Introduction
Recently, emerging studies have demonstrated that
70–90% of the human genome is capable of being pervasively
transcribed. Surprisingly, <2% of the total genome is found to
have the ability to encode proteins, indicating that non-coding
RNAs account for most of the human transcriptome. In fact, recent
evidence has suggested that the human non-coding RNAs include
approximately 9,000 small RNAs, 10,000–32,000 long non-coding RNAs
(lncRNAs) and 11,000 pseudogenes (1,2).
Small non-coding RNAs have been well characterized and categorized
by transfer RNAs, microRNAs (miRNAs), small-interfering RNAs,
PIWI-interacting RNAs, small nuclear RNAs, small nucleolar RNAs and
transcription initiation RNAs.
lncRNAs can vary in length from 200 nucleotides to
100 kilobases. A previous study demonstrated that lncRNAs play an
important role in a diverse range of biological processes (3–5),
such as participating in epigenetics, nuclear import, alternative
splicing, RNA decay and translation. More importantly, due to their
widespread distribution in nucleus, lncRNAs could regulate the
transcription of adjacent genes by binding to the specific
transcription factors and polymerases (6,7), for
example, the expression of microRNAs and their bio-function are
mainly regulated by lncRNAs. On the other hand, they can serve as
precursors to microRNAs, ceRNA of microRNAs, and natural microRNA
sponges. Therefore, aberrant lncRNA expression can cause various
human diseases and disorders.
microRNAs, 23-nucleotide-long endogenous RNAs, can
bind with their target RNA transcripts by pairing with microRNA
response elements (MREs) and then result in degradation or
translational repression of the target transcripts (8,9).
Recent studies show that microRNAs regulate a number of the
transcriptions, including the coding and noncoding transcriptome
(10). Therefore, a better
understanding of microRNA profiling associated with lncRNA in
cancer is imperative for the development of novel therapeutic
strategies. However, many details remain unknown regarding microRNA
profiling regulated by lncRNA.
In this study, we used Illumina deep sequencing to
sequence miRNA libraries prepared from the 5637 cells of normal
high-expression and knockdown of UCA1, which are specifically
upregulated in bladder cancer. Overall, expression of 75 miRNAs
showed significant difference associated with UCA1: 38 were
upregulated and 37 downregulated. GO analysis of the host target
genes revealed that these dysregulated miRNAs are involved in
complex cellular pathways, including biological process, cellular
component and molecular function. After confirming with RT-qPCR, 8
miRNAs associated with UCA1 were selected to draw the TF-miRNA-mRNA
network and enrich KEGG pathway analysis. miR-196a was one of these
8 candidate microRNAs and its targeted gene p27kip1
participated in PI3K-Akt signaling pathway. Then the correlation
among UCA1, miR-196a and p27kip1 in bladder cancer cells
and patients was analyzed.
Material and methods
Tissue specimen collection
In this study, 35 bladder cancer tissues and 16
bladder non-tumor tissues were obtained from August 2012 to
September 2013 at the Department of Clinical Laboratory, the First
Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China. All
samples were identified by two independent pathologists. Informed
consent was provided by each patient, and this study was approved
by the Ethics Committee of the First Affiliated Hospital of Xi'an
Jiaotong University and accordance with the Declaration of
Helsinki.
Cell culture and treatment
Human bladder cancer cell lines (5637 cell line and
UMUC2 cell line) obtained from the American Type Culture Collection
(ATCC) were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA)
supplemented with 10% heat inactivated bovine calf serum and 150
μg/ml G418 at 37°C in a humidified atmosphere with 5%
CO2. Stable cell lines with UCA1 knockdown in 5637 cells
and UCA1 over-expression in UMUC2 cells were a kind gift of our
colleague Zhengkun Li, the Center for Translational Medicine of the
First Affiliated Hospital, School of Medicine, Xi'an Jiaotong
University (Xi'an, China).
RNA isolation and Illumina
sequencing
The total RNA was extracted from UCA1 knockdown 5637
cells and control cells using TRIzol reagent (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer's instructions. The RNA
concentration was detected by measuring the absorbance at OD 260 nm
(A260) and OD 280 nm (A280). The purity of RNA was evaluated by the
A260/A280 ratio using NanoDrop ND-1000 (NanoDrop Technologies,
Wilmington, DE, USA). The integrity of RNA samples were further
measured by 28S/18S ratio using Agilent 2100 Bioanalyzer (Agilent
Technologies, Santa Clara, CA, USA) and all RNA samples were
verified as intact with the RNA integrity number (RIN) >7. The
two RNA samples were then sent to Huada BGI-Tech (Shanghai, China)
for Illumina sequencing of the small RNAs.
Analysis of sequencing data
The small RNA sequencing reads obtained were then
subjected to the following: i) reads were selected by removing the
low-quality reads with the Solexa Chastity quality filter; ii) the
adapter sequences were trimmed and the reads longer than 15 were
retained; and iii) low two-copy sequences were discarded. The
filtered short reads were then mapped to the Repbase database
(available online: http://www.girinst.org/repbase/), Rfam database, NCBI
database (available online: http://www.ncbi.nlm.nih.gov/) (available online:
http://rfam.janelia.org/). The unmappable
sequences not mapped to miRBase 19.0 were used to predict
potentially novel candidate miRNAs with the Mfold RNA folding
prediction web server (available online: http://mfold.rna.albany.edu/).
Identification of
differentially-expressed miRNAs
To compare the differential miRNA expression in 5637
cells of normal high-expression and knockdown of UCA1, the number
of raw tags in each library was normalized to tags per million of
the total miRNA reads. Differentially-expressed miRNAs were
determined according to the criterion of a fold change of ≥2 in the
sequence counts between libraries.
