Introduction
Gastric cancer (GC) remains a major health problem
and is a leading cause of morbidity and mortality worldwide,
representing the second most common cause of cancer-related deaths
(1). In 2012, GC caused 723,000
new deaths in the world (2)
because of its poor prognosis and the limited efficacy of the
treatment. It is widely accepted that environmental factors, such
as Helicobacter pylori infections (3), Epstein-Barr viruses, diet, smoking
and obesity, contribute to gastric carcinogenesis (4). Additionally, genetic factors have
been implicated in GC development, including somatic mutations,
gene amplifications and deletions, epigenetic inactivation of
several genes, and aberrant DNA methylation (5,6).
Molecular profiling of GCs has been performed using gene expression
or DNA sequencing, which has facilitated the identification of
putative biomarkers for subtype classification, prognosis and
therapeutic targets (7–9). However, the molecular mechanisms
underlying the progression of gastric carcinoma are still poorly
understood. Besides protein coding genes, recent evidence has shown
that there is an important role for microRNAs (miRs) in human
cancers (10). miRs are short (~22
nucleotides), non-coding RNAs that regulate gene expression
primarily by translational repression or transcriptional
degradation, and are involved in cellular processes such as cell
proliferation, cell death, and tumorigenesis (11–13).
Studies have suggested that there are oncogenic and tumor
suppressive roles for miRs in cancers (14,15).
miRs have great potential as cancer biomarkers in terms of their
tissue-specific expression and aberrant expression in cancer cells
(16) based on data from
high-throughput microarray analysis. In GC, aberrant miR expression
profiles have been associated with GC progression, prognosis
(17,18) and pathogenesis by perturbing the
function of its target genes (19–22).
miR-148a is one of the most significantly
downregulated miRs in GC cell lines, GC tissue samples, and in GC
patients compared with adjacent normal gastric tissues (23–25).
Therefore, the aberrant expression of miR-148a suggests that it may
be involved in GC progression. In this study, we used MNNG
(N-methyl-N9-nitroN-nitrosoguanidine) treatment to establish a rat
GC model, and used the human GC cell lines AGS, YCC3 and BGC-823 to
investigate the effects of miR-148a on the development and
progression of GC.
Material and methods
Animals
Forty-four one-week-old, grade 3, male Wistar rats
(approximately 50 g in weight) were supplied by the Experimental
Animal Center (Hubei, China) and kept in a room under a 12 h
artificial light/dark cycle with free access to food and water.
Acclimatization period of one week was observed prior to the
initiation of experimentation. The procedures were approved by the
Animal Care Committee of Wuhan University and complied with the
recommendations of the Chinese guidelines for the care and use of
laboratory animals.
Cell culture
GC cell lines (AGS, YCC3, SCH and BGC-823 cells)
were grown in DMEM (Hyclone, HyClone Laboratories, Logan, UT, USA)
containing 10% fetal bovine serum (FBS; Gibco, Waltham, MA, USA),
100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a
humidified chamber. YCC3 and SCH cells were maintained in Roswell
Park Memorial Institute medium (RPMI-1640; Hyclone, HyClone
Laboratories) containing 10% FBS, 100 U/ml penicillin and 100 μg/ml
streptomycin at 37°C in a humidified chamber.
Drug treatments
MNNG (Fluka, Germany) solution was prepared three
times per week with distilled water at a concentration of 100
mg/ml. The solution was protected from light and given ad
libitum to the animals in their drinking water. In addition to
MNNG, all animals were given 1 ml of 10% sodium chloride weekly by
oral gavage in the initial six weeks of the study to enhance GC
development. MNNG and control PBS were administered by oral gavage
daily from the age of six weeks for a total of 40 weeks.
Plasmid construction and
transfection
The fragments encoding primary miRs were cloned into
the pcDNA-3.1(-) vector (Invitrogen; Carlsbad, CA, USA) following
digestion with Xhol/BamHI (Thermo Scientific;
Waltham, MA, USA). The forward and reverse primers for the human
primary miR-148a (pri-miR-148a) are listed in Table I. To construct the luciferase
reporter plasmids, the 3′-UTR sequences from +2,286 to +2,308 of
TGFβ2 and sequence from +8,335 to +8,357 of SMAD2 were
inserted at the HindIII/XhoI site downstream of the
luciferase gene in the pGL3-promoter vector. All final constructs
were verified by DNA sequence analysis.
 | Table IPrimer sequences used for real-time
RT-PCR. |
Table I
Primer sequences used for real-time
RT-PCR.
| Gene |
Forward/Reverse | Primer (5′-3′) | Tm (°C) | Cycles |
|---|
| hTGFβ2 | Forward |
TGGTGAAAGCAGAGTTCAGAG | 60 | 40 |
| Reverse |
CACAACTTTGCTGTCGATGTAG | | |
| hSMAD2 | Forward |
AAGCAGTGAAAAGTCTGGTG | 60 | 40 |
| Reverse |
GTTGTATCCCACTGATCTAT | | |
| hSMAD4 | Forward |
TGGGAAGAGATCACCCTGTC | 60 | 40 |
| Reverse |
CCCAACGGTAAAAGACCTCA | | |
| hGAPDH | Forward |
GAGTCAACGGATTTGGTCGT | 60 | 40 |
| Reverse |
AATGAAGGGGTCATTGATGG | | |
| rTGFα | Forward |
GTATCCTGGTAGCTGTGTGT | 60 | 40 |
| Reverse |
ATACTGAGTGTGGGAATCTGGG | | |
| rTGFβ2 | Forward |
CATGCCCTTATCTGTGGAGTTC | 60 | 40 |
| Reverse |
TGGGCGTATTGCCAATGT | | |
| rSMAD2 | Forward |
AGCAGTGAAAAGTCTGGTGA | 60 | 40 |
| Reverse |
AGAGCAAGTGCTTGGTATGG | | |
| rSMAD3 | Forward |
CAGAACGTGAACACCAAGT | 60 | 40 |
| Reverse |
CAAGCGGCAGTAGATGAC | | |
| rSMAD4 | Forward | CGCCTGTCTGAGCA
TTGTAC | 60 | 40 |
| Reverse |
CGATTACTTGGTGGATGTTGG | | |
| rGAPDH | Forward |
TGACTCTACCCACGGCAAGTTCAA | 60 | 40 |
| Reverse |
ACGACATACTCAGCACCAGCATCA | | |
| miR-148a | |
GTCGTATCCAGTGCGTGTCGTGGA | | |
| RT | |
GTCGGCAATTGCACTGGATACGACACAAAG | | |
| miR-148a | Forward |
GTTCCATTATCGGTCGCAT | 60 | 40 |
| Reverse |
TCTACAGTCAGGAGTCCACC | | |
| U6 | Forward |
CTCGCTTCGGCAGCACA | 60 | 40 |
| Reverse |
AACGCTTCACGAATTTGCGT | | |
|
pcDNA3.1-miR-148a | Forward | CCTCGAGGTTCCATTATCGGTCGCAT | | |
| Reverse | CGGGATCCACCAGGGTAAGATGAAGAGA | | |
Real-time quantitative reverse
transcriptase PCR
Gastric tissues and total cellular RNA was isolated
using TRIzol reagent (Invitrogen) according to the manufacturer's
protocol. RNA (1 μg) was reverse transcribed into cDNA using dNTPs
(1 mM), 5X reverse transcription buffer (500 mM Tris-HCl, pH 8.3,
250 mM KCl, 50 mM MgCl2, and 50 mM DTT), 16 units
RNasin, and 2.5 units of M-MLV reverse transcriptase (Promega;
Madison, WI, USA). SYBR Green (Toyobo; Osaka, Japan) was used for
all real-time PCR reactions. All real-time PCR reactions were
performed and analyzed with the CFX connect real-time PCR detection
system (Bio-Rad; Hercules, CA, USA). The primers and PCR conditions
for miR-148a, human and rat TGFα, TGFβ2, SMAD2, SMAD3 and SMAD4 are
listed in Table I. GAPDH was used
to normalize the relative expression levels of TGFα, TGFβ2, SMAD2,
SMAD3 and SMAD4. U6 was used to normalize the relative expression
levels of miRs. Gene expression levels were calculated using the
2−ΔΔCT method relative to the internal control.
