Introduction
Non-small cell lung cancer (NSCLC) is the most
common type of lung cancer, which accounts for >80% of all lung
cancer cases. The 5-year overall survival rate remains <15% in
patients with NSCLC, despite advances in integrative therapies,
including surgery, chemotherapy and radiotherapy (1). The administration in a clinical
setting of epidermal growth factor receptor (EGFR) tyrosine kinase
inhibitors (TKIs), such as gefitinib and erlotinib, have
significantly improved the overall survival of cancer patients that
harbor somatic mutations in the EGFR gene, such as an in-frame
deletion mutation in exon 19 (2).
Nevertheless, most tumors develop acquired resistance to EGFR-TKIs
after a period of time (a median of 6–12 months) (3) in which the tumors exhibit a dramatic
response to gefitinib and erlotinib. Acquired resistance to
EGFR-TKIs is usually inevitable, leading to a limitation in
treatment efficacy. However, the mechanisms underlying acquired
resistance to EGFR-TKIs have not been fully understood.
Since acquired drug resistance is a major problem in
cancer treatment, different mechanisms have been proposed for
developing novel treatment strategies, one example being the
progress in the targeting of lung cancer stem cells (CSCs) in
recent years. Eramo et al reported (4) isolation and identification of a CSC
population that showed extensive drug resistance from tumor
specimens of patients with lung cancer. Another study found that
the stem cell factor (SCF) and its receptor c-kit (CD117) were
expressed to relative degrees in CSCs. The signal transduction
pathways of phosphatidylinositol 3-kinase (PI3K) are involved in
SCF/c-kit (CD117) activation. Therefore, the proliferation of CSCs
can be inhibited by receptor TKIs (5). However, the markers of CSCs are still
controversial. A large number of studies have shown that the cell
population of CD133+ has more characteristics of CSCs
than that of CD133− (4,6). CD133 is currently recognized
as a well-known marker for CSCs. This marker has been widely used
in the isolation and purification of CSCs.
Furthermore, evidence has recently shown that
microRNAs (miRNAs) also regulate certain genes associated with
resistance to chemotherapy and EGFR-TKIs (7–9).
Among miRNAs related to drug resistance, miRNA-223 (miR-223) was
reported to regulate multiple cellular functions via PI3K/Akt
signaling pathways in most literature. Our previous studies also
showed that miR-223 expression is reduced in a Lewis lung carcinoma
cell line and that insulin-like growth factor 1 receptor (IGF1R)
served as a target gene of miR-223. The expression of IGF1R and the
activity of Akt, its downstream target, were decreased, while
miR-223 was overexpressed, indicating that miR-223 inhibited the
invasion and metastasis of Lewis lung carcinoma cells by targeting
IGF1R-Akt pathway (10). Because
of the Akt activity regulated by P13K, the aberrant activation of
IGF1R/P13K/Akt signaling pathway may be the mechanism underlying
resistance to EGFR-TKIs. Although several studies showed that IGF1R
is implicated in the resistance to chemotherapy, including the
targeted therapies, such as EGFR-TKIs (11,12),
the correlation between miR-223 and the IGF1R/P13K/Akt pathway in
the resistance of EGFR-TKIs has yet to be determined.
In this study, we developed an EGFR-TKI-resistant
PC9/ER cell line, in which the percentage of CD133+
cells was so high that isolation of stem cells from
CD133+ (PC9/CD133+ cells) was performed. Our
study revealed that CD133+ was resistant to erlotinib.
The expression of miR-223 in ER and CD133+ cells was
downregulated, compared to their parent cells. IGF1R was also
verified as a target gene of miR-223 in our study. According to
these findings, we hypothesized that downregulation of miR-223
expression may induce the activation of the IGF1R/PI3K/Akt
signaling pathway, leading to erlotinib resistance. Here, we
provide evidence to verify our hypothesis.
Materials and methods
Cells and reagents
The human lung cancer HCC827 cell line was purchased
from ATCC (ATCC® CRL-2868™). The PC9 cell line, which
was derived from a human adenocarcinoma of lung tissue, was
preserved in our laboratory. The lung cancer cells were cultured in
RPMI-1640 medium containing 10% fetal bovine serum (Gibco BRL,
Carlsbad, CA, USA) and 100 U/ml penicillin/streptomycin at 37°C in
a humidified incubator containing 5% CO2. Erlotinib
(OSI-744) was purchased from Selleck Chemicals (Houston, TX, USA).
Insulin-like growth factor 1 human recombinant was obtained from
ProSpec (ProSpec, Rehovot, Israel). Two erlotinib-resistant lines,
namely HCC827/ER and PC9/ER, were developed by applying high-dose
(1–5 μM) pulses of erlotinib combined with continuous low-dose
(0.01 μM) administration for >8 months (13). To avoid the effects of the drugs,
resistant cell lines were cultured in a drug-free medium for ≥2
weeks prior to further experiments.
Isolation of CD133+ cells from
the PC9 cell line with paclitaxel treatment
Approximately 106/ml PC9 cells were
suspended in F12 serum-free medium (Hyclone, USA) supplement with
0.4% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA),
insulin 5 g/ml (Sigma-Aldrich), human recombinant epidermal growth
factor, 20 ng/ml (PeproTech, Rehovot, Israel) and basic fibroblast
growth factor, 10 ng/ml (PeproTech). When spheroids emerged, cells
were treated for 48 h with paclitaxel injection (Powerdone, China)
at a concentration of 100 nmol/l. The culture medium was replaced
with fresh complete medium twice per week until new spheroids
emerged. To isolate CD133+ cells, spheroids were
dissociated into single cells, washed in phosphate-buffered saline
(PBS) 3 times and incubated with PE-conjugated monoclonal antibody
against human CD133/1 (Miltenyi Biotec), according to the
manufacturer's instructions. After incubation for 30 min at 4°C,
cells were washed in PBS twice and CD133+ cells were
sorted by flow cytometry (BD Biosciences).
Construction of stable cell lines with
overexpressed miR-223
To stably upregulate miR-223 expression in PC9/ER
cells or PC9/CD133+, lentivirus carrying the miR-223 or
negative control vector (empty viral vector; EV) was packaged using
the lentiviral packaging kit (Shanghai Genechem Co., Ltd.,
Shanghai, China), according to the manufacturer's instructions. The
green fluorescent protein (GFP) gene was inserted into the
packaging system, which allows co-expression of GFP with miR-223.
To establish stable cell lines, PC9/ER and PC9/CD133+
cells were infected with lentivirus carrying miR-223 or EV in the
presence of polybrene (Sigma-Aldrich).
Luciferase reporter assay and related
plasmids
The plasmids of firefly luciferase reporter,
IGF1R-WT (wild-type of miR-223 binding site in 3′-UTR of IGF1R) and
IGF1R-MU (mutated miR-223 binding site in 3′-UTR of IGF1R) were
constructed by Genechem (Shanghai Genechem Co., Ltd., Shanghai,
China). The miR-223 mimic and the mimic negative control were
obtained from Ribobio (Guangzhou RiboBio Co., Ltd., Guangzhou,
China). HEK293 cells were co-transfected with firefly luciferase
reporter (0.05 μg) and IGF1R plasmid (0.05 μg), as well as 0.01 μg
Renilla luciferase control vector using calcium phosphate
transfection. Luciferase activity was measured 36 h after
transfection. To obtain data, Renilla luciferase activity was
normalized to firefly luciferase expression, according to the
manufacturer's instructions (Dual-Luciferase Reporter Assay System;
Promega Corp., Madison, WI, USA).
Cell proliferation assay
Cells that reached mid-log phase growth were spilt
and plated into 96-well plates at different densities: 5,000
cells/well (in triplicate) for HCC827 and HCC827/ER cells, but
3,000 cells/well (in triplicate) for PC9, PC9/ER, PC9/ER-miR-223,
PC9/ER-EV, PC9/CD133+, PC9/CD133+-miR-223,
and PC9/CD133+-EV cells. Then cells were cultured for 48
h in complete medium containing increasing concentrations of 0,
0.001, 0.01, 0.1, 1, 5 or 10 μM erlotinib. The number of viable
cells was quantified using a Cell Counting Kit-8 (CCK-8) (Dojindo
Laboratories, Kumamoto, Japan). A total of 10 μl CCK-8 solution was
added to each well at the end of treatment, followed by another 2-h
incubation. Optical density values at 450 nm were measured using a
microplate reader. Each experiment was independently performed 3
times.
Western blot analysis
Cells were harvested from the experiments by
transfection and treatment with inhibitor, washed twice with cold
PBS and lysed in RIPA lysis buffer containing protease inhibitors.
The protein concentration of the lysate was measured. Lysate with a
loading buffer was denatured, and then separated by SDS-PAGE, and
then electrotransferred to a polyvinylidene difluoride membrane.