Real-time PCR
The total RNA was extracted from cells or bladder
cancer patient tissues using TRIzol reagent (Invitrogen) as
described above. RNA was subjected to reverse transcription
reactions using the PrimeScript RT reagent kit (Invitrogen).
Selected miRNAs were subjected to RT-qPCR using SYBR Premis Ex Tag
II (Takara, Dalian, China) and monitored with the CFX96 Touch
Real-time PCR detection system (Bio-Rad, Hercules, CA, USA). The
PCR reaction mixture (20 μl) consisted of 2 μl of cDNA, 10 μl of 2X
SYBR-Green PCR master mix, 0.8 μl each of the microRNA-specific
forward primer, a universal reverse primer (10 μM) and 6.4 μl of
nuclease-free water. The cycling parameters were 94°C for 5 min
followed by 40 cycles of 94°C for 30 sec and 60°C for 30 sec. U6
was used as the endogenous control for normalization. The relative
expression levels were calculated with the 2−ΔCt method.
All the miRNA primers were obtained from Ribobio (Guangzhou,
China). Three independent biological replicates were performed.
miRNA target prediction
The potential target genes of the
differentially-expressed miRNAs were predicted with four miRNA
target prediction algorithms, miRanda (available online: http://www.microrna.org/), miRDB (available online:
http://www.mirdb.org/), RNAhybrid (available
online: http://bibiserv.techfak.uni-bielefeld.de/) and
TargetScan (available online: http://www.targetscan.org/).
Gene ontology and KEGG pathway
analysis
The Gene Ontology program (available online:
http://www.geneontology.org) was used
for GO annotation and enrichment analysis of the miRNA target genes
based on: cell component, biological process and molecular
function. The false discovery rate (FDR) was evaluated using the
default parameters to adjust the P-values. Genes with FDR ≤0.05
were considered to be significantly enriched in the target
candidate genes. Enriched KEGG pathway analyses of 8 candidate
miRNA-targeted genes were performed by bioinformatics tool DAVID
6.7. P-value of significance by Fisher's test was set at
P<0.05.
Western blot analysis
The 5637 or UMUC2 cells were pelleted and then lysed
by RIPA buffer (Thermo Scientific, Waltham, MA, USA) containing
protease inhibitors (Roche). After SDS-PAGE resolution and membrane
transfer, the target proteins were probed with primary antibody
against human p27kip1 or β-actin (Abcam, Cambridge, UK)
followed by incubation with HRP-conjugated secondary antibodies.
Then, the bands were visualized using a chemiluminescence detection
kit (Pierce, Rockford, IL, USA) and the specific bands were
recorded on X-ray film.
Immunohistochemistry (IHC)
Tissue sections were deparaffinized and rehydrated.
Endogenous peroxidase activity was blocked with 0.3% hydrogen
peroxide for 20 min. For antigen retrieval, slides were microwave
treated and boiled in a 10 mM citrate buffer (pH 6.0) for 10 min.
Nonspecific binding was blocked with 10% normal rabbit serum for 20
min. The slides were incubated with monoclonal mouse anti-human
p27kip1 (1:150 dilution; Abcam) overnight at 4°C in a
moist chamber. The slides were sequentially incubated with
biotinylated rabbit anti-mouse immunoglobulin at a concentration of
1:150 for 30 min at 37°C and then reacted with a
streptavidin-peroxdase conjugate for 30 min at 37°C and
diaminobenzidine as a chromogen substrate. The nucleus was
counterstained using Meyer's hematoxylin.
Statistical analysis
Statistical analysis was performed with SPSS
software (SPSS standard version 11.5; SPSS Inc., Chicago, IL, USA).
The association analysis was assessed by the Chi-square test. Data
are represented as mean ± SD. P-value of <0.05 was considered
significant.
Results
Analysis of miRNAs from deep
sequencing
We sequenced two small RNA libraries built from 5637
cells of normal high-expression and knockdown of UCA1, which
contained 17,747,882 and 18,522,937 raw reads, respectively. In
5637 cells, 82.40% of total reads and 48.06% of unique reads could
be mapped to the human reference genome, and in 5637 cells of UCA1
knockdown, the proportions were 85.46 and 38.49%, respectively.
After quality control procedures, 11,446,960 and 11,462,035 short
reads were kept for further analyses for 5637 cells of normal
high-expression or knockdown of UCA1, respectively (Table I). We found that 12.68 and 13.15%
of the clean reads correspond to miRNAs for 5637 cells of normal
high-expression or knockdown of UCA1, respectively. The remaining
clean reads could be mapped to other genomic locations,
corresponding to other different kinds of small RNAs, including
repeats, snRNA, snoRNA, rRNA and tRNA. The chromosomal
distributions of these sRNAs from 5637 cells of normal
high-expression or knockdown of UCA1 are shown in Fig. 1A and B, respectively. Most of the
small RNAs (91.30 and 89.66%, respectively) from both libraries
were 21–24 nt in length (Fig. 1C and
D). Importantly, 22-nt RNAs were the most abundant, accounting
for 65.29 and 62.26% of the small RNAs in 5637 cells of normal
high-expression or knockdown of UCA1, respectively, which is
consistent with the typical size of miRNA from Dicer-derived
products. Analysis of the first nucleotide bias in these miRNAs is
presented in Fig. 1E and F. The
results showed that uridine (U) dominated the first position,
whereas guanine (G) was the least favored first base, which is a
characteristic feature of miRNAs. Furthermore, Fig. 1G and H show the analysis of the
each nucleotide bias in these miRNAs, suggesting that uridine (U)
had higher frequence, especially in position 1, 6, 9, 13, 14, 16,
18, 21 and 22.
 | Table IThe summary of data produced by small
RNA sequencing. |
Table I
The summary of data produced by small
RNA sequencing.
