Introduction
Cell adhesion involves adhesions comprised of
intercellular cell-cell attachments and of cell extracellular
matrix protein (ECM) interactions (1). Proper tuning of the cell adhesion can
lead to efficient migration and invasion for a successful
metastasis (2). Complicated
processes leading to metastasis, migration and invasion should be
effectively coordinated by the functions of spatial and temporal
cues between cellular and microenvironmental regions (3). Furthermore, regulation of cell
adhesion properties may be targeted to control the migration and
invasion for cancer metastasis. Diverse signaling or adaptor
proteins play critical roles in promoting cancer cells to
metastasize (4). Although the
signaling molecules contributing to control of the metastatic
potentials are very complicated and interconnected, more molecules
still need to be identified to understand the control of cellular
migration.
Lysyl-tRNA synthetase, KRS, is involved in protein
translation. It catalyzes the addition of amino-lysyl to peptides
that are being synthesized along the mRNA codons. Although
cytosolic KRS is involved in this housekeeping role, KRS at the
plasma membranes functions in immune responses (5,6) and
tumor metastasis (7,8), clearly indicating non-canonical
biological functions besides protein translation. Phosphorylation
at the Thr52 residue of KRS by p38 mitogen-activated protein kinase
(MAPK) causes dissociation from the cytosolic multi-tRNA synthetase
complex (MSC) and translocation to the plasma membrane, where it
associates with and stabilizes a 67-kDa laminin receptor (p67LR)
for migration and metastasis (7).
The p67LR associates with integrin α6β1 (9) or α6β4 (10). The integrins are important for
cell-extracellular matrix (ECM) adhesion involved in cell migration
and invasion (11), as well as
signal transduction activation leading to tyrosine phosphorylations
of focal adhesion kinase (FAK), paxillin and c-Src involved in
cell-ECM adhesion (12). After
subcutaneous injection of cells into mice, the interaction between
KRS and p67LR can be targeted to inhibit the KRS-dependent
metastasis upon a subcutaneous injection of cells to mouse,
indicating that KRS can be targeted to block cancer metastasis
(8). Although targeting KRS has
been shown to inhibit metastasis, how KRS regulates cell-cell
adhesion and cell-ECM adhesion is not clearly known.
Cell-cell adhesion involves tight junctions,
adherence junctions, gap junctions and desmosomes, whereas cell-ECM
adhesion occurs at focal adhesions (FAs) and hemidesmosomes
(2). Homophilic or heterophilic
interactions at the junctions between membrane proteins are
important in the epithelial-mesenchymal transition (EMT), and loss
of cell-cell adhesion and further polarity of the epithelial cells
can lead to migratory cells with mesenchymal properties (13). Cell-ECM adhesion is also important
in the migration and invasion processes. Acquiring new areas for
attachment to the leading edges and detachment from the rear
regions must be coordinated for efficient translocation of cell
bodies (14). During the
coordinated regulation of cell-ECM adhesion, FA dynamics are
important (15). During cell
adhesion involving integrin to the ECM, cells can activate
intracellular signaling molecules such as RhoA GTPases, FAK, c-Src,
ERKs and paxillin (16). These
activations can lead to the reorganization of F-actin networks to
form new integrin-ECM interactions and subsequent tractive force
generation (17).
In the present study, the KRS housekeeping protein
was characterized for possible regulatory roles in cell-cell
adhesion and cell-ECM adhesion related to its pro-migratory
functions in HCT116 colon cancer cells. KRS regulated the
expression of epithelial markers, including E-cadherin and
β-catenin, involved in cell-cell adhesion. It also regulated
cell-laminin adhesion-mediated signaling activities for FAK, ERKs
and paxillin. Suppression of KRS resulted in loss of E-cadherin and
β-catenin without a disruption of cell-cell adhesion, and of
cell-laminin adhesion-mediated signaling activities, leading to
impaired migratory abilities. The results of the present study
showed that KRS was able to regulate cell adhesion properties
involved in cellular migration, suggesting that it could be a
therapeutic target for treatment of colon cancer metastasis.
Materials and methods
Cells
HCT116 colon cancer cells (American Type Culture
Collection, Manassas, VA, USA) were stably transfected with shRNA
against KRS, as previously described (18). HCT116 cells overexpressing KRS were
established via the stable transfection of a myc-tagged KRS plasmid
(7). The cells were maintained in
RPMI-1640 medium (Welgene, Daegu, Korea) containing 10% FBS and
antibiotics (Invitrogen, Grand Island, NY, USA).
Extract preparation and western
blots
Colon cancer cells were washed with ice-cold
phosphate-buffered saline (PBS) and lysed in a modified RIPA buffer
(50 mM Tris-HCl, 150 mM NaCl, 1% NP-40 and 0.25% sodium
deoxycholate) with a protease inhibitor cocktail (GenDepot, Barker,
TX, USA). The extracts were centrifuged for 30 min at 15,000 × g at
4°C, and then the lysates were collected from the supernatant. The
protein amounts were normalized, before further immunoblotting with
different antibodies. The primary antibodies used in the study were
as follows and were against: α-tubulin (Sigma-Aldrich, St. Louis,
MO, USA); pY416c-Src, ERKs, and phospho-ERKs (Cell
Signaling Technology, Danvers, MA, USA); paxillin and FAK (BD
Biosciences, San Jose, CA, USA); pY397FAK (Abcam,
Cambridge, UK); c-Src, pY118pax-illin,
pY577FAK, pY861FAK, pY925FAK,
Snail1, E-cadherin and β-catenin (Santa Cruz Biotechnology, Santa
Cruz, CA, USA), integrin α6 and β1 (Millipore, Billerica, MA, USA);
and KRS (Atlas Antibodies, Stockholm, Sweden).