Transient transfection and luciferase
assays
GC cell lines (AGS, YCC3 and SCH) were used for the
luciferase assays. Cells were transiently co-transfected with
pcDNA-3.1(-) expression vectors or with an empty pGL-3 vector. A
dual-luciferase reporter assay system was used according to the
manufacturer's instructions (Promega). Luciferase activity was
quantified 48 h after transfection using a luminometer and the Stop
& GloH Dual Luciferase kit (Promega). To control for
transfection efficiency, firefly luciferase activity was corrected
according to Renilla luciferase activity.
Western blotting
Western blots were used to detect the protein levels
in different rat groups. Briefly, the tissues of the control and GC
groups were weighed and homogenized with an ultrasound homogenizer
and gastric tissues were subsequently lysed in lysis buffer (1 ml
of 1% NP-40, 25 mM Tris-HCl, 150 mM NaCl, 10 mM EDTA, pH 8.0,
containing a 2.5% protease inhibitor cocktail P2714 (Sigma, St.
Louis, MO, USA) for 30 min on ice. Samples were centrifuged at
14,000 x g for 15 min. To detect the TGFα, TGFβ2, SMAD2, SMAD3 and
SMAD4 proteins, the proteins (30 μg) were separated by
SDS-polyacrylamide gel electrophoresis. The membranes were
incubated for 2 h with a monoclonal mouse anti-human TGFα antibody
(1:1500, Santa Cruz Biotechnology; Santa Cruz, CA, USA), a
polyclonal rabbit anti-human TGFβ2 antibody (1:2000, Millipore;
Billerica, MA, USA), or a polyclonal goat anti-human SMAD2 and
SMAD4 (1:1500, Santa Cruz Biotechnology). All of the blots were
incubated with a peroxidase-conjugated secondary antibody (1:6000,
Millipore) for 1 h. β-actin was used as an internal control. The
blots were then developed using the ECL detection kit (Thermo
Scientific) to produce a chemiluminescent signal that was captured
on X-ray film.
Immunofluorescence cytochemistry
The GC cell line AGS was washed twice with cold-PBS
and then fixed with 4% paraformaldehyde for 10 min, permeabilized
and blocked with 0.1% Triton X-100 and 1% BSA for 10 min. The cells
were incubated with a polyclonal rabbit anti-human TGFβ2 antibody
(1:100, Millipore) or a polyclonal goat anti-human SMAD2 antibody
(1:100, Santa Cruz Biotechnology) overnight at 4°C and then
incubated with DYLight 549 (1:100, Boster Biological Technology;
Wuhan, China) in 1% BSA for 1.5 h at 37°C. After being washed
three-times with ice-cold PBS, the cells were incubated with DAPI
for 10 min. The images were captured and analyzed using
fluorescence microscopy (Olympus Corp., Tokyo, Japan).
Cell proliferation, migration, and
invasion assays
All experiments were performed according to the
manufacturer's instructions. Briefly, cell proliferation assays
were performed using a Cell Titer 96 Aqueous non-radioactive Cell
Proliferation Assay kit (Promega) and measured using a Perkin Elmer
plate reader (EnVision™ Multilabel Plate Reader). Triplicate assays
were performed. Colony formation assay was used to further study
the effect of miR-148a on cell proliferation. Three cells were
plated in 6-well plates and cultured for 10 days. Colonies were
fixed with 10% formaldehyde for 5 min and stained with 1.0% crystal
violet for 30 sec.
Cell migration assays were performed using BioCoat™
24-well chambers with 8-μm pore filter inserts (BD Biosciences, San
Diego, CA, USA). After chamber rehydration, 5×104 cells
were transferred to the upper chamber in 500 μl of serum-free
medium. Complete medium with 10% FBS was used as a chemoattractant.
Cells were incubated for 48 h, and the migrated cells on the lower
surface of the insert or in the wells were trypsinized so that cell
numbers could be quantified. Each assay was performed in
triplicate, and the results were averaged over three independent
experiments.
Cell invasion assays were performed similarly using
BioCoat Matrigel™ invasion chambers with 8-μm pore polycarbonate
membranes that were precoated with Matrigel Matrix (BD
Biosciences).
Tumor growth in nude mice
An equal number (1×107) of BGC-823 cells
transfected with pcDNA-3.1-miR-148a or pcDNA-3.1 were harvested
after 72 h. Three groups (each group with six mice) of four- to
six-week-old male Balb/c nude mice were given subcutaneous
injections with BGC-823-miR-148a cells or BGC-vector cells. The
mice were monitored every three days for tumor formation. The tumor
sizes were measured every three days using calipers. The tumor size
at 31 days post-injection was used as the endpoint reading. The
data are presented as the mean volume mean ± SD. TGFβ2 and SMAD2
proteins in tumors were detected by qPCR.