| N | S |
|---|
|
|
|
|---|
| Type | Count | Percentage | Count | Percentage |
|---|
| Total reads | 17,747,882 | - | 18,522,937 | - |
| High quality | 11,932,390 | 100.00 | 11,941,633 | 100.00 |
| 3′ adapter
null | 7,725 | 0.06 | 5,284 | 0.04 |
| Insert null | 14,454 | 0.12 | 10,770 | 0.09 |
| 5′ adapter
contaminants | 409,756 | 3.43 | 391,361 | 3.28 |
| Smaller than 18
nt | 47,127 | 0.39 | 67,854 | 0.57 |
| PolyA | 6,368 | 0.05 | 4,329 | 0.04 |
| Clean reads | 11,446,960 | 95.93 | 11,462,035 | 95.98 |
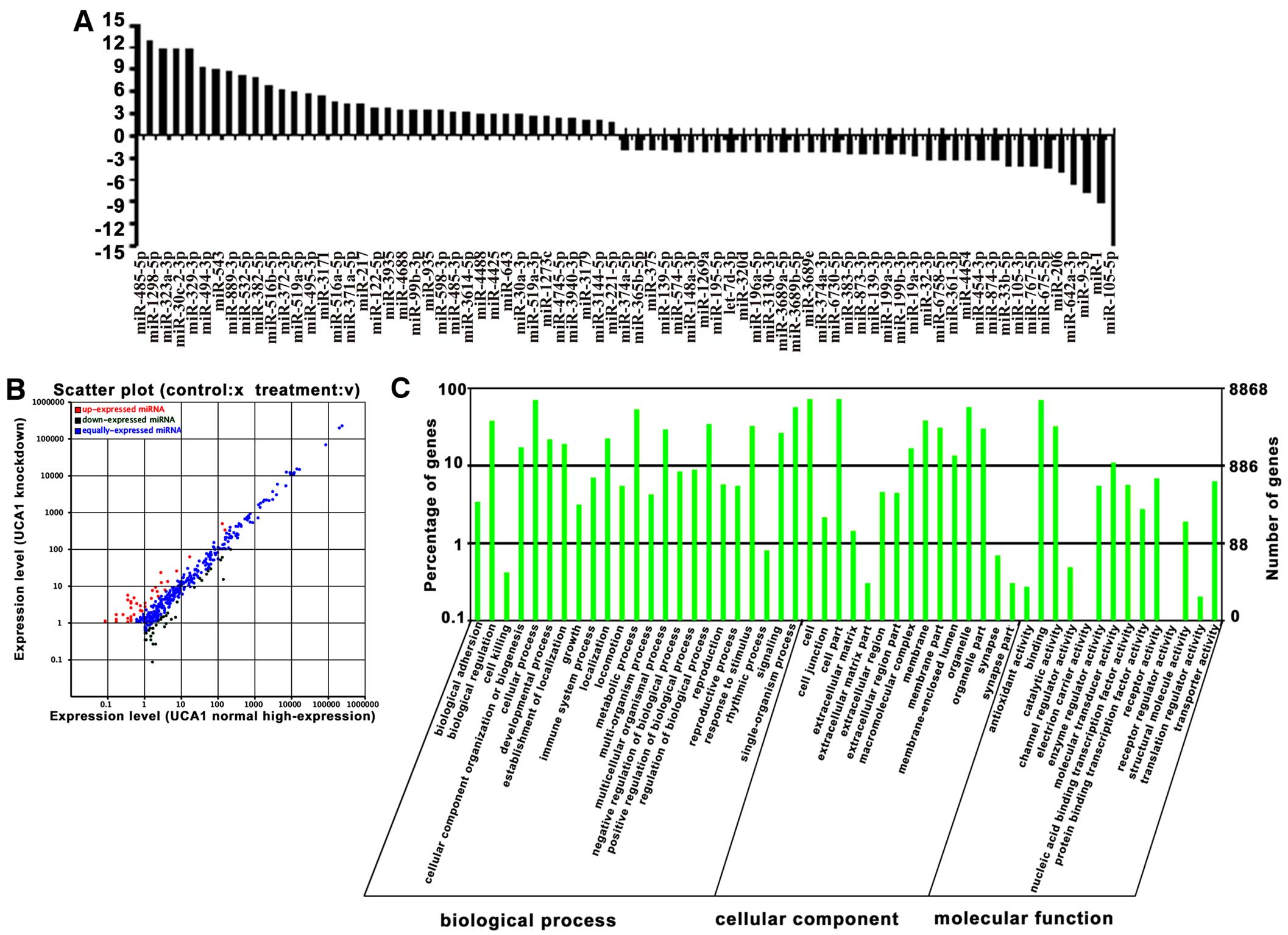
Analysis of miRNAs associated with
UCA1
Of the 829 human mature miRNAs deposited in miRBase
were identified in our two libraries (235 in UCA1 normal
high-expression cells and 225 in UCA1 knockdown cells). Among
these, 75 miRNAs were found in both libraries and significantly
differentially expressed by ≥2-fold from 5637 cells of normal
high-expression or knockdown of UCA1 (P<0.05), in which 38
miRNAs were highly expressed (upregulated), and 37 miRNAs had low
expression (downregulated) in UCA1 knockdown cells (Fig. 2A and B), suggesting that these
miRNAs are associated with UCA1 expression in bladder cancer cells.
To further understand the physiological functions of the miRNAs
associated with UCA1, the potential targets of the up- and
downregulated miRNAs were predicted with four miRNA target
prediction software programs: TargetScan, miRanda, Clip-Seq and
miRDB. We found tens of thousands of putative targets in the
combined outputs of the programs. All of the predicted target genes
of miRNAs were then subjected separately to GO analysis. The GO
category analysis revealed that these genes fell into three major
categories: biological process, cellular component and molecular
function (Fig. 2C).
