4
h after transfection. Whole-cell extracts were prepared and western
blotting was performed to analyze expression of LANA1 (20 μg),
v-Cyclin (20 μg), v-Flip (20 μg), caspase-3 (10 μg), cleaved
caspase-3 (20 μg), STAT3 (10 μg), β-actin (5 μg) or GAPDH (5 μg).
(C) BC-3, BCBL-1 and JSC-1 cells were treated with vehicle control
or triptolide (100 nM) for 24 and 48 h. The levels of LANA1
transcript were determined by qRT-PCR. The level of LANA1
transcript in cells treated with vehicle control is set as 1. |
Since triptolide reduced expression of LANA1, a
real-time quantitative reverse transcript polymerase chain reaction
(qRT-PCR) analysis was used to determine if triptolide suppressed
LANA1 at the mRNA level. BC-3, BCBL-1 and JSC-1 cells were treated
with vehicle control or triptolide (100 nM) for 24 and 48 h,
respectively. The total RNAs were harvested and subjected to
qRT-PCR. The relative expression was detected by comparison to the
housekeeping gene GAPDH. It was indicated that the mRNA levels of
LANA1 in BCBL-1, BC-3 and JSC-1 cells were undetectable upon
triptolide treatment (Fig.
3C).
Triptolide induces proteasomal
degradation of LANA1
To examine whether triptolide decreased expression
of LANA1 via the proteasome pathway, HeLa cells were transiently
transfected with the pSG5-LANA1 and treated with vehicle control or
triptolide in the absence or presence of proteosomal inhibitor
MG-132. As shown in Fig. 4A,
MG-132 obviously attenuated the effect of triptolide on LANA1
expression. Similarly, MG-132 also resulted in endogenous LANA1
protein level accumulation in BCBL-1 (Fig. 4B) and BC-3 (Fig. 4C) cells after treatment with
triptolide.
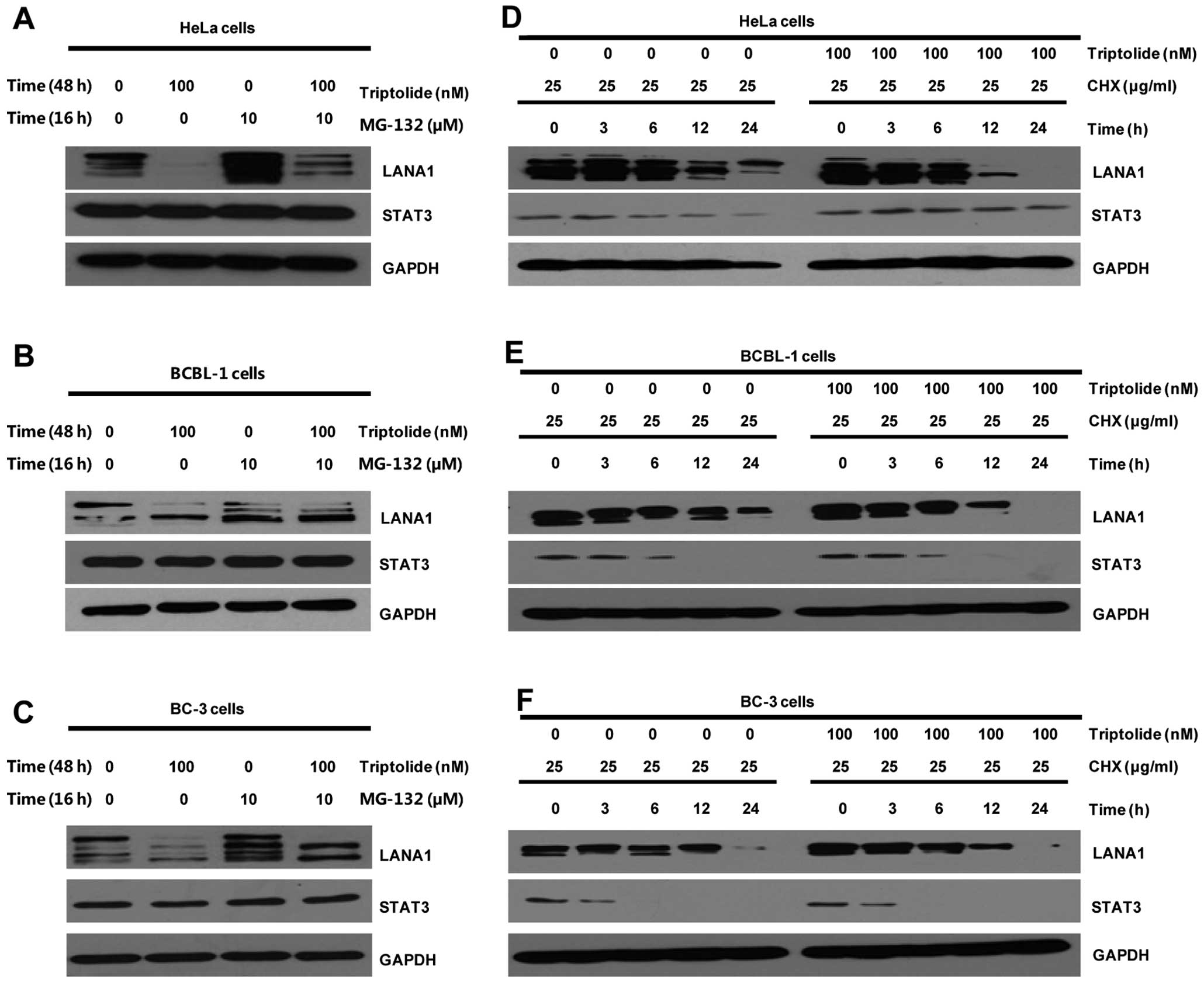 | Figure 4Triptolide induces proteasomal
degradation of LANA1, and reduces half-life of LANA1. (A–C)
Proteasomal degradation of LANA1. (A) HeLa cells were transiently
transfected with pSG5-LANA1, followed by a 44-h treatment with