Flow cytometry assay
BGC-823-miR-148a cells or BGC-vector cells were
isolated and made into single cell suspensions, washed with PBS and
then fixed in 3 ml cold 70% ethanol for 1 h at −20°C. The cells
were then washed three times with BioLegend Cell Staining Buffer
and resuspended in 100 μl of PBS. APC-conjugated Ki-67 antibody (10
μl, BioLegend) was added and incubated at room temperature in the
dark for 30 min. Cells were washed with BioLegend Cell Staining
Buffer and then resuspended in 0.5 ml of PBS for flow cytometric
analysis.
Statistical analyses
The results were expressed as the mean ± SD.
Statistical analyses were performed using one-way ANOVAs followed
by least significant difference (LSD) post-hoc tests, with a
p-value <0.05 being considered to indicate a statistically
significant difference.
Results
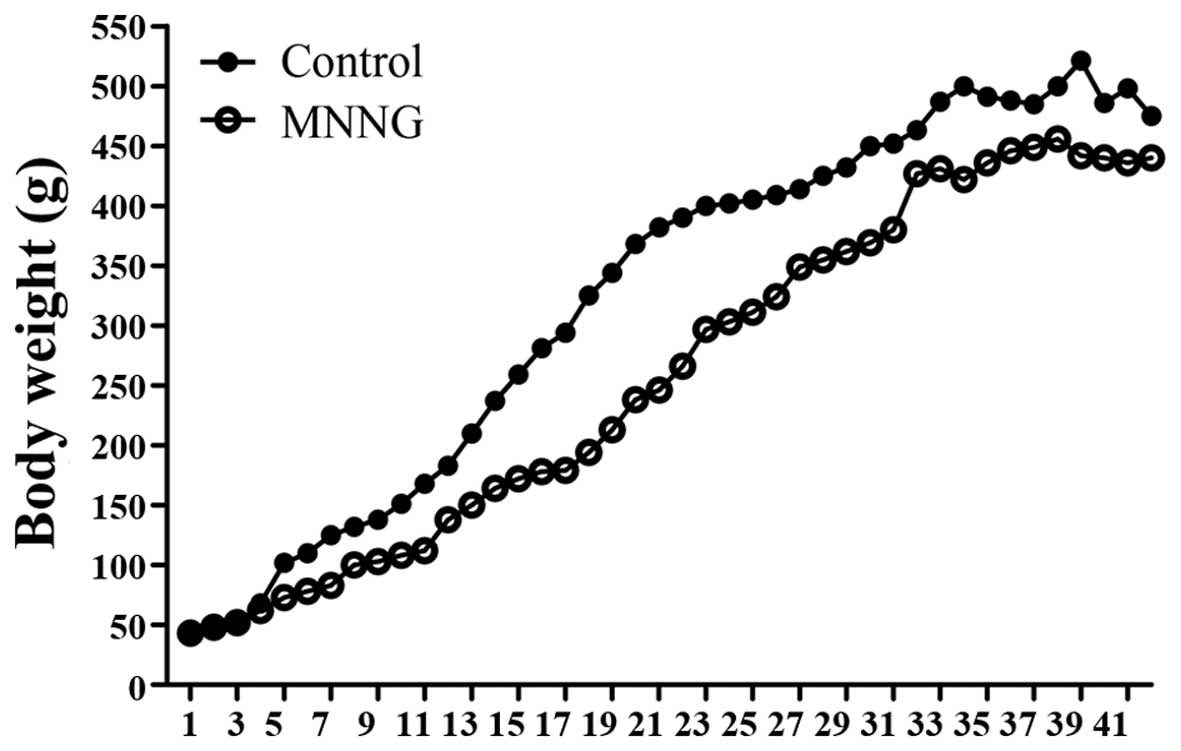
MNNG-induced gastric cancer in rats
To determine the effects of the MNNG-initiated GC
rat model, all animals were monitored closely for general health
during the study period and their body weights were recorded
weekly. The results demonstrated that the body weights of the
control animals were higher than that in the MNNG-treated group
throughout the study. In the early phase of the study (from the
14th-31st week), the differences in body weights between the
control and MNNG-treated groups were significant (Fig. 1). There were a total of five deaths
during this study period: none in the control group and five in the
MNNG-treated group. The causes of death are listed in Table II. The incidence of MNNG-initiated
GC are shown in Table II.
| Table IITumor incidence and the cause of
death in the groups of study animals. |
Table II
Tumor incidence and the cause of
death in the groups of study animals.
| Group | Control | MNNG |
|---|
| No. of rats | 20 | 20 |
| No. of death
(%) | 0 (0) | 5 (25) |
| Cause of death | | Gastric cancer (3),
small bowel cancer (1), unknown (1) |
| No. of rats with
gastric tumors (%) | 0 (0) | 18 (90) |
MNNG increases TGFβ2, SMAD2, and SMAD4 in
rat gastric tissues
The mRNA and protein levels of TGFβ2 were 2.1-fold
and 2.1-fold higher in the MNNG-treated group compared with the
control group, respectively (Fig. 2B
and G). SMAD2 mRNA and protein levels were 1.8- and 2.0-fold
higher in the MNNG-treated group compared with the control group,
respectively (Fig. 2C and H).
SMAD4 mRNA and protein levels were 1.3- and 1.5-fold higher in the
MNNG-treated group compared with the control group, respectively
(Fig. 2E and J). However, there
were no differences in TGFα and SMAD3 in either the mRNA or protein
level (Fig. 2A, D, F and I). These
data suggested that MNNG could upregulate TGFβ2, SMAD2 and SMAD4,
subsequently leading to GC. However, the results demonstrated that
TGFα and SMAD3 were not involved in the mechanism of MNNG initiated
GC in the rat model.