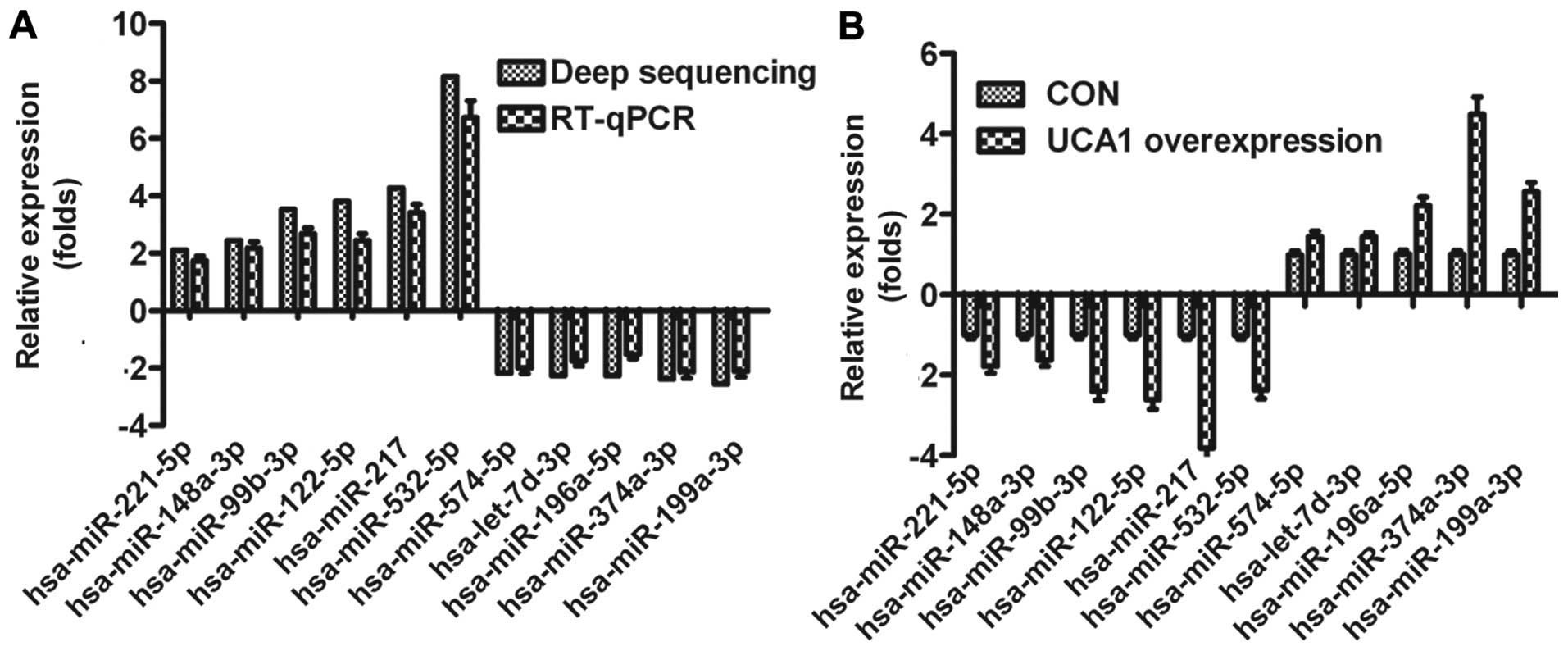
Validation of deep sequencing results by
RT-qPCR
To validate the results obtained from deep
sequencing, we selected 11 miRNAs already known in miRBase
associated with bladder cancer in this study for validation by
qRT-PCR, respectively. The expression levels of the miRNAs
determined with deep sequencing and RT-qPCR were compared (Fig. 3A). On the whole, the relative
expression levels of the selected miRNAs detected with RT-qPCR were
lower than the relative expression levels determined with deep
sequencing, which may result from the difference in the sensitivity
of the two methods. To further confirm that the sequencing miRNAs
were associated with UCA1 in bladder cancer, we detected the
expression of the above 11 miRNAs in UMUC2 cells transfected by
UCA1. As shown in Fig. 3B,
expression of 11 miRNAs was positively correlated to UCA1, which
also validated the sequencing results.