The membranes were blocked at room temperature for 1 h in
Tris-buffered saline and Tween-20 (TBST) containing 5% (w/v) BSA
(Wuhan Boster Bio-Engineering Co., Ltd., Wuhan, China), before
being incubated with the following antibodies: EGFR, p-EGFR, IGF1R,
p-IGF1R (1:500, Cell Signaling Technology, Beverly, MA, USA),
P70S6K (P70S6 kinase), 70S6K, Akt, p-Akt (Ser473) (1:1,000,
Signalway Antibody, Boston, MA, USA), and PTEN (1:500, Wuhan Boster
Bio-Engineering Co., Ltd.). After being washed, the membranes were
incubated with secondary antibodies conjugated to horseradish
peroxidase (Wuhan Boster Bio-Engineering Co., Ltd.). Protein bands
were visualized using an enhanced chemiluminescence detection kit
(Pierce Biotechnology, Inc., Rockford, IL, USA). β-actin (Wuhan
Boster Bio-Engineering Co., Ltd.) was used as an internal
control.
Real-time RT-PCR
Total RNA was extracted from the tissues and
harvested cells using RNAiso Reagent Plus (Takara Biotechnology,
Dalian, China). Reverse transcription reactions were performed
using an RT kit from Takara, according to the manufacturer's
protocol. The relative expression level of miR-223 was normalized
to that of U6 expression, but the expression levels of other
protein coding genes was normalized to that of β-actin. The primer
sets used in quantitative real-time polymerase chain reaction
(qRT-PCR) for hsa-miR-223-3p Primer Set and U6 small nuclear
ribonucleic acid (snRNA) were purchased from Guangzhou RiboBio
Bio-Technique Co., Ltd. The sequences of other primers used in this
experiment were as follows: IGF1R: forward,
5′-GGACAGGTCAGAGGGTTTC-3; and reverse, 5′-CTCGTAACTCTTCTCTGTGCC-3′.
β-actin: forward, 5′-GAGCTACGAGCTGCCTGACG-3′; and reverse,
5′-CCTAGAAGCATTTGCGGTGG-3′ (10);
PTEN: forward, 5′-ACCCCTTCATTGACCTCAACTA-3′; and reverse,
5′-TCTCGCTCCTGGAAGATGGTGA-3′; C-Met: forward,
5′-TCATTGGTTCCAATCACAGCTCA-3′, reverse,
5′-GCCACCGAGACAGAGGCTAATC-3′. Real-time RT-PCR was performed using
the ABI7500 Sequence Detection System. All reactions were performed
in triplicate, and all experiments were conducted 3 times
independently.
Xenografts
Male BALB/c nude mice (SPF, 6–8-week-old), obtained
from the Animal Research Center of Xinqiao Hospital of The Third
Military Medical University in China (Chongqing, China), were
housed in groups of 5 per cage in a temperature (30±3°C) and
humidity (55±5%) controlled room. All mice were given water and
chow ad libitum at all times. Our animal study was approved
by the Institutional Animal Care and Use Committee of Xinqiao
Hospital of The Third Military Medical University. The experimental
mice were randomized into four groups (5 mice/group). For the
tumorigenesis assay, mice were subcutaneously injected with a total
of 5×106 cells of PC9, or PC9/ER, or PC9/ER transfected
with control lentivirus (PC9/ER-EV) or PC9/ER transfected with the
miR-223-overexpressing lentivirus (PC9/ER-miR-223), in the right
front leg. The tumor volume (V) was calculated according to the
formula V = (length × width2)/2. When the tumors reached
a mean volume of ~100 mm3 (14), the mice (n=5) began to receive the
erlotinib treatment (30 mg/kg/d, via gavage for two weeks)
(15). To test the tumor formation
ability of PC9/CD133+ cells, nude mice were randomized
into three groups (3 mice per group). A total of 5×104
PC9/CD133+ or PC9 cells as well as 5×106 PC9
cells were subcutaneously injected into nude mice (6–8-week-old)
under the front left legs for the tumorigenesis assay. Nude mice
were euthanized at the experimental end-point (3 weeks after
inoculation of tumor cells).
Statistical analysis
Data analysis was performed using SPSS 17.0 software
(SPSS, Inc., Chicago, IL, USA). All assays were repeated three
times, and the results are expressed as the means ± SD. The
statistical significance of the results between each group was
determined using one-way ANOVA. P<0.05 was considered to be
significant.
Results
Establishment of erlotinib-resistant PC9
and HCC827 cells
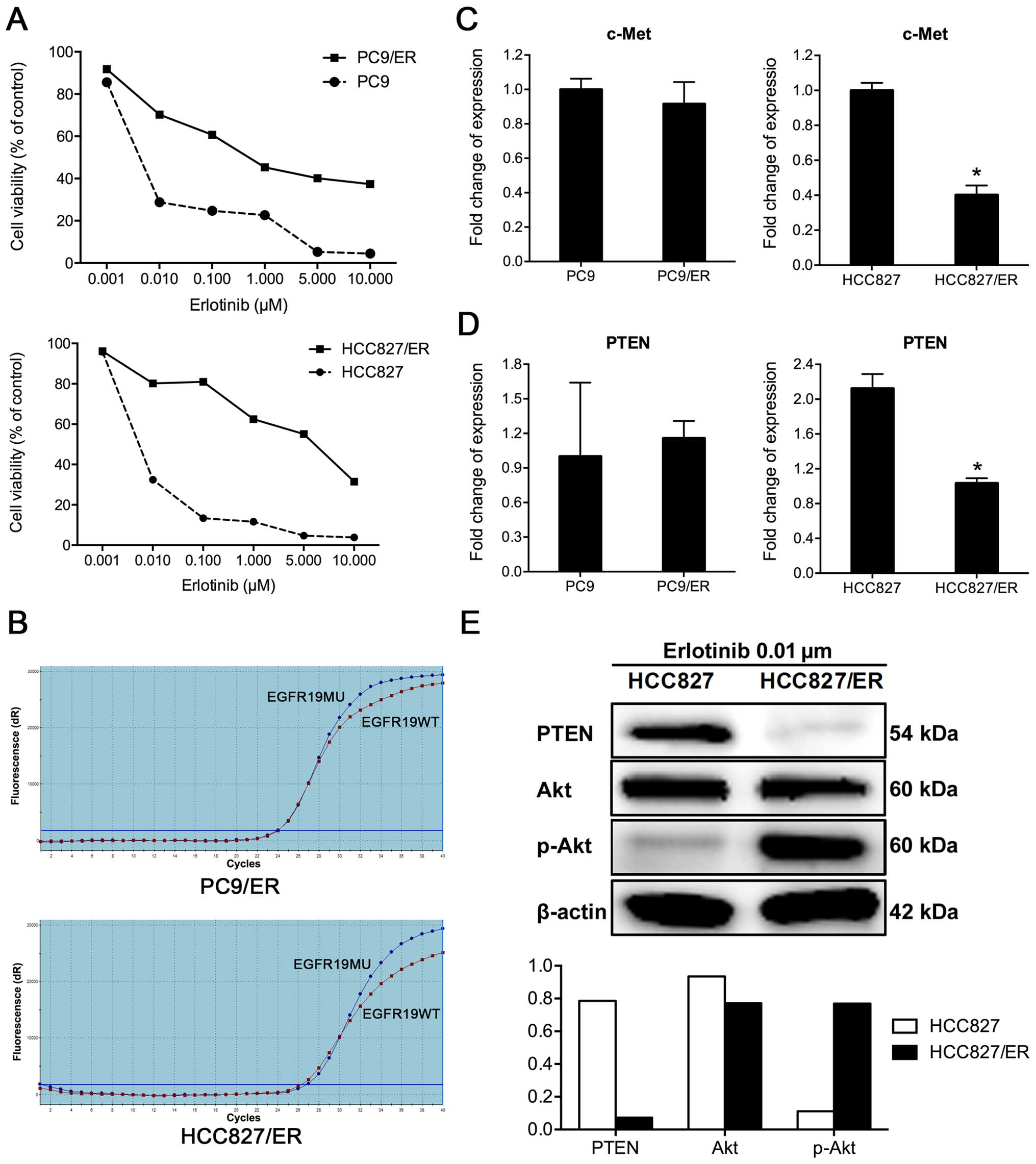
Two cell lines resistant to EGFR-TKIs, namely PC9/ER
and HCC827/ER, were derived from their parental cells through the
treatment with high-dose (1–5 μM) pulses of erlotinib, combined
with continuous low-dose (0.01 μM) for >8 months. Resistance in
the treated cell lines was determined via CCK8 assay, according to
the effects of increasing concentrations of erlotinib on cell
viability. The half maximal inhibitory concentration
(IC50) values for erlotinib were ~37.4 times higher in
PC9/ER cells and 155.4-fold higher in HCC827/ER cells, than in the
corresponding parental cells (Fig.
1A). To check the characteristics of those ER resistant cells,
several tests were performed, in which the secondary T790M EGFR
mutation was not detected in PC9/ER or HCC827/ER cells in the
amplification refractory mutation system (ARMS) assay (Fig. 1B). Amplification of the c-Met gene
was not found in either PC9/ER or HCC827/ER cells, based on the
results of real-time RT-PCR (Fig.