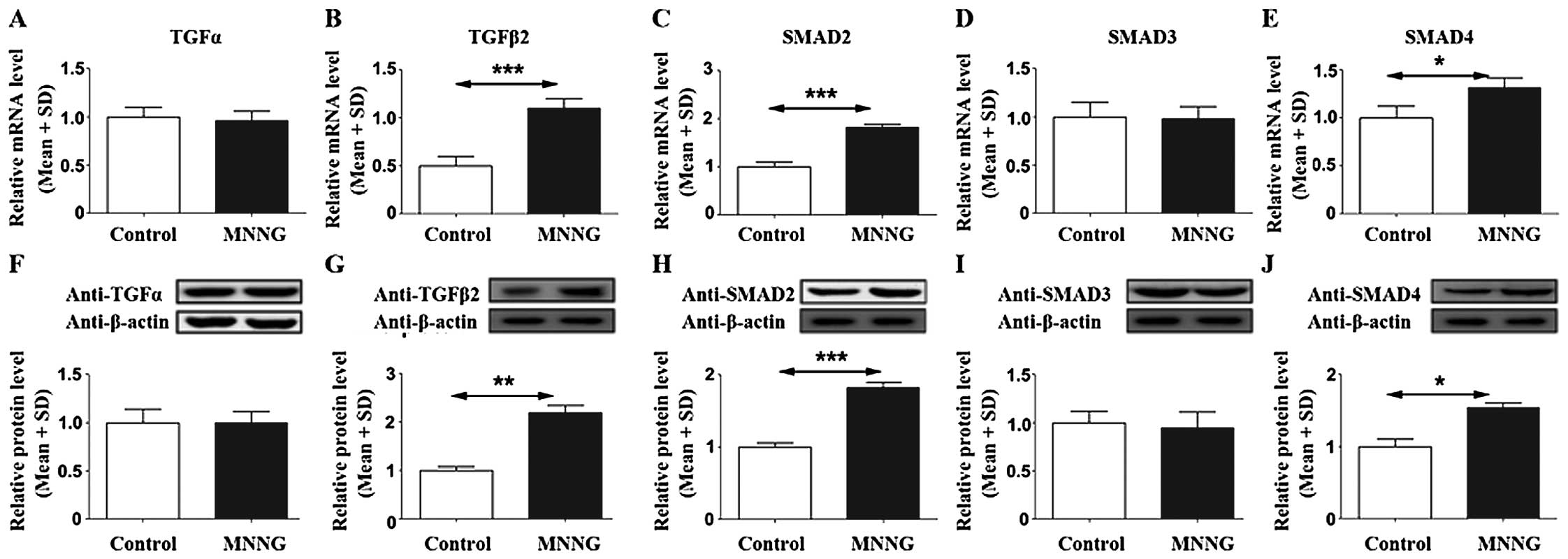 | Figure 2Changes in TGFα, TGFβ2, SMAD2, SMAD3,
and SMAD4 in MNNG-treated rats. MNNG induced TGFβ2, SMAD2, and
SMAD4 mRNA levels (B, C and E) and protein levels (G, H and J), but
did not affect TGFα and SMAD3 mRNA levels (A and E) and protein
levels (F and I). The upper part shows mRNA levels in different
groups (A–E), and lower part shows protein levels by western
blotting. A representative immunoblot is shown. β-actin was used as
a loading control (F–J). n=20 (control), and n=15 (MNNG).
*p<0.05, **p<0.01 and
***p<0.001 vs. the control. |
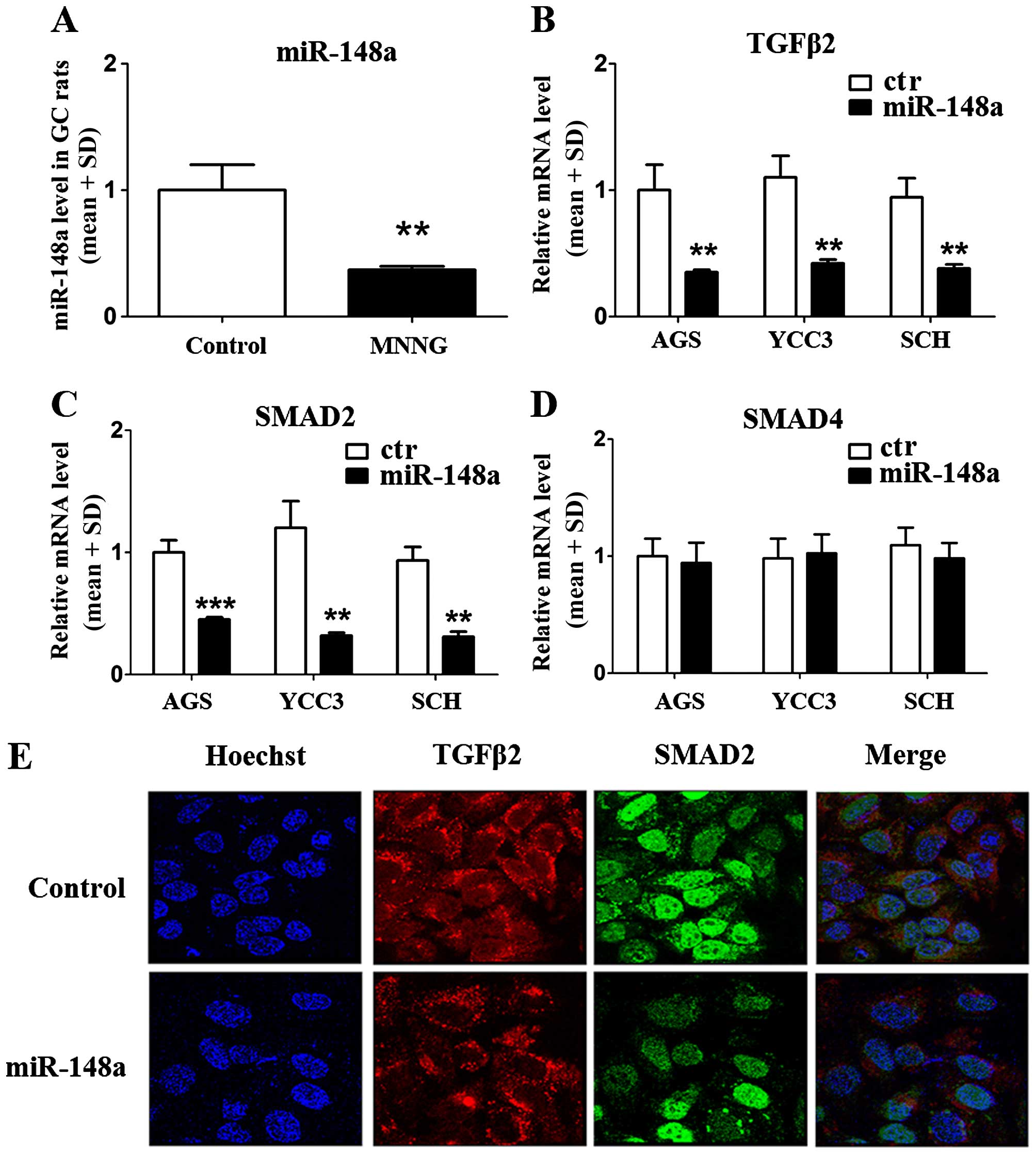
miR-148a decreases the expression of
TGFβ2 and SMAD2 in GC cells
To verify whether miR-148a regulated the expression
of TGFβ2, SMAD2, and SMAD4, we used a QPCR assay to quantify the
level of miR-148a in the GC rat model. The results showed that
miR-148a was inhibited significantly in MNNG-treated rats compared
with normal rats (reduced 63%) (Fig.
