Introduction
Gastric cancer was the fourth most common malignancy
in the world in 2008 with approximately one million new cases
(1). Although gastric cancer
incidence has steadily declined by more than 80% over the past 50
years in most developed countries, such as North America and Europe
(1–3), gastric cancer is one of the most
significant health problems in developing countries. If diagnosed
in the early-stage, 90% of gastric cancer patients can survive for
more than 5 years after surgery (2), while the survival rate is
dramatically reduced for patients with local recurrent and
metastasized disease (3).
Currently, most gastric cancer patients are diagnosed in the
advanced stages of disease, which contribute to the high rate of
gastric cancer lethality in China as well as in most other
developing countries (4). Thus,
novel strategies in prevention, early diagnosis, and effective
control of gastric cancer progression is urgently needed. To this
end, better understanding of the molecular mechanisms of gastric
cancer development and progression could provide insightful
information to develop novel approaches for gastric cancer
treatment.
Chemokines are a family of small proteins (8–11 kDa)
that are divided into four groups according to the number and
spacing of the first two cysteine residues in their amino-terminal
end (C, CC, CXC and CX3C) (5,6).
They represent a large family of polypeptide signaling molecules,
originally characterized by their ability to promote the directed
chemo-taxis of leukocytes, and are known to play important roles in
inflammation and cancer (7). Of
these chemokines, CXC chemokines can be further subdivided into two
groups on the basis of presence or absence of an ELR motif
(glutamic acid, leucine and arginine) which precedes the first
cysteine residues in the protein. These CXC chemokines have been
implicated in the initiation and amplification of inflammatory
diseases (8). CXC chemokines are
known to bind to G-protein-coupled receptors (GPCR) mainly CXCR1
and CXCR2 which plays an important role in cancer progression and
metastasis (9). Many studies have
shown that chemokine receptor CXCR1/2 and their ligands played an
important regulatory role in the occurrence, proliferation, growth,
invasion, metastasis, angiogenesis and drug resistance of various
tumors (10), such as malignant
melanoma (11), breast (12), pancreatic (13), colon (14) and ovarian cancer (15).
CXCR1 is a cell surface receptor of interleukin-8
(IL-8 called CXCL8) (16,17). This molecule is shown to be
upregulated and associated with poor prognosis of gastric cancer
(18). Specifically, CXCR1 is a
class A, rhodopsin-like G protein-coupled receptor, the largest
class of integral membrane proteins responsible for cellular signal
transduction and targeted as drug receptors (19). Despite the potential importance in
cells, the molecular mechanism of CXCR1 signal transduction is
poorly understood due to the limited structural information
available (19). Furthermore, the
role and mechanism of CXCR1 in gastric cancer are still unclear.
Thus, in the present study, we aimed to detect the influence of
CXCR1 stable knockdown or overexpression on the gastric cancer cell
proliferation, migration and invasion, and the protein expression
related to the proliferation, growth, apoptosis (Bcl-2, Bax, cyclin
D1, EGFR and Ki-67), angiogenesis (VEGF), invasion and metastasis
(MMP-9, MMP-2 and E-cadherin) and the phosphorylation of AKT and
ERK1/2 before and after the silencing of CXCR1.
Materials and methods
Cell culture
Gastric cancer cell lines MKN45 and BGC823 were
obtained from the Type Culture Collection of Chinese Academy of
Sciences (Shanghai, China). The cells were cultured with Dulbecco's
modified Eagle's medium (DMEM; Gibco-BRL, Life Technologies,
Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS;
Gibco-BRL) and 1% penicillin/streptomycin (Invitrogen, Carlsbad,
CA, USA) at 37°C in 5% CO2 atmosphere.
CXCR1 RNA interference and stable
lines
RNA interference (RNAi) specific for CXCR1 were
designed with GenScript software (https://www.genscript.com/ssl-bin/app/RNAi, GenScript,
USA) using the human CXCR1 mRNA sequence (GenBank registration
number: NM_000634. 2). Three sequences were identified as specific
CXCR1-shRNA targeted sequences and confirmed by the homologous
analysis using the BLAST algorithm. In addition, scrambled shRNA
sequence was designed, which did not target specific gene coding
regions. Primers were synthesized by Invitrogen (Shanghai,
China).
CXCR1 shRNA and scramble shRNA sequence were cloned
into the pYr 1.1 plasmid vector (Yingrun Biotechnologies, Inc.,
Changsha, China). This vector contains human U6 promoter, encoded
green fluorescent protein (GFP) report gene, kanamycin resistance
gene as a marker in prokaryotic screening and neomycin resistance
gene as a marker in eukaryotic screening. MKN45 cells at 90–95%
confluence were transiently transfected using Lipofectamine™ 2000
(Invitrogen) with plasmids pYr-1.1-CXCR1-shRNA-1,
pYr-1.1-CXCR1-shRNA-2, pYr-1.1-CXCR1-shRNA-3 and
pYr-1.1-scramble-shRNA. After a 48-h culture in complete medium,
cells were further incubated in a conditioned medium contained G418
(800 μg/ml) for one month to select positive clones. Finally, cells
were screened and cultured in medium with 400 μg/ml of G418 to
establish the stable transfected cell lines (CXCR1-shRNA-MKN45 and
scramble-shRNA-MKN45). The positive clones were analyzed by
real-time RT-PCR and western blot analysis.
CXCR1 overexpression and stable
lines
The 5′-GAGACACTCAACAAGTATGTT-3′ (mature sense for
CXCR1-shRNA-3) sequence was inserted into the plasmid
pIRES2-ZsGreen1. The CXCR1 overexpression plasmid,
pIRES2-ZsGreen1-CXCR1, was purchased from Yingrun Biotechnologies.
Plasmid pIRES2-ZsGreen1 also contained the kanamycin and neomycin
resistance genes. BGC823 cells at 90–95% confluence were
transiently transfected using Lipofectamine 2000 with the plasmids
pIRES2-ZsGreen1-CXCR1 and pIRES2-ZsGreen1. After a 48-h culture in
complete medium, cells were further incubated in a conditioned
medium containing G418 (800 μg/ml) for one month to select positive
clones. Finally, cells were screened and cultured in medium with 40
μg/ml of G418 to establish the stable transfected cell lines
[pIRES2-ZsGreen1-CXCR1-BGC823 (CXCR1-BGC823) and
pIRES2-ZsGreen1-BGC823 (vacant-BGC823)]. The positive clones were
analyzed by real-time RT-PCR and western blot analysis.
CXCR1 RNAi rescue experiment
To verify that the knockdown of CXCR1 expression by
RNAi was efficient and that there were no off-target effects, we
constructed a vector containing a mutated CXCR1 gene
(pIRES2-ZsGreen1-CXCR1-Mut). Site-directed mutagenesis was used to
alter three bases (underline) in the CXCR1-shRNA-3 target sequence.
The sequence before the mutation was 5′-GAG ACA CTC AAC AAG TAT GTT-3, and the
sequence after the mutation was 5′-GAG ACA TTG AAT AAG TAT GTT-3′. Two primers
were synthesized as follows: CXCR1-Mut-forward:
5′-GTATGCTAGAAACTGAGACATTGAATAAGTATGTTGTGATCATCGC-3′.
CXCR1-Mut-rerverse:
5′-GCGATGATCACAACATACTTATTCAATGTCTCAGTTTCTAGCATAC-3′. The mutation
sequence was inserted into plasmid pIRES2-ZsGreen1.
pIRES2-ZsGreen1-CXCR1-Mut was sequenced and identified by
Invitrogen. pYr-1.1-CXCR1-shRNA-3 and pIRES2-ZsGreen1-CXCR1-mut
were both transfected into MKN45 cells, and the expression of CXCR1
was detected by western blot analysis.
MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cell
proliferation assay
Stably transfected cells were seeded into 96-well
plates at a density of 5,000 cells/well. MTT solution (20 μl) (5
mg/ml; Sigma Aldrich, St. Louis, MO, USA) was added after 24, 48
and 72 h. The culture was terminated 4 h of addition MMT solution,
and the supernatant in each well was carefully aspirated and
discarded. DMSO (150 μl) was added into each well, and the plate
was rotated for 10 min at room temperature. Absorbance was measured
at 568 nm using a microplate reader (Bio-Rad Laboratories,
Hercules, CA, USA).
Colony forming assay
Stable transfected cells were seeded into 6-well
culture plates (200 cells/well) and cultured for 2 to 3 weeks at
37°C in a 5% CO2 incubator. The medium was changed every
3 days. At the end of the experiment, cells were fixed for 15 min
with methanol, stained for 25 min with crystal violet, and the
number of clones was counted. Cloning efficiency = (colonies
formed/cells seeded) x 100%.
