Introduction
In the last three decades, cancer research focused
predominantly on breast cancer cell characteristics. Recently,
compelling evidence has emerged that metastatic breast cancer
development depends upon essential contributions from the tumor
microenvironment, which is composed of a wide range of
hematopoietic cells, adipocytes, fibroblasts, as well as the
important immune effector cells, and soluble signal factors
secreted by these cells (1–4).
Adipocyte, initially considered only to be a fat-storing cell, is
now recognized as a key components in tumor microenvironment. It
plays complex functions mainly mediated by a variety of
adipocyte-derived molecules, named adipokines. The adipokines are
deemed to be key mediators linking obesity with breast cancer, and
among which, leptin, whose synthesis and plasma levels increase in
parallel to total adipose tissue mass, has been extensively
examined in this regard.
Leptin is a 16 kDa protein encoded by the ob gene.
It is a pleiotropic molecule that is related to food intake,
inflammation, cell differentiation, and proliferation of different
cell types including breast cancer cells (5). The activities of leptin are mediated
through the transmembrane leptin receptor (ObR). Leptin and leptin
receptor are overexpressed in breast tumors, but not in the normal
cases. Its binding to ObR induces activation of canonical
(JAK2/STATs; MAPK/ERK 1/2, PI3K/AKT) and non-canonical (PKC, JNK,
p38 MAPK and AMPK) signaling pathways that increase breast cancer
cell proliferation and transformation, exert anti-apoptotic
effects, induce the expression of cell cycle modulators, reduce
efficacy of breast cancer treatment, and influence cancer
initiation processes (6,7).
As is well known, inflammation is involved in almost
all tumor pathological processes. Notably, leptin, through
induction of pro-inflammatory cytokines and stimulation of
macrophage function, has pro-inflammatory effects (8). Leptin receptor is widely expressed in
immune system and activates a number of independently regulated
intra-cellular signalling pathways which are important in cytokine
gene regulation, including JAK2/STAT, PI3K and MAPK (9). Thus, in vitro, leptin
influences cytokine secretion in a variety of cells including the
monocytes/macrophage lineage (10–12).
For example, treatment with leptin enhances the production of
proinflammatory cytokines such as tumor necrosis factor-α (TNF-α)
and IL-6 by macrophages (13–15).
Here, macrophages, also known as tumor-associated macrophages
(TAMs), exhibit marked phenotypic heterogeneity and functional
diversity results from a differentiation program that is subject to
environmental imprinting (16–18).
It comprises of classically activated macrophages (M1 macrophages)
and alternatively activated macrophages (M2 macrophages). Clinical
evidence has revealed a strong correlation between a high density
of TAMs and poor prognosis in breast cancer. Analysis of the
transcriptome of TAMs in mouse models of breast cancer has also
provided evidence that an enrichment in macrophage transcripts is
predictive of poor prognosis and reduced survival (19). However, the molecular mechanisms
underlying these observations remain unclear.
IL-18, a member of the IL-1 family, is a
pro-inflammatory cytokine that initially was thought to be produced
by activated macrophages. Now it has been confirmed being expressed
by both immune and non-immune cells (20,21).
IL-18 was originally described as an interferon-γ-inducing factor
with an anti-tumor activity through the activation of NK and/or T
cell responses (22,23). However, some studies demonstrated
that IL-18 has a pro-tumor effect in different cancers. Increased
IL-18 levels in the serum of cancer patients correlated with
malignancy, and IL-18 acts as a crucial factor for cell migration
in gastric cancer and melanoma (24,25).
In the present study, we examined the effect of
leptin on breast cancer progression. We showed that leptin promotes
the migration and invasion of breast cancer cells via activation of
IL-18 signalling, which is TAM-dependent or -independent. Overall,
our findings suggest that leptin and TAMs, acting together or
independently, were involved in the mammary tumor cell migration
and invasion.
Materials and methods
Cell lines
THP1 human monocytes, and MCF-7, SK-BR-3 and
MDA-MB-231 human breast tumor cell lines were obtained from
American Type Culture Collection (ATCC). THP1 cells, and MDA-MB-231
cells were cultured in RPMI-1640, and MCF-7 and SK-BR-3 cells were
kept in Dulbecco's modified Eagle's medium supplemented with 10%
fetal bovine serum, penicillin (100 U/ml) and streptomycin (100
μg/ml).
THP1-derived macrophages
To generate THP-1 macrophages, 5×105
THP-1 cells were seeded into a six-well plate and treated with
Phorbol-12-myristate-13-acetate (PMA) 100 nM for 6 h and then
cultured with PMA plus 20 ng/ml IL-4 for further 66 h (26,27).
PMA treatment, which activates protein kinase C (PKC), also induces
a greater degree of differentiation in THP-1 cells as reflected by
increased adherence and expression of surface markers associated
with macrophage differentiation. Furthermore, cells were stimulated
by IL-4 to differentiate to M2 macrophages, which are in general
more prone to protumoral activities.
Specimens
The 48 surgical specimens were obtained from women
undergoing surgery for breast carcinoma or reduction mammoplasty
after informed consent was given by the Department of Pathology of
the First Affiliated Hospital of Chongqing Medical University
(Chongqing, China). None of the patients had received pre-operative
chemotherapy. The experiments were approved by the Ethics Committee
of the First Affiliated Hospital of Chongqing Medical University
and were conducted in accordance with the Helsinki Declaration.