3A). The data suggested that miR-148a might be involved in the
occurrence and development of GC via MNNG treatment. Furthermore,
we constructed a primary miR-148a expression plasmid and
transfected it into GC cells (AGS, YCC3, and SCH) and verified the
mRNA levels of TGFβ2, SMAD2, and SMAD4. The results demonstrated
that the mRNA levels for both TGFβ2 and SMAD2 were reduced in all
three GC cell lines (Fig. 3B and
C), but SMAD4 mRNA levels were not affected by miR-148a
(Fig. 3D). These data suggested
that miR-148a could regulate the mRNA levels of TGFβ2 and SMAD2,
but not SMAD4. We used immunofluorescence cytochemistry to
demonstrate the effects of miR-148a overexpression in AGS cells.
The results confirmed that miR-148a could inhibit the expression of
TGFβ2 and SMAD2 (Fig. 3E). As
shown in Fig. 3E, the expression
of TGFβ2 (red) was predominantly located in the cytoplasm, and
SMAD2 was expressed in both the cytoplasm and nucleus (green).
Overexpression of miR-148a inhibited the levels of TGFβ2 and SMAD2
significantly (Fig. 3E, down).
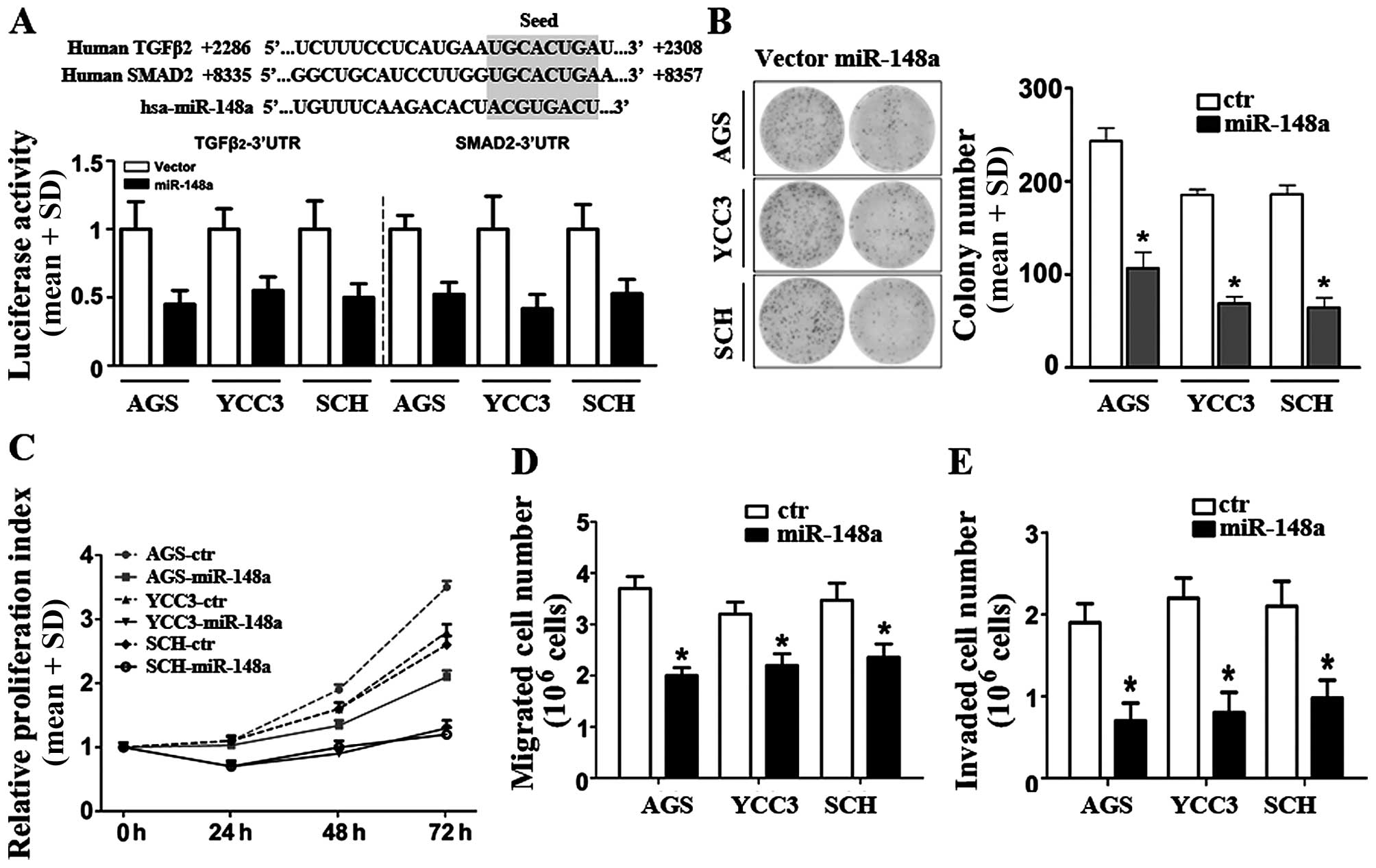
miR-148a binds to TGFβ2- and SMAD2-3′UTRs
in GC cells
To verify the regulation mechanism of TGFβ2 and
SMAD2 by miR-148a, we analyzed the 3′-UTR sequences of the
TGFβ2 and SMAD2 genes and performed luciferase assays
to confirm whether miR-148a could bind to the TGFβ2 and
SMAD2 genes. The luciferase activities in AGS, YCC3, and SCH
cells transfected with the TGFβ2- and SMAD2-3′-UTR
reporter plasmids were inhibited following co-transfection with a
miR-148a plasmid (Fig. 4A). These
data suggested that the binding sites of miR-148a in both the
TGFβ2- and SMAD2-3′UTRs were functional.
miR-148a reduces the proliferation,
migration, and invasion of GC cells
To investigate the functional significance of
miR-148a downregulation in GC cells, we selected three GC cell
lines (AGS, YCC3, and SCH). The miR-148a primary plasmid was
constructed and transfected into these cell lines, and the miR-148a
expression levels in these cells were confirmed by qRT-PCR (data
not shown). To investigate the biological effect of miR-148a
inhibition on gastric cancer progression, the AGS, YCC3, and SCH
gastric cancer cell lines were transfected with miR-148a. Colony
formation assay showed that miR-148a decreased the rate of cell
proliferation significantly (Fig.