Cell cycle and apoptosis assay
Stable transfected cells were collected by
centrifugation and washed twice with ice-cold PBS. Cells were then
fixed with 70% cold ethanol overnight at 4°C. The next day, cells
were collected by centrifugation and washed with PBS before
incubation in 500 μl of PBS containing 50 μg/ml of propidium iodide
(PI; Sigma), 100 μg/ml of RNase A and 0.2% Triton X-100 in the dark
for 30 min at 4°C. Flow cytometric analysis was performed using a
flow cytometry system and ModFit software (BD Biosciences, San
Jose, CA, USA). The proportion of cells in the G0/G1, S, G2/M
phases of the cell cycle was obtained. Sub-G1 indicated cells that
were undergoing apoptosis.
Cell migration and invasion assays
In vitro cell migration and invasion
experiments were performed using Transwell inserts (Corning Costar,
Corning, NY, USA) with the polycarbonate membranes (8 μm pore size)
in 24-well plates. For the invasion assay, the filter was coated
with 20 μg/ml of Matrigel (Becton-Dickinson) overnight at 4°C.
Stable transfected cells (100 μl) at a concentration of
1.0×105/ml in serum-free medium were seeded into the
upper chamber, and 800 μl of culture medium containing 10% FBS was
placed in the lower chamber. After a 12-h incubation at 37°C with
5% CO2, non-invaded cells were removed from the upper
surface of the membrane by gently scraping with a cotton swab, and
the invading cells on the lower surface of the membrane were fixed
with methanol, stained with crystal violet solution, rinsed with
water and air-dried. The invading cells were viewed under an
inverted microscope (Olympus, Tokyo, Japan) and ten fields
(magnification, x200) photographed and counted. The same
experimental conditions were used for the migration assay with the
exception that the chambers were not coated with Matrigel.
Wound-healing assay
Stable transfected cells were seeded into 6-well
plates at a density of 1×105/well and cultured until a
confluent monolayer formed. The tip of a 200-μl pipette was then
used to draw a wound at the bottom of each well. The cells were
washed with PBS and then further cultured for 24, 48 and 72 h. The
wounded areas were photographed at x10 magnification using
computer-assisted microscopy (Olympus IX70; Olympus). Images were
captured at 0, 24, 48 and 72 h after the scratch was made. The
distance cells migrated was measured and analyzed using
CellProfiler 2.0 cell image analysis software (http://www.cellprofiler.org).
Real-time RT-PCR and RT-PCR
Total RNA was isolated from cells using TRIzol
(Invitrogen), and the purity and concentration was measured
spectrophotometrically. Single-stranded cDNA was synthetized using
a First Strand cDNA Synthesis kit (Fermentas, Amherst NY, USA) with
Oligo(dT)18 and M-MuLV reverse transcriptase. The primers are shown
in Table I. The primers specific
for CXCR1 were purchased from the GeneCopoeia, Inc. (Guangzhou,
China; cat. no: HQP009679). All real-time PCR reactions were
performed using SYBR-Green Master Mix kit (Thermo Fisher
Scientific, Rockville, MD, USA) using an ABI real-time PCR system.
GADPH was used as the internal reference. Reactions were performed
using the following conditions: denaturation at 95°C for 10 min, 40
cycles at 95°C for 10 sec and 60°C for 20 sec. The
2−ΔΔCt method was used to calculate the relative mRNA
expression of target genes.
 | Table IPrimer sequences for RT-PCR or
real-time RT-PCR. |
Table I
Primer sequences for RT-PCR or
real-time RT-PCR.
| Gene | Forward primer
sequence | Reverse primer
sequence | Product length
(bp) |
|---|
| β-actin |
5′-TGACGTGGACATCCGCAAAG-3′ |
5′-CTGGAAGGTGGACAGCGAGG-3′ | 205 |
| GAPDH |
5′-TGAACGGGAAGCTCACTGG-3′ |
5′-TCCACCACCCTGTTGCTGTA-3′ | 307 |
| Bax |
5′-CCCGAGAGGTCTTTTTCCGAG-3′ |
5′-CCAGCCCATGATGGTTCTGAT-3′ | 155 |
| Bcl-2 |
5′-CCTGGGCAATTCCGCATT-3′ |
5′-AACAGGCCACGTAAAGCAAC-3′ | 158 |
| Cyclin D1 |
5′-GCTGCGAAGTGGAAACCATC-3′ |
5′-CCTCCTTCTGCACACATTTGAA-3′ | 135 |
| EGFR |
5′-AGGCACGAGTAACAAGCTCAC-3′ |
5′-ATGAGGACATAACCAGCCACC-3′ | 177 |
| VEGF |
5′-ATTATGCGGATCAAACCTC-3′ |
5′-ATTTCTTGCGCTTTCGTT-3′ | 157 |
| MMP-9 |
5′-ACTACTGTGCCTTTGAGTCC-3′ |
5′-AGAATCGCCAGTACTTCCCA-3′ | 115 |
| MMP-2 |
5′-ACTCTGGACTTAGACCGCTTG-3′ |
5′-ACAGGTTGCAGCTCTCCTTG-3′ | 217 |
| E-cadherin |
5′-GCTAACGTCGTAATCACCAC-3′ |
5′-AATGCCATCGTTGTTCACTG-3′ | 141 |
| Ki-67 |
5′-AGAAGACCTGCTACTCCAAAGA-3′ |
5′-AGTTTGCGTGGCCTGTACTAA-3′ | 70 |
For RT-PCR, the cDNA template was amplified with Taq
DNA polymerase (Tiangen Biotech Co., Ltd., Beijing, China). The
reaction conditions used were denaturation at 94°C for 3 min; 35
cycles of denaturation at 94°C for 30 sec, annealing at 55–60°C for
30 sec, extension at 72°C for 30 sec; followed by an extension step
at 72°C for 3 min. PCR products were electrophoresed on a 1.5% of
agarose gel. β-actin was used as the internal control.
Western blot analysis
Stable transfected cells were lysed with lysis
buffer (20 mM of Tris, pH 7.4, 150 mM of NaCl, 2 mM of EDTA and 1%
Triton X-100) containing the protease inhibitor,
phenylmethylsulfonyl fluoride (PMSF). Cells were centrifuged at
12,000 rpm for 12 min at 4°C, and the supernatant collected. Equal
amounts of protein (50 μg), quantified by BCA protein assay kit
(Pierce Biotechnology, Rockford, IL, USA), were separated by sodium
dodecyl sulfate-polyacrylamide electrophoresis (SDS-PAGE, 8–12%).
After electrophoresis, the separated proteins were
electrophoretically transferred to polyvinylidene fluoride (PVDF)
membranes (Millipore). Membranes were incubated in blocking
solution containing 5% non-fat milk for 1.5 h followed by
incubation with the following primary antibodies against
p-ERK-Thr202/Tyr204, p-AKT-Ser473 and AKT, (Anbo Biotechnology,
Co., Ltd., San Francisco, CA, USA), and β-actin, CXCR1, Bcl-2, Bax,
cyclin D1, EGFR, Ki-67, VEGF, MMP-9, MMP-2, E-cadherin, and GAPDH
(Santa Cruz Biotechnology, Santa Cruz, CA, USA). Antibodies were
used at a 1:500 dilution in TBST buffer overnight at 4°C. After
three washes, the blots were subsequently incubated with the
appropriate horseradish peroxidase-conjugated secondary antibody
(1:2,000 diluted in TBST buffer; Beijing Zhongshan Golden Bridge
Biotechnology, Co., Ltd., Beijing, China) for 1 h at room
temperature. GADPH used as a loading control. Proteins were
detected using the enhanced chemiluminescence (ECL substrate;
Pierce Biotechnology).
Gastric carcinoma xenograft mouse
model
Thirty-six female BALB/c nude mice (4–6 weeks old
and 18–22 g) were purchased from the Institute of Zoology, Chinese
Academy of Sciences (Beijing, China) and housed at the Animal Care
Center of the Third Hospital of Xiang-Ya, Central South University
(Changsha, China). All experimental procedures were approved by the
Animal Care and Ethics Committee of the Third Hospital of Xiang-Ya
(no. 2012–10).
Mice were randomly divided into 6 groups:
CXCR1-shRNA-MKN45 group, scramble-shRNA-MKN45 group, MKN45 group,
CXCR1-BGC823 group, vacant-BGC823 group and BGC823 group using a
random number table. Cell suspension containing 2×107
cells in 0.2 ml was injected into the right back region of each
nude mouse. Tumor volume was measured using digital calipers once
every two days: V = (a × b2) × 0.5 (a for length
diameter, b for short diameter). The health and activity of the
nude mice was monitored. Three weeks after the cell
transplantation, the mice were euthanized under anesthesia and the
transplantation tumor was cut and weighed. The tumor was fixed in
10% formaldehyde, embedded in paraffin, stained with H&E and
observed under an inverted microscope.