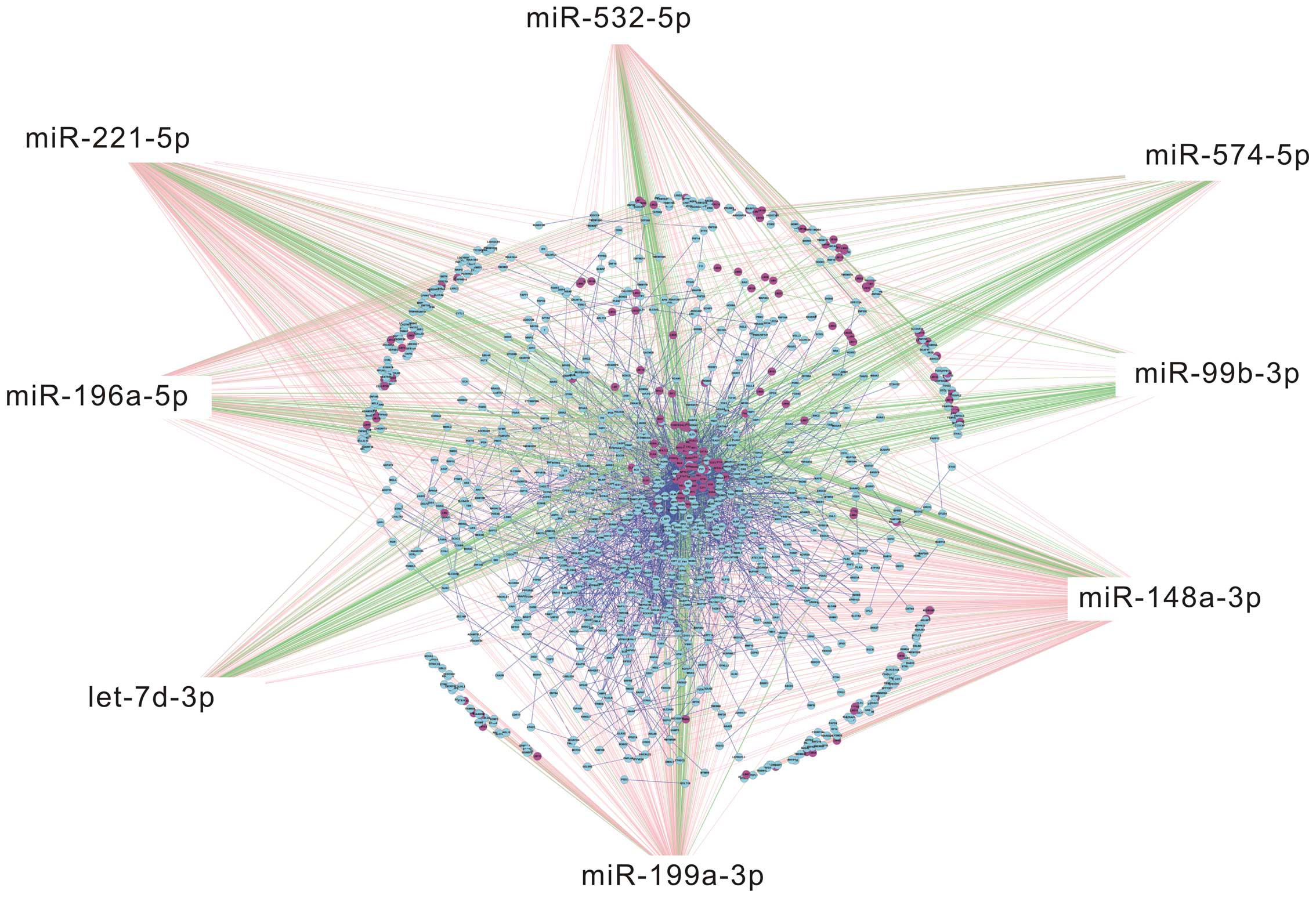
Bioinformatics analysis of 8 candidate
miRNAs
Eight miRNAs associated with UCA1 selected from the
above 11 miRNAs were selected to draw TF-miRNA-mRNA network
(Fig. 4). The top 50 genes and
transcription factors in TF-miRNA-mRNA network are listed based on
the relevance (Fig. 5A). KEGG
pathway analysis was employed to predict target genes of the 8
miRNAs and six signaling pathways were found statistically
significant (P<0.05) (Table
II). p27kip1 was found as a crucial target gene for
the 8 candidate miRNAs. In addition, enriched KEGG pathway analysis
showed that the target gene p27kip1 regulated by one of
the 8 candidate miRNAs, miR-196a, was involved in PI3K-Akt
signaling pathway (Fig. 5B),
suggesting that PI3K-Akt signaling played an important role in the
regulation of the 8 candidate miRNAs.
| Table IIKEGG pathway analysis of 8 candidate
miRNAs. |
Table II
KEGG pathway analysis of 8 candidate
miRNAs.
| Pathway | Id | Sample no. | Background no. | P-value | Genes |
|---|
| Proteoglycans in
cancer - Homo sapiens (human) | hsa05205 | 31 | 225 | 8.59E-06 |
MTOR|SMAD2|NRAS|SDC4|ITGA5|PAK1|SDC2|ESR1|
FZD6|PXN|PRKACB|IGF1|CAMK2G|PPP1CB|WNT1|
FRS2|IGF1R|HPSE2|CDC42|WNT10B|MRAS|ROCK1|
CBLB|ITGB1|FN1|PDCD4|SOS2|CBL| KRAS|ERBB3|ERBB4| |
| ErbB signaling
pathway | hsa04012 | 14 | 88 | 0.000581 |
CBLB|CDKN1B|PAK4|NRAS|SOS2|MTOR|CBL|CAMK2G|KRAS|PAK1|TGFA|PAK7|ERBB3|ERBB4| |
| ECM-receptor
interaction | hsa04512 | 14 | 88 | 0.000581 |
ITGA11|THBS2|ITGB1|COL3A1|CD47|COL4A1|COL2A1|COL4A5|ITGB8|SDC4|ITGA5|SDC2|ITGA3|FN1| |
| Glioma | hsa05214 | 11 | 65 | 0.001305 |
PTEN|NRAS|IGF1R|SOS2|IGF1|CAMK2G|RB1|CALM3|KRAS|MTOR|TGFA| |
| PI3K-Akt signaling
pathway - Homo sapiens (human) | hsa04151 | 34 | 346 | 0.002319 |
JAK2|PDGFC|NRAS|SGK3|MTOR|PPP2R2A|COL4A5|ITGA5|PRKAA1|PPP2R5E|ITGA3|THBS2|EPO|IGF1|ITGB8|LPAR4|YWHAE|IGF1R|DDIT4|PTEN|ITGA11|BCL2L11|COL2A1|IL3RA|CDKN1B|ITGB1|COL3A1|FN1|COL4A1|SOS2|G6PC|KRAS|YWHAB|HSP90B1| |
| Focal adhesion | hsa04510 | 23 | 206 | 0.002327 |
PAK4|PDGFC|ITGA5|PAK1|PPP1CB|ITGA3|THBS2|PXN|IGF1|ITGB8|PAK7|IGF1R|CDC42|ITGA11|ROCK1|COL2A1|COL4A5|ITGB1|COL3A1|FN1|COL4A1|SOS2|PTEN| |
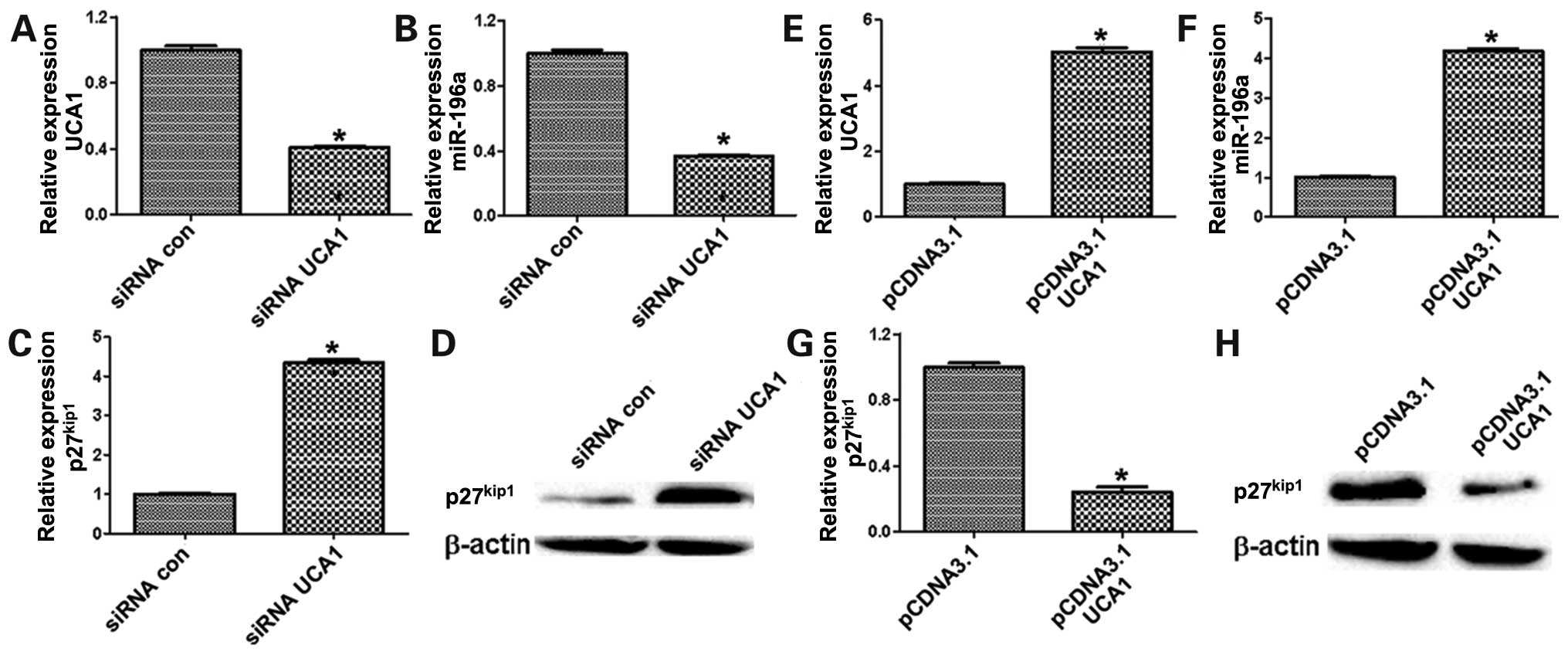
The correlation among UCA1, miR-196a and
p27kip1 in bladder cancer cells and patients