4B).
Then, we compared the cell proliferation rates of
the control- and miR-148a-transfected cells at various time points.
In all three cell lines, the growth of miR-148a-transfected cells
was reduced significantly compared to cells transfected with
negative-control miRs (Fig. 4C).
These results suggested that miR-148a overexpression was sufficient
to inhibit cellular proliferation in GC cells. To assess the
effects of miR-148a on GC migration and invasion, we tested the
three cell lines that stably expressed miR-148a or empty vectors.
All cell expressing vector controls migrated robustly in the
Transwell assays (Fig. 4D),
whereas those overexpressing miR-148a exhibited a significant
reduction in migration capacity in GC cells (reduced approximately
46, 32 and 33%, respectively). Similarly, in the invasion assays,
overexpression of miR-148a exhibited inhibition of the invasion
capacity compared with controls in three GC cell lines (the
inhibition rates were 63% in AGS, 64% in YCC3, and 53% in SCH
cells) (Fig. 4E). Taken
collectively, these results indicated that miR-148a over-expression
was sufficient to suppress several pro-oncogenic traits in
vitro, consistent with miR-148a playing a tumor-suppressive
role in GC cells.
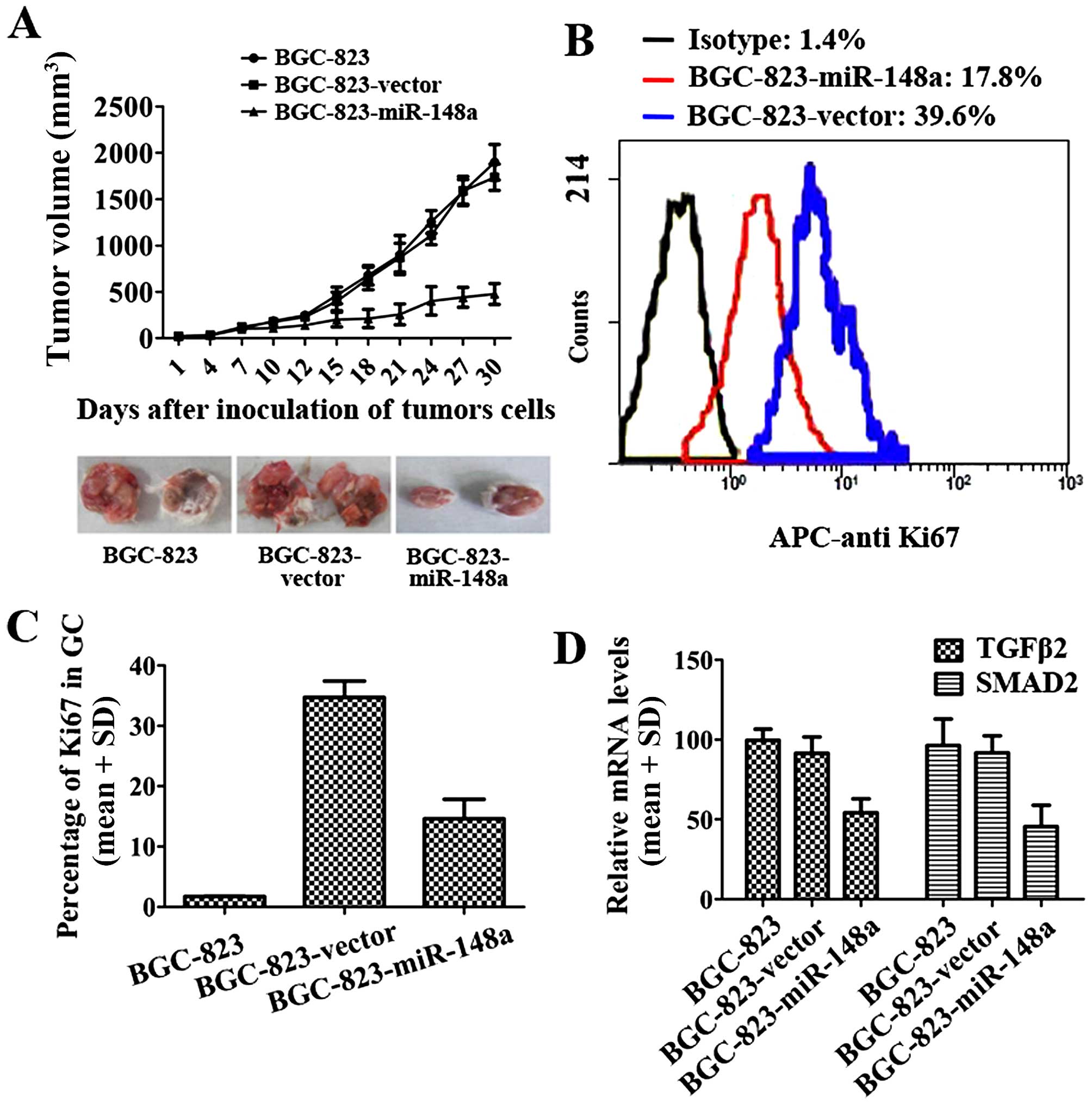
miR-148a inhibits GC development by
inhibiting TGFβ2 and SMAD2
To further investigate the function of miR-148a in a
mouse GC model, BALB/c nude mice were given subcutaneous injections
withBGC-823-miR-148a cells or BGC-vector cells. The tumor volumes
were calculated every three days. The results demonstrated that
miR-148a could decrease BCG-823 growth in vivo (Fig. 5A). The Ki-67 protein is a cellular
marker for proliferation. In this study, flow cytometry results
indicated that the percentage of Ki-67 in BGC-823-miR-148a tumor
tissues was lower than that in BGC-823-vector (Fig. 5B and C). In the gastric tumor
tissues, TGFβ2 and SMAD2 expression levels were determined by
qRT-PCR. In BGC-miR-148a, TGFβ2 and SMAD2 expression levels were
much lower than in BCG-vector cells (Fig. 5D). These results suggested that
miR-148a could efficiently reduce gastric cancer proliferation and
Ki-67 expression was reduced in BGC-823-miR-148a cells (Fig. 5A–C). miR-148a also inhibited the
expression of TGFβ2 and SMAD2 (Fig.