Statistical analysis
The SPSS 13.0 software system (SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis. Data are expressed as
mean ± standard deviation (SD) from at least 3 independent
experiments. Data were analyzed using one-way analysis of variance
(ANOVA) with Turkey's test for post hoc analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
Establishment of gastric cancer cell
lines with stable knockdown or overexpression of the CXCR1
gene
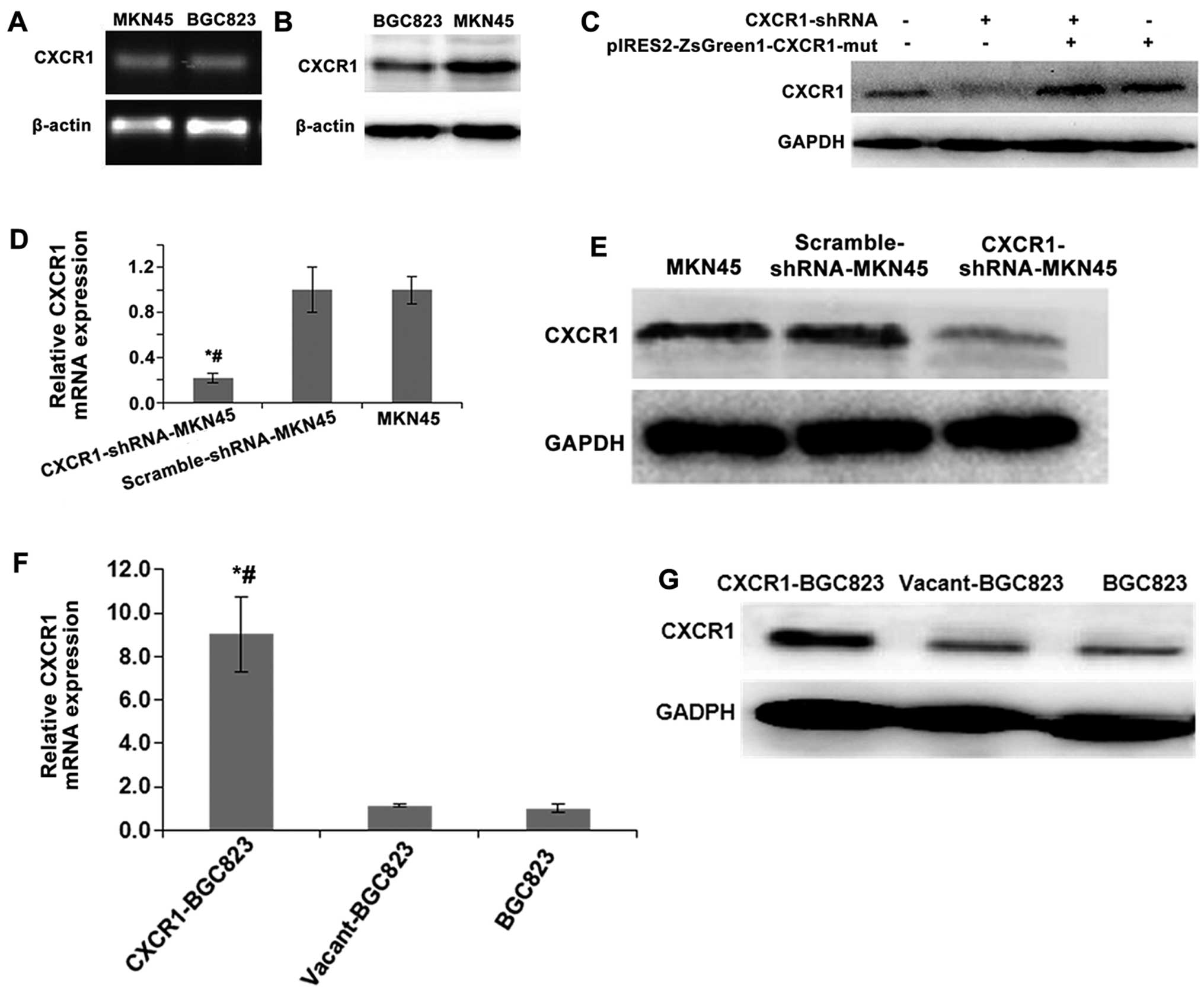
CXCR1 mRNA and protein expression levels revealed
relatively higher expression in MKN45 cells and lower expression in
BGC823 cells (Fig. 1A and B). In
the subsequent experiments, the MKN45 cells were stably transfected
with the CXCR1 shRNA, and BGC823 cells were used to overexpress the
CXCR1 gene.
MKN45 cells were transfected with the three plasmid
vectors pYr-1.1-CXCR1-shRNA1 (CXCR1-shRNA1), CXCR1-shRNA2 and
CXCR1-shRNA3 to test the efficiency of CXCR1 knockdown by RT-PCR
and western blot analysis. Based on the results plasmid vector
pYr-1.1-CXCR1 shRNA-3 (CXCR1-shRNA3) was chosen for the subsequent
experiments.
To verify that the knockdown of CXCR1 expression by
RNAi was efficient and that no off-target effects existed, we
performed CXCR1 RNAi rescue experiments. CXCR1 protein levels in
cells transfected with the pIRES2-ZsGreen1-CXCR1-Mut were not
different from those transfected with both the mutant and
pYr-1.1-CXCR1-shRNA3 (CXCR1-shRNA), but CXCR1 protein levels in
cells transfected with CXCR1-shRNA alone were obviously reduced
compared to the non-transfected MKN45 cells (Fig. 1C). These results suggested that the
CXCR1-shRNA-mediated RNA interference of the CXCR1 gene in MKN45
cells did not have off-target effects.
CXCR1-shRNA and the control plasmid vector,
pYr-1.1-CXCR1-scramble shRNA (scramble-shRNA), were stably
transfected into MKN45 cells, respectively. One month after the
screening with G418, CXCR1 mRNA and protein levels were measured in
CXCR1-shRNA and scramble-shRNA cell clones by real-time RT-PCR and
western blot analysis (Fig. 1D and
E). CXCR1 expression was lower in CXCR1-shRNA cells than
non-transfected MKN45 cells and scramble-shRNA cells (all
P<0.05). CXCR1mRNA and protein levels were also measured in
BGC823 cells after stable transfection with the expression plasmid,
pIRES2-ZsGreen1-CXCR1 (CXCR1-BGC823) and pIRES2-ZsGreen1
(vacant-BGC823) (Fig. 1F and G).
CXCR1 mRNA and protein levels in CXCR1-BGC823 cell clones were
higher than those in the vacant-BGC823 or non-transfected BGC823
cells (all P<0.05).
Effects of CXCR1 stable knockdown or
overexpression on the proliferation and growth of gastric carcinoma
cells
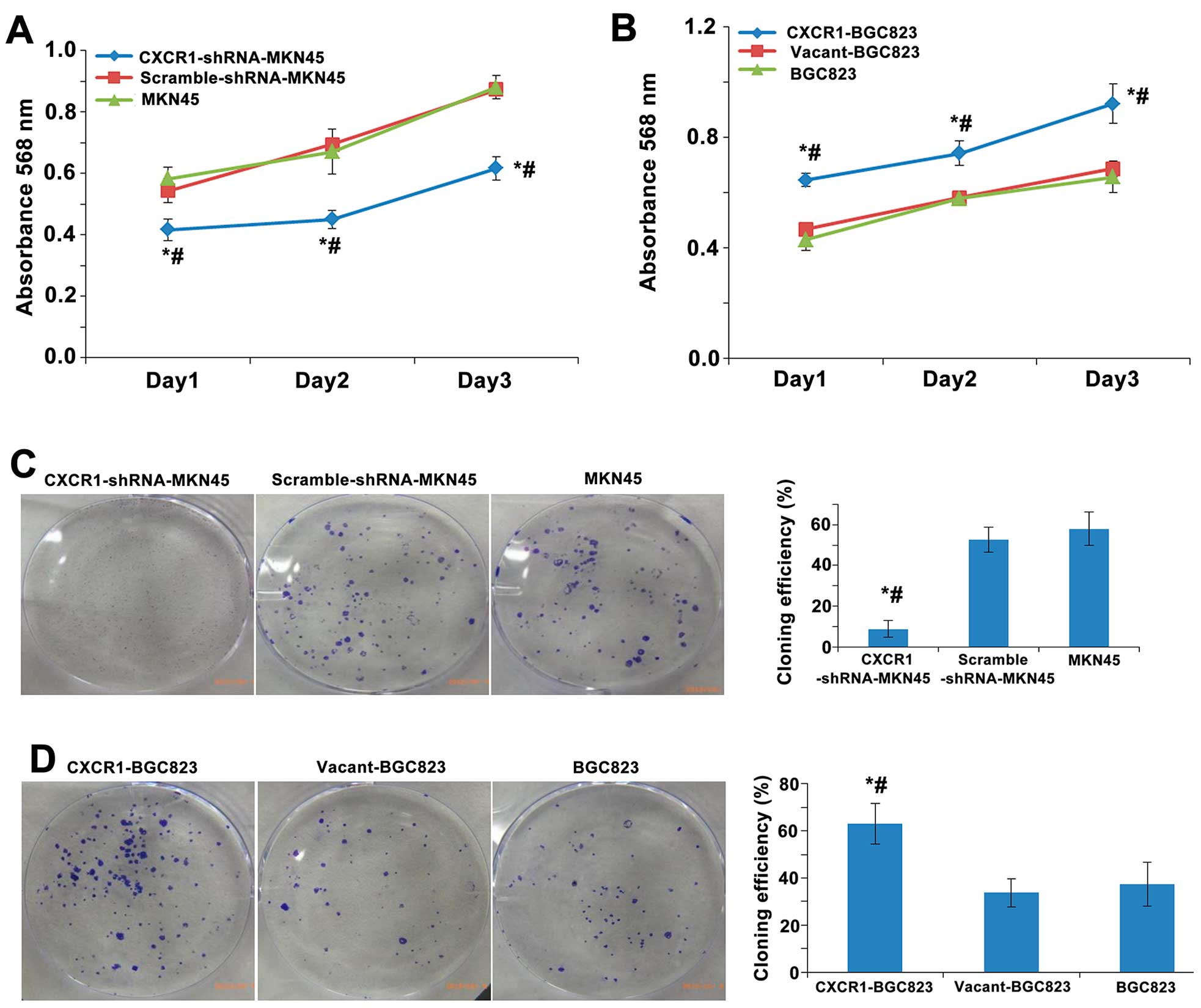
Cell proliferation indicated as absorbance over a
72-h period was measured using the MTT assay in MKN45 cells stably
transfected with CXCR1-shRNA, scrambled-shRNA or non-transfected
cells. Cell proliferation was lower in the CXCR1-shRNA-MKN45 group
than the MKN45 and scramble-shRNA-MKN45 groups over a 72-h period
(all P<0.05) (Fig. 2A).