Normal culture or replating on ECM
layer
Cells were kept in suspension or replated on serum
(10%) or ECM (10 μg/ml laminin; BD Biosciences) -precoated dishes
or coverglasses in the presence of 2% FBS for 1 h, before being
analyzed for cell-serum or -laminin adhesion signaling by standard
western blotting, as previously described (19). Pharmacological inhibitors were
added to the culture media for 24 h or to replating media at the
reseeding time. Cells were harvested for whole cell lysates, prior
to immunoblottings for the indicated proteins, or imaging to
monitor cell growth patterns. Confluent cells were wounded with a
pipette tip and washed twice with PBS. After treatment with DMSO,
U0126 (10 μM) LC Laboratories, Woburn, MA, USA), or YH16899 (10 μM)
and incubation in a CO2 incubator for 24 h, the marginal
edges of the wounds were imaged (CKX41; Olympus, Tokyo, Japan).
Coimmunoprecipitations
HCT116 cells stably expressing myc-KRS WT in
standard media containing 10% FBS in the presence of control
vehicle or YH16899 treatment (10 μM) for 24 h were harvested for
whole cell lysates, and lysates were immunoprecipitated overnight
using anti-myc tagged antibody-coated agarose beads
(Sigma-Aldrich). The immunoprecipitated proteins were boiled in 2X
SDS-PAGE sample buffer before standard western blot analyses.
Indirect immunofluorescence
Cells reseeded in normal culture media- or
laminin-precoated glass coverslips in 2% FBS-containing media for 2
or 24 h were stained for F-actin using phalloidin, or immunostained
using antibodies against E-cadherin, β-catenin, N-cadherin, pERK,
phospho-Tyr397, or phospho-Tyr118 paxillin, in addition to
4′,6-diamidino-2-phe-nylindole (DAPI) staining for the nucleus.
Immunofluorescence images were acquired on a fluorescence
microscope (BX51TR; Olympus) or on a confocal laser scanning
microscope with a Nikon Plan Apochromat 60x/1.4 N.A. oil objective
(Nikon Eclipse Ti microscope; Nikon, Tokyo, Japan).
RT-PCR
Total RNA was extracted from cells under diverse
experimental conditions, using TRIzol® (Invitrogen)
according to the manufacturer's protocol. Total RNA (1 μg) was
reverse transcribed using the amfiRivert Platinum cDNA Synthesis
Master Mix (GenDepot). Primers were designed using Primer3 software
as follows: human E-cadherin (CDH1) mRNA, forward
5′-TGCCCAGAAAATGAAAAAGG-3′ and reverse 5′-GTGTATGTGGCAATGCGTTC-3′;
human N-cadherin (CDH2) mRNA, forward 5′-ACAGTGGCCACCTACAAA
GG-3′ and reverse 5′-CCGAGATGGGGTTGATAATG-3′; human vimentin
(VIM) mRNA, forward 5′-GAGAACTTTG CCGTTGAAGC-3′ and reverse
5′-GCTTCCTGTAGGTGGC AATC-3′; human Twist (TWIST) mRNA,
forward 5′-GGAGT CCGCAGTCTTACGAG-3′ and reverse 5′-TCTGGAGGACC
TGGTAGAGG-3′; human paxillin (PXN) mRNA, forward
5′-GAAATCAGCTGAGCCTTCAC-3′ and reverse 5′-TTAG
GCTTCTCTTTCGTCAGG-3′, Snail1, forward 5′-GGTTC
TTCTGCGCTACTGCT-3′ and reverse 5′-TAGGGCTGCTG GAAGGTAAA-3′;
slug, forward 5′-GGGGAGAAGCCTTT TTCTTG-3′ and reverse
5′-TCTCATGTTTGTGCAGGAG-3′; human KRS (KARS) mRNA, forward
5′-CAATGCCCATGC CCCAGCCA-3′ and reverse 5′-ACCCCACCCTTCCGGCG
AAT-3′; human β-actin (ACTB) mRNA, forward 5′-TGAC
GGGGTCACCCACACTGTGCCCATCTA-3′ and reverse
5′-CTAGAAGCATTTGCGGTGGACGACGGAGGG-3′.
Statistical methods
The Student's t-test was performed for statistical
comparisons of mean values to determine significance. A P-value
<0.05 was considered to indicate a statistically significant
result.
Results
KRS suppression caused an incomplete EMT
phenotype
To explore the roles of KRS in metastatic potential,
HCT116 colon cancer cells with endogenous, suppressed, or
overexpressed KRS levels were first analyzed for EMT phenotypes.