Materials
Cytokine leptin (300-27) was obtained from
Peprotech, Inc. (Rocky Hill, NJ, USA), antibody leptin (ab3583) and
IL18 (ab117342) were purchased from Abcam (Cambridge, UK), IL-18 bp
(10357-H08H) was purchased from BioVision, Inc. (Milpitas, CA,
USA), antibody for pAKTSer473, AKT, pNF-κB, NF-κB and
NF-κB1 were obtained from Cell Signaling Technology, Inc. (Danvers
or Beverly, MA, USA), antibody for ATF-2 (AB55011) CD68 (ZM0060)
and CD163 (AB60965a) were purchased from Sangon Biotech (Shanghai,
China), β-actin (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA), HRP anti-mouse or rabbit IgG (Beyotime, China) was used as
secondary antibody, ELISA kit was purchased from USCN Life Science
Inc. (Los Angeles, CA, USA). IL18 siRNA: (IL-18-459, sense,
5′-CCUAGAGGUAUGGCUGUAATT-3′; and antisense,
5′-UUACAGCCAUACCUCUAGGTT-3′) were purchased from GenePharma
(Shanghai, China). PI3k inhibitor Ly294002 and the NF-κB inhibitor
Bay11-7082 were obtained from Beyotime.
In vitro cell migration assay
The conditioned medium (CM) was collected. The
monocyte THP1 differentiated to TAMs according to the projected
protocol, and then the TAMs were treated with different factors for
various time periods. The cells were washed with serum-free medium
and then with fresh medium with 5% FBS, the supernatants were
collected at 36 h after cell culture and then stored at −20°C for
future use.
A 24-well plate of 8.0 μm pore-size diameter
polycarbonate (PC) membrane Transwell inserts (Corning Inc.;
Corning, NY, USA) were coated with Matrigel (Sigma; St. Louis, MO,
USA). Breast cancer cells (50,000) suspended in 100 μl of DMEM
medium (supplemented with 1% FBS) was added to the upper
compartment of the Transwell and placed into the lower chamber
containing the collected CM with 1% FBS. After 24 h, Transwells of
each condition assessed in triplicate were removed, the upper side
of the compartment of the PC membrane was wiped clean using a
cotton swab and the lower compartment was fixed with 1%
paraformaldehyde and stained with 0.1% crystal violet for 10 min.
The number of cells passing through the Matrigel was counted in
five random fields under a microscope and the average number of
cells per experimental condition is reported.
Scratch assay
The breast cancer cells were counted and plated at
4–6×105 cells/ml in 12-well dishes. Cells were incubated
in the collected CM with 1% FBS overnight yielding confluent
monolayers for wounding. Wounds were made using a pipette tip and
photographs were taken immediately (time zero) and 24 or 36 h after
wounding, respectively. The distance migrated by the cell monolayer
to close the wounded area during this period was measured. Results
were expressed as a migration index. Experiments were carried out
in triplicate and repeated at least five times.
Real-time PCR
The expression of IL-18 mRNA was analyzed over a
12-h period for all four cell lines to determine if leptin
regulates IL-18 gene expression. Total RNA was isolated using the
TRIzol reagent (Invitrogen) and was reversely transcribed into
first-strand complementary DNA (Takara) according to the
manufacturer's instructions. Quantitative real-time reverse
transcription PCR (qRT-PCR) was performed using SYBR Green Premix
Ex Taq™ (Takara). The following primers: IL-18 forward:
5′-CCAGCCTGACCAACA-3′ and IL-18 reverse: 5′-CCACAACCTCTACCTCC-3′
were designed using Primer primer 5.0 and synthesized by
Invitrogen. Data were analyzed according to the comparative
threshold method and normalized against the actin internal control
transcript.
ELISA
Supernatants obtained from breast cancer cells
cultured alone or treated with leptin were subjected to ELISA,
according to the manufacturer's instructions.
In vivo orthotopic animal study
Female nude BALB/c mice, aged 6–8 weeks, were
purchased from Center of Laboratory Animals, Chongqing Medical
University. Selective macrophage depletion, by means of the
macrophage suicide technique utilizing liposome-mediated
intracellular delivery of dichloromethylene-biphosphonate [Cl2MBP
(clodronate)] is a well-established experimental protocol (28,29).
MCF-7 cells (1×106) were inoculated into the mammary fat
pads of nude mice, and cancer evolution and metastasis to the lungs
were evaluated. From the 15 days after tumor cells inoculation,
mice were injected with PBS or leptin at 0.1 μg/g biweekly, leptin
with clophosome-clodronate liposomes (CCL) or control neutral
liposome (CNL), as initial dose of 0.2 ml per mouse, followed by
0.1 ml per mouse once a week for 5 weeks, respectively.
Immunohistochemistry
Nude mouse tumor tissues were fixed in 10% neutral
buffered formalin. Sections (5 μm) of formalin-fixed and
paraffin-embedded tissue were placed on slides. The procedure
operated as the basic protocol of immunohistochemistry, all the
antibodies were diluted at 1:50. The tissue sections were scored
quantitatively according to the percentage of positive cells and
staining intensity in five random fields under a microscope.
Statistical analysis
All quantitative results are displayed as the
average value ± the standard deviation. To determine statistical
significance, SPSS for Windows version 17.0 software was employed.
Using a 95% confidence interval, ANOVA was conducted for each
variable independently and as interacting factors, p-values
<0.05 resulted in a null-hypothesis rejection.