However, stable overexpression of CXCR1 in the CXCR1-BGC823 cells
promoted cell proliferation and growth significantly above the
BGC823 and vacant-BGC823 groups over a 72-h period (all P<0.05)
(Fig. 2B). There was no
significant difference in the proliferation or growth between the
MKN45 and vcramble-shRNA-MKN45 cells, or between the BGC823 and
vacant-BGC823 cells (all P>0.05) (Fig. 2A and B).
In order to further establish the role of CXCR1
stable knockdown or overexpression on the proliferation and growth
of gastric carcinoma cells, the proliferative capacity of single
cells was measured using a plate colony formation assay. The
cloning efficiency in the CXCR1-shRNA-MKN45 group was significantly
lower than the MKN45 and scramble-shRNA-MKN45 groups (both
P<0.05). No significant difference in cloning efficiency was
detected between scramble-shRNA-MKN45 and non-transfected MKN45
cells (Fig. 2C). The cloning
efficiency of CXCR1-BGC823 cells was significantly higher than the
BGC823 and vacant-BGC823 cells (both P<0.05) and no significant
differences were found in the cloning efficiency between
vacant-BGC823 and non-transfected BGC823 cells (Fig. 2D).
Effects of CXCR1 stable knockdown or
overexpression on cell cycle distribution and apoptosis of gastric
carcinoma cells
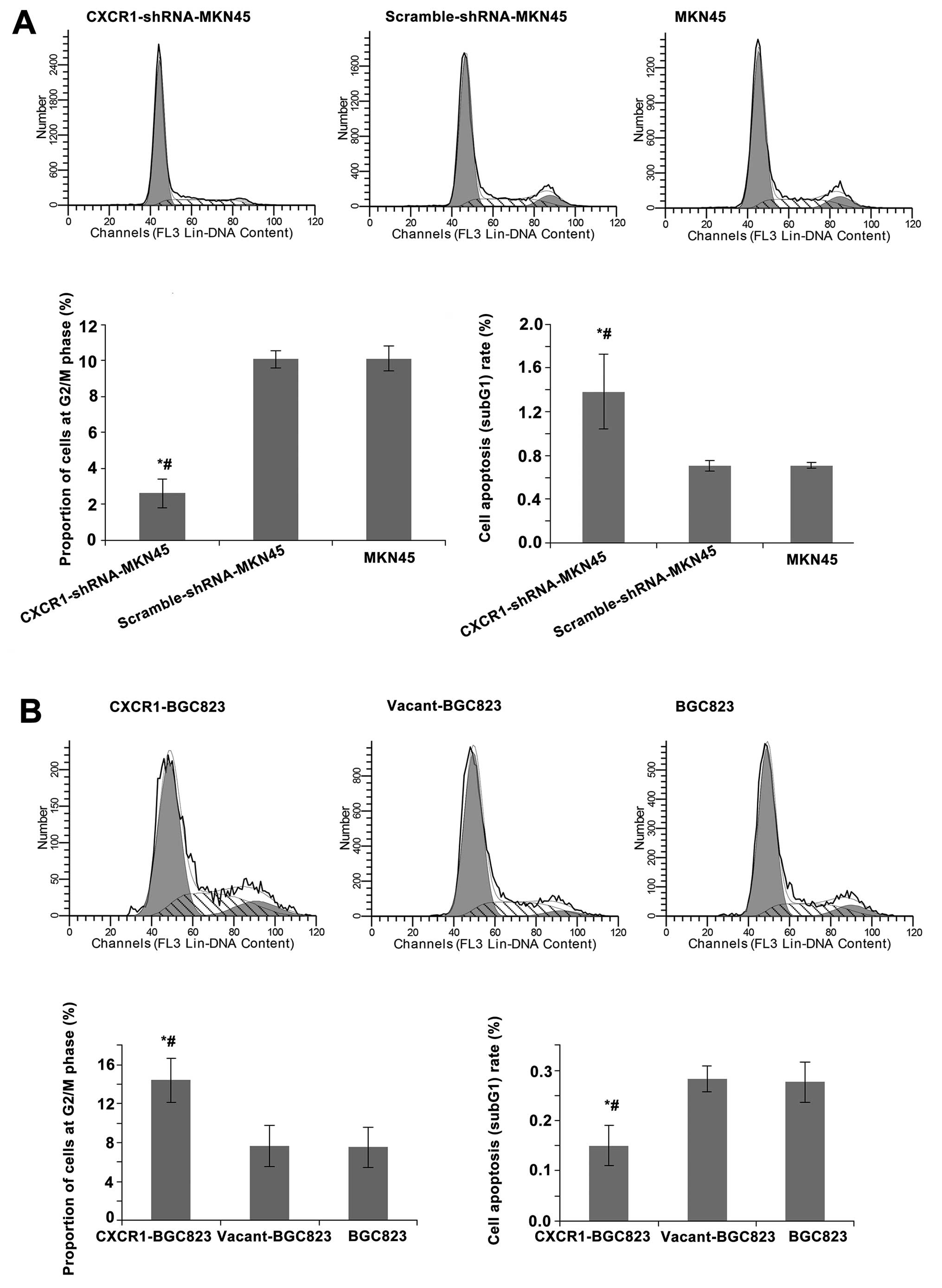
To investigate the effects of CXCR1 stable knockdown
or overexpression on cell cycle distribution and apoptosis, cells
were analyzed by flow cytometry using PI. The proportion of MKN45
cells at the G2/M phase significantly decreased in the
CXCR1-shRNA-MKN45 group (both P<0.05) and the percentage of
apoptotic cells increased significantly (both P<0.05) relative
to the control groups, MKN45 and scramble-shRNA-MKN45. There was no
significant difference in the proportion of cells at the G2/M phase
or percentage of apoptotic cells between the scramble-shRNA-MKN45
and MKN45 groups (Fig. 3A). The
proportion of BGC823 cells at the G2/M phase significantly
increased in the CXCR1-BGC823 group (both P<0.05) and the
percentage of apoptotic cells significantly decreased (both
P<0.05) relative to the control groups, vacant-BGC823 and
BGC823. There was no significant difference in the proportion of
cells at the G2/M phase or percentage of apoptotic cells between
the vacant-BGC823 and BGC823 groups (Fig. 3B).