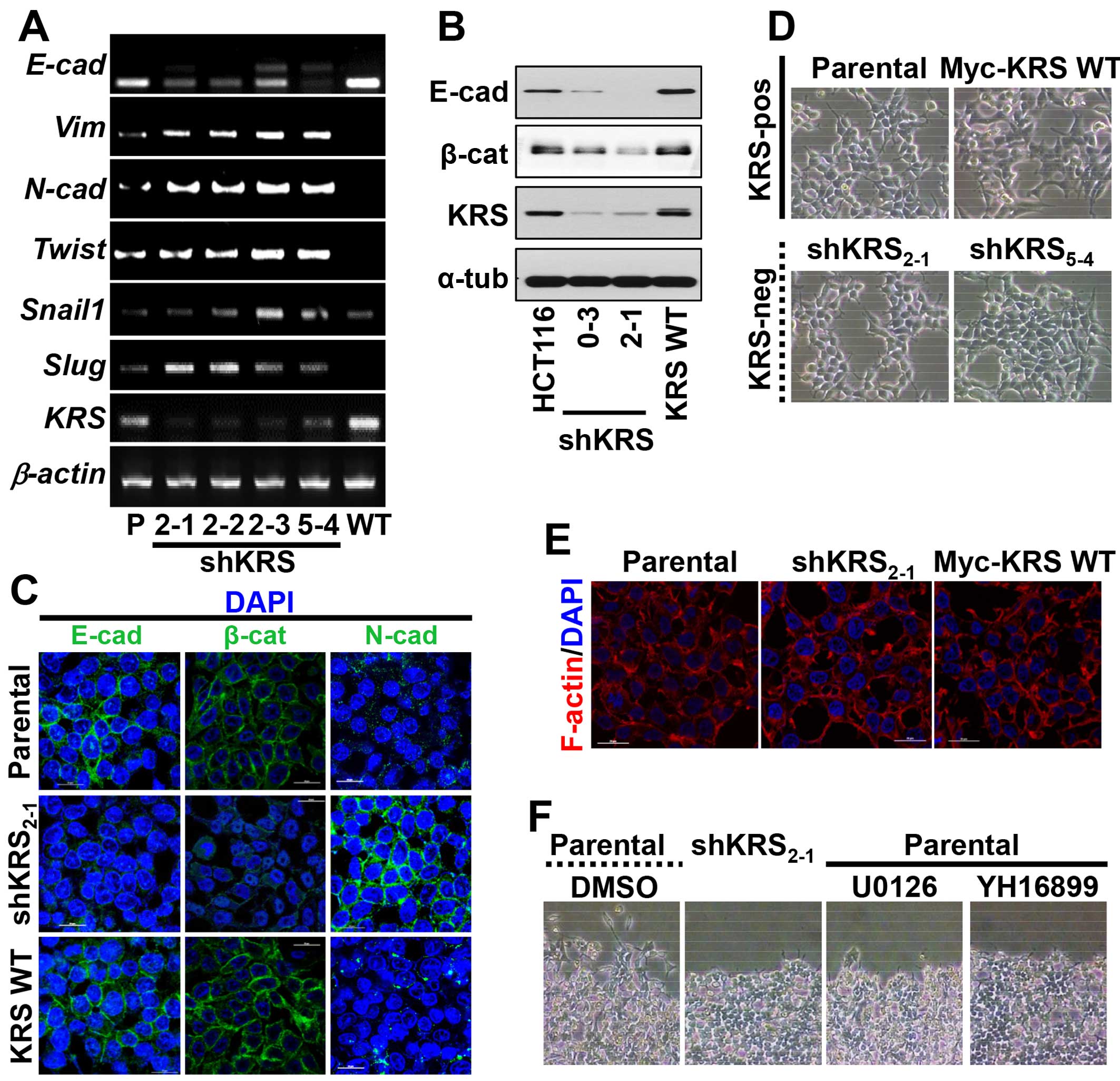
Using an RT-PCR approach, we found that KRS suppression decreased
E-cadherin mRNA but increased the mRNAs of mesenchymal markers
(Fig. 1A). Furthermore, these KRS
suppression-dependent changes in EMT markers also applied to
protein levels; epithelial markers, including E-cadherin, were
expressed at higher levels in KRS-expressing parental and
KRS-overexpressing HCT116 cells, whereas KRS-suppressed cell clones
had decreased E-cadherin and β-catenin expression (Fig. 1B). We then checked whether KRS
downregulation could affect general protein translation and/or
apoptosis, and there were no differences between KRS-expressing and
KRS-suppressed cell clones (18).
E-cadherin was not clearly observed in cell-cell junctions but
N-cadherin was expressed in the junctions with minimal β-catenin
when KRS was suppressed, as compared with KRS-expressing cells
(Fig. 1C). Although the inverse
relationship between KRS and epithelial marker expression might
suggest the induction of EMT phenotypes, cell-cell contacts were
not disrupted, presumably due to N-cadherin at the junctions after
KRS suppression (Fig. 1C and D).
Furthermore, cortical actin, but not stress fibers, was observed at
the cell junctions independent of KRS expression (Fig. 1E). These observations may suggest
that KRS suppression caused a partial EMT phenotype.
However, when wounds were made in confluent cells in
normal 10% fetal bovine serum (FBS)-containing conditions, the
KRS-expressing parental cells exhibited clear outbound movement at
the wound boundary, whereas KRS-suppressed cells did not (Fig. 1F). These KRS-dependent differential