vehicle control or triptolide (100 nM) beginning at 4 h after
transfection in the absence or presence of proteasome inhibitor
MG-132 (10 μM) for the last 16 h. (B) BCBL-1 and (C) BC-3 cells
were treated for 48 h with vehicle control or triptolide (100 nM)
in the absence or presence of proteasome inhibitor MG-132 (10 μM)
for the last 16 h. (D–F) Half-life analysis of LANA1 in HeLa, BC-3
and BCBL-1 cells. (D) HeLa cells were transiently transfected with
pSG5-LANA1 overnight, followed by incubation with vehicle control
or triptolide (100 nM) in the presence of CHX (25 μg/ml) for 0, 3,
6, 12 and 24 h, respectively. (E) BCBL-1 and (F) BC-3 cells were
treated with vehicle control or triptolide (100 nM) in the presence
of CHX (25 μg/ml) for 0, 3, 6, 12 and 24 h, respectively.
Whole-cell lysates were immunoblotted with anti-LANA1, anti-STAT3
or anti-GAPDH antibody. |
Triptolide reduces stability or half-life
of LANA1
Previous studies had indicated that triptolide
decreased the expression of LANA1 and induced proteasomal
degradation of LANA1. To determine whether triptolide affect LANA1
stability through some other mechanisms, HeLa cells were
transiently transfected with the pSG5-LANA1 and treated with
vehicle control or triptolide in the presence of cycloheximide
(CHX). As shown in Fig. 4D,
triptolide reduced the half-life of LANA1 when compared to vehicle
control, while not affecting GAPDH levels. Similarly, triptolide
also reduced the half-life of endogenous LANA1 protein in the
CHX-treated BCBL-1 (Fig. 4E) and
BC-3 (Fig. 4F) cells. Notably,
triptolide did not significantly affect STAT3 stability (Fig. 4D–F).
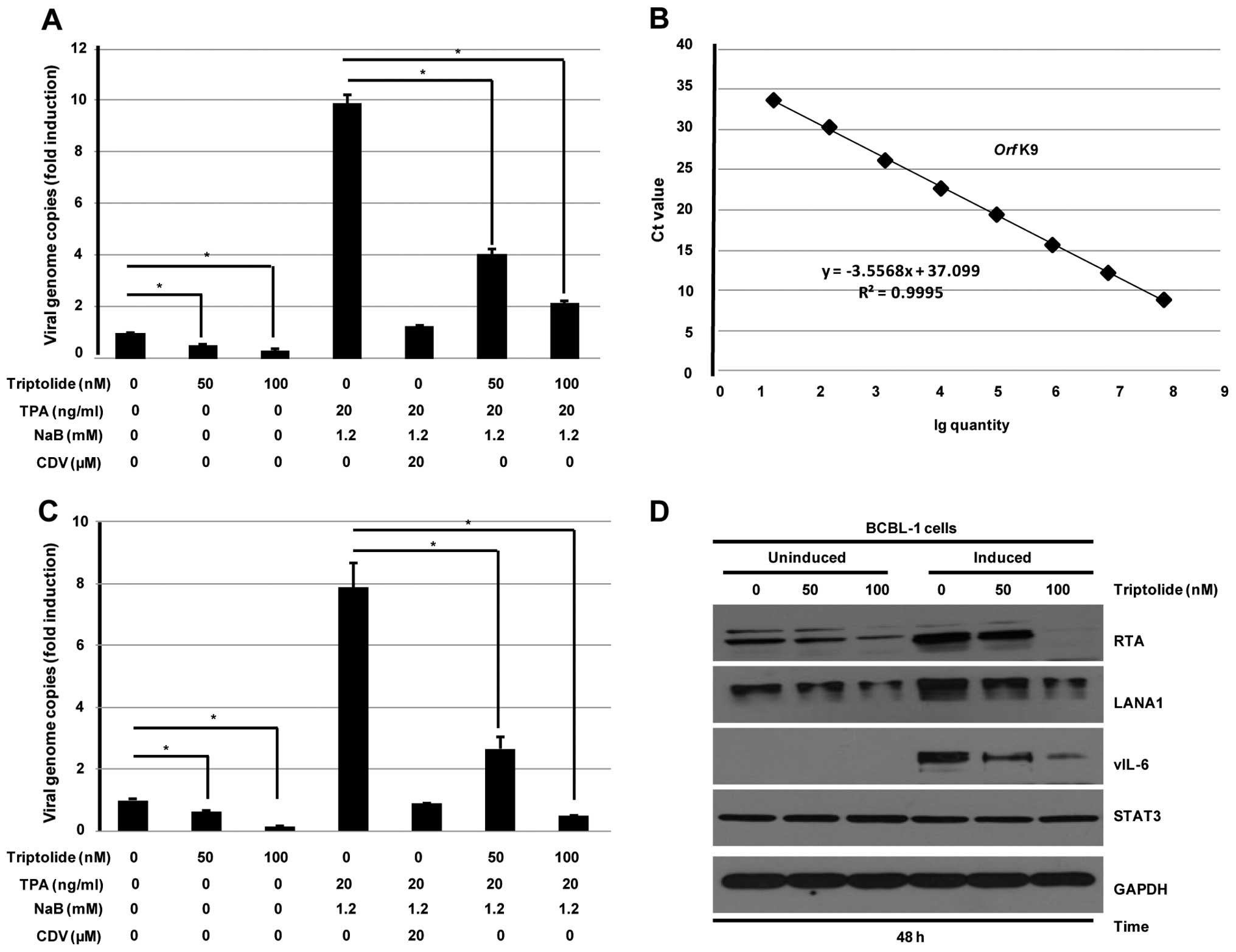
Triptolide reduces lytic and latent
replication of KSHV in BCBL-1 cells
To examine the effect of triptolide on antiviral
activity, BCBL-1 cells uninduced or induced by TPA and NaB for 3 h
were treated with indicated concentrations of triptolide for 48 h.