Effects of CXCR1 stable knockdown or
overexpression on the migration and invasion of gastric carcinoma
cells
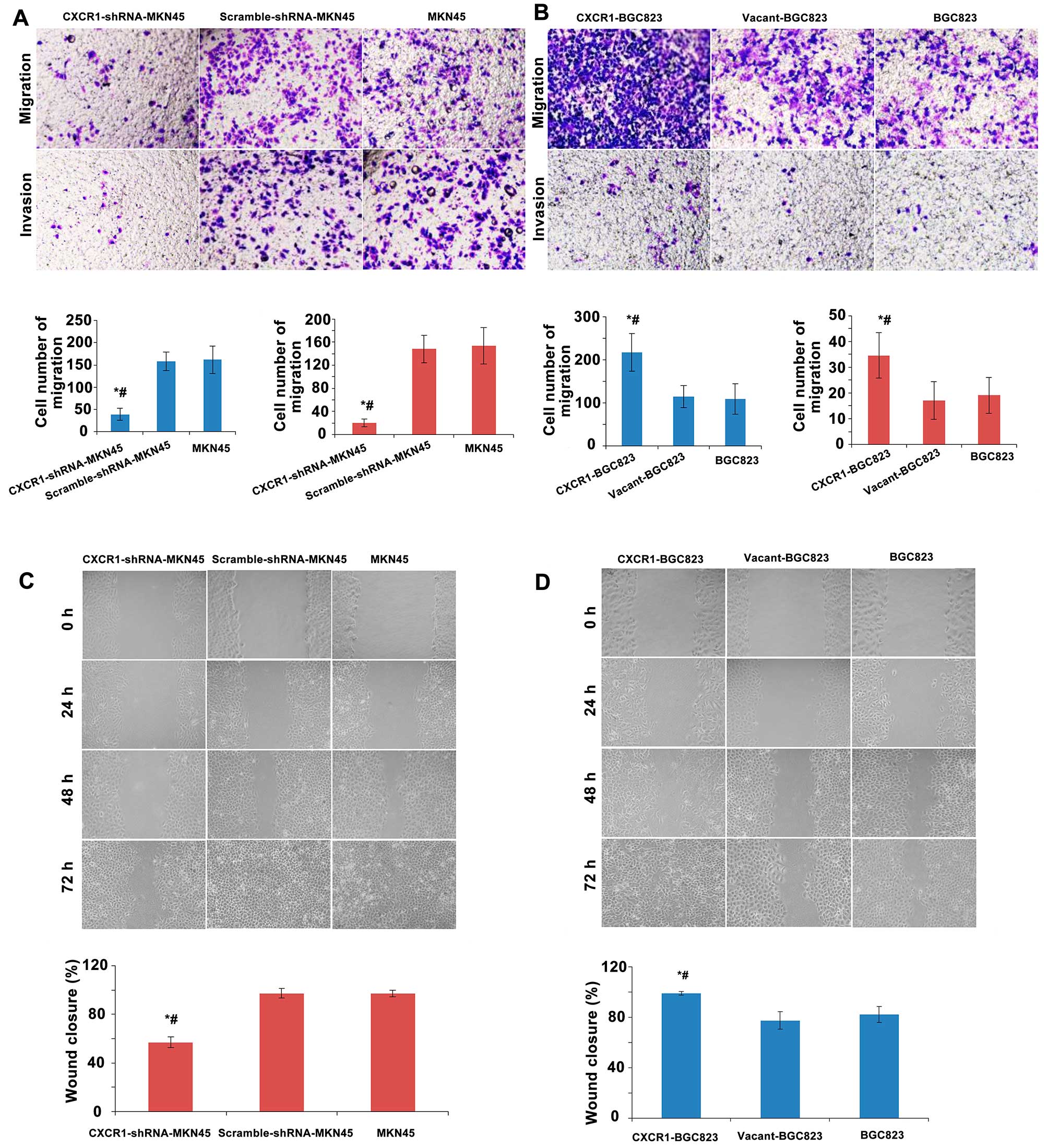
To investigate the effects of CXCR1 stable knockdown
or overexpression on the migration and invasion of gastric
carcinoma cells, a Transwell migration/invasion assay was used.
CXCR1-shRNA-MKN45 cells revealed a decrease in the number of cells
that migrated (both P<0.05) or invaded (both P<0.05) the
Transwell membrane relative to non-transfected MKN45 or
scramble-shRNA-MKN45 cells (Fig.
4A). There was no significant difference in the number of cells
that migrated and invaded through the Transwell membrane between
the scramble-shRNA-MKN45 and the MKN45 groups (Fig. 4A). BGC823 cells overexpressed CXCR1
(CXCR1-BGC823 group) showed an increase in the number of cells that
migrated (both P<0.05) or invaded (both P<0.05) the Transwell
membrane relative to non-transfected BGC823 cells or the
vacant-BGC823 cells (Fig. 4B).
There was no significant difference in the number of cells that
migrated or invaded the Transwell membrane between the BGC823 and
vacant-BGC823 groups (Fig.
4B).
In order to further investigate the effect of CXCR1
stable knockdown or overexpression on the migration and invasion of
gastric carcinoma cells, wound-healing assay was conducted. The
wound closure rate of CXCR1-shRNA-MKN45 cells was significantly
less than non-transfected MKN45 cells and scramble-shRNA-MKN45
cells (both P<0.05). No obvious differences in the wound closure
rates between the scramble-shRNA-MKN45 cells and non-transfected
MKN45 cells were detected (Fig.
4C). The rate of wound closure of CXCR1-BGC823 cells was
significantly higher than non-transfected BGC823 cells and the
vacant-BGC823 cells (both P<0.05). Wound closure rates of the
vacant-BGC823 and BGC823 groups were not different (Fig. 4D).
Effect of CXCR1 stable knockdown or
overexpression on the in vivo tumor growth of gastric carcinoma
cells
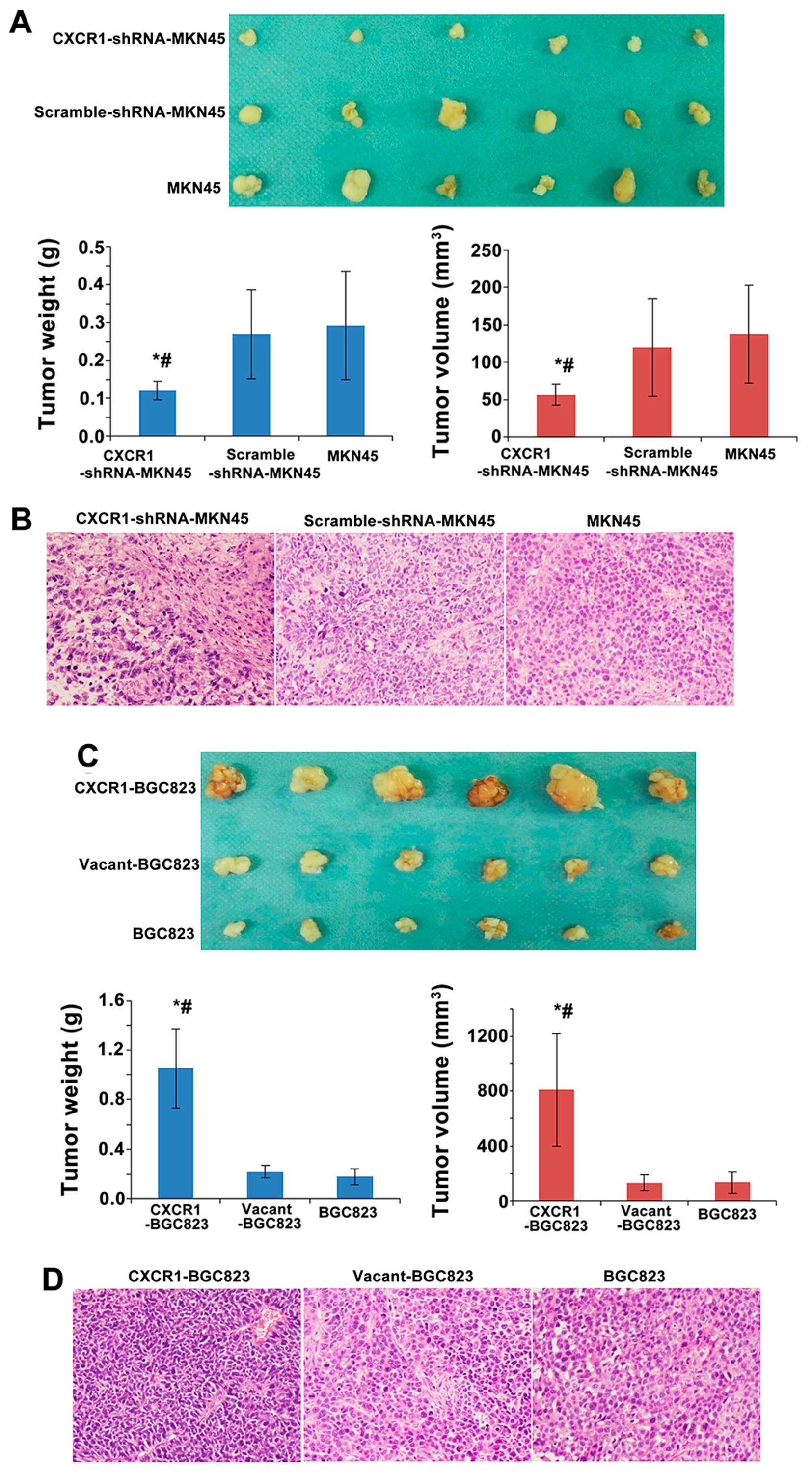
To investigate the effect of CXCR1 stable knockdown
or over-expression on the ability of gastric carcinoma cells to
form tumors, gastric carcinoma cells were transplanted into nude
mice. The weight and volume of the tumors formed in nude mice
injected with CXCR1-shRNA-MKN45 cells was significantly smaller
than mice transplanted with control, MKN45 cells or
scramble-shRNA-MKN45 cells (both P<0.05). The weight and volume
of the xenografts in nude mice were not different between the MKN45
and the scramble-shRNA-MKN45 groups (Fig. 5A). H&E staining confirmed that
the xenografts were composed of cancer cells rather than abscesses
(Fig. 5B). Furthermore, in nude
mice transplanted with CXCR1-BGC823 cells, the weight and volume of
the resulting tumors was significantly larger (both P<0.05) than
mice transplanted with BGC823 cells or vacant-BGC823 cells. There
was no significant difference in the weight and volume of
xenografts in nude mice between the BGC823 and vacant-BGC823 groups
(Fig. 5C). H&E staining
confirmed that the xenografts resulting from CXCR1 overexpressing
cells were composed of cancer cells rather than abscesses (Fig. 5D).