5D). These results suggested that miR-148a inhibited GC
development by inhibiting TGFβ2 and SMAD2.
Discussion
In this study, we determined the changes in
miR-148a, TGFα, TGFβ2, SMAD2, SMAD3, and SMAD4 levels in
MNNG-initiated gastric carcinoma in Wistar rats, and examined the
role of miR-148a in inhibiting the expression of TGFβ2 and SMAD2 in
GC cell lines. We found that miR-148a could inhibit cellular
proliferation, migration, and invasion in GC cells. MNNG-induced GC
is a well-established animal model for gastric carcinogenesis
(26,27). GC is one of the most devastating
cancers in humans and causes a particularly high mortality
worldwide (28). Achieving a
better understanding of the molecular mechanisms associated with GC
carcinogenesis might identify new diagnostic and treatment
strategies for this disease. Numerous genes have been established
to be involved in this disease, including tumor-suppressor genes
and oncogenes (p53, β-catenin), gene amplifications and deletions
(c-Meta, ERBB2), and anti-inflammatory factors (TGFα, TGFβ2)
(29,30), but the function of miRs in this
disease remains unclear.
We found that, compared with the control group, the
mRNA and protein levels of TGFβ2, SMAD2, and SMAD4 were increased
significantly in MNNG-initiated GC rats, but miR-148a levels were
reduced. However, the TGFα and SMAD3 expression levels were not
influenced by MNNG. Previously, studies have shown that SMAD2 is
the primary intracellular signaling pathway downstream of TGF-β
(29). TGF-β can act as an
oncogenic factor, and it is involved in proliferation,
angiogenesis, invasion, and metastasis (31). In glioma patients, high TGFβ-SMAD
activity was present in aggressive, highly proliferative gliomas,
and it conferred poor prognosis (32). Another research group reported that
activation of SMAD2 could induce the migration of mouse squamous
carcinoma cells (33).
Overexpression of SMAD2 was also suggested to be associated with
metastasis and was correlated with poor prognosis of GCs,
especially diffuse-type gastric carcinomas (34). In our study, TGFβ2 and SMAD2
expression levels were both elevated in MNNG-treated rats,
suggesting that TGFβ2 and SMAD2 were promising target genes that
were related to metastasis in GC cells.
Recently, evidence has convincingly shown that miRs
play an important role in many human cancers (8,14). A
previous miR profiling study that used tumor and normal GC tissues
found that approximately 80 miRs exhibited expression imbalance.
Among them, the tumor-suppressor miR-148a was one of the most
significantly downregulated miRNAs in GC cell lines and GC tissues
from GC patients compared with the adjacent normal gastric tissues
(35). However, the downstream
targets of this miRNA are still unclear. We identified the role of
miR-148a in regulating the expression of TGFβ2, SMAD2, and SMAD4.
The results indicated that overexpression of miR-148a could reduce
the mRNA levels of TGFβ2 and SMAD2 significantly, but not SMAD4.
These results were consistent with the immunofluorescence
cytochemistry data. Furthermore, we assayed luciferase activity
using human GC cells and found that overexpression of miR-148a
could inhibit the activities of the TGFβ2- and
SMAD2-3′UTRs. These data suggested that the binding sites of
miR-148a were functional. In this study, we demonstrated the
effects of miR-148a on proliferation, migration, and invasion in GC
cells. The growth of miR-148a-transfected cells was reduced
significantly compared with cells that were transfected with
negative-control miRs. Simultaneously, the miR-148a-transfected
cells exhibited the capacity for migration and invasion compared
with the controls in three GC cell lines. In vivo, miR-148a
also displayed an inhibited function to GC, which suggested that
miR-148a inhibited GC development via inhibiting TGFβ2 and
SMAD2.
In conclusion, to our knowledge, our study is the
first to functionally explore the role of miR-148a in GC
development and progression. Our data suggests that miR-148a may
act as a novel tumor-suppressor miR in GC. In addition to providing
the probable mechanism involved in the regulation of the expression
of TGFβ2 and SMAD2, our findings may also have translational
relevance as miRNAs have also been proposed to be potential
therapeutic candidates. By understanding the mechanism and function
of miR-148a as a tumor suppressor, it may eventually be possible to
develop miR-148a as a therapeutic agent in GC treatment.
Acknowledgements
This work was supported by the Key Project of the
National Natural Science Foundation of China (81230031/H18), the
National Science Foundation of China (61272274), and the Hubei
Province's Outstanding Medical Academic Leader Program.
Abbreviations:
|
TGFα
|
transforming growth factor α
|
|
GC
|
gastric cancer
|
|
TGFβ2
|
transforming growth factor-β2
|
|
MNNG
|
N-methyl-N9-nitro-N-nitrosoguanidine
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
UTR
|
untranslated region
|
|
M-MLV
|
moloney murine leukemia virus reverse
transcriptase
|
|
miR
|
microRNA
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Uemura N, Okamoto S, Yamamoto S, Matsumura
N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N and Schlemper RJ:
Helicobacter pylori infection and the development of gastric
cancer. N Engl J Med. 345:784–789. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li G, Wulan H, Song Z, Paik PA, Tsao ML,
Goodman GM, MacEachern PT, Downey RS, Jankowska AJ, Rabinowitz YM,
et al: Regulatory B cell function is suppressed by smoking and
obesity in H pylori-infected subjects and is correlated with
elevated risk of gastric cancer. PLoS One. 10:e01345912015.
View Article : Google Scholar
|
|
5
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bass AJ, Thorsson V, Shmulevich I,
Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C,
Shen H, et al; Cancer Genome Atlas Research Network. Comprehensive
molecular characterization of gastric adenocarcinoma. Nature.