Whole cell lysates, supernatants, and genomic DNA were prepared at
48 h post-incubation. qRT-PCR and western blotting were used to
analyze the effect of triptolide on lytic and latent replication of
KSHV. As shown in Fig. 5A,
triptolide decreased viral genome copies in both uninduced and
induced BCBL-1 cells. When compared to the vehicle control in
induced BCBL-1 cells, intracellular viral DNA replication was
reduced by 5-fold after triptolide (100 nM) treatment for 48 h.
Production of progeny virion extracted from the BCBL-1 cell
supernatants was calculated according to the standard curve derived
from the Orf K9 construct (Fig.
5B). Results indicated that the production of progeny virion
was dose-dependently reduced after triptolide treatment (Fig. 5C). As expected, CDV, which was used
as a positive control, efficiently reduced the lytic replication
and virion production of KSHV as previously described (33). Replication and transcription
activator (RTA), a lytic switch protein, is necessary for KSHV
reactivation and lytic DNA replication. Western blotting was used
to detect the effect of triptolide on the expression of RTA in
BCBL-1 cells. As shown in Fig. 5D,
triptolide reduced expression of LANA1 and RTA in both uninduced
and induced BCBL-1 cells. In addition, vIL-6, encoded by KSHV
Orf K2, contributed to the growth, survival and spread of
PEL. Our results as shown in Fig.
5D also indicated that triptolide decreased vIL-6 expression in
induced BCBL-1 cells.
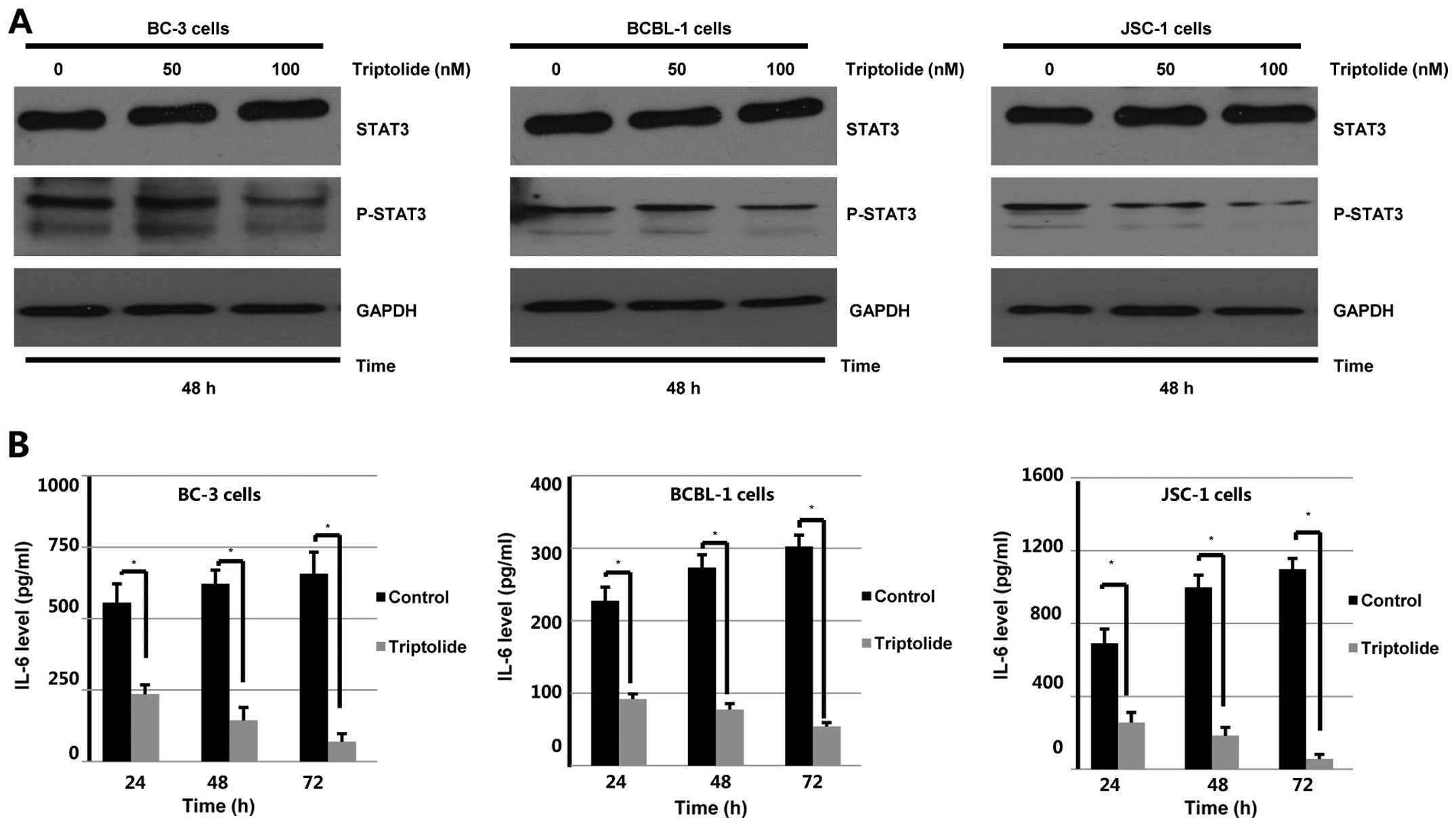
Triptolide suppresses STAT3 activity in
PEL cells
LANA1, a transcriptional co-activator of STAT3,
modulates STAT3 activity in PEL cells (20). STAT3 is constitutively
phosphorylated and is essential to cell survival in PEL cells
(34). Since STAT3 represents a
potential target for treatment of PEL, the STAT3 activity affected
by triptolide was determined. Results shown in Fig. 6A indicated that triptolide
dephosphorylated STAT3 in BCBL-1, BC-3 and JSC-1 cells. It has been
reported that KSHV encoded LANA1 upregulated IL-6 expression by
inducing transcription of the IL-6 promoter (35). IL-6 and vIL-6 secreted by
KSHV-positive cells are multifunctional cytokines that contribute
to the pathogenesis of PEL (35).