movements toward free spaces might be correlated with ineffective
cell-substrate adhesion processes.
KRS suppression impaired cellular
signaling and focal adhesion formation
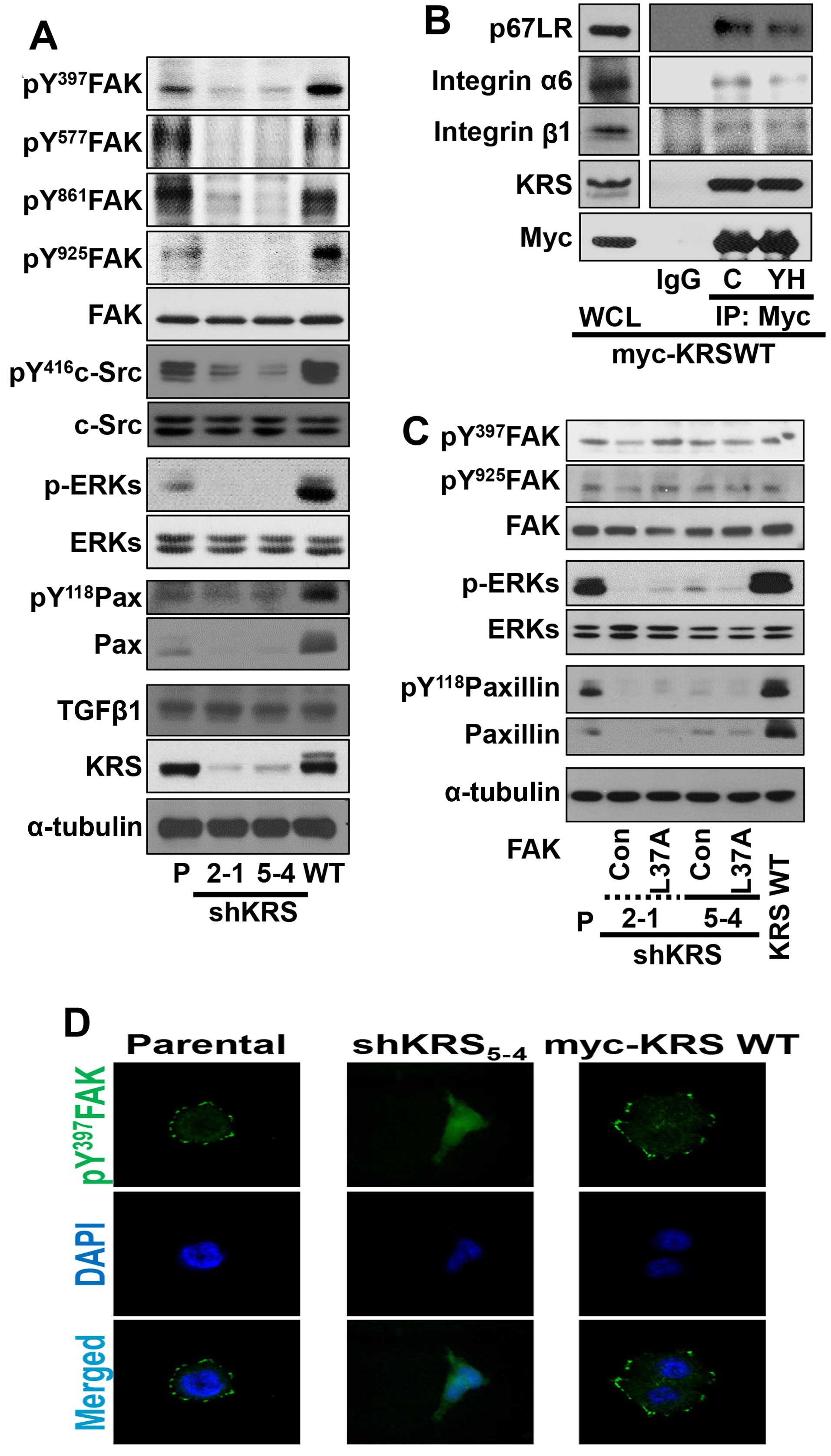
We then examined the relationship between KRS
expression and cell-ECM adhesion properties. First, cells with
various KRS expression levels were allowed to adhere under normal
10% FBS-containing culture conditions and were examined for
adhesion-related signaling activities. Under normal 10%
FBS-containing conditions, KRS suppression abolished the signaling
activities of FAK, ERK, c-Src and paxillin, and the expression of
paxillin, compared with KRS-expressing (or -overexpressing) cells
(Fig. 2A). Integrins activate FAK,
ERK and paxillin for diverse cellular functions in different cell
systems (16). KRS is translocated
to the plasma membrane when it associates and collaborates with
p67LR (a non-integrin laminin receptor) in the presence of
extracellular laminin, following Thr52-phosphorylation-dependent
disassociation from cytosolic MSC (7). Integrin α6β1 and α6β4 respond to
laminin and bind p67LR (9,10). Therefore, to determine whether the
interaction between KRS, p67LR and integrins could be correlated to
signaling activation for FAK, c-Src, ERKs and paxillin, we examined
possible physical interactions between these proteins via a
coimmunoprecipitation study. Myc-KRS in extracts prepared from
normal 10% FBS-containing culture conditions co-precipitated
integrins α6, and β1 and this interaction was blocked by the
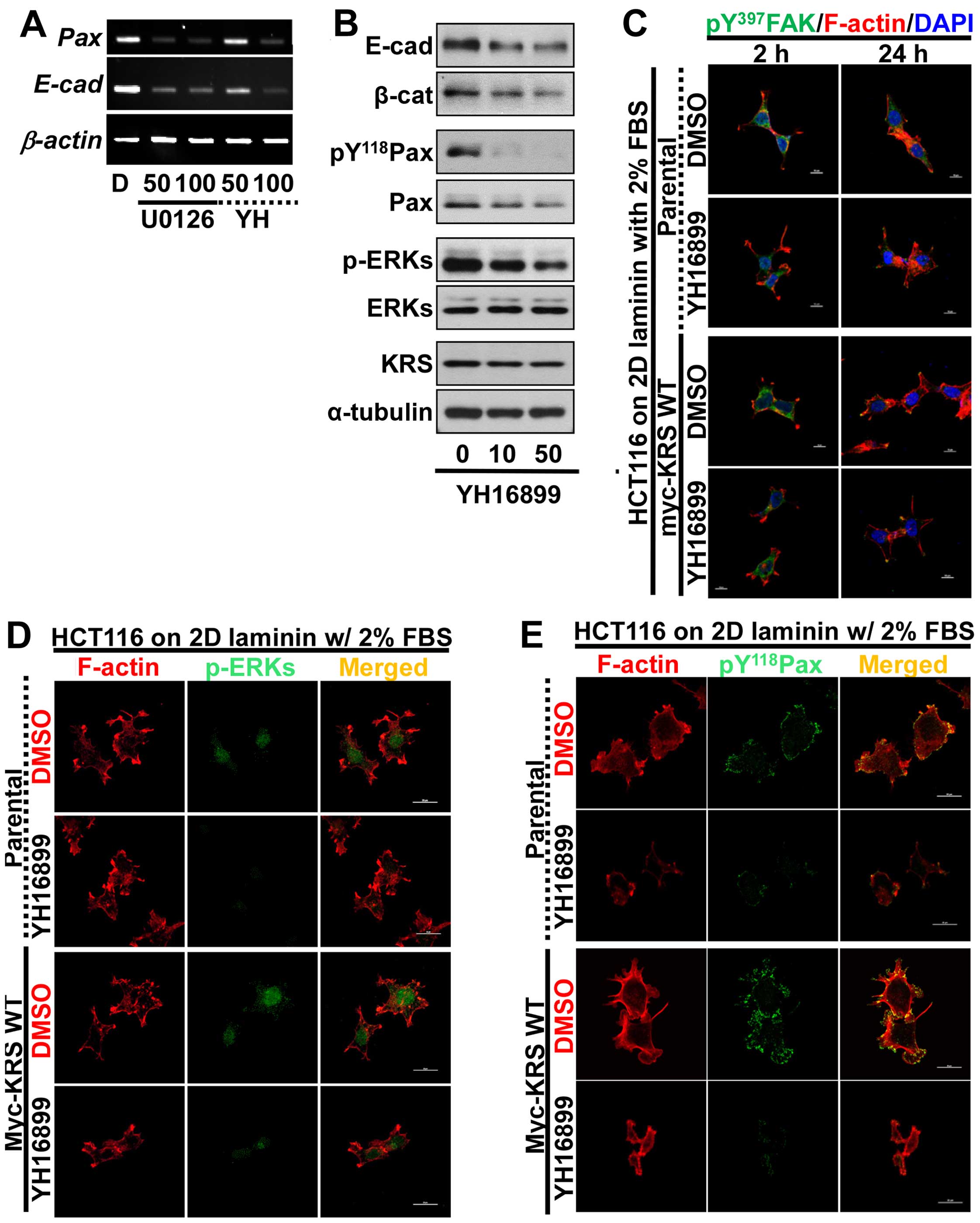
anti-KRS inhibitor (YH16899) treatment (Fig. 4C) (8).