Results
Leptin stimulates breast cancer MCF-7
cell invision and migration via IL-18 secreted in TAMs
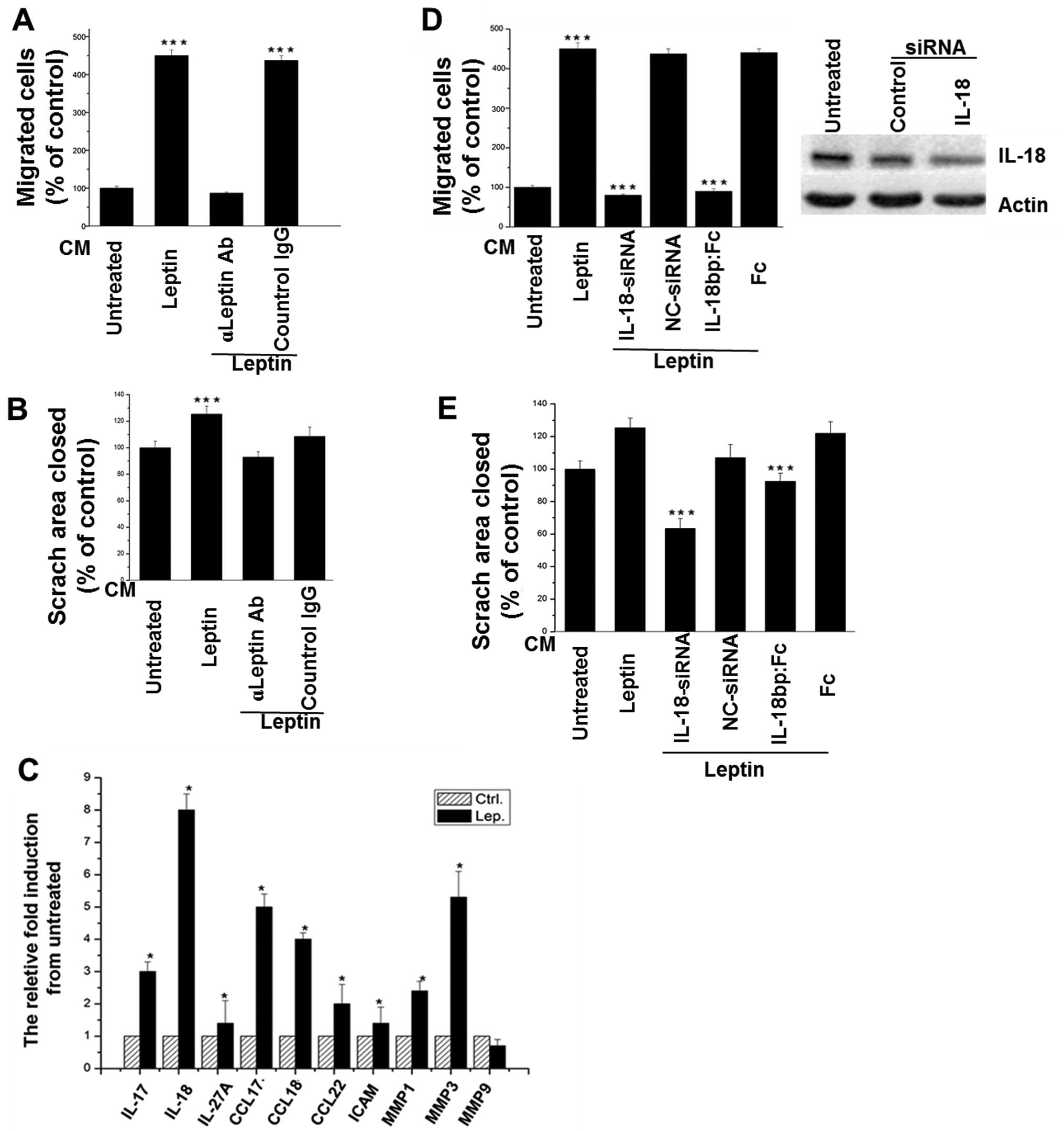
As shown in Fig. 1,
after treated by leptin, the conditioned medium (CM) of TAMs
significantly enhanced the migration and invasion of breast cancer
cells. However, the effect was abolished by the leptin-neutralizing
antibody (Fig. 1A and B). In
addition, it was found that an array of cytokines was increased in
the CM of TAMs, such as IL-17, IL-18, IL-27A, as well as CCL-17,
CCL-18, and CCL-22 (Fig. 1C).
IL-18 was the most elevated, and involved in promoting migration
(Fig. 1D) and invasion (Fig. 1E) of breast cancer cells, confirmed
by the addition of IL-18 siRNA or IL-18BP-Fc chimeras.
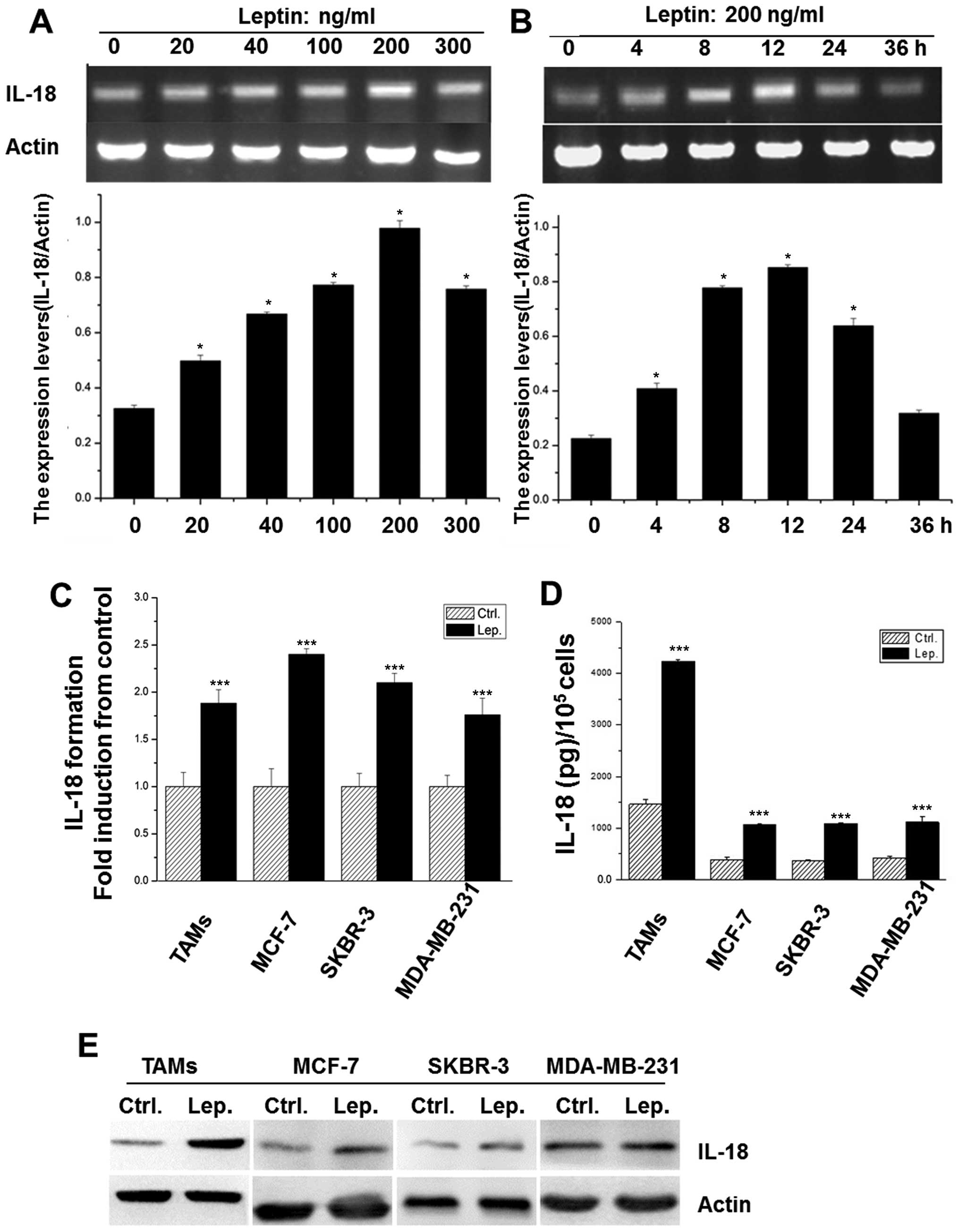
Leptin induces IL-18 expression in TAMs
and breast cancer cells
Treatment with leptin visibly increased IL-18