To determine effect of triptolide on secretion of IL-6,
KSHV-associated PEL cells were treated with vehicle control or 100
nM triptolide for 24, 48 and 72 h. Cell supernatants were collected
and subjected to ELISA. As shown in Fig. 6B, triptolide resulted in reduced
secretion of IL-6 in the supernanants of BCBL-1, BC-3 and JSC-1
cells. These results suggests that downregulation of LANA1 by
triptolide results in inhibition of IL-6/STAT3 pathway in PEL
cells.
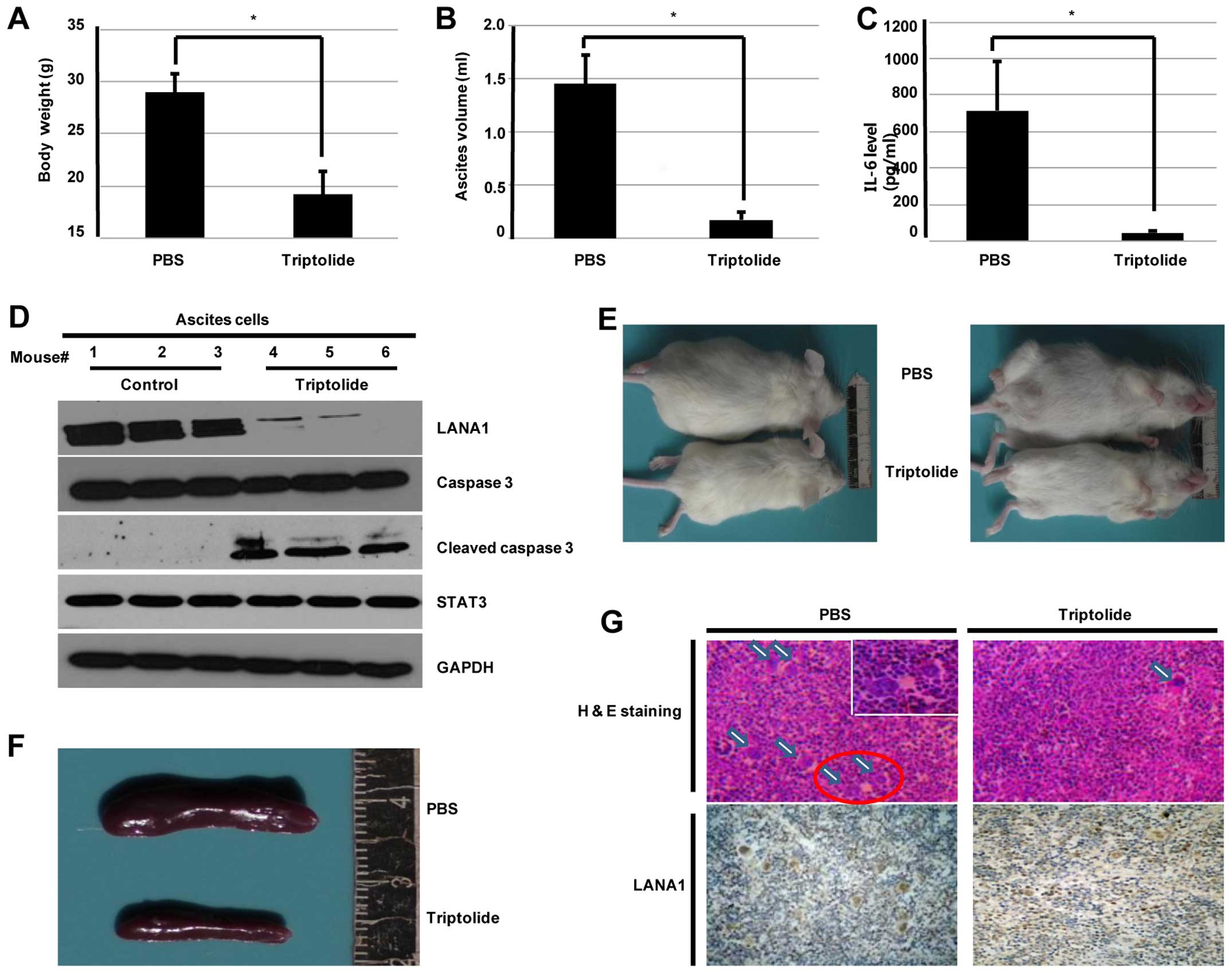
In vivo effects of triptolide on NOD/SCID
mice inoculated with BCBL-1 cells
Since our previous results indicated efficacy of
triptolide for treatment of PEL in vitro, effects of
triptolide in vivo were also assessed in an immunodeficient
xenograft mouse model. NOD/SCID mice were inoculated
intraperitoneally with 1×107 BCBL-1 cells. A dose of 0.4
mg/kg triptolide or PBS alone was administrated via intraperitoneal
injection daily for 3 weeks from day 3 after cell inoculation. As
the dose of triptolide from intraperitoneal injection had already
been reported (23), the dosage of
triptolide in vivo in the present study was expected to be
safe. The body weight of the PBS alone treated mice significantly
increased compared to that of triptolide treated mice (28.9±1.79
vs. 19.2±2.21 g, n=6, P<0.01; Fig.
7A). Moreover, mice treated with triptolide had a significantly
lower volume of ascites (1.45±0.27 vs. 0.17±0.07 ml, n=6,
P<0.01; Fig. 7B). In addition,
IL-6 level in the supernatants of ascites was significantly
decreased in comparison to that of triptolide treatment (Fig. 7C). Western blotting results of
ascites cells indicated that triptolide significantly cut down the
expression of LANA1 and induced ascites cell apoptosis via
caspase-3 pathways (Fig. 7D).
Representative photos of mice treated with PBS alone and triptolide
treated are shown in Fig. 7E.
Organ invasion by BCBL-1 cells on day 24 was
evaluated by hematoxylin and eosin staining, as well as LANA1
immu-nostaining. As shown in Fig.
7F, spleens of mice treated with triptolide were significantly
smaller compared to the group treated with PBS alone. The mice
inoculated with BCBL-1 cells exhibited infiltration in the spleen
and the number of LANA1 positive cells in triptolide treated mice