In addition, stable transfection of L37A mutant FAK,
which was shown to be active (20), into KRS-suppressed cells did not
cause ERK activation, paxillin expression and Tyr118
phosphorylation (Fig. 2C),
suggesting that KRS might regulate ERKs/paxillin signaling activity
and expression independently of FAK activation. Furthermore, when
phospho-Tyr397 FAK was imaged to visualize the focal adhesions
(FAs), KRS-suppressed cells did not show FAs, whereas
KRS-expressing cells showed well-formed FAs (Fig. 2D).
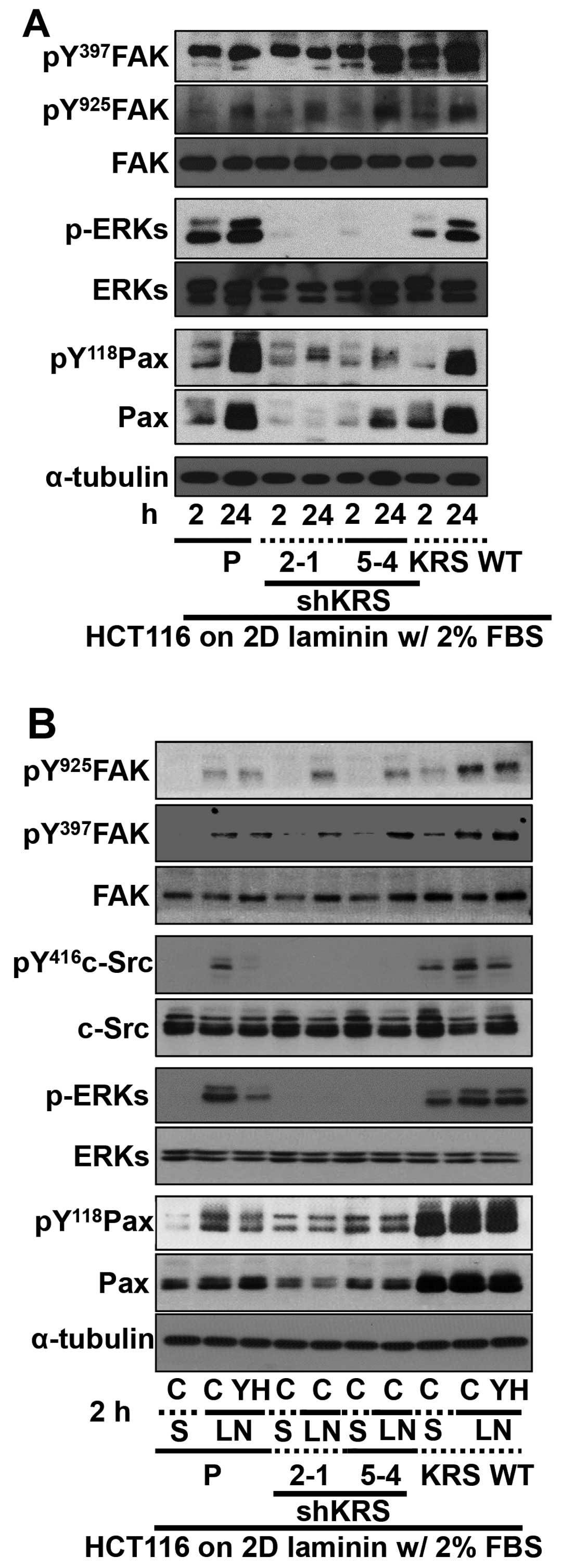
Cell adhesion-related signaling activities were then
analyzed after the cells were kept in suspension or reseeded onto
ECM-precoated culture dishes. Because KRS has been shown to
translocate to the plasma membrane in a laminin-dependent manner
(7), and because KRS suppression
did not change laminin expression (18), we explored the biological
significance of KRS expression after reseeding onto
laminin-precoated plates under reduced (2%) serum-containing
conditions. FAK phosphorylation depended on KRS expression in cases
of parental or overexpressing cells, as reported previously showing
that KRS overexpression in A549 cells further increased Tyr397 FAK
phosphorylation upon being replated onto laminin-precoated dishes
in the absence of serum (7).