1C). PTEN mRNA expression was decreased by 50% in HCC827/ER
cells, compared to its parental HCC827 cells. The expression level
of PTEN protein was also downregulated in HCC827/ER cells (Fig. 1E). The downregulation of PTEN may
contribute to secondary resistance in HCC827/ER cells. However, no
significant differences in PTEN expression were observed between
PC9 and PC9/ER cells (Fig.
1D).
Activation of IGF1R/PI3K/Akt pathway in
PC9/ER cells
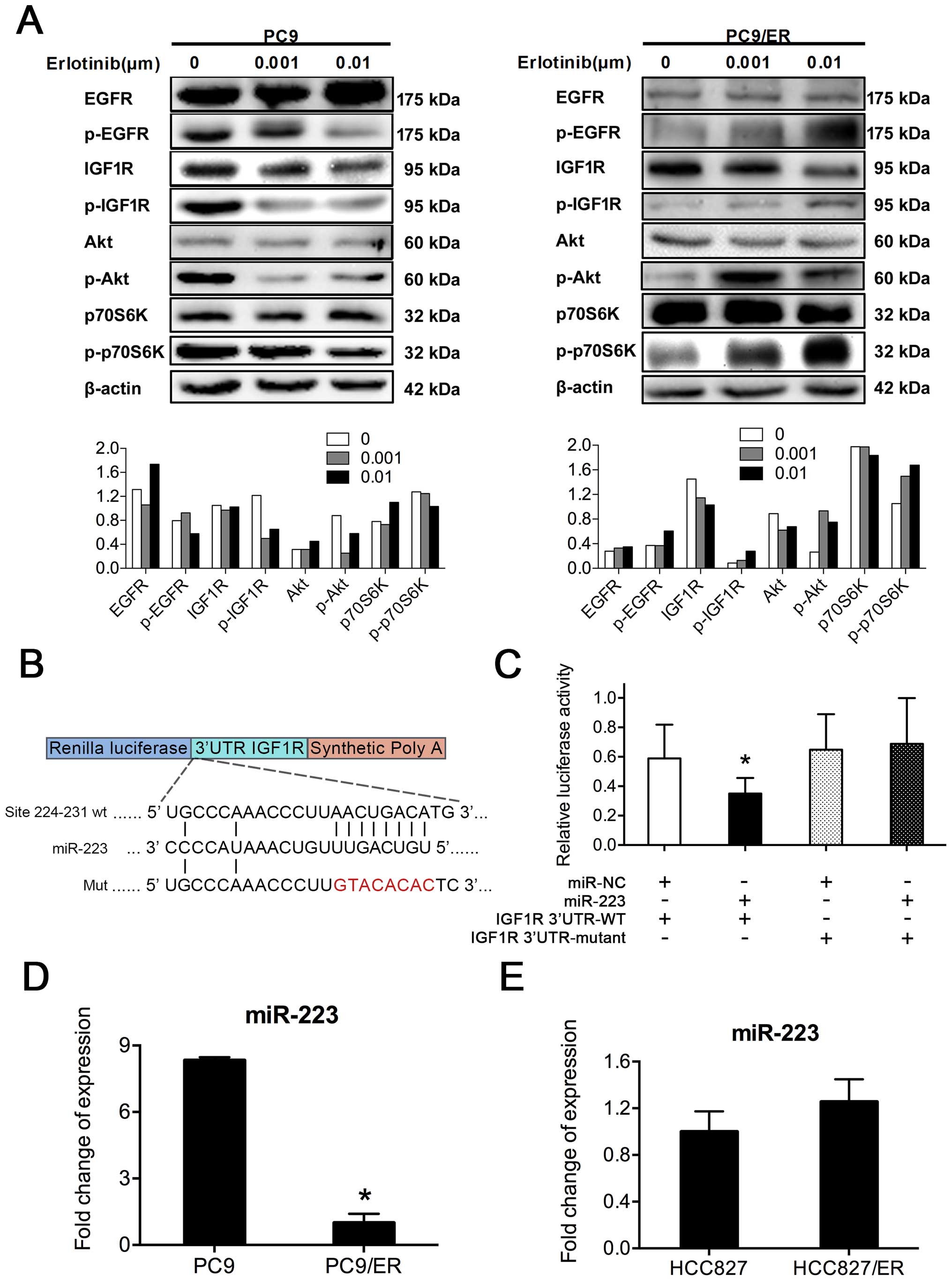
Our study revealed that the acquired resistance of
erlotinib in PC9/ER cells was not associated with any known
mechanisms related to erlotinib resistance, such as the T790M EGFR
mutation, the amplification of the proto-oncogene, receptor
tyrosine kinase MET oncogene, or the downregulation of PTEN. As
some reports showed that the activation of IGF1R is linked to
acquired resistance to EGFR-TKIs (16), we investigated the role of IGF1R in
the development of acquired erlotinib resistance in PC9/ER cells.
Our results showed that when PC9 and PC9/ER cells were treated with
erlotinib at doses of 0, 0.001, and 0.01 μM for 72 h (17), the phosphorylation of EGFR in the
parent cells was inhibited by erlotinib, but it was not affected in
PC9/ER cells (Fig. 2A). Moreover,
downstream molecules of the IGF1R/Akt signaling pathway were
persistently activated in PC9/ER cells. Therefore, the activation
of IGF1R/PI3K/Akt signaling cascades was involved in the acquired
erlotinib resistance of PC9/ER cells. According to this
observation, we considered IGF1R as a direct target of miR-223. To
verify this speculation, HEK293 cells were transfected with
miR-223, mimic control, the wild form 3′-UTR of IGF1R gene
(3′-UTR-WT), and its mutant form (3′-UTR-MU). The results of
luciferase reporter assay showed that the luciferase activity in
the group co-transfected with miR-223 and IGF1R-3′-UTR-WT was
significantly lower than that in the group with miR-NC and
IGF1R3′-UTR-MU (P<0.05), implying the inhibitory effect of
miR-223 on 3′-UTR of the IGF1R gene, which might be caused by the
binding of miR-223 to the 3′-UTR of IGF1R. Thus, IGF1R was
identified as a direct target gene of miR-223 (Fig. 2B and C). To explore the
relationship between miR-223 expression and erlotinib resistance,
we also examined the expression level of miR-223 in both PC9/ER and
HCC827/ER as well as in their parental cells. The analysis of data
from real-time RT-PCR revealed that miR-223 level was decreased by
88% in PC9/ER cells, compared to that in PC9 cells (Fig. 2D). However, no significant
difference in the expression levels of miR-223 was observed between
HCC827 and HCC827/ER cells (Fig.
2E). Thus, we speculated that the downregulation of miR-223
expression may be involved in the acquired erlotinib resistance in
PC9/ER cells.
Overexpression of miR-223 in PC9/ER
cells
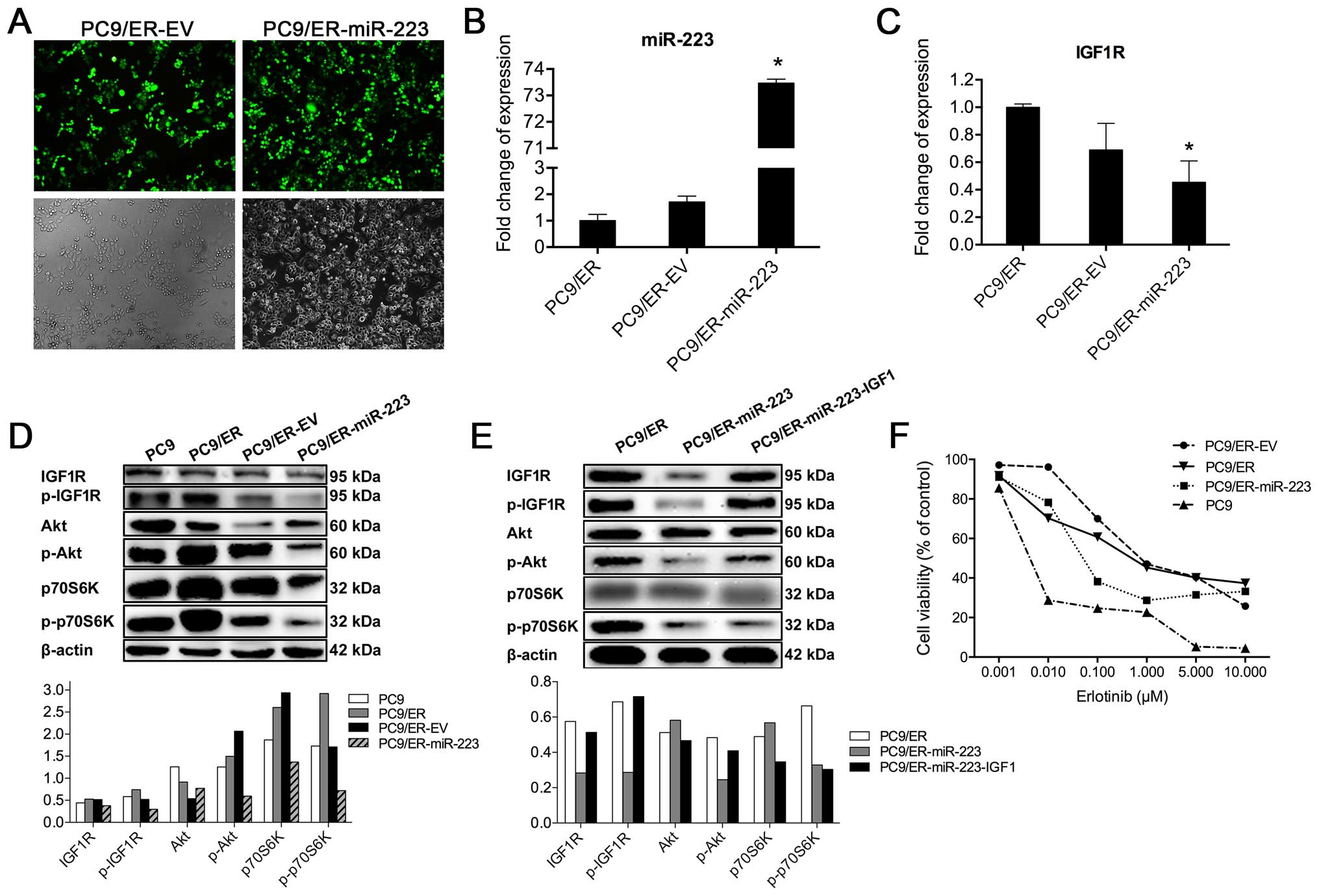
To evaluate the role of miR-223 in the development
of acquired erlotinib resistance in PC9/ER cells, we introduced
miR-223 mimic into this cell line. Because the lentiviral vector is
a potent experimental tool for inducing the stable gain- and
loss-of-function phenotypes caused by individual miRNAs, PC9/ER
cells were infected with the lentiviral vector GV259 carrying
hsa-miR-223. The infection efficiency, which can be estimated by
GFP signals of the GV259 vector, was ~85% in the infected PC9/ER
cells with either EV or miR-223 (Fig.