was significantly reduced (Fig.
7G). These results suggest that triptolide efficiently inhibits
growth and infiltration of PEL cells in vivo.
Discussion
LANA1 is a pivotal latent protein, which is
consistently expressed in all proliferating KSHV-positive cells and
is essential for cell survival. Moreover, LANA1 plays an important
role in regulating segregation, replication and persistence of
viral episome (36), providing an
attractive target for the treatment of KSHV-induced diseases. In
the present study, the direct effects of triptolide on
KSHV-associated PEL cells in vitro and in vivo were
evaluated. Firstly, it was demonstrated that triptolide effectively
inhibited KSHV-associated PEL cells proliferation with a nanomolar
concentration. Moreover, triptolide induced cell cycle arrest and
apoptosis in the KSHV-associated PEL cells.
Our previous study demonstrated that triptolide
reduced the expression of LMP1 in latency III type infected
EBV-positive B lymphocytes (29).
Since LANA1 is reported to be a client protein of Hsp90, 17-DMAG is
efficacious against KS by promoting the degradation of LANA1
(30). Of note, our findings
indicate that triptolide decreases LANA1 expression in a variety of
cell types, including three different KSHV-associated PEL cells and
KSHV-negative HeLa cells transiently transfected with pSG5-LANA1.
Further study indicated that triptolide did not reduce LANA1
expression at the mRNA levels. In addition, LANA1 accumulated after
treatment with MG-132 in the presence of triptolide. Triptolide
also decreased the stability or half-life of LANA1. In addition to
LANA1, v-Cyclin and v-Flip are two KSHV-encoded latent proteins.
KSHV-encoded v-Cyclin contributed to tumorigenesis through
inhibiting cell cycle inhibitor p27Kip1 in PEL (37). KSHV-encoded v-Flip contributed to
activation of NF-kappaB pathway, which is essential for survival in
a majority of PEL cells (38). Our
results also indicated that triptolide inhibited expression of
v-Cyclin and v-Flip in KSHV-associated PEL cells. These results
suggest that triptolide reduces the expression of three major
latent proteins, together with inducing proteasomal degradation of
LANA1 and impairing its stability.