However, FAK phosphorylations in KRS-suppressed cells were somewhat
comparable to that in parental HCT116 cells (Fig. 3A). KRS-dependent FAK activity in
10% serum-containing normal conditions (Fig. 2A) appeared different from HCT116
cells manipulated and newly adhered onto laminin under a lower
serum conditions (Fig. 3A). The
adhesion-dependent phosphorylation of FAK in KRS-expressing cells
under these conditions was not affected by treatment with a KRS
inhibitor YH16899 (Fig. 3B), which
blocks the interaction between KRS and p67LR (Fig. 2B). However, the phosphorylation of
ERK, paxillin and c-Src in KRS-expressing cells increased upon
adhesion to laminin and was abolished by additional YH16899
treatment, whereas no effects were observed in KRS-suppressed cells
(Fig. 3B).
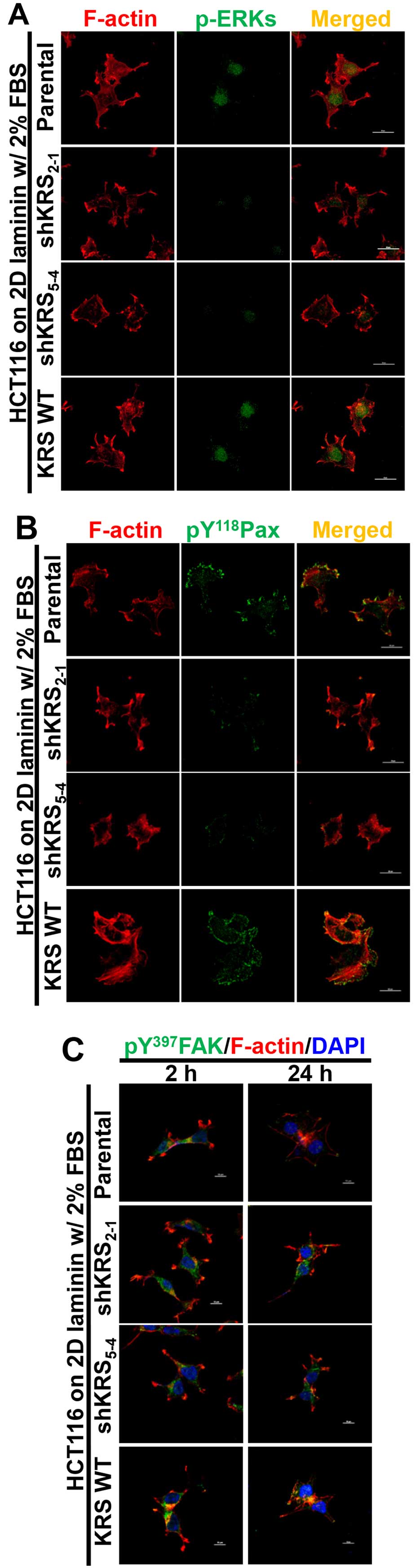
We next examined the subcellular localization of
phospho-ERK, phospho-Tyr118 paxillin, or phospho-Tyr397 FAK in
cells with various KRS expression levels. When the cells were
reseeded onto laminin-precoated coverglasses in 2% serum-containing
media, KRS-positive (but not KRS-suppressed) cells showed obvious
phospho-ERK staining in the nucleus (Fig. 4A). Phospho-Tyr118 paxillin was also
clearly localized to FAs in a KRS-dependent manner (Fig. 4B). Notably, replating the cells
onto laminin for 2 or 24 h resulted in comparable protrusions and
spreading with comparable phospho-Tyr397 FAK levels, independent of
KRS suppression (Fig. 4C). This
suggested that FAK activity in response to laminin stimulation
might not depend on KRS expression. However, ERK1/2 phosphorylation
and paxillin expression and phosphorylation depended on KRS
expression levels (Figs. 3 and
4).
Anti-KRS reagent abolishes KRS-dependent
cell adhesion properties
We then explored whether KRS functioned on cell
adhesion properties, using the anti-KRS reagent YH16899. Treating
KRS-expressing cells with either U0126 or YH16899 decreased both
paxillin and E-cadherin mRNA levels (Fig. 5A). Upon KRS inhibition by the
YH16899 treatment, E-cadherin and β-catenin proteins also
decreased, similarly to ERK and paxillin phosphorylations (Fig. 5B). However, YH16899 treatment of
KRS-expressing cells did not result in changes in protrusion and
spreading as visualized by Tyr397-phosphorylated FAK staining
(Fig. 5C), but did result in
decreased phospho-ERK (Fig. 5D)
and phospho-Tyr118 paxillin (Fig.
5E), under the laminin-precoated conditions containing 2% FBS.
These observations, therefore, suggested that the KRS-dependent
cell adhesion activity could involve ERK and paxillin expression
and activity.
Discussion
The present study suggests that KRS functions
regulating cell-cell adhesion properties, leading to incomplete
EMT, and cell-laminin adhesion-dependent signaling via an
association with p67LR and integrin α6β1 receptors. The roles of
KRS in regulating cell-cell intercellular and cell-substrate
adhesions by regulating E-cadherin expression, ERK activity, and
paxillin expression and activity could promote cellular migration.
During the KRS-dependent activation of the cell-substrate adhesion
signaling pathway, FAK appeared unrelated to ERKs, paxillin
activation and paxillin expression. This KRS/ERK/paxillin signaling
axis may be a useful target against KRS-dependent cancer
metastases.
KRS suppression modulated the epithelial-mesenchymal
properties of cells; KRS suppression decreased the expression of
epithelial markers and concomitantly increased mesenchymal markers.
However, how KRS causes these alterations should be explored
further. Because mesenchymal markers increased upon KRS
suppression, a possible global effect on protein translational
capacity from the downregulated level of KRS was not the cause.