expression of TAMs in a dose- and time-dependent manner (Fig. 2A and B), and 200 ng/ml leptin at 12
h exerted the maximum effect and was chosen for further
experiments. Additionally, the direct effect of leptin on IL-18
expression was also investigated in different breast cancer cell
lines, including MCF-7, MDA-MB-231, and SK-BR-3. As shown in
Fig. 2C and E, after treated with
leptin, the mRNA and protein levels of IL-18 were significantly
increased in these cells, which was strengthened by the detection
of IL-18 in all the supernatants by ELISA assay (Fig. 2D).
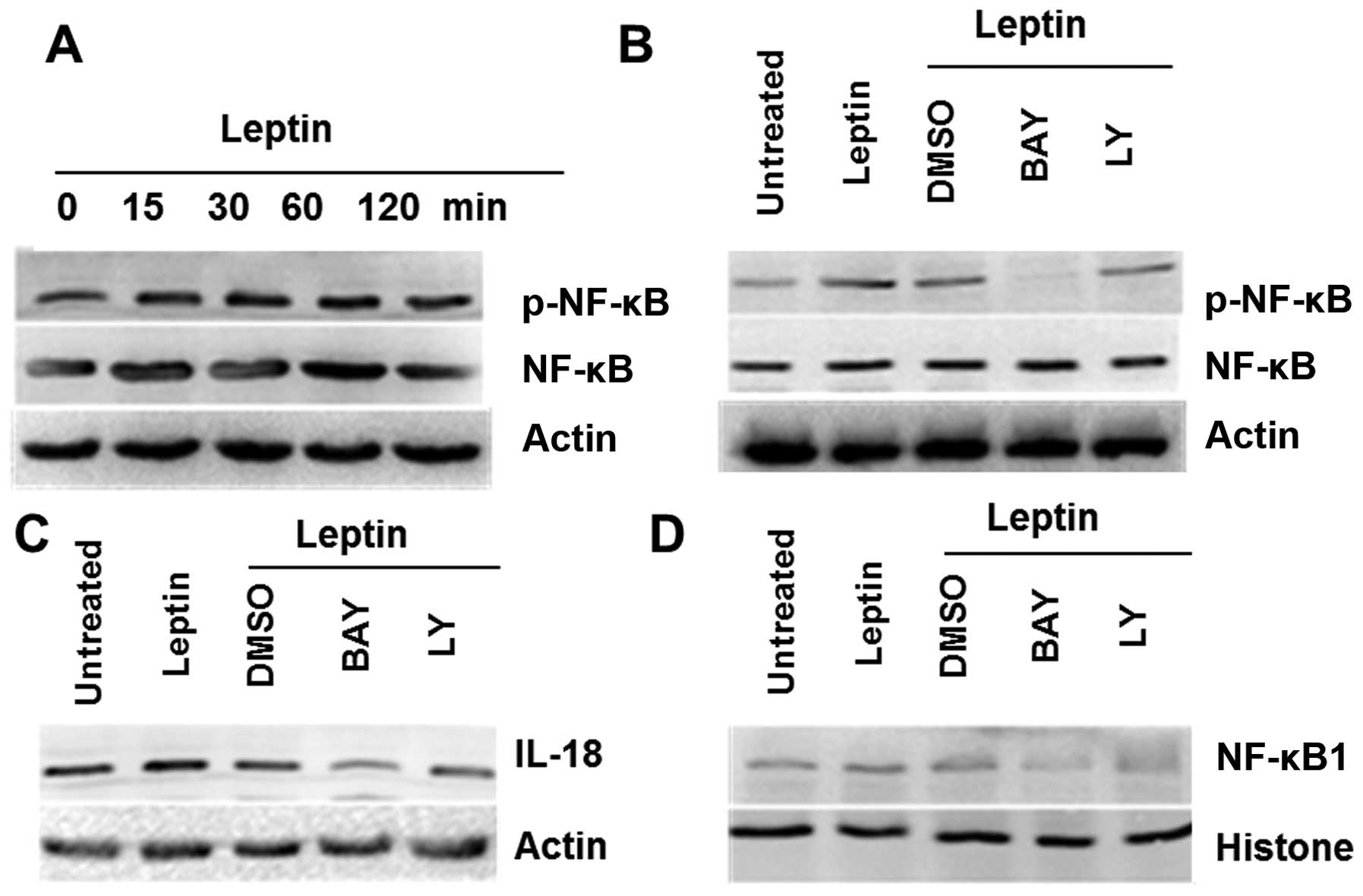
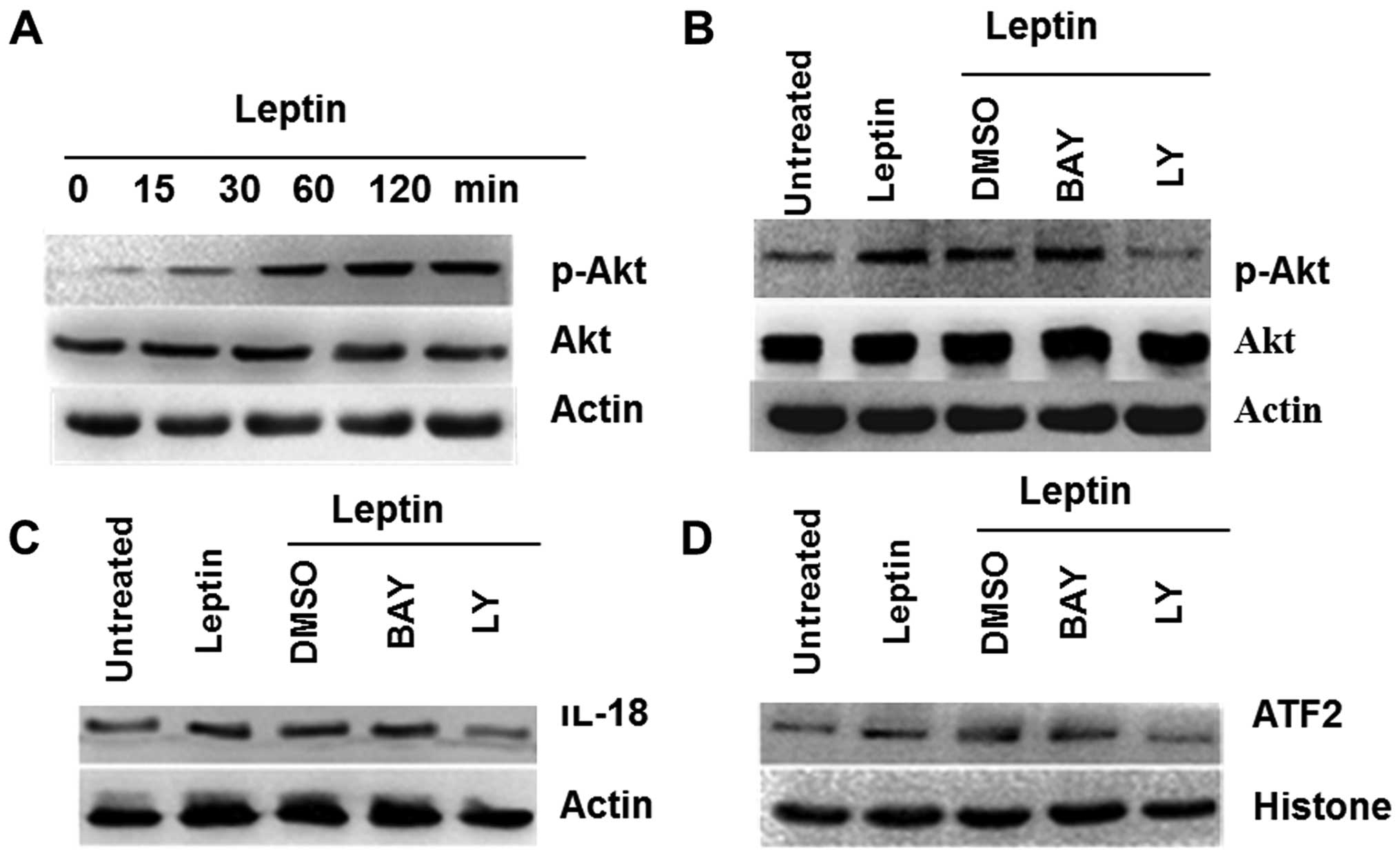
Leptin-induced IL-18 expression varies in
TAMs and MCF-7 breast cancer cells
On the basis of leptin-induced IL-18 overexpression
both in TAMs and breast cancer cells, some of inhibitors,
Bay11-7082 (inhibitor of NF-κB) and Ly294002 (inhibitor of PI3K),
were used to elucidate further molecular mechanisms. The results
showed that NF-κB in TAMs and PI3K in MCF-7 cells were activated by
leptin, respectively (Figs. 3A and
4A). Furthermore, the effects of
leptin on induction of NF-κB phosphorylation in TAMs were also
significantly attenuated by a pharmacological NF-κB inhibitor
(Fig. 3B), and for breast cancer
cells MCF-7, PI3K inhibitor affected the decrease of Akt
phosphorylation (Fig. 4B).
Bay11-7082 decreased the overexpression of IL-18 in TAMs induced by
leptin (Fig. 3C), and Ly294002
played a similar role in MCF-7 cells (Fig. 4C). These results indicated that
leptin induced IL-18 expression in TAMs and in MCF-7 via NF-κB, the
PI3K signaling, respectively.
Moreover, western blotting of nuclear protein
extracts revealed a significant increase of NF-κB1 in TAMs and
ATF-2 in MCF-7 (Figs. 3D and
4D) treated by leptin, and the
effects were blocked by its pharmacologic inhibitors.