3A). While the levels of miR-223 were increased by 73.5-fold in
PC9/ER-miR-223 cells, compared to those in PC9/ER-EV cells,
according to the results of real-time RT-PCR analysis (Fig. 3B). In our further experiments,
PC9/ER-miR-223 was used as a stable cell line with overexpressed
miR-223.
Inhibition of IGF1R/PI3K/Akt signaling
pathway by overexpression of miR-223
To evaluate the effect of miR-223 on IGF1R
expression, we examined the levels of IGF1R mRNA in PC9/ER-miR-223
cells via real-time RT-PCR. Results showed that IGF1R mRNA
expression was reduced by 50% in PC9/ER-miR-223 cells, compared to
PC9/ER cells (Fig. 3C). The
overexpression of miR-223 also inhibited either the expression or
the phosphorylation of IGF1R protein. It is well known that Akt is
an essential protein kinase in the PI3K/Akt signaling pathway and
it is also an important downstream molecule of IGF1R. Therefore,
next, we determined the effect of miR-223 on Akt expression and its
phosphorylation. We found that phosphorylation of Akt was reduced
in PC9/ER-miR-223 cells. We further examined the expression of
p70S6K, a key protein kinase in the mechanistic target of rapamycin
(mTOR) signaling pathway, and its phosphorylation in PC9/ER-miR-223
cells. As shown in Fig. 3D, the
phosphorylation levels of p70S6K was greatly reduced in
PC9/ER-miR-223 cells, but significant changes in total p70S6K
protein were not observed. Furthermore, our study also found that
miR-223 mediated inhibition of the phosphorylation of either
insulin-like growth factor 1 receptor (IGF1R) and its
phosphorylation form (p-IGF1R) or Akt (p-Akt), except for 70S6K,
which can be abolished by treatment with IGF1, an IGF1R agonist at
dose of 10 ng/ml for 15 min (Fig.
3E). This result indicates that inactivation of
IGF1R/PI3K/Akt/mTOR signaling pathway by miR-223 might be due to
miR-223-mediated downregulation of IGF1R expression at both the
mRNA and protein levels. Moreover, the cell viability of PC9,
PC9/ER, PC9/ER-EV, and PC9/ER-miR-223 was compared, according to
the results of CCK8 assay. We found that after erlotinib treatment,
the cell viability of PC9/ER-miR-223 was lower than that of PC9/ER
and PC9/ER-EV cells, but higher than that of PC9 cells (Fig. 3F), indicating that the blockade of
the IGF1R/PI3K/Akt/mTOR signaling pathway is able to partially
restore the sensitivity of the erlotinib-resistant cell lines. This
partial reversion suggested that the activation of PI3K is not the
sole mechanism for the resistance to EGFR-TKIs.
miR-223 partially reversed the acquired
resistance to erlotinib by inhibiting the IGF1R/PI3K/Akt pathway in
vivo
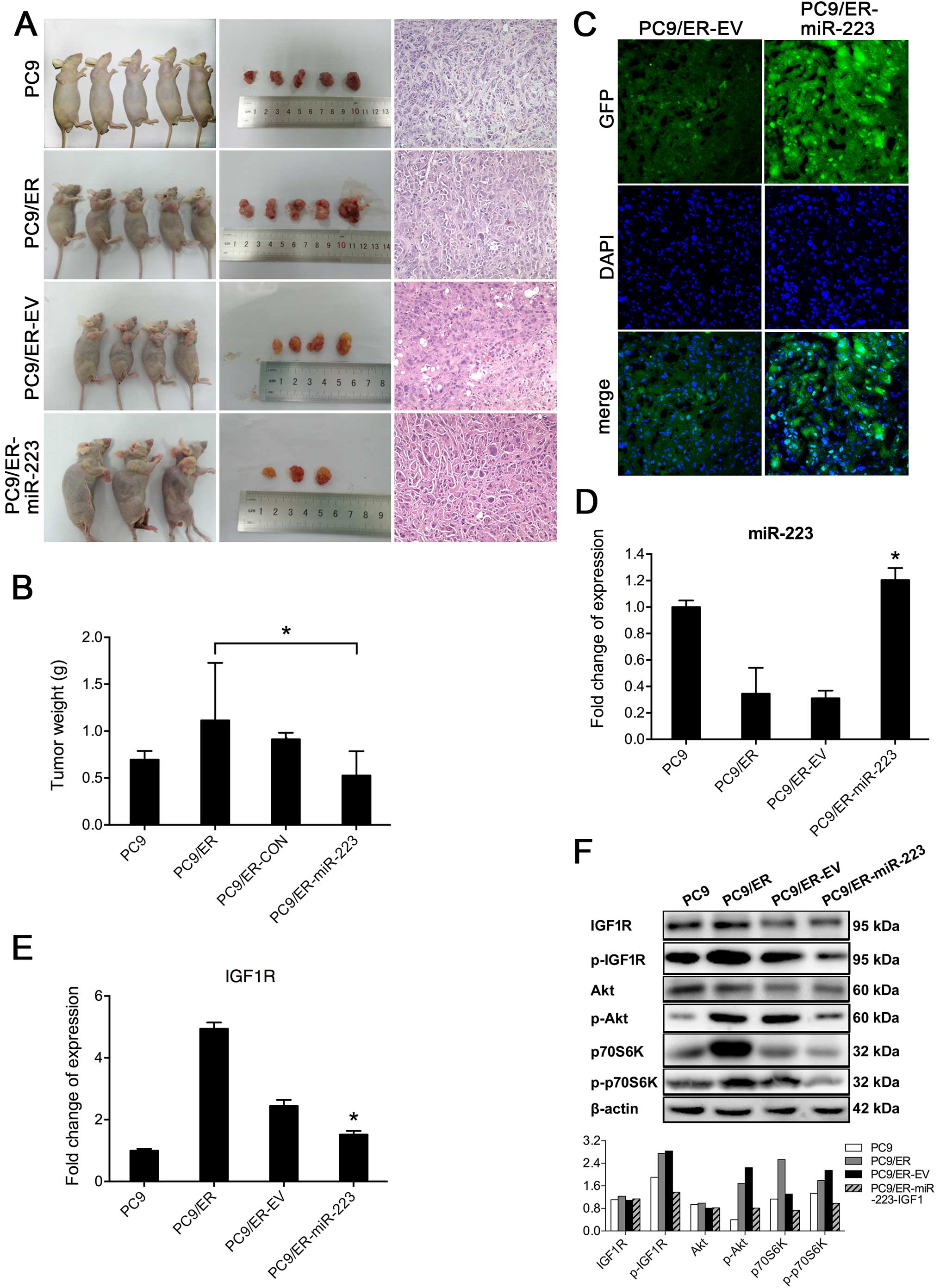
To verify the effect of miR-223 on the acquired
resistance to erlotinib in vivo, a total of 5×106
PC9, PC9/ER, PC9/ER-EV or PC9/ER-miR-223 cells were subcutaneously
injected into the front left legs of nude mice. The tumors appeared
to be viable within one week after injection. When tumor volume
reached 100 mm3 (14),
nude mice bearing the xenograft tumors were treated with erlotinib
(30 mg/kg/d) (15) for two weeks.