Furthermore, KRS suppression did not cause cellular loss of
adherence even after E-cadherin loss, presumably because
mesenchymal N-cadherin replaced epithelial E-cadherin at the
adherence junctions of KRS-suppressed cells. In addition to its
roles in cell-cell contacts, KRS promoted cell-substrate adhesion
signaling, which is important for myosin contractility-dependent
adhesion strength and traction force (21) and for cellular contractility
(22). Thus, KRS-mediated
migration is involved in cell-substrate adhesion for
contractility/traction force, so that cells can ‘crawl out’, even
in the cases where cell-cell adhesion is well-formed via adherence
junctions. The early phases of colon cancer metastasis are
suggested to have adapted EMT-like de-differentiation at the
invasive edges, in order to detach and migrate (23). HCT116 cells quite highly express
E-cadherin and are categorized as the most epithelial-like cells in
the EMT score among the diverse tumor cell types (24). Presumably, E-cadherin for cell-cell
adhesions and cell-substrate adhesion in KRS-positive colon cancer
cells may coordinately contribute to migration for efficient
metastasis involving a transient EMT-like process. We observed that
KRS-dependent migration did not depend solely on either epithelial
or mesenchymal characteristics, because KRS-suppressed cells could
not efficiently migrate, although they obviously lost E-cadherin
and other epithelial markers. It was recently reported that
circulating tumor cells (CTCs) originated from primary breast
tumors consisting of cells with epithelial, mesenchymal, or both
characteristics (25,26). Therefore, it is likely that
KRS-expressing cells with certain epithelial characteristics have
efficient migration.
KRS-suppressed cells did not move away from the
boundaries of cell masses, therefore, the loss of epithelial
markers was not sufficient for the outward movement. In addition to
regulating cell-cell adhesion, KRS could also regulate
cell-substrate adhesion as part of its pro-metastatic functions.
KRS translocates from the cytosolic MSC to the plasma membrane
after p38 MAPK-dependent Thr52 phosphorylation upon extracellular
laminin stimulation, where it protects p67LR from
ubiquitination-mediated degradation (7). Furthermore, the interaction between
KRS and p67LR is critically involved in the lung metastasis of
subcutaneously-injected mouse breast carcinoma 4T1 cells with KRS
overexpression (8). The KRS/p67LR
complex also included integrin α6β1, allowing KRS-expressing cells
to transduce intracellular signals under extracellular
laminin-stimulated conditions. Moreover, extra-cellular laminin,
but not collagen I, causes KRS translocation to the plasma membrane
for binding to p67LR (7). Thus, it
is likely that KRS may mediate ERK activation for pro-metastatic
roles following specific adhesion-mediated signals from the
extracellular microenvironment.
In the present study, KRS-dependent migration was
shown to require ERK activity and paxillin expression and activity,
leading to efficient FA formation; inactivating ERK or disrupting
complex formation among KRS, p67LR and integrin α6β1 blocked
KRS-dependent effects. FAK, c-Src family kinases (SFKs), paxillin,
and ERK1/2 are key regulators of focal adhesion dynamics,
especially during cell adhesion and migration (27,28).
Cell-substrate adhesion activates ERK via a FAK Tyr925
phosphorylation-mediated signaling pathway from FAK to the Ras
cascade upon integrin/ECM interaction (29). However, FAK activity was not
relevant for the KRS-dependent ERK and paxillin activations and
paxillin expression, because manipulation of FAK activity did not
lead to ERK activity and paxillin expression/Tyr118
phosphorylation. FAK-independent ERK activation may occur during
cell adhesion events. The association of integrins with caveolin-1
and Fyn (a SFK member) recruits Shc for ERK activation (30–32).
Furthermore, the cell adhesion-dependent activation of CaMKII in
vascular smooth muscle cells leads to ERK activation independent of
FAK activity (33).
The signaling transduced from the KRS/p67LR/integrin
α6β1 complex to paxillin expression/phosphorylation via ERK
activity could be targeted to treat colon cancer metastasis
depending on KRS expression and functions.
Acknowledgements
The present study was supported by the National
Research Foundation of Korea (NRF) grant for the Tumor
Microenvironment Global Core Research Center (GCRC) funded by the
Korea government (Ministry of Science, ICT & Future Planning)
(2011-0030001 to J.W.L.), for the Senior Researchers Program (Leap
research, 2012-0005606/2013-035235 to J.W.L.), and for the
Medicinal Bioconvergence Research Center (NRF-M1AXA002-2010-0029785
to S.K. and NRF-2012M3A6A4054271 to J.W.L.).
References
|
1
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collins C and Nelson WJ: Running with
neighbors: Coordinating cell migration and cell-cell adhesion. Curr
Opin Cell Biol. 36:62–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Geiger B, Spatz JP and Bershadsky AD:
Environmental sensing through focal adhesions. Nat Rev Mol Cell
Biol. 10:21–33. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Geiger T and Geiger B: Towards elucidation
of functional molecular signatures of the adhesive-migratory
phenotype of malignant cells. Semin Cancer Biol. 20:146–152. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park SG, Kim HJ, Min YH, Choi EC, Shin YK,
Park BJ, Lee SW and Kim S: Human lysyl-tRNA synthetase is secreted
to trigger proinflammatory response. Proc Natl Acad Sci USA.
102:6356–6361. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yannay-Cohen N, Carmi-Levy I, Kay G, Yang
CM, Han JM, Kemeny DM, Kim S, Nechushtan H and Razin E: LysRS
serves as a key signaling molecule in the immune response by
regulating gene expression. Mol Cell. 34:603–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim DG, Choi JW, Lee JY, Kim H, Oh YS, Lee
JW, Tak YK, Song JM, Razin E, Yun SH, et al: Interaction of two
translational components, lysyl-tRNA synthetase and p40/37LRP, in
plasma membrane promotes laminin-dependent cell migration. FASEB J.