Next, we observed miR-196a expression in bladder
cancer and focused on the regulation of UCA1 on miR-196a. The
result revealed that miR-196a was significantly decreased in 5637
cells after transfection by siUCA1 (Fig. 6A and B), whereas the levels of
p27kip1 was remarkably increased (Fig. 6C and D). In contrast,
overexpression of UCA1 could significantly upregulate miR-196a in
UMUC2 cells (Fig. 6E and F),
whereas p27kip1 was downregulated in UMUC2 cells of
ectopic UCA1 expression (Fig. 6G and
H). Besides, the expression of UCA1 and miR-196a were
statistically significantly higher in bladder cancer tissues than
those in non-tumor bladder tissues (Fig. 7A and B), whereas p27kip1
was downregulated in bladder cancer tissues compared with non-tumor
bladder tissues (Fig. 7C).
Furthermore, p27kip1 expression was also detected in
bladder cancer tissues and non-tumor bladder tissues using IHC. As
shown in Fig. 7D,
p27kip1 staining was remarkably decreased in bladder
cancer tissues than that in non-tumor bladder tissues. Whether UCA1
expression was correlated with miR-196a and p27kip1 in
bladder cancer was investigated as well. As expected, UCA1 was
positively correlated with miR-196a in bladder cancer patients
(Fig. 7E), whereas the levels of
UCA1 and miR-196a were negatively correlated with
p27kip1 in bladder cancer, respectively (Fig. 7F and G). These data suggested that
miR-196a was regulated by UCA1 in bladder cancer and implemented
its bio-function via inhibiting p27kip1 expression,
which was also confirmed by the valuable information of miRNA
profiling associated with UCA1 in bladder cancer.
Discussion
Diseases, especially cancer, are often associated
with the dysregulation of gene and aberrant transcriptomes, which
includes the production of abnormal levels of protein-coding mRNAs
and deregulated expression of the noncoding dimension of the human
genome. This is especially surprising since recent findings of the
human transcriptome have revealed that over three quarters of the
genome is transcribed (2,11), including mRNA, small non-coding RNA
and lncRNAs. The non-coding transcriptome and their transcriptions
play an important role in determining the greater complexity of
higher eukaryotes and in disease pathogenesis (12–14).