Effects of CXCR1 stable knockdown on the
levels of AKT and ERK1/2 phosphorylation and markers of apoptosis,
proliferation and growth, angiogenesis, invasion and metastasis
related signaling molecules in MKN45 cells
In order to determine whether the AKT and ERK1/2
signal pathways were activated by CXCR1 receptor/ligand
interaction, phosphorylation of AKT and ERK1/2 was measured in the
MKN45 cells stably transfected with a CXCR1 specific shRNA.
Phosphorylated AKT (p-AKT) and p-ERK1/2 levels, detected by western
blot analysis, were lower in the CXCR1-shRNA-MKN45 group than the
MKN45 and scramble-shRNA-MKN45 groups, but total AKT levels were
not different among the three groups. There was no difference in
the phosphorylated and total AKT and ERK1/2 levels between scramble
shRNA-MKN45 and MKN45 groups (Fig.
6A).
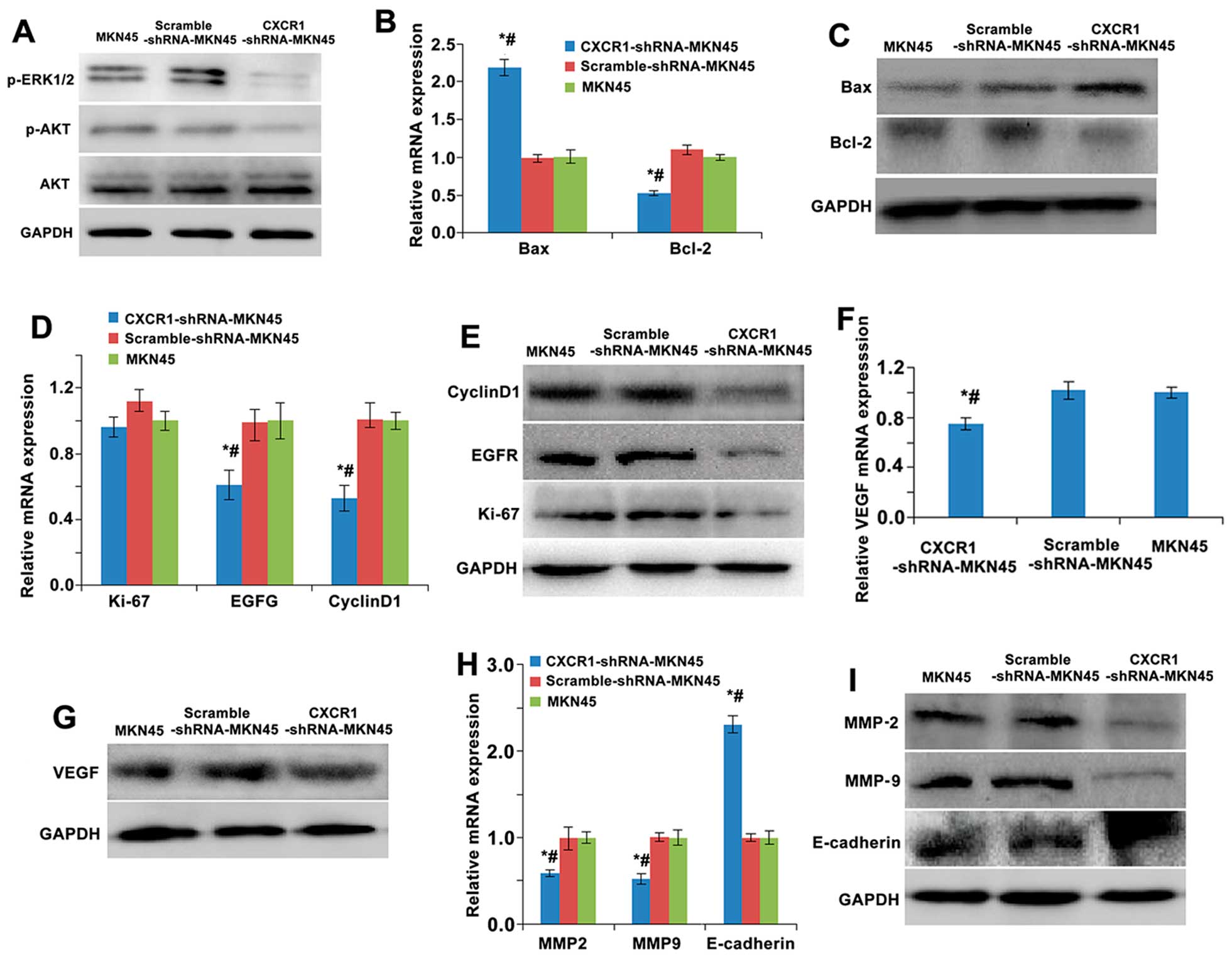 | Figure 6Effects of CXCR1 stable knockdown in
MKN45 cells on cell proliferation, cell cycle, cell apoptosis, cell
migration and invasion-related signaling molecule expression. (A)
Phosphorylation of AKT and ERK1/2 was determined by western blot
analysis. mRNA expression of apoptosis (Bcl-2 and Bax) (B),
proliferation and growth (cyclin D1, EGFR and Ki-67) (D),
angiogenesis (VEGF) (F), invasion and metastasis (MMP-9, MMP-2 and
E-cadherin) (H) was determined by real-time RT-PCR. Protein
expression of apoptosis (Bcl-2 and Bax) (C), proliferation and
growth (cyclin D1, EGFR and Ki-67) (E), angiogenesis (VEGF) (G),
invasion and metastasis (MMP-9, MMP-2 and E-cadherin) (I) were
determined by western blotting. GAPDH was used as a loading
control. Data are shown as mean ± SD. *P<0.05 vs.
MKN45; #P<0.05 vs. scramble-shRNA-MKN45. |
To investigate the mechanism by which CXCR1
regulates apoptosis, we measured the expression of apoptotic genes
(Bax and BCL-2) by real-time RT-PCR and western blot analysis. The
expression of Bcl-2 in the CXCR1-shRNA-MKN45 group was lower than
the levels in the MKN45 and scramble-shRNA-MKN45 groups. In
contrast the CXCR1 shRNA increased the expression of Bax. No
difference in the expression of Bax and Bcl-2 were detected between
the scramble-shRNA-MKN45 and MKN45 groups (Fig. 6B and C).
To elucidate the potential mechanism by which CXCR1
shRNA inhibits the proliferation of gastric carcinoma cells, the
expression of proliferation, and growth- and cell cycle-related
genes (cyclin D1, EGFR and Ki-67) were evaluated. Real-time RT-PCR
revealed lower EGFR and cyclin D1 mRNA levels in CXCR1-shRNA-MKN45
cells compared to MKN45 and scramble-shRNA-MKN45 cell groups (all
P<0.05). The level of Ki-67 was not altered by CXCR1 RNA
interference. Furthermore, cyclin D1, EGFR and Ki-67 mRNA levels
were not different between scramble-shRNA-MKN45 and MKN45 cells
(Fig. 6D and E). Western blots
showed that the expression of cyclin D1, EGFR and Ki-67 in
CXCR1-shRNA-MKN45 cells were significantly lower than in the MKN45
and scramble-shRNA-MKN45 groups. Cyclin D1, EGFR and Ki-67 protein
levels were not different between the scramble-shRNA-MKN45 and
MKN45 groups (Fig. 6D and E).
As an indicator of tumor angiogenesis, VEGF mRNA and
protein levels were measured by real-time RT-PCR and western blot
analysis, respectively, and were found lower in CXCR1-shRNA-MKN45
cells than in the MKN45 and scramble-shRNA-MKN45 groups. No
differences were found in the expression of VEGF between the
scramble-shRNA-MKN45 and MKN45 groups (Fig. 6F and G).