513:202–209. 2014. View Article : Google Scholar :
|
|
7
|
Tan IB, Ivanova T, Lim KH, Ong CW, Deng N,
Lee J, Tan SH, Wu J, Lee MH, Ooi CH, et al: Intrinsic subtypes of
gastric cancer, based on gene expression pattern, predict survival
and respond differently to chemotherapy. Gastroenterology.
141:476–485. 485.e1–485.e11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lei Z, Tan IB, Das K, Deng N, Zouridis H,
Pattison S, Chua C, Feng Z, Guan YK, Ooi CH, et al: Identification
of molecular subtypes of gastric cancer with different responses to
PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology.
145:554–565. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang K, Kan J, Yuen ST, Shi ST, Chu KM,
Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al: Exome sequencing
identifies frequent mutation of ARID1A in molecular subtypes of
gastric cancer. Nat Genet. 43:1219–1223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hwang HW and Mendell JT: MicroRNAs in cell
proliferation, cell death, and tumorigenesis. Br J Cancer.
96(Suppl): R40–R44. 2007.PubMed/NCBI
|
|
14
|
Kent OA and Mendell JT: A small piece in
the cancer puzzle: microRNAs as tumor suppressors and oncogenes.
Oncogene. 25:6188–6196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gaur A, Jewell DA, Liang Y, Ridzon D,
Moore JH, Chen C, Ambros VR and Israel MA: Characterization of
microRNA expression levels and their biological correlates in human
cancer cell lines. Cancer Res. 67:2456–2468. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueda T, Volinia S, Okumura H, Shimizu M,
Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al:
Relation between microRNA expression and progression and prognosis
of gastric cancer: A microRNA expression analysis. Lancet Oncol.
11:136–146. 2010. View Article : Google Scholar
|
|
18
|
Li X, Zhang Y, Zhang Y, Ding J, Wu K and
Fan D: Survival prediction of gastric cancer by a seven-microRNA
signature. Gut. 59:579–585. 2010. View Article : Google Scholar
|
|
19
|
Petrocca F, Visone R, Onelli MR, Shah MH,
Nicoloso MS, de Martino I, Iliopoulos D, Pilozzi E, Liu CG, Negrini
M, et al: E2F1-regulated microRNAs impair TGFbeta-dependent
cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell.
13:272–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bandres E, Bitarte N, Arias F, Agorreta J,
Fortes P, Agirre X, Zarate R, Diaz-Gonzalez JA, Ramirez N, Sola JJ,
et al: microRNA-451 regulates macrophage migration inhibitory
factor production and proliferation of gastrointestinal cancer
cells. Clin Cancer Res. 15:2281–2290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oh HK, Tan AL, Das K, Ooi CH, Deng NT, Tan
IB, Beillard E, Lee J, Ramnarayanan K, Rha SY, et al: Genomic loss
of miR-486 regulates tumor progression and the OLFM4 antiapoptotic
factor in gastric cancer. Clin Cancer Res. 17:2657–2667. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carvalho J, van Grieken NC, Pereira PM,
Sousa S, Tijssen M, Buffart TE, Diosdado B, Grabsch H, Santos MA,
Meijer G, et al: Lack of microRNA-101 causes E-cadherin functional
deregulation through EZH2 up-regulation in intestinal gastric
cancer. J Pathol. 228:31–44. 2012.PubMed/NCBI
|
|
23
|
Chen Z, Saad R, Jia P, Peng D, Zhu S,
Washington MK, Zhao Z, Xu Z and El-Rifai W: Gastric adenocarcinoma
has a unique microRNA signature not present in esophageal
adenocarcinoma. Cancer. 119:1985–1993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng G, Xiong Y, Xu W, Wang Y, Chen F,
Wang Z and Yan Z: A two-microRNA signature as a potential biomarker
for early gastric cancer. Oncol Lett. 7:679–684. 2014.PubMed/NCBI
|
|
25
|
Sakamoto N, Naito Y, Oue N, Sentani K,
Uraoka N, Zarni Oo H, Yanagihara K, Aoyagi K, Sasaki H and Yasui W:
MicroRNA-148a is downregulated in gastric cancer, targets MMP7, and
indicates tumor invasiveness and poor prognosis. Cancer Sci.
105:236–243. 2014. View Article : Google Scholar
|
|
26
|
Yu S, Yang M and Nam KT: Mouse models of
gastric carcinogenesis. J Gastric Cancer. 14:67–86. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Q, Jie Z, Ye S, Li Z, Han Z, Wu J,
Yang C and Jiang Y: Genetic variations in miR-27a gene decrease
mature miR-27a level and reduce gastric cancer susceptibility.
Oncogene. 33:193–202. 2014. View Article : Google Scholar
|
|
28
|
Ang TL, Khor CJ and Gotoda T: Diagnosis
and endoscopic resection of early gastric cancer. Singapore Med J.
51:93–100. 2010.PubMed/NCBI
|
|
29
|
Li QL, Ito K, Sakakura C, Fukamachi H,
Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, et al: Causal
relationship between the loss of RUNX3 expression and gastric
cancer. Cell. 109:113–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kang SH, Bang YJ, Im YH, Yang HK, Lee DA,
Lee HY, Lee HS, Kim NK and Kim SJ: Transcriptional repression of
the transforming growth factor-beta type I receptor gene by DNA
methylation results in the development of TGF-beta resistance in
human gastric cancer. Oncogene. 18:7280–7286. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Derynck R, Akhurst RJ and Balmain A:
TGF-beta signaling in tumor suppression and cancer progression. Nat
Genet. 29:117–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bruna A, Darken RS, Rojo F, Ocaña A,
Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, et al:
High TGFbeta-Smad activity confers poor prognosis in glioma
patients and promotes cell proliferation depending on the
methylation of the PDGF-B gene. Cancer Cell. 11:147–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Oft M, Akhurst RJ and Balmain A:
Metastasis is driven by sequential elevation of H-ras and Smad2
levels. Nat Cell Biol. 4:487–494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shinto O, Yashiro M, Toyokawa T, Nishii T,
Kaizaki R, Matsuzaki T, Noda S, Kubo N, Tanaka H, Doi Y, et al:
Phosphorylated smad2 in advanced stage gastric carcinoma. BMC
Cancer. 10:6522010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xia J, Guo X, Yan J and Deng K: The role
of miR-148a in gastric cancer. J Cancer Res Clin Oncol.
140:1451–1456. 2014. View Article : Google Scholar : PubMed/NCBI
|