Lytic replication and gene expression of KSHV are
essential to promote tumorigenesis (39). Previous studies have demonstrated
that higher KSHV viral load has been associated with increased risk
of KSHV-induced malignancies (40). Consistent with the ability of
triptolide to decrease LANA1 expression in KSHV latency infected
cells, our results demonstrated that triptolide impaired lytic
replication of KSHV, as well as virion production released from
induced BCBL-1 cells, suggesting that triptolide treatment is
efficacious against KSHV replication. Triptolide also reduced the
expression of lytic genes RTA, and inhibited their expression
induced by TPA and NaB, thereby allaying these safety concerns.
Essentially similar effects of BIIB021 and 17-AAG on the expression
of RTA have been reported recently (38,41).
In addition, triptolide reduced expression of vIL-6, which
contributed to pathogenesis of PEL, in TPA and NaB induced BCBL-1
cells. At present, the exact mechanism for the inhibition of lytic
replication and gene expression of KSHV by triptolide is not clear
and need further study. Taken together, triptolide can efficiently
induce apoptosis and cell cycle arrest, together with
down-regulation of LANA1 expression and viral replication in the
KSHV-associated PEL cells.
STAT3 activity plays an essential role in the
survival of PEL cells. STAT3 is constitutively active and
inhibition of STAT3 results in apoptosis in PEL cells (33). Previous reports have indicated that
LANA1, a transcriptional co-activator of STAT3, modulates STAT3
activity in PEL cells (20).
Overexpression of LANA1 enhanced STAT3 activity, and inhibition of
LANA1 expression decreased the STAT3-dependent transcription
(20). Consistent with the effects
of AG490, a tyrosine kinase inhibitor, on STAT3 activity in PEL
cells (42), triptolide reduced
STAT3 activity in PEL cells. In addition, IL-6 is a pivotal
cytokine contributing to cell survival. LANA1 enhanced IL-6
expression by inducing transcription of the IL-6 promoter (34). Our results demonstrated that
triptolide reduced secretion of IL-6 in BCBL-1, BC-3 and JSC-1.
Therefore, inhibition of STAT3 activity and IL-6 secretion may
result from the downregulation of LANA1 by triptolide.
In addition to the effect of triptolide on the
KSHV-positive malignant cells in vitro, the present study
also evaluated the therapeutic effect of triptolide against PEL
in vivo using a mouse xenograft model. PEL xenograft mice
are characterized by lymphomatous effusions in body cavities and
rarely present with a definable tumor mass, which are similar to
the clinical feature of human PEL patients (43). Consistent with the therapeutic
effect of other drugs against PEL xenograft mouse models (44), triptolide treatment significantly
repressed the formation of malignant ascites and lymphomatous
effusions caused by BCBL-1 cells in NOD/SCID mice. Moreover,
triptolide treatment decreased the expression of LANA1 in ascites
cells and induced apoptosis of ascites cells in a caspase-dependent
manner.
Although KSHV was discovered 20 years ago, there is
little progress in the management of KSHV-related diseases. Due to
the toxic effect or incomplete efficiency of classical antiviral
agents, such as acyclovir, ganciclovir and adefovir, antiviral
treatments for KSHV-infected diseases are limited. To date,
standard therapeutic guidelines are not drawn for the treatment of
KSHV-associated malignancies. Currently antiviral drugs mainly
target lytic replication of virus, which is essential to disease
progression and virus dissemination. Nevertheless, the majority of
KSHV-infected tumor cells are in latency, and antiviral drugs
targeting KSHV lytic replication have not shown consistent results
for the treatment of KSHV-associated KS in clinic (6). Therefore, targeting viral latency may
be a novel strategy for the treatments of KSHV-infected diseases.
In this study, our results have demonstrated the ability of
triptolide in vitro and in vivo to inhibit cell
proliferation and induce cell apoptosis in the KSHV-associated PEL
cells. In addition, triptolide can efficiently reduce the
expression of LANA1, inhibit viral replication, and impair
production of progeny virion in the KSHV-associated PEL cells. Our
results show that triptolide may be a promising compound for the
treatment of KSHV-associated diseases.
Acknowledgements
The present study was supported by the Initiative
Research Program of Wuhan University (no. 410100020), the Advanced
Talent Independent Research Program of Wuhan University (no.
410500011), and the National Natural Science Foundation of China
(no. 210700228).
References
|
1
|
Chang Y, Cesarman E, Pessin MS, Lee F,
Culpepper J, Knowles DM and Moore PS: Identification of
herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma.
Science. 266:1865–1869. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Boulanger E, Gérard L, Gabarre J, Molina
JM, Rapp C, Abino JF, Cadranel J, Chevret S and Oksenhendler E:
Prognostic factors and outcome of human herpesvirus 8-associated
primary effusion lymphoma in patients with AIDS. J Clin Oncol.