Then all mice were euthanized, except for those that had died
early. Tumors were removed and weighed. Results showed that the
average weight of tumors in the group with PC9/ER-miR-223 cells was
0.53±0.26 g, which was significantly less than that of the mice
injected with PC9/ER cells (1.11±0.61 g) (Fig. 4A and B). These results implied that
the transfection of PC9/ER cells with miR-223 could partially
restore the sensitivity of these cells to erlotinib. The histology
of tumor xenografts was evaluated, using hematoxylin and eosin
(H&E) staining (Fig. 4A) and
confocal laser-scanning microscopy. Both PC9/ER-miR-223 and
PC9/ER-EV cells, which were visualized upon signals of GFP, can be
clearly detected in frozen tumor sections via confocal laser
scanning microscopy (Fig. 4C). The
expression of miR-223 and IGF1R was determined via real-time
RT-PCR. The expression levels of miR-223 in tumor xenografts from
mice inoculated with PC9/ER-miR-223 cells were significantly higher
than those from mice with PC9/ER and PC9/ER-EV cells (Fig. 4D). The expression levels of IGF1R
mRNA in tumor xenografts from mice with PC9/ER-miR-223 were lower
than those of mice with PC9/ER and PC9/ER-EV (Fig. 4E). These in vivo results
were consistent with the results of in vitro
experiments.
To investigate whether the inhibition of IGF1R can
induce the inactivation of PI3K/Akt signaling pathway in
vivo, we examined the activity of key molecules of
IGF1R/PI3K/Akt signaling pathway in tumor tissues via western blot
analysis. Our results showed that compared to the levels of
p-IGF1R, p-Akt, and 70S6K in tumors derived from PC9/ER, the
activity/phosphorylation of those molecules was inhibited in tumors
derived from PC9/ER-miR-223 cells, but not in PC9/ER-EV derived
tumors. The levels of p-IGF1R and p-Akt were decreased, and 70S6K
was marginally reduced in the tumors derived from PC9 or PC9/ER
cells after the treatment with erlotinib (Fig. 4F). Our results indicated that
miR-223, rather than erlotinib, inhibits the activation of p70S6K,
which is the downstream molecule of the PI3K/Akt pathway.
Furthermore, miR-223 may partially reverse the resistance of PC9/ER
cells to erlotinib by inhibiting the IGF1R/PI3K/Akt signaling
pathway.
Detection of CD133+ population
in cell lines of PC9ER and PC9
Since the CD133+ subpopulation with
cancer stem cell characteristics is associated with
chemoresistance, we measured the percentage of CD133+
cells in the cell lines of PC9/ER and PC9 by flow cytometry
analysis. In PC9/ER, the percentage of CD133+ cells was
18.56% (Fig. 5A-b) but was only
3.74% in the PC9 cell line (Fig.
5A-a). The ratio of CD133+ cells (%) between these
two cell lines was 4.96, with a statistically significant
difference.
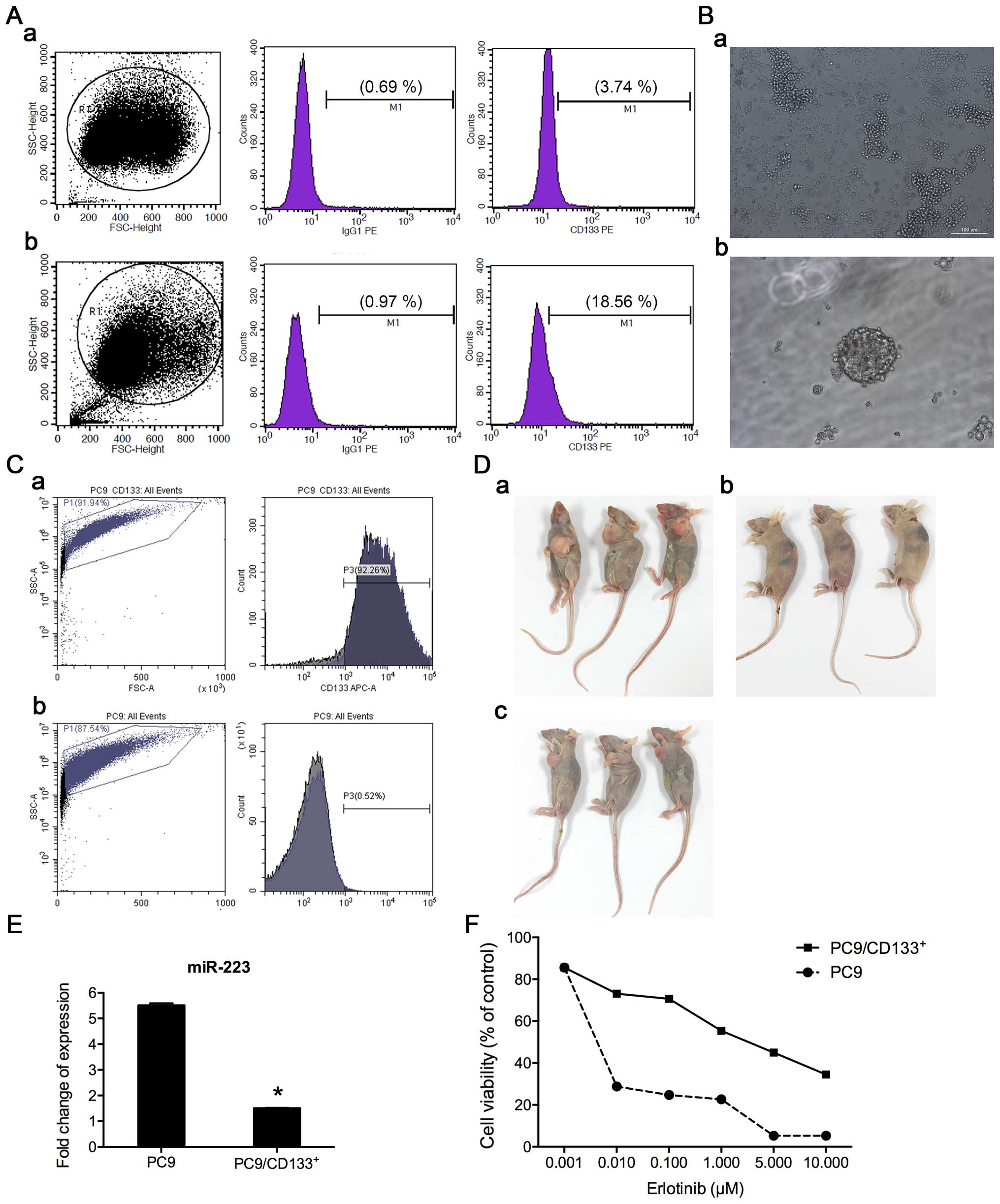 | Figure 5Isolation and expansion of
CD133+ subpopulation for drug sensitivity testing. (A)
Flow cytometry analysis of the percentage of CD133+
cells in PC9 (a) and PC9/ER (b) cell lines. (B-a) The morphology of
the cells cultured in medium for stem cells, prior to paclitaxel
treatment. Most were adherent cells; few were spheroids cells, with
magnification, ×100; (b) after paclitaxel treatment, most adherent
cells died, and surviving cells regrew as spheroids, approximately
two weeks after treatment, with magnification, ×200. (C) Enrichment
of CD133+ subpopulation by paclitaxel treatment, which
was confirmed by flow cytometry analysis (92.26% CD133+
cells in second generation of PC9 spheroids cells). (D) Ability of
tumor formation in vivo between PC9/CD133+
5×104 cells (a) and PC9 5×106 cells (c),
while no tumor formed by injection of 5×104 PC9 cells
(b). (E) Examination of miR-223 expression in PC9 and
PC9/CD133+ cells by real-time RT-PCR. Level of miR-223
was decreased by 72.6% in PC9/CD133+ cells, compared to
that in PC9 cells. (F) Cytotoxicity and IC50 values of
erlotinib in PC9 and PC9/CD133+ cells. Cells were
treated with indicated concentrations of erlotinib for 72 h in
medium containing 1% FBS. Cell viability and IC50 values
were determined using the CCK8 assay. |
Inverse-induction with paclitaxel to
enrich CD133+ subpopulation from PC9 cell line
In this study, we combined paclitaxel with a
serum-free medium culture (inverse induction) to enrich
CD133+ subpopulation from PC9 cells. Briefly, PC9 cells
were resuspended in a serum-free medium and grown into spheres
(Fig. 5B-a). At the second
passage, paclitaxel (100 nmol/l) was added to the culture. The
medium was replaced with fresh medium 48 h later. After paclitaxel
treatment, most of the treated cells died. Approximately two weeks
after treatment, the surviving cells were able to gradually regrow
into spheres (Fig. 5B-b).