Especially in cancer, the noncoding transcriptome is often
dysregulated (15).
Over the past decade, many studies focused on the
microRNAs in cancer and have demonstrated that microRNAs are
involved in cancer biology through controlling expression of their
target mRNAs. Thus, microRNAs play an important role in
facilitating tumor growth, invasion, angiogenesis, and even immune
evasion (16,17). Additionally, tumor microRNA
profiles in cancer are explored and the different expression of
microRNAs in tumor is related with subtypes, patient survival and
treatment response (18–20). Importantly, cancer-associated
microRNA biomarkers can be detected in biological fluids, allowing
less-invasive monitoring (21).
Accumulating evidence suggests that lncRNAs play an
important role in tumorigenesis (22) and in the regulation at chromatin
organization, at transcriptional, and post-transcriptional levels
(23). LncRNAs have been
considered as essential regulators in diverse aspects of biology.
LncRNAs can serve as important players in post-transcriptional
regulation, such as mRNA editors, mRNA splicing regulators, and
reservoir of small ncRNAs. For example, lncRNAs can regulate miRNAs
by competing endogenous RNAs (ceRNAs) or natural microRNA sponges
(14). Considering the roles of
lncRNAs and microRNAs in cancer, a better understanding of microRNA
profiling associated with lncRNA in cancer is imperative for the
development of novel therapeutic strategies.
Our previous studies demonstrated that lncRNA and
UCA1 played an important role in cell proliferation, cell cycle,
invasion and metastasis in bladder cancer (24,25).
However, it remains unknown whether microRNAs participate in these
processes regulated by UCA1. Deep sequencing is considered as a
powerful strategy for identifying novel miRNAs and studying the
expression profiles of miRNA in different species at various
developmental stages and in diseased states (26,27).
In the present investigation, we analyzed the microRNA profiling
associated with UCA1 in bladder cancer by deep sequencing.
The miRNAs frequently varied from their miRBase
reference sequences, displaying multiple mature isoforms. Thus, we
compared all of these isoforms in our libraries, finding that 3′
heterogeneity was more common than 5′ heterogeneity in the miRNAs
from deep sequencing. This is consistent with the findings of other
miRNA deep sequencing studies and the observation that most
variability occurs in either the Dicer1 or Drosha cleavage position
within the pre-miRNA hairpin (28–30).
We further analyzed the first nucleotide bias and the each
nucleotide bias in the miRNAs from sequencing. Uridine (U)
dominated the first position and the each nucleotide bias, whereas
guanine (G) was the least favored in this study, which is a
characteristic feature of miRNAs.
According to the deep sequencing results, we found
expression of 75 miRNAs significantly associated with UCA1: 38 were
upregulated and 37 downregulated. To explore the potential
physiological functions of the miRNAs associated with UCA1, we
analyzed the potential targets of the up- and downregulated miRNAs
with four miRNA target prediction software programs, and performed
Gene ontology analysis. GO analysis revealed that 75 significantly
differentially expressed miRNAs participated in a variety of
biological processes potentially important to bladder cancer
progression, including biological process, cellular component and
molecular function.
To confirm the sequencing data, 11 candidate miRNAs
were randomly selected and detected by real-time PCR. On the whole,
the relative expression levels of the selected miRNAs detected with
RT-qPCR were lower than the relative expression levels determined
with deep sequencing. The difference may be due to the difference
in the sensitivity of the methods.
In addition, we constructed a network regulated by 8
candidate miRNAs selected from the above 11 miRNAs, predicted their
transcription factors and targeting mRNAs. In TF-miRNA-mRNA
network, p27kip1 was a crucial gene. Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathway analysis found all these 8
candidate miRNAs participate in PI3K-Akt signaling, suggesting that
PI3K-Akt signaling was responsible for the bio-function of these 8
candidate miRNAs. Indeed, PI3K-Akt signaling plays an important
role in cancer progression by regulating cell cycle, apoptosis and
metastasis. miR-196a was one of these 8 candidate miRNAs.
Previous studies showed that the miR-196a expression
was dysregulated in non-small cell lung carcinoma, colorectal
cancer, and glioblastoma (31–33).
Besides, the high expression of miR-196a promoted esophageal cell
proliferation, anchorage-independent growth and suppressed
apoptosis by directly targeting Annexin A1 (34). Recent study indicated that miR-196a
could enhance gastric cancer cell proliferation by directly binding
and downregulating p27kip1 (35).
In this study, we found that miR-196a expression was
associated with UCA1 in bladder cancer by deep sequencing. Our
results demonstrated that the tissue levels of UCA1 and miR-196a
were statistically significantly higher in bladder cancer patients
than those in non-tumor bladder tissues, whereas p27kip1
expression was remarkably decreased in bladder cancer patients.
Similar results were observed in bladder cancer cells when
transfecting with the plasmids of pCDNA3.1-UCA1 or shUCA1. Further,
UCA1 was positively correlated with miR-196a in bladder cancer
pateints, whereas the levels of UCA1 and miR-196a were negatively
correlated with p27kip1 in bladder cancer, respectively.
p27kip1 was an important downstream molecule of PI3K-Akt
signaling. Taken together, the results of deep sequencing
demonstrated that miRNAs associated with UCA1 play an important
role in bladder cancer progression through PI3K-Akt signaling.
Further studies are still needed to reveal the underlying molecular
mechanism involved in this signaling pathway.