23:4372–4380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boshoff C and Chang Y: Kaposi's
sarcoma-associated herpes-virus: A new DNA tumor virus. Annu Rev
Med. 52:453–470. 2001. View Article : Google Scholar
|
|
4
|
Bottero V, Sadagopan S, Johnson KE, Dutta
S, Veettil MV and Chandran B: Kaposi's sarcoma-associated
herpesvirus-positive primary effusion lymphoma tumor formation in
NOD/SCID mice is inhibited by neomycin and neamine blocking
angiogenin's nuclear translocation. J Virol. 87:11806–11820. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sarosiek KA, Cavallin LE, Bhatt S, Toomey
NL, Natkunam Y, Blasini W, Gentles AJ, Ramos JC, Mesri EA and
Lossos IS: Efficacy of bortezomib in a direct xenograft model of
primary effusion lymphoma. Proc Natl Acad Sci USA. 107:13069–13074.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Coen N, Duraffour S, Snoeck R and Andrei
G: KSHV targeted therapy: An update on inhibitors of viral lytic
replication. Viruses. 6:4731–4759. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dyson OF, Walker LR, Whitehouse A, Cook PP
and Akula SM: Resveratrol inhibits KSHV reactivation by lowering
the levels of cellular EGR-1. PLoS One. 7:e333642012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee GM, Shahian T, Baharuddin A, Gable JE
and Craik CS: Enzyme inhibition by allosteric capture of an
inactive conformation. J Mol Biol. 411:999–1016. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Verma SC, Lan K and Robertson E: Structure
and function of latency-associated nuclear antigen. Curr Top
Microbiol Immunol. 312:101–136. 2007.
|
|
10
|
Sun Q, Tsurimoto T, Juillard F, Li L, Li
S, De León Vázquez E, Chen S and Kaye K: Kaposi's
sarcoma-associated herpesvirus LANA recruits the DNA polymerase
clamp loader to mediate efficient replication and virus
persistence. Proc Natl Acad Sci USA. 111:11816–11821. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Uppal T, Banerjee S, Sun Z, Verma SC and
Robertson ES: KSHV LANA - the master regulator of KSHV latency.
Viruses. 6:4961–4998. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujimuro M, Wu FY, ApRhys C, Kajumbula H,
Young DB, Hayward GS and Hayward SD: A novel viral mechanism for
dysregulation of beta-catenin in Kaposi's sarcoma-associated
herpesvirus latency. Nat Med. 9:300–306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu J, Jha HC, Verma SC, Sun Z, Banerjee S,
Dzeng R and Robertson ES: Kaposi's sarcoma-associated
herpesvirus-encoded LANA contributes to viral latent replication by
activating phosphorylation of survivin. J Virol. 88:4204–4217.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paudel N, Sadagopan S, Chakraborty S,
Sarek G, Ojala PM and Chandran B: Kaposi's sarcoma-associated
herpesvirus latency-associated nuclear antigen interacts with
multifunctional angiogenin to utilize its antiapoptotic functions.
J Virol. 86:5974–5991. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Verma SC, Borah S and Robertson ES:
Latency-associated nuclear antigen of Kaposi's sarcoma-associated
herpesvirus up-regulates transcription of human telomerase reverse
transcriptase promoter through interaction with transcription
factor Sp1. J Virol. 78:10348–10359. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun R, Liang D, Gao Y and Lan K: Kaposi's
sarcoma-associated herpesvirus-encoded LANA interacts with host
KAP1 to facilitate establishment of viral latency. J Virol.
88:7331–7344. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Z, Xiao B, Jha HC, Lu J, Banerjee S
and Robertson ES: Kaposi's sarcoma-associated herpesvirus-encoded
LANA can induce chromosomal instability through targeted
degradation of the mitotic checkpoint kinase Bub1. J Virol.
88:7367–7378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Friborg J Jr, Kong W, Hottiger MO and
Nabel GJ: p53 inhibition by the LANA protein of KSHV protects
against cell death. Nature. 402:889–894. 1999.
|
|
19
|
Radkov SA, Kellam P and Boshoff C: The
latent nuclear antigen of Kaposi sarcoma-associated herpesvirus
targets the retino-blastoma-E2F pathway and with the oncogene Hras
transforms primary rat cells. Nat Med. 6:1121–1127. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Muromoto R, Okabe K, Fujimuro M, Sugiyama
K, Yokosawa H, Seya T and Matsuda T: Physical and functional
interactions between STAT3 and Kaposi's sarcoma-associated
herpesvirus-encoded LANA. FEBS Lett. 580:93–98. 2006. View Article : Google Scholar
|
|
21
|
Kupchan SM, Court WA, Dailey RG Jr,
Gilmore CJ and Bryan RF: Triptolide and tripdiolide, novel
antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J
Am Chem Soc. 94:7194–7195. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu Q: Triptolide and its expanding
multiple pharmacological functions. Int Immunopharmacol.
11:377–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou ZL, Yang YX, Ding J, Li YC and Miao
ZH: Triptolide: Structural modifications, structure-activity
relationships, bioactivities, clinical development and mechanisms.