CD133+ cells in spheroids were then measured by flow
cytometry. We found that the expression of CD133+ was
~90% in the fourth generation of spheroids after inverse induction
combined with paclitaxel treatment, suggesting that the surviving
cell subpopulation after the inverse induction with paclitaxel
treatment might highly express the CD133+ marker
(Fig. 5C).
To further verify whether the enriched
CD133+ cell subpopulation represents cancer stem cells
(CSCs), the ability of tumor formation of these cells in
vivo was examined. In vivo subcutaneous xenografts on
nude mice showed that as few as a total of 5×104
PC9/CD133+ cells were able to generate a tumor the size
of ~2 mm in diameter, 5 days after inoculation, while an
inoculation of 5×104 PC9 cells did not form a tumor,
observed after 2 weeks. It took about one week to see a visible
tumor after an inoculation of 5×106 PC9 cells into nude
mice. The period of the time for observation in our study was 3
weeks, after which all mice were euthanized (Fig. 5D). Our animal data demonstrated
that PC9/CD133+ cells have features of stemness with a
stronger capability of tumorigenesis than PC9 cells.
Determination of miR-223 expression in
PC9/CD133+ and PC9 cells as well as their erlotinib
resistance
The expression level of miR-223 in
PC9/CD133+ cells sorted by flow cytometry was determined
using real-time RT-PCR. Results showed that miR-223 was
downregulated by 72.6% in PC9/CD133+ cells, compared to
that in PC9 cells (Fig. 5E). The
dose-dependent effect of erlotinib on cell viability was determined
via the CCK8 assay. The IC50 value of erlotinib in
PC9/CD133+ cells was 69.90-fold higher than that in PC9
cells (Fig. 5F), implying that
PC9/CD133+ cells are more resistant than PC9 cells to
erlotinib.
Effects of miR-223 on IGF1R/PI3K/Akt
signaling pathway in PC9/CD133+ cells
To make a stably transfected cell line with miR-223,
PC9/CD133+ cells, which were sorted by flow cytometry
were grown in the culture medium of stem cells for 2 days. Then
cells with good viability were selected for transfection of miR-223
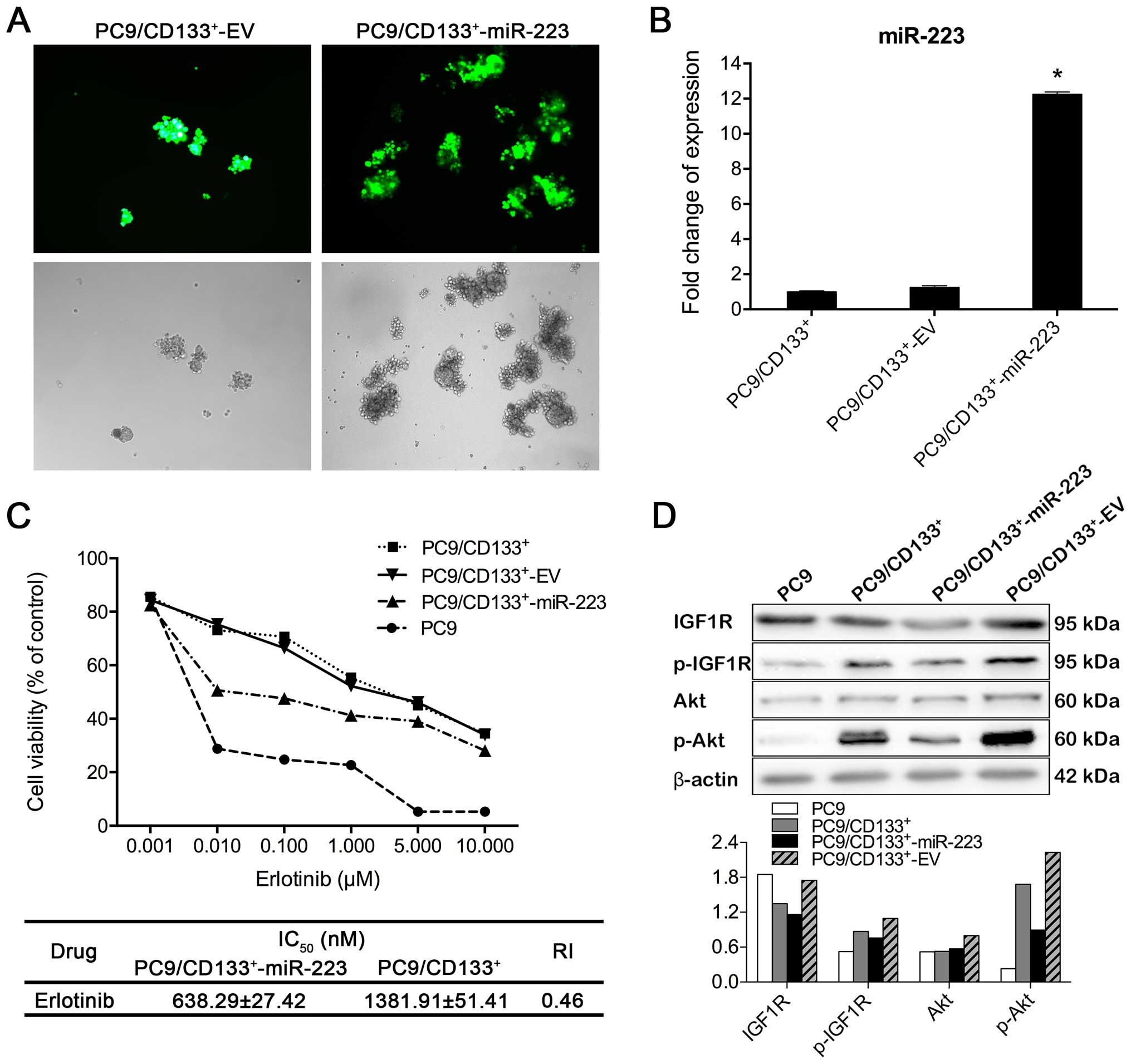
or control EV (Fig. 6A).
Transfection efficacy was confirmed using real-time RT-PCR. PCR
data showed that miR-223 expression level in
PC9/CD133+-miR-223 cells was 12.25-fold higher than that
in PC9/CD133+ cells (Fig.
6B). IC50 values of erlotinib in both
PC9/CD133+-miR-223 and PC9/CD133+ cells were
calculated, based on data from CCK8 assay. Compared to
PC9/CD133+ cells, IC50 of erlotinib was
decreased by 45% in PC9/CD133+-miR-223 cells (Fig. 6C), from which the resistance index
was calculated as 0.46. As shown in Fig. 6D, the phosphorylation of IGF1R and
Akt was inhibited in PC9/CD133+-miR-223 cells, compared
to that in both PC9/CD133+ and PC9/CD133+-EV
cells, implicating that upregulation of miR-223 expression in
PC9/CD133+ cells is able to inhibit the IGF1R/PI3K/Akt
signaling pathway.
Discussion
Besides the mutation of the EGFR gene or
amplification of the MET proto-oncogene, other mechanisms for the
acquired resistance to erlotinib have recently been found. Our
study showed that Akt, a key downstream molecule of the PI3K
pathway, was constitutively active in PC9/ER and HCC827/ER cells.
The important role of PI3K/Akt signaling pathway in the mechanisms
underlying acquired resistance in lung cancer has been reported
(18). In vitro experiments
showed that the inhibitor of PI3K/Akt pathway can inhibit
proliferation of cancer cells and induced apoptosis (19,20).
PI3K inhibitors also markedly enhanced the sensitivity of NSCLC
cells with high level of phosphorylated Akt to drug-induced
apoptosis. As apoptosis deficiency is the principal mechanism of
drug resistance in cancer cells, it has become a major goal to
re-sensitize drug-resistant cells and kill them through overcoming
defective apoptosis. PI3K-Akt signaling is known to promote
survival under apoptotic stresses, so that the inhibition of
PI3K/Akt signaling can block growth of cancer cells and promote
their apoptosis (21–23). It has been reported that
continuously activated PI3K signaling induces the resistance to
EGFR-TKIs. Thus, the blockage of PI3K/Akt signaling overcame the
resistance to EGFR-TKIs by inducing apoptosis of cancer cells via
in vitro and in vivo models (24). Moreover, most cell models of
acquired resistance exhibited continuous activity of PI3K signaling
pathway (13,25–27).
Our study also confirmed that PI3K/Akt signaling pathway is
activated in either PC9/ER or HCC827/ER cells, indicating that
PI3K/Akt signaling pathway may be associated with acquired
erlotinib resistance in both cell lines.
Aberrant expression of miRNA is a common
characteristic of human cancers, including NSCLC (28,29).