Abbreviations:
|
lncRNA
|
long noncoding RNA
|
|
UCA1
|
urothelial cancer-associated 1
|
References
|
1
|
Volders PJ, Helsens K, Wang X, Menten B,
Martens L, Gevaert K, Vandesompele J and Mestdagh P: LNCipedia: A
database for annotated human lncRNA transcript sequences and
structures. Nucleic Acids Res. 41:D246–D251. 2013. View Article : Google Scholar :
|
|
2
|
ENCODE Project Consortium. An integrated
encyclopedia of DNA elements in the human genome. Nature.
489:57–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:382011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ponting CP, Oliver PL and Reik W:
Evolution and functions of long noncoding RNAs. Cell. 136:629–641.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Song X, Glass CK and Rosenfeld MG:
The long arm of long noncoding RNAs: Roles as sensors regulating
gene transcriptional programs. Cold Spring Harb Perspect Biol.
3:a0037562011. View Article : Google Scholar
|
|
8
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas M, Lieberman J and Lal A:
Desperately seeking microRNA targets. Nat Struct Mol Biol.
17:1169–1174. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Djebali S, Davis CA, Merkel A, Dobin A,
Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F,
et al: Landscape of transcription in human cells. Nature.
489:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mattick JS: Non-coding RNAs: The
architects of eukaryotic complexity. EMBO Rep. 2:986–991. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mattick JS and Gagen MJ: The evolution of
controlled multitasked gene networks: The role of introns and other
noncoding RNAs in the development of complex organisms. Mol Biol
Evol. 18:1611–1630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gutschner T and Diederichs S: The
hallmarks of cancer: A long non-coding RNA point of view. RNA Biol.
9:703–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stahlhut C and Slack FJ: MicroRNAs and the
cancer phenotype: Profiling, signatures and clinical implications.
Genome Med. 5:111–122. 2013. View
Article : Google Scholar
|
|
18
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dvinge H, Git A, Gräf S, Salmon-Divon M,
Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska EA, et al:
The shaping and functional consequences of the microRNA landscape
in breast cancer. Nature. 497:378–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim TM, Huang W, Park R, Park PJ and
Johnson MD: A developmental taxonomy of glioblastoma defined and
maintained by MicroRNAs. Cancer Res. 71:3387–3399. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Manterola L, Guruceaga E, Gállego
Pérez-Larraya J, González-Huarriz M, Jauregui P, Tejada S,
Diez-Valle R, Segura V, Samprón N, Barrena C, et al: A small
noncoding RNA signature found in exosomes of GBM patient serum as a
diagnostic tool. Neuro-oncol. 16:520–527. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsai MC, Spitale RC and Chang HY: Long
intergenic noncoding RNAs: New links in cancer progression. Cancer
Res. 71:3–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang F, Li X, Xie X, Zhao L and Chen W:
UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma
and embryo, influencing cell growth and promoting invasion. FEBS
Lett. 582:1919–1927. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang C, Li X, Wang Y, Zhao L and Chen W:
Long non-coding RNA UCA1 regulated cell cycle distribution via CREB
through PI3-K dependent pathway in bladder carcinoma cells. Gene.
496:8–16. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Metzker ML: Sequencing technologies - the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar
|
|
27
|
Buermans HP, Ariyurek Y, van Ommen G, den
Dunnen JT and ‘t Hoen PA: New methods for next generation
sequencing based microRNA expression profiling. BMC Genomics.
11:716–731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anselmo A, Flori L, Jaffrezic F,
Rutigliano T, Cecere M, Cortes-Perez N, Lefèvre F, Rogel-Gaillard C
and Giuffra E: Co-expression of host and viral microRNAs in porcine
dendritic cells infected by the pseudorabies virus. PLoS One.
6:e173742011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stark MS, Tyagi S, Nancarrow DJ, Boyle GM,
Cook AL, Whiteman DC, Parsons PG, Schmidt C, Sturm RA and Hayward
NK: Characterization of the melanoma miRNAome by deep sequencing.
PLoS One. 5:e96852010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li M, Liu Y, Wang T, Guan J, Luo Z, Chen
H, Wang X, Chen L, Ma J, Mu Z, et al: Repertoire of porcine
microRNAs in adult ovary and testis by deep sequencing. Int J Biol
Sci. 7:1045–1055. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang R, Wang ZX, Yang JS, Pan X, De W and
Chen LB: MicroRNA-451 functions as a tumor suppressor in human
non-small cell lung cancer by targeting ras-related protein 14
(RAB14). Oncogene. 30:2644–2658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guan Y, Mizoguchi M, Yoshimoto K, Hata N,
Shono T, Suzuki SO, Araki Y, Kuga D, Nakamizo A, Amano T, et al:
MiRNA-196 is upregulated in glioblastoma but not in anaplastic
astrocytoma and has prognostic significance. Clin Cancer Res.
16:4289–4297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schimanski CC, Frerichs K, Rahman F,
Berger M, Lang H, Galle PR, Moehler M and Gockel I: High miR-196a
levels promote the oncogenic phenotype of colorectal cancer cells.
World J Gastroenterol. 15:2089–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Luthra R, Singh RR, Luthra MG, Li YX,
Hannah C, Romans AM, Barkoh BA, Chen SS, Ensor J, Maru DM, et al:
MicroRNA-196a targets annexin A1: A microRNA-mediated mechanism of
annexin A1 downregulation in cancers. Oncogene. 27:6667–6678. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun M, Liu XH, Li JH, Yang JS, Zhang EB,
Yin DD, Liu ZL, Zhou J, Ding Y, Li SQ, et al: MiR-196a is
upregulated in gastric cancer and promotes cell proliferation by
downregulating p27(kip1). Mol Cancer Ther. 11:842–852. 2012.
View Article : Google Scholar : PubMed/NCBI
|