Nat Prod Rep. 29:457–475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Banerjee S, Sangwan V, McGinn O, Chugh R,
Dudeja V, Vickers SM and Saluja AK: Triptolide-induced cell death
in pancreatic cancer is mediated by O-GlcNAc modification of
transcription factor Sp1. J Biol Chem. 288:33927–33938. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Phillips PA, Dudeja V, McCarroll JA,
Borja-Cacho D, Dawra RK, Grizzle WE, Vickers SM and Saluja AK:
Triptolide induces pancreatic cancer cell death via inhibition of
heat shock protein 70. Cancer Res. 67:9407–9416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chugh R, Sangwan V, Patil SP, Dudeja V,
Dawra RK, Banerjee S, Schumacher RJ, Blazar BR, Georg GI, Vickers
SM, et al: A preclinical evaluation of Minnelide as a therapeutic
agent against pancreatic cancer. Sci Transl Med. 4:156ra1392012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sun X, Barlow EA, Ma S, Hagemeier SR,
Duellman SJ, Burgess RR, Tellam J, Khanna R and Kenney SC: Hsp90
inhibitors block outgrowth of EBV-infected malignant cells in vitro
and in vivo through an EBNA1-dependent mechanism. Proc Natl Acad
Sci USA. 107:3146–3151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun X, Bristol JA, Iwahori S, Hagemeier
SR, Meng Q, Barlow EA, Fingeroth JD, Tarakanova VL, Kalejta RF and
Kenney SC: Hsp90 inhibitor 17-DMAG decreases expression of
conserved herpesvirus protein kinases and reduces virus production
in Epstein-Barr virus-infected cells. J Virol. 87:10126–10138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou H, Guo W, Long C, Wang H, Wang J and
Sun X: Triptolide inhibits proliferation of Epstein-Barr
virus-positive B lymphocytes by down-regulating expression of a
viral protein LMP1. Biochem Biophys Res Commun. 456:815–820. 2015.
View Article : Google Scholar
|
|
30
|
Chen W, Sin SH, Wen KW, Damania B and
Dittmer DP: Hsp90 inhibitors are efficacious against Kaposi Sarcoma
by enhancing the degradation of the essential viral gene LANA, of
the viral co-receptor EphA2 as well as other client proteins. PLoS
Pathog. 8:e10030482012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin X, Liang D, He Z, Deng Q, Robertson ES
and Lan K: miR-K12-7-5p encoded by Kaposi's sarcoma-associated
herpesvirus stabilizes the latent state by targeting viral
ORF50/RTA. PLoS One. 6:e162242011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Choi ES, Nam JS, Jung JY, Cho NP and Cho
SD: Modulation of specificity protein 1 by mithramycin A as a novel
therapeutic strategy for cervical cancer. Sci Rep. 4:71622014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pica F, Serafino A, Garaci E and Volpi A:
Cidofovir on HHV-8 in BCBL-1 cells. Antivir Ther. 9:823–825.
2004.PubMed/NCBI
|
|
34
|
Aoki Y, Feldman GM and Tosato G:
Inhibition of STAT3 signaling induces apoptosis and decreases
survivin expression in primary effusion lymphoma. Blood.
101:1535–1542. 2003. View Article : Google Scholar
|
|
35
|
An J, Lichtenstein AK, Brent G and Rettig
MB: The Kaposi sarcoma-associated herpesvirus (KSHV) induces
cellular interleukin 6 expression: Role of the KSHV
latency-associated nuclear antigen and the AP1 response element.
Blood. 99:649–654. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vázquez EL, Carey VJ and Kaye KM:
Identification of Kaposi's sarcoma-associated herpesvirus LANA
regions important for episome segregation, replication, and
persistence. J Virol. 87:12270–12283. 2013. View Article : Google Scholar
|
|
37
|
Järviluoma A, Koopal S, Räsänen S, Mäkelä
TP and Ojala PM: KSHV viral cyclin binds to p27KIP1 in primary
effusion lymphomas. Blood. 104:3349–3354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gopalakrishnan R, Matta H and Chaudhary
PM: A purine scaffold HSP90 inhibitor BIIB021 has selective
activity against KSHV-associated primary effusion lymphoma and
blocks vFLIP K13-induced NF-κB. Clin Cancer Res. 19:5016–5026.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nicholas J: Human herpesvirus 8-encoded
proteins with potential roles in virus-associated neoplasia. Front
Biosci. 12:265–281. 2007. View
Article : Google Scholar
|
|
40
|
Quinlivan EB, Zhang C, Stewart PW,
Komoltri C, Davis MG and Wehbie RS: Elevated virus loads of
Kaposi's sarcoma-associated human herpesvirus 8 predict Kaposi's
sarcoma disease progression, but elevated levels of human
immunodeficiency virus type 1 do not. J Infect Dis. 185:1736–1744.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Higashi C, Saji C, Yamada K, Kagawa H,
Ohga R, Taira T and Fujimuro M: The effects of heat shock protein
90 inhibitors on apoptosis and viral replication in primary
effusion lymphoma cells. Biol Pharm Bull. 35:725–730. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Granato M, Chiozzi B, Filardi MR, Lotti
LV, Di Renzo L, Faggioni A and Cirone M: Tyrosine kinase inhibitor
tyrphostin AG490 triggers both apoptosis and autophagy by reducing
HSF1 and Mcl-1 in PEL cells. Cancer Lett. 366:191–197. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nador RG, Cesarman E, Chadburn A, Dawson
DB, Ansari MQ, Sald J and Knowles DM: Primary effusion lymphoma: A
distinct clinicopathologic entity associated with the Kaposi's
sarcoma-associated herpes virus. Blood. 88:645–656. 1996.PubMed/NCBI
|
|
44
|
Goto H, Kariya R, Shimamoto M, Kudo E,
Taura M, Katano H and Okada S: Antitumor effect of berberine
against primary effusion lymphoma via inhibition of NF-κB pathway.
Cancer Sci. 103:775–781. 2012. View Article : Google Scholar : PubMed/NCBI
|