Leptin promotes metastasis of breast
tumor cells in vivo by the secretion of IL-18 in TAMs
The expression of IL-18 in the leptin-induced TAMs
were correlated closely with malignant breast cancer, suggesting
that the CD163, a sensitive and accurate marker of TAMs (30), and IL-18 might be related to the
evolvement of breast cancer directly. CD68 served as a marker of
macrophages (30).
To test this notion, 48 human pathological specimens
of breast cancer, previously scored for tumor grade, were tested to
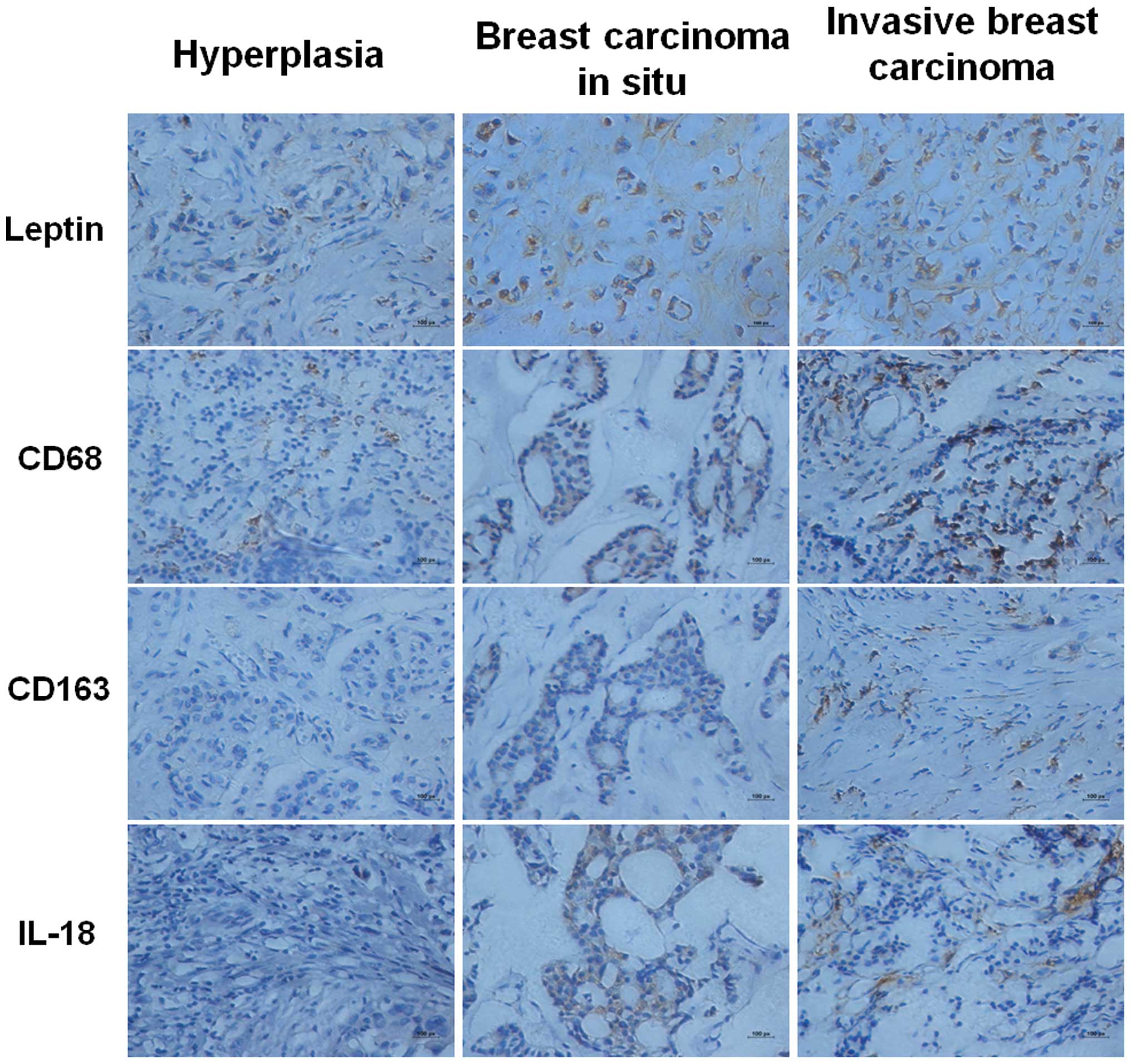
investigate the expression of leptin, IL-18, CD68 and CD163.
Immunohistochemistry results revealed that the staining intensity
of leptin in breast carcinoma without lymph node metastases (LNM)
(17/20) was lower than in LNM (13/16), while stronger compared with
that of benign breast tissue (8/12); CD163 and IL-18 were observed
scattered in the tumor stroma cells of invasive breast carcinomas,
few were identified in breast carcinomas in situ; the CD68
results were similar to CD163 (Fig.
5).