Recent studies have confirmed that miRNAs are involved in the
modulation of the resistance of lung cancer cells to EGFR-TKIs. For
example, miR-21 participates in the process of the acquired
resistance to EGFR-TKIs in NSCLC, through downregulating PTEN and
PDCD4 as well as activating PI3K/Akt pathway (30). The miR-134/487b/655 cluster is
involved in the regulation of drug resistance to gefitinib by
targeting MAGI2 in lung adenocarcinoma cells (31). Another example showed that
depletion of miR-205 induces erlotinib resistance (32). As for miR-223: in the literature,
this miRNA has been confirmed to modulate multidrug resistance via
the downregulation of ABCB1 in hepatocellular carcinoma (33). The suppression of miR-223
expression restored FBXW7 expression and the sensitivity of GC
cells to trastuzumab (34).
However, the role of miR-223 in resistance to EGFR-TKIs is rarely
reported. IGF1R has been identified as a functional target of
miR-223 and upregulation of miR-223 expression leads to inhibition
of IGF1R expression (10,35). In NSCLC, IGF1R acts as an important
receptor tyrosine kinase and an upstream regulator of Akt (36), but no additional evidence supports
the involvement of miR-223 in the acquired resistance of EGFR-TKIs
in NSCLC. In this study, we assessed the expression levels of
miR-223 in PC9/ER and HCC827/ER cells and found that miR-223
expression was reduced by 88% in PC9/ER cells, compared to PC9
cells. However, the expression of miR-223 was 1.5 times greater in
HCC827/ER cells, compared to HCC827 cells. Moreover, the mRNA level
of IGF1R measured by real-time RT-PCR was greatly increased in
PC9/ER cells, compared to that in PC9 cells. Western blot analysis
also demonstrated an increase in p-IGF1R induced by the treatment
with 0.01 μM erlotinib in PC9/ER cells, while the phosphorylation
of IGF1R was inhibited in erlotinib-treated PC9 cells. In our
study, overexpression of miR-223 in PC9/ER cells via lentiviral
transduction inhibited the IGF1R/PI3K/Akt pathway and partially
reversed the resistance of PC9/ER cells to erlotinib. Similar
results were found in our in vivo study. Furthermore,
miR-223-mediated inhibition of the phosphorylation of either IGF1R
(p-IGF1R) or Akt (p-Akt), can be abolished by treatment with IGF1R
agonist, suggesting that miR-223-mediated downregulation of IGF1R
expression at mRNA and protein levels resulted in the inhibition of
the IGF1R/PI3K/Akt/mTOR signaling pathway. These findings confirmed
that the downregulation of miR-223 activates the IGF1R/PI3K/Akt
pathway in PC9/ER cells and induces resistance to erlotinib.
Recently, CD133 has been identified as a biomarker
of cancer stem cells in a variety of human tumors, including lung
carcinoma (37–39). Eramo et al (4) confirmed that CD133+
subpopulation isolated from the tissue samples of lung carcinoma
processed more cancer stem-cell-like characteristics, such as
formation of tumor spheres in vitro and generation of tumors
in nude mice, compared to CD133− cells. We found that
the percentage of CD133+ subpopulation in PC9ER cells
was 4.96 times that in PC9 cells. We used the method of inverse
induction combined with paclitaxel to enrich the CD133+
subpopulation from the PC9 cell line. The IC50 values of
erlotinib between PC9 and PC9/CD133+ cells were
compared. We found that PC9/CD133+ cells were more
resistant to erlotinib, than PC9 cells. To clarify whether there is
downregulation of miR-223 expression and activation of the
IGF1R/PI3K/Akt signaling pathway in the CD133+ cell
population, we examined miR-223 levels and the phosphorylation of
key molecules of the IGF1R/PI3K/Akt pathway.
Our data revealed that downregulation of the miR-223
expression in PC9/CD133+ cells led to the activation of
the IGF1R/PI3K/Akt signaling pathway, which may be the reason for
the greater capacity of CD133+ cells for erlotinib
resistance. It is still debated today whether CD133+
alone is the marker of CSCs. In our study, the CD133+
subpopulation, which was isolated from PC9 cells, displayed cancer
stem cell-like characteristics, including continuous expansion of
tumor spheres (self-renewal) and pluripotent differentiation
potential (differentiating into multiple cell types), a large
capacity for drug resistance, and greater tumorigenic potential
in vivo. Our findings of erlotinib resistance in the
CD133+ subpopulation suggest that erlotinib treatment
may not eradicate cancer stem cells in lung carcinoma, leading to a
failure of molecular target therapy. Our study also revealed that
the erlotinib resistance in CD133+ cells with cancer
stem cell-like characteristics is associated with downregulation of
miR-223 and activation of the IGF1R/PI3K/Akt signaling pathway.
These results provide new insight into the mechanism underlying the
resistance to EGFR-TKIs.
In this study, we developed the erlotinib-resistant
cell lines HCC827/ER and PC9/ER by administering high-dose (1–5 μM)
pulses combined with continuous low-dose applications of erlotinib
(0.01 μM) for >8 months, which mimicked the mode of
administration of erlotinib in the clinic. In most previous
reports, cell models of NSCLC resistance to EGFR-TKIs were
established via stepwise escalation of EGFR-TKI concentrations.
Acquired resistance associated with genetic changes, such as T790M
EGFR mutation or MET amplification, was gained via the stepwise
escalation method of EGFR-TKIs exposure after prolonged treatment
(13,40). In our study, neither T790M EGFR
mutation nor MET amplification was observed in two
erlotinib-resistant cell lines: PC9/ER and HCC827/ER, in which
PI3K/Akt signaling pathway was activated and an in-frame deletion
in exon 19 of EGFR was detected. We also found that downregulation
of miR-223 resulted in the activation of IGF1R/PI3K/Akt pathway in
PC9/ER cells, leading to a secondary resistance of these cells to
erlotinib. In contrast, the expression of miR-223 was 1.5 times
greater in HCC827/ER cells than in HCC827 cells. To explain these
contradictory findings, we examined PTEN expression in HCC827/ER
and PC9/ER cells. Results showed that PTEN mRNA was decreased in
HCC827/ER cells, but slightly increased in PC9/ER cells, compared
to the corresponding parental cells. Similar results for the PTEN
protein were also obtained. Therefore, the resistance of HCC827/ER
cells to erlotinib may be considered a result of the loss of PTEN,
thereby activating the PI3K/Akt pathway. Our findings revealed
different mechanisms of resistance to EGFR-TKIs in lung
adenocarcinoma, harboring mutated oncogene or loss of tumor
suppressor gene, although the establishment of these resistant cell
lines used the same culture conditions and methods. The mechanisms
underlying the secondary resistance to EGFR-TKIs in patients with
lung cancer may be more complex, because of the complexity of the
tumor microenvironment and differences between individuals. As a
result, for precise individualized therapy, it is necessary to
explore additional mechanisms for resistance to EGFR-TKIs.
In conclusion, miR-223 expression was downregulated
in PC9/ER cells and PC9/CD133+ cells, leading to the
activation of the IGF1R/PI3K/Akt signaling pathway in these cells,
which may be one mechanism responsible for the resistance of PC9/ER
cells and PC9/CD133+ cells to erlotinib.
Acknowledgements
This study was supported in part by grants from the
National Natural Science Foundation of China (81172070 and
81071786) and the National High Technology Research and Development
Program of China (2008AA02Z104).
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J,
Murray T and Thun MJ: Cancer statistics, 2008. CA Cancer J Clin.
58:71–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bezjak A, Tu D, Seymour L, Clark G,
Trajkovic A, Zukin M, Ayoub J, Lago S, de Albuquerque Ribeiro R,
Gerogianni A, et al; National Cancer Institute of Canada Clinical
Trials Group Study BR.21. Symptom improvement in lung cancer
patients treated with erlotinib: Quality of life analysis of the
National Cancer Institute of Canada Clinical Trials Group Study
BR.21. J Clin Oncol. 24:3831–3837. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eramo A, Lotti F, Sette G, Pilozzi E,
Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C and De
Maria R: Identification and expansion of the tumorigenic lung
cancer stem cell population. Cell Death Differ. 15:504–514. 2008.
View Article : Google Scholar
|
|
5
|
Levina V, Marrangoni A, Wang T, Parikh S,
Su Y, Herberman R, Lokshin A and Gorelik E: Elimination of human
lung cancer stem cells through targeting of the stem cell
factor-c-kit autocrine signaling loop. Cancer Res. 70:338–346.
2010. View Article : Google Scholar
|
|
6
|
Sun FF, Hu YH, Xiong LP, Tu XY, Zhao JH,
Chen SS, Song J and Ye XQ: Enhanced expression of stem cell markers
and drug resistance in sphere-forming non-small cell lung cancer
cells. Int J Clin Exp Pathol. 8:6287–6300. 2015.PubMed/NCBI
|
|
7
|
Hassan SS, Romero R, Pineles B, Tarca AL,
Montenegro D, Erez O, Mittal P, Kusanovic JP, Mazaki-Tovi S,
Espinoza J, et al: MicroRNA expression profiling of the human
uterine cervix after term labor and delivery. Am J Obstet Gynecol.