26:4142–4159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim DG, Lee JY, Kwon NH, Fang P, Zhang Q,
Wang J, Young NL, Guo M, Cho HY, Mushtaq AU, et al: Chemical
inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor
interaction. Nat Chem Biol. 10:29–34. 2014. View Article : Google Scholar :
|
|
9
|
Canfield SM and Khakoo AY: The nonintegrin
laminin binding protein (p67 LBP) is expressed on a subset of
activated human T lymphocytes and, together with the integrin very
late activation antigen-6, mediates avid cellular adherence to
laminin. J Immunol. 163:3430–3440. 1999.PubMed/NCBI
|
|
10
|
Ardini E, Tagliabue E, Magnifico A, Butò
S, Castronovo V, Colnaghi MI and Ménard S: Co-regulation and
physical association of the 67-kDa monomeric laminin receptor and
the alpha6beta4 integrin. J Biol Chem. 272:2342–2345. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berno V, Porrini D, Castiglioni F,
Campiglio M, Casalini P, Pupa SM, Balsari A, Ménard S and Tagliabue
E: The 67 kDa laminin receptor increases tumor aggressiveness by
remodeling laminin-1. Endocr Relat Cancer. 12:393–406. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carragher NO and Frame MC: Focal adhesion
and actin dynamics: A place where kinases and proteases meet to
promote invasion. Trends Cell Biol. 14:241–249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scales TM and Parsons M: Spatial and
temporal regulation of integrin signalling during cell migration.
Curr Opin Cell Biol. 23:562–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Albiges-Rizo C, Destaing O, Fourcade B,
Planus E and Block MR: Actin machinery and mechanosensitivity in
invado-podia, podosomes and focal adhesions. J Cell Sci.
122:3037–3049. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee JW and Juliano R: Mitogenic signal
transduction by integrin-and growth factor receptor-mediated
pathways. Mol Cells. 17:188–202. 2004.PubMed/NCBI
|
|
17
|
Valdembri D and Serini G: Regulation of
adhesion site dynamics by integrin traffic. Curr Opin Cell Biol.
24:582–591. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nam SH, Kim D, Lee MS, Lee D, Kwak TK,
Kang M, Ryu J, Kim HJ, Song HE, Choi J, et al: Noncanonical roles
of membranous lysyl-tRNA synthetase in transducing cell-substrate
signaling for invasive dissemination of colon cancer spheroids in
3D collagen I gels. Oncotarget. 6:21655–21674. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee SA, Kim YM, Kwak TK, Kim HJ, Kim S, Ko
W, Kim SH, Park KH, Kim HJ, Cho M, et al: The extracellular loop 2
of TM4SF5 inhibits integrin alpha2 on hepatocytes under collagen
type I environment. Carcinogenesis. 30:1872–1879. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jung O, Choi S, Jang SB, Lee SA, Lim ST,
Choi YJ, Kim HJ, Kim DH, Kwak TK, Kim H, et al: Tetraspan
TM4SF5-dependent direct activation of FAK and metastatic potential
of hepatocarcinoma cells. J Cell Sci. 125:5960–5973. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dumbauld DW, Lee TT, Singh A, Scrimgeour
J, Gersbach CA, Zamir EA, Fu J, Chen CS, Curtis JE, Craig SW, et
al: How vinculin regulates force transmission. Proc Natl Acad Sci
USA. 110:9788–9793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clark K, Howe JD, Pullar CE, Green JA,
Artym VV, Yamada KM and Critchley DR: Tensin 2 modulates cell
contractility in 3D collagen gels through the RhoGAP DLC1. J Cell
Biochem. 109:808–817. 2010.PubMed/NCBI
|
|
23
|
Chua KN, Poon KL, Lim J, Sim WJ, Huang RY
and Thiery JP: Target cell movement in tumor and cardiovascular
diseases based on the epithelial-mesenchymal transition concept.
Adv Drug Deliv Rev. 63:558–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang RY, Wong MK, Tan TZ, Kuay KT, Ng AH,
Chung VY, Chu YS, Matsumura N, Lai HC, Lee YF, et al: An EMT
spectrum defines an anoikis-resistant and spheroidogenic
intermediate mesenchymal state that is sensitive to e-cadherin
restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell
Death Dis. 4:e9152013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thiery JP and Lim CT: Tumor dissemination:
An EMT affair. Cancer Cell. 23:272–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu M, Bardia A, Wittner BS, Stott SL, Smas
ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al:
Circulating breast tumor cells exhibit dynamic changes in
epithelial and mesenchymal composition. Science. 339:580–584. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Panetti TS: Tyrosine phosphorylation of
paxillin, FAK, and p130CAS: Effects on cell spreading and
migration. Front Biosci. 7:d143–d150. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Webb DJ, Donais K, Whitmore LA, Thomas SM,
Turner CE, Parsons JT and Horwitz AF: FAK-Src signalling through
paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell
Biol. 6:154–161. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schlaepfer DD, Hanks SK, Hunter T and van
der Geer P: Integrin-mediated signal transduction linked to Ras
pathway by GRB2 binding to focal adhesion kinase. Nature.
372:786–791. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin TH, Aplin AE, Shen Y, Chen Q, Schaller
M, Romer L, Aukhil I and Juliano RL: Integrin-mediated activation
of MAP kinase is independent of FAK: Evidence for dual integrin
signaling pathways in fibroblasts. J Cell Biol. 136:1385–1395.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wary KK, Mainiero F, Isakoff SJ,
Marcantonio EE and Giancotti FG: The adaptor protein Shc couples a
class of integrins to the control of cell cycle progression. Cell.
87:733–743. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wary KK, Mariotti A, Zurzolo C and
Giancotti FG: A requirement for caveolin-1 and associated kinase
Fyn in integrin signaling and anchorage-dependent cell growth.
Cell. 94:625–634. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu KK, Armstrong SE, Ginnan R and Singer
HA: Adhesion-dependent activation of CaMKII and regulation of ERK
activation in vascular smooth muscle. Am J Physiol Cell Physiol.
289:C1343–C1350. 2005. View Article : Google Scholar : PubMed/NCBI
|