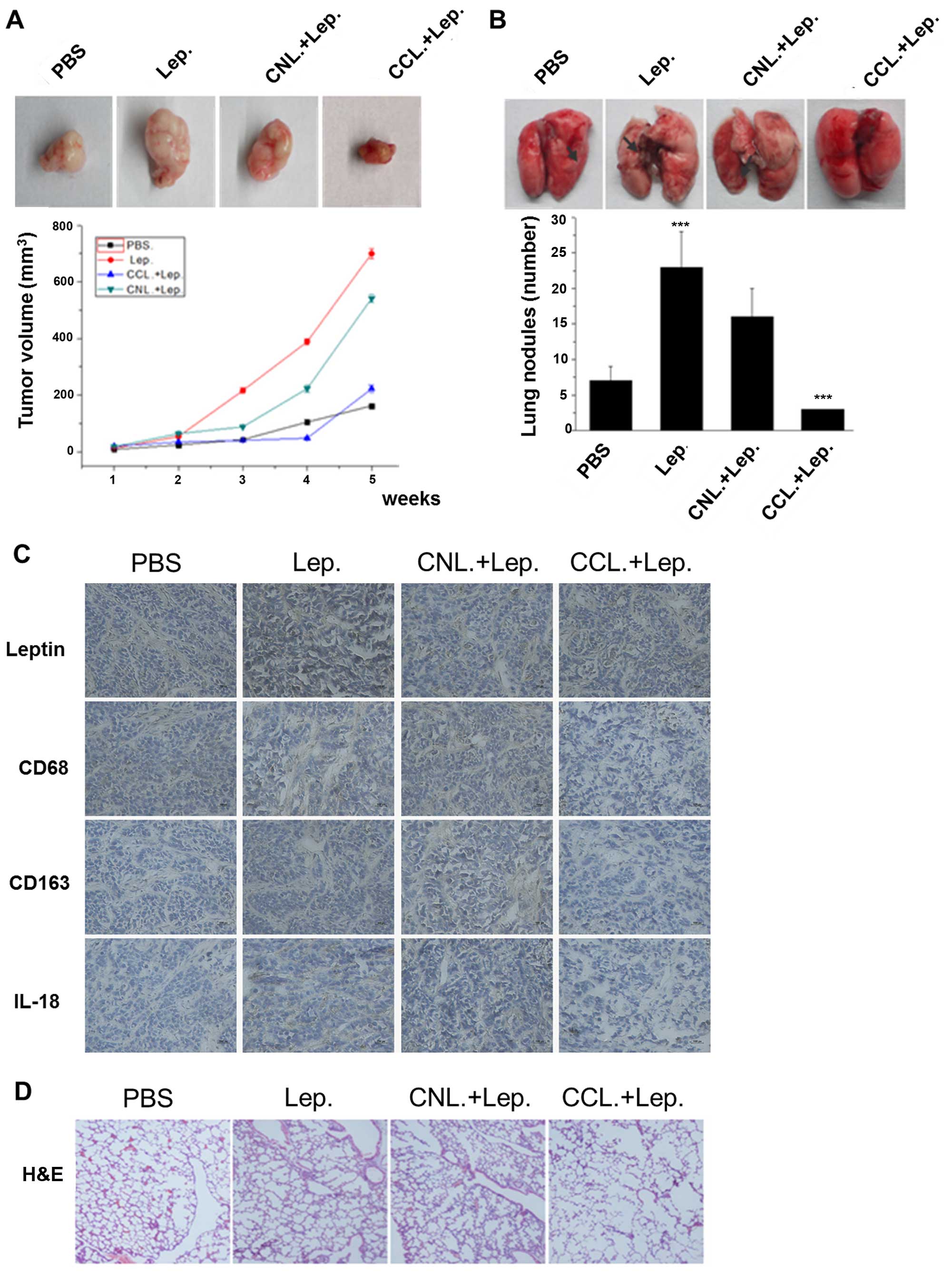
To determine the effect of IL-18 secreted in TAMs
induced by leptin in vivo, intraperitoneal (i.p.) injection
of CCL was used to deplete macrophages, and CNL as control. It was
observed that the volume of tumor in leptin groups was
significantly larger than that of PBS groups, and the tumor growth
was reduced in CCL groups compared with that of CNL control group
(p<0.05) (Fig. 6A). Pulmonary
metastases were also observed in xenograft models. It was found
that leptin greatly accelerated lung metastases, and macrophage
depletion could suppress the metastasis (Fig. 6B). The expression of IL-18 was
highly correlated with the expression of leptin, however, IL-18 was
decreased under the macrophage depletion with CCL (Fig. 6C). We also investigated lung
metastasis of breast cancer xenografts by H&E staining
(Fig. 6D). Thus, these results in
human breast cancer tissues and in animal models confirmed our
observations in vitro, lending further support to our
hypothesis that IL-18 of TAMs was required for cancer cell
migration and invasion induced by leptin.
Together, our results indicated that the expression
of IL-18 in TAMs induced by leptin may affect breast cancer cell
migration and invasion.
Discussion
Leptin is a pleiotropic adipokine that regulates
inflammatory cytokines, including IL-1 family, in different cell
types and pathological conditions (31). However, published studies
concerning the relationships between leptin and IL-1 family in
breast cancer are scarce. In addition, leptin could probably
regulate the function of TAMs, which is the main source of
inflammatory cytokines in tumor microenvironment, nevertheless, the
underlying mechnism is not fully understood. Here, we showed that
leptin induced IL-18 expression both in TAMs and breast cancer
cells. In our studies, leptin-induced IL-18 expression was
regulated via NF-κB/NF-κB1 signaling in TAMs, while via
PI3K-AKT/ATF-2 signaling in breast cancer cells, which, eventually
lead to invasion and metastasis of breast cancer cells.
TAMs, derived from circulating peripheral blood
monocytes, are a key component of the tumor microenvironment in
aggressive tumors. Studies have demonstrated that increasing
infiltration of TAMs is directly linked with advanced tumor
prognosis and metastasis (13).