To further understand the possible mechanism by
which inhibition of CXCR1 expression could inhibit the invasion and
metastasis of gastric carcinoma cells, the expression of several
markers of cell invasion and metastasis (MMP-9, MMP-2 and
E-cadherin) were measured. In CXCR1-shRNA-MKN45 group the
expression of MMP-2 and MMP-9 at both the mRNA and protein levels
were lower and E-cadherin higher than the MKN45 and
scramble-shRNA-MKN45 groups. No differences in the expression of
MMP-9, MMP-2 or E-cadherin were observed between the
scramble-shRNA-MKN45 and MKN45 groups (Fig. 6H and I).
Discussion
The association of high CXCR1 levels with several
types of aggressive and invasive cancers warrants a more complete
understanding of the function of this pro-inflammatory chemokine in
permissive tumor cell environments (10). This is especially true in the
gastrointestinal tract where chronic states of inflammation may
promote the transformation of a cancer cell phenotype. In order to
elucidate the functional role of CXCR1 in the development of
gastric cancer, both loss of function and gain of function
approaches were used to characterize gastric carcinoma cell
proliferation, invasion and migration in vitro. This was
accomplished by developing stable transfected cell lines containing
a CXCR1-shRNA to inhibited CXCR1 expression, or CXCR1 coding
sequence to overexpress the gene. These well characterized high and
low CXCR1 expressing gastric carcinoma cells lines were then used
as xenografts in nude mice to test their potential for tumor
formation. The data collected from these experiments clearly
indicate that CXCR1 expression promotes the proliferation and
growth, and enhances the migration and invasion properties of
gastric carcinoma cells.
To establish a cell line with CXCR1 loss-of-function
using RNAi technology, several steps were incorporated to design
and test shRNA that both were efficient and specific. The first
step was to choose a cell system with high CXCR1 expression to
characterize the shRNA. MKN45 cells with relatively higher CXCR1
expression were chosen for the RNA interference experiments. Next,
three shRNA fragments were successfully sub-cloned into the plasmid
pYr 1.1. All three shRNA tested in transient transfection assays
suppressed CXCR1 expression, and pYr-1.1-CXCR1-shRNA-3 was chosen
for stable transfections. Subsequent G418 screening, real-time
RT-PCR and western blot testing were used to establish a stable
CXCR1-shRNA-MKN45 cell line, which provided a tool for the
subsequent research on the function of CXCR1.
To verify that the knockdown of CXCR1 expression by
RNAi was efficient and that no off-target effects existed, we
carried out CXCR1 RNAi rescue experiments, as first proposed by
Jackson and colleagues (20).
Cells transfected with a single gene-specific siRNA have been
reported to interfere with the expression of a large number of
non-specific genes in genome wide screens with altered expression
levels of 1.5- to 3-fold (21). To
verify there were no off-target effects in our experiment, we
mutated 3 bases according to the target sites of the CXCR1 gene to
prevent the interference sequence of pYr-1.1-CXCR1-shRNA-3. The
pYr-1.1-CXCR1-shRNA-3 (CXCR1-shRNA) and pIRES2-ZsGreen1-CXCR1-Mut
were transfected at the same time into MKN45 cells. Compared with
the pIRES2-ZsGreen1-CXCR1-Mut alone group, the expression of CXCR1
in MKN45 cells transfected with both the pIRES2-ZsGreen1-CXCR1-Mut
and CXCR1-shRNA did not interfere with CXCR1 expression and
suggested that there were no off-target effects in our
experiment.
The occurrence and development of gastric cancer is
complex and involves aberrant regulation of proliferation, invasion
and metastasis of cancer cells. To investigate the role of CXCR1 in
the development of gastric cancer, we observed the effect of CXCR1
knockdown or overexpression on the proliferation, cell cycle
distribution, and apoptosis of gastric carcinoma cells by MTT,
plate colony formation and flow cytometry assays. The results
showed CXCR1 knockdown inhibited cell proliferation and growth.
This was accompanied by G2/M phase arrest and cell apoptosis.
Importantly, CXCR1 stable knockdown prevented the in vivo
formation of tumors when cells were transplanted into mice. The
opposite result was observed when CXCR1 overexpressing cells were
used as xenografts, and much larger tumors were observed in the
nude mice. Thus, CXCR1 participates in the development of gastric
tumors by inhibiting apoptosis and promoting the proliferation and
growth of gastric cancer cells. The results were consistent with
the role of CXCR1 in other cancers (11–15).
Invasion and metastasis are also the important manifestations of
gastric cancer and often the reason for poor patient prognosis.
Many factors also regulate the invasion and metastasis of cancer
cells. In order to explore the role of CXCR1 in gastric cancer cell
invasion and metastasis, the ability of cells to transverse
non-coated and Matrigel-coated membranes was tested in a Transwell
system. This experiment was complimented with wound-healing assay
to monitor cell migration rates. Results from these in vitro
measurements, again using knockdown and overexpression cell lines,
showed that CXCR1 knockdown inhibited the migration and invasion
properties of gastric carcinoma cells while the overexpression of
CXCR1 increased the migration and invasion potential of cancer
cells. These results are in line with the findings from the recent
study (22).
To further explore the mechanism by which CXCR1
regulates altered proliferation, growth, apoptosis, invasion and
migration in loss-of-function and gain-of-function culture models,
the expression of several key genes involved in these cellular
functions was measured. Previous studies have found that the
abnormal phosphorylation of AKT and ERK1/2 was associated with the
proliferation, growth, angiogenesis, invasion and metastasis of
gastric cancer (23,24), and was regarded as the bridge
between CXCR1/2 and downstream molecules (10,25,26).
Our results confirm this finding and show that CXCR1 knockdown
suppressed the phosphorylation of AKT and ERK1/2. This finding
suggests CXCR1 receptor/ligand interactions might regulate the AKT
and ERK1/2 signaling pathway. The ratio of Bcl-2 to Bax of Bcl-2
family are key indicators of tumor apoptosis with high expression
of Bcl-2 and low expression of Bax considered to inhibit apoptosis
of cancer cells (27). In the
present experiments CXCR1 knockdown induced apoptosis of gastric
cancer cells and was accompanied by the upregulation of Bax and
downregulation of Bcl-2. In addition to enhancing apoptosis,
inhibition of CXCR1 decreased proliferation and this was
accompanied by the downregulation of several proliferation genes
(cyclin D1, and EGFR) associated with gastrointestinal tumors as
well as many other types of cancers (28–31).
The next set of molecules characterized in these
CXCR1 in vitro models was the balance between MMP-9, MMP-2
and E-cadherin. MMPs are well known to play an indispensable role
in the invasion and metastasis of cancers through their ability to
degrade extracellular matrix allowing the invasion and metastasis
of cancer cells (32–35). E-cadherin is a cell surface
adhesion molecule involved in cell-cell and cell-matrix
interactions, and studies support a role for the loss of E-cadherin
in the invasion of cancer cells (36,37).
In the present experimental system knockdown of CXCR1 decreased
MMP-2 and MMP-9 and increased E-cadherin expression, and is
consistent with a less invasive cancer cell phenotype.
Another characteristic of an aggressive, invasive
tumor is the ability to form new blood vessels that supply the
tumor with nutrition for the growth and provided a conduit for
metastasis (38). The levels of
VEGF, an angiogenic factor, are closely correlated with
microvascular density in many types of tumors (39). Our experiments showed that CXCR1
knockdown resulted in decreasing VEGF expression, and thus further
suggests that high CXCR1 levels could be associated with a highly
aggressive, vascularized tumor.
CXCR1 silence inhibited proliferation and growth,
induced cell cycle arrest and apoptosis and prevented migration and
invasion ability of gastric cancer cells. However, overexpression
of CXCR1 could reverse the above malignant behavior of gastric
cancer cells, possibly relating to the phosphorylation level of AKT
and ERK1/2 and the expression of BCL-2, cyclin D1, EGFR and VEGF,
MMP-2, MMP-9, Bax and E-cadherin. Further study is necessary to
fully clarify the interaction and regulation of these genes in
gastric cancer cells. This study provides preclinical data to
support CXCR1 as a novel therapeutic target for gastric cancer.
Acknowledgements
The present study was partially supported by the
China Postdoctoral Science Foundation (no. 2014M562137), the Hunan
Provincial Innovation Foundation for Postgraduate (no. CX2011B046),
the Science and Technology Program Foundation of Changsha City
(nos. K1005005-31 and K1106041-31).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Herszényi L and Tulassay Z: Epidemiology
of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci.
14:249–258. 2010.PubMed/NCBI
|
|
3
|
Brenner H, Rothenbacher D and Arndt V:
Epidemiology of stomach cancer. Methods Mol Biol. 472:467–477.
2009. View Article : Google Scholar
|
|
4
|
Kusano T, Shiraishi N, Shiroshita H, Etoh
T, Inomata M and Kitano S: Poor prognosis of advanced gastric
cancer with metastatic suprapancreatic lymph nodes. Ann Surg Oncol.