202:80.e1–80.e8. 2010. View Article : Google Scholar
|
|
8
|
Geng Q, Fan T, Zhang B, Wang W, Xu Y and
Hu H: Five microRNAs in plasma as novel biomarkers for screening of
early-stage non-small cell lung cancer. Respir Res. 15:1492014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Wang L, Wu Z, Sun R, Jin H, Ma J,
Liu L, Ling R, Yi J, Wang L, et al: Three dysregulated microRNAs in
serum as novel biomarkers for gastric cancer screening. Med Oncol.
31:2982014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nian W, Ao X, Wu Y, Huang Y, Shao J, Wang
Y, Chen Z, Chen F and Wang D: miR-223 functions as a potent tumor
suppressor of the Lewis lung carcinoma cell line by targeting
insulin-like growth factor-1 receptor and cyclin-dependent kinase
2. Oncol Lett. 6:359–366. 2013.PubMed/NCBI
|
|
11
|
Miller TE, Ghoshal K, Ramaswamy B, Roy S,
Datta J, Shapiro CL, Jacob S and Majumder S: MicroRNA-221/222
confers tamoxifen resistance in breast cancer by targeting p27Kip1.
J Biol Chem. 283:29897–29903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R,
Qian Y, Li Q, Wang L, Shi Z, et al: Alteration in Mir-21/PTEN
expression modulates gefitinib resistance in non-small cell lung
cancer. PLoS One. 9:e1033052014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma H, Yao Q, Zhang AM, Lin S, Wang XX, Wu
L, Sun JG and Chen ZT: The effects of artesunate on the expression
of EGFR and ABCG2 in A549 human lung cancer cells and a xenograft
model. Molecules. 16:10556–10569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mu XY, Dong XL, Sun J, Ni YH, Dong Z, Li
XL, Sun EL, Yi Z and Li G: Simultaneous blockage of epidermal
growth factor receptor and cyclooxygenase-2 in a human
xenotransplanted lung cancer model. Asian Pac J Cancer Prev.
15:69–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guix M, Faber AC, Wang SE, Olivares MG,
Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, et al:
Acquired resistance to EGFR tyrosine kinase inhibitors in cancer
cells is mediated by loss of IGF-binding proteins. J Clin Invest.
118:2609–2619. 2008.PubMed/NCBI
|
|
17
|
Choi YJ, Park GM, Rho JK, Kim SY, So GS,
Kim HR, Choi CM and Lee JC: Role of IGF-binding protein 3 in the
resistance of EGFR mutant lung cancer cells to EGFR-tyrosine kinase
inhibitors. PLoS One. 8:e813932013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu LZ, Zhou XD, Qian G, Shi X, Fang J and
Jiang BH: AKT1 amplification regulates cisplatin resistance in
human lung cancer cells through the mammalian target of
rapamycin/p70S6K1 pathway. Cancer Res. 67:6325–6332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alexia C, Bras M, Fallot G, Vadrot N,
Daniel F, Lasfer M, Tamouza H and Groyer A: Pleiotropic effects of
PI-3′ kinase/Akt signaling in human hepatoma cell proliferation and
drug-induced apoptosis. Ann NY Acad Sci. 1090:1–17. 2006.
View Article : Google Scholar
|
|
20
|
Poh TW and Pervaiz S: LY294002 and
LY303511 sensitize tumor cells to drug-induced apoptosis via
intracellular hydrogen peroxide production independent of the
phosphoinositide 3-kinase-Akt pathway. Cancer Res. 65:6264–6274.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brozovic A and Osmak M: Activation of
mitogen-activated protein kinases by cisplatin and their role in
cisplatin-resistance. Cancer Lett. 251:1–16. 2007. View Article : Google Scholar
|
|
22
|
Corradetti MN and Guan KL: Upstream of the
mammalian target of rapamycin: Do all roads pass through mTOR?
Oncogene. 25:6347–6360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lekmine F, Uddin S, Sassano A, Parmar S,
Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN and
Platanias LC: Activation of the p70 S6 kinase and phosphorylation
of the 4E-BP1 repressor of mRNA translation by type I interferons.
J Biol Chem. 278:27772–27780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Donev IS, Wang W, Yamada T, Li Q, Takeuchi
S, Matsumoto K, Yamori T, Nishioka Y, Sone S and Yano S: Transient
PI3K inhibition induces apoptosis and overcomes HGF-mediated
resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer
Res. 17:2260–2269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ogino A, Kitao H, Hirano S, Uchida A,
Ishiai M, Kozuki T, Takigawa N, Takata M, Kiura K and Tanimoto M:
Emergence of epidermal growth factor receptor T790M mutation during
chronic exposure to gefitinib in a non small cell lung cancer cell
line. Cancer Res. 67:7807–7814. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Engelman JA, Mukohara T, Zejnullahu K,
Lifshits E, Borrás AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun
J, et al: Allelic dilution obscures detection of a biologically
significant resistance mutation in EGFR-amplified lung cancer. J
Clin Invest. 116:2695–2706. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li H, Schmid-Bindert G, Wang D, Zhao Y,
Yang X, Su B and Zhou C: Blocking the PI3K/AKT and MEK/ERK
signaling pathways can overcome gefitinib-resistance in non-small
cell lung cancer cell lines. Adv Med Sci. 56:275–284. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Du L and Pertsemlidis A: microRNAs and
lung cancer: Tumors and 22-mers. Cancer Metastasis Rev. 29:109–122.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gibson NW: Engineered microRNA
therapeutics. J R Coll Physicians Edinb. 44:196–200. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li B, Ren S, Li X, Wang Y, Garfield D,
Zhou S, Chen X, Su C, Chen M, Kuang P, et al: MiR-21 overexpression
is associated with acquired resistance of EGFR-TKI in non-small
cell lung cancer. Lung Cancer. 83:146–153. 2014. View Article : Google Scholar
|
|
31
|
Kitamura K, Seike M, Okano T, Matsuda K,
Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K and Gemma A:
MiR-134/487b/655 cluster regulates TGF-β-induced
epithelial-mesenchymal transition and drug resistance to gefitinib
by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther.
13:444–453. 2014. View Article : Google Scholar
|
|
32
|
Park KS, Raffeld M, Moon YW, Xi L, Bianco
C, Pham T, Lee LC, Mitsudomi T, Yatabe Y, Okamoto I, et al: CRIPTO1
expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor
resistance. J Clin Invest. 124:3003–3015. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang T, Zheng ZM, Li XN, Li ZF, Wang Y,
Geng YF, Bai L and Zhang XB: MiR-223 modulates multidrug resistance
via downregulation of ABCB1 in hepatocellular carcinoma cells. Exp
Biol Med (Maywood). 238:1024–1032. 2013. View Article : Google Scholar
|
|
34
|
Eto K, Iwatsuki M, Watanabe M, Ishimoto T,
Ida S, Imamura Y, Iwagami S, Baba Y, Sakamoto Y, Miyamoto Y, et al:
The sensitivity of gastric cancer to trastuzumab is regulated by
the miR-223/FBXW7 pathway. Int J Cancer. 136:1537–1545. 2015.
View Article : Google Scholar
|
|
35
|
Jia CY, Li HH, Zhu XC, Dong YW, Fu D, Zhao
QL, Wu W and Wu XZ: MiR-223 suppresses cell proliferation by
targeting IGF-1R. PLoS One. 6:e270082011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Allen GW, Saba C, Armstrong EA, Huang SM,
Benavente S, Ludwig DL, Hicklin DJ and Harari PM: Insulin-like
growth factor-I receptor signaling blockade combined with
radiation. Cancer Res. 67:1155–1162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Atashpour S, Fouladdel S, Movahhed TK,
Barzegar E, Ghahremani MH, Ostad SN and Azizi E: Quercetin induces
cell cycle arrest and apoptosis in CD133(+) cancer stem cells of
human colorectal HT29 cancer cell line and enhances anticancer
effects of doxorubicin. Iran J Basic Med Sci. 18:635–643.
2015.PubMed/NCBI
|
|
38
|
Miki J, Furusato B, Li H, Gu Y, Takahashi
H, Egawa S, Sesterhenn IA, McLeod DG, Srivastava S and Rhim JS:
Identification of putative stem cell markers, CD133 and CXCR4, in
hTERT-immortalized primary nonmalignant and malignant tumor-derived
human prostate epithelial cell lines and in prostate cancer
specimens. Cancer Res. 67:3153–3161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim CK, Kim SK, Yang YH, Lee MS, Yoon JH
and Park CI: A case of recurrent infantile polycystic kidney
associated with hydrops fetalis. Yonsei Med J. 30:95–103. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suda K, Murakami I, Katayama T, Tomizawa
K, Osada H, Sekido Y, Maehara Y, Yatabe Y and Mitsudomi T:
Reciprocal and complementary role of MET amplification and EGFR
T790M mutation in acquired resistance to kinase inhibitors in lung
cancer. Clin Cancer Res. 16:5489–5498. 2010. View Article : Google Scholar : PubMed/NCBI
|