20:2290–2295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Murphy PM, Baggiolini M, Charo IF, Hébert
CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ and Power CA:
International Union of Pharmacology. XXII Nomenclature for
chemokine receptors. Pharmacol Rev. 52:145–176. 2000.PubMed/NCBI
|
|
6
|
Zlotnik A and Yoshie O: Chemokines: A new
classification system and their role in immunity. Immunity.
12:121–127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wells TN, Lusti-Narasimhan M, Chung CW,
Cooke R, Power CA, Peitsch MC and Proudfoot AE: The molecular basis
of selectivity between CC and CXC chemokines: The possibility of
chemokine antagonists as anti-inflammatory agents. Ann NY Acad Sci.
796:1 Cytokines. 245–256. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vandercappellen J, Van Damme J and Struyf
S: The role of CXC chemokines and their receptors in cancer. Cancer
Lett. 267:226–244. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Waugh DJ and Wilson C: The interleukin-8
pathway in cancer. Clin Cancer Res. 14:6735–6741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh S, Nannuru KC, Sadanandam A, Varney
ML and Singh RK: CXCR1 and CXCR2 enhances human melanoma
tumourigenesis, growth and invasion. Br J Cancer. 100:1638–1646.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ginestier C, Liu S, Diebel ME, Korkaya H,
Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum
D, et al: CXCR1 blockade selectively targets human breast cancer
stem cells in vitro and in xenografts. J Clin Invest. 120:485–497.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Y, Shi M, Yu GZ, Qin XR, Jin G, Chen
P and Zhu MH: Interleukin-8, a promising predictor for prognosis of
pancreatic cancer. World J Gastroenterol. 18:1123–1129. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li A, Varney ML and Singh RK: Expression
of interleukin 8 and its receptors in human colon carcinoma cells
with different metastatic potentials. Clin Cancer Res. 7:3298–3304.
2001.PubMed/NCBI
|
|
15
|
Yang G, Rosen DG, Liu G, Yang F, Guo X,
Xiao X, Xue F, Mercado-Uribe I, Huang J, Lin SH, et al: CXCR2
promotes ovarian cancer growth through dysregulated cell cycle,
diminished apoptosis, and enhanced angiogenesis. Clin Cancer Res.
16:3875–3886. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leonard DA, Merhige ME, Williams BA and
Greene RS: Elevated expression of the interleukin-8 receptors CXCR1
and CXCR2 in peripheral blood cells in obstructive coronary artery
disease. Coron Artery Dis. 22:491–496. 2011.PubMed/NCBI
|
|
17
|
Singh S, Wu S, Varney M, Singh AP and
Singh RK: CXCR1 and CXCR2 silencing modulates CXCL8-dependent
endothelial cell proliferation, migration and capillary-like
structure formation. Microvasc Res. 82:318–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang JP, Hu WM, Wang KS, Yu J, Luo BH, Wu
C, Chen ZH, Luo GQ, Liu YW, Liu QL, et al: Expression of C-X-C
chemokine receptor types 1/2 in patients with gastric carcinoma:
Clinicopathological correlations and significance. Oncol Lett.
5:574–582. 2013.PubMed/NCBI
|
|
19
|
Park SH, Das BB, Casagrande F, Tian Y,
Nothnagel HJ, Chu M, Kiefer H, Maier K, De Angelis AA, Marassi FM,
et al: Structure of the chemokine receptor CXCR1 in phospholipid
bilayers. Nature. 491:779–783. 2012.PubMed/NCBI
|
|
20
|
Jackson AL, Bartz SR, Schelter J,
Kobayashi SV, Burchard J, Mao M, Li B, Cavet G and Linsley PS:
Expression profiling reveals off-target gene regulation by RNAi.
Nat Biotechnol. 21:635–637. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fedorov Y, Anderson EM, Birmingham A,
Reynolds A, Karpilow J, Robinson K, Leake D, Marshall WS and
Khvorova A: Off-target effects by siRNA can induce toxic phenotype.
RNA. 12:1188–1196. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Z, Wang Y, Dong S, Ge C, Xiao Y, Li R,
Ma X, Xue Y, Zhang Q, Lv J, et al: Association of CXCR1 and 2
expressions with gastric cancer metastasis in ex vivo and tumor
cell invasion in vitro. Cytokine. 69:6–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Almhanna K, Strosberg J and Malafa M:
Targeting AKT protein kinase in gastric cancer. Anticancer Res.
31:4387–4392. 2011.PubMed/NCBI
|
|
24
|
Sasaki T and Kuniyasu H: Significance of
AKT in gastric cancer (Review). Int J Oncol. 45:2187–2192.
2014.PubMed/NCBI
|
|
25
|
Zhang Y, Wang L, Zhang M, Jin M, Bai C and
Wang X: Potential mechanism of interleukin-8 production from lung
cancer cells: An involvement of EGF-EGFR-PI3K-Akt-Erk pathway. J
Cell Physiol. 227:35–43. 2012. View Article : Google Scholar
|
|
26
|
Knall C, Worthen GS and Johnson GL:
Interleukin 8-stimulated phosphatidylinositol-3-kinase activity
regulates the migration of human neutrophils independent of
extracellular signal-regulated kinase and p38 mitogen-activated
protein kinases. Proc Natl Acad Sci USA. 94:3052–3057. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Correia C, Lee SH, Meng XW, Vincelette ND,
Knorr KL, Ding H, Nowakowski GS, Dai H and Kaufmann SH: Emerging
understanding of Bcl-2 biology: Implications for neoplastic
progression and treatment. Biochim Biophys Acta. 1853:1658–1671.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ye X, Guo Y, Zhang Q, Chen W, Hua X, Liu
W, Yang Y and Chen G: βKlotho suppresses tumor growth in
hepatocellular carcinoma by regulating Akt/GSK-3β/cyclin D1
signaling pathway. PLoS One. 8:e556152013. View Article : Google Scholar
|
|
29
|
Tsai HL, Yeh YS, Chang YT, Yang IP, Lin
CH, Kuo CH, Juo SH and Wang JY: Co-existence of cyclin D1 and
vascular endothe-lial growth factor protein expression is a poor
prognostic factor for UICC stage I-III colorectal cancer patients
after curative resection. J Surg Oncol. 107:148–154. 2013.
View Article : Google Scholar
|
|
30
|
Venkatakrishnan G, Salgia R and Groopman
JE: Chemokine receptors CXCR-1/2 activate mitogen-activated protein
kinase via the epidermal growth factor receptor in ovarian cancer
cells. J Biol Chem. 275:6868–6875. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luppi F, Longo AM, de Boer WI, Rabe KF and
Hiemstra PS: Interleukin-8 stimulates cell proliferation in
non-small cell lung cancer through epidermal growth factor receptor
transactivation. Lung Cancer. 56:25–33. 2007. View Article : Google Scholar
|
|
32
|
Roomi MW, Monterrey JC, Kalinovsky T, Rath
M and Niedzwiecki A: Comparative effects of EGCG, green tea and a
nutrient mixture on the patterns of MMP-2 and MMP-9 expression in
cancer cell lines. Oncol Rep. 24:747–757. 2010.PubMed/NCBI
|
|
33
|
Roomi MW, Monterrey JC, Kalinovsky T, Rath
M and Niedzwiecki A: Patterns of MMP-2 and MMP-9 expression in
human cancer cell lines. Oncol Rep. 21:1323–1333. 2009.PubMed/NCBI
|
|
34
|
Sancéau J, Truchet S and Bauvois B: Matrix
metalloproteinase-9 silencing by RNA interference triggers the
migratory-adhesive switch in Ewing's sarcoma cells. J Biol Chem.
278:36537–36546. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bergers G, Brekken R, McMahon G, Vu TH,
Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, et al:
Matrix metalloproteinase-9 triggers the angiogenic switch during
carcinogenesis. Nat Cell Biol. 2:737–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu X and Chu KM: E-cadherin and gastric
cancer: Cause, consequence, and applications. Biomed Res Int.
2014:6373082014.PubMed/NCBI
|
|
37
|
Carneiro P, Figueiredo J, Bordeira-Carriço
R, Fernandes MS, Carvalho J, Oliveira C and Seruca R: Therapeutic
targets associated to E-cadherin dysfunction in gastric cancer.
Expert Opin Ther Targets. 17:1187–1201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kitadai Y: Cancer-stromal cell interaction
and tumor angiogenesis in gastric cancer. Cancer Microenviron.
3:109–116. 2010. View Article : Google Scholar
|
|
39
|
Bădescu A, Georgescu CV, Vere CC, Crăiţoiu
S and Grigore D: Correlations between Her2 oncoprotein, VEGF
expression, MVD and clinicopathological parameters in gastric
cancer. Rom J Morphol Embryol. 53:997–1005. 2012.
|