These effects are regulated by multiple cues from tumor cells and
the tumor microenvironment. Upon direct or indirect interaction
with TAMs and tumor microenvironments, TAMs synthesized and
released a vast diversity of growth factors, cytokines, chemokines,
ECM components, and protease enzymes. These TAM-derived factors
then promoted matrix remodeling, angiogenesis, anti-immune
responses, and tumor progression (13). Data from the present investigation
show for the first time that leptin upregulates the transcriptional
expression of different cytokines of TAMs, including IL-18 as the
most elevated. Additionally, after treated with leptin, TAMs
significantly promoted the migration and invasion of breast cancer
cells. Indeed, previously studies has demonstrated that IL-18 is a
critical factor in the metastasis and pathogenesis of breast or
other cancers, such as gastric cancer and melanoma (32). Consistently, in our study,
co-incubation of IL-18 siRNA or IL-18BP-Fc chimeras abolished the
effect of leptin-incubated TAMs in promoting the migration and
invasion of breast cancer cells, indicating that IL-18 of TAMs was
required for the cancer cell migration and invasion induced by
leptin. To understand the mechanisms underlying the effect of
leptin, we examined leptin-induced activation of NF-κB/NF-κB1
signaling pathway in TAMs. Our results showed that leptin
stimulated the expression of NF-κB and its phosphorylation, with a
significant increase of NF-κB1 induction, which, however, were
inhibited by the addition of the pharmacological NF-κB inhibitor,
Bay11-7082. Notably, leptin could also directly stimulate IL-18
expression in breast cancer cells, which, differently, was via the
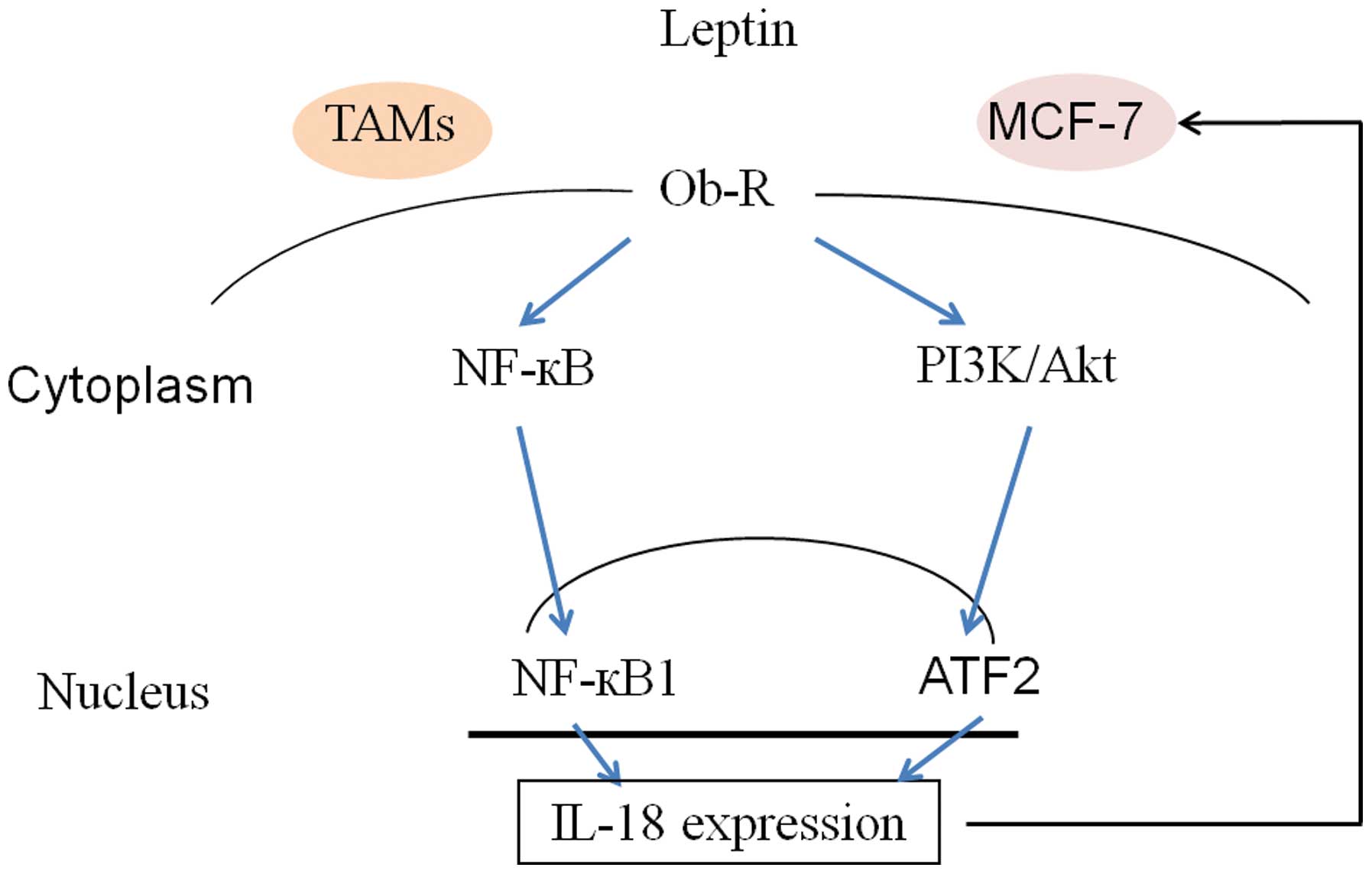
PI3K/AKT-ATF-2 signaling pathway. Herein, as shown in Fig. 7, our results suggest that
interaction between TAMs and breast cancer cells, and as well as
leptin- IL-18 crosstalk may promote cancer cells invasion and
metastasis in autocrine and paracrine manner (Fig. 7).
Accumulating evidence suggests that the high level
of TAMs infiltration in tumors, which correlates with poor
prognosis, is advantageous to the spread of cancers via enhancement
of tumor angiogenesis and tumor cell migration and invasion
(33–35). To further verify our hypothesis
that leptin-induced production of IL-18 from TAMs is responsible
for the metastasis and pathogenesis of breast cancer cells,
immunohistochemistry analysis of leptin, IL-18, CD68 and CD163 in
human pathological specimens was carried out. The results showed
that malignant breast carcinoma with lymph node metastases (LNM),
which represents poor prognosis, exhibited stronger expression of
leptin, IL-8, and TAM markers. Moreover, xenograft tumor-bearing
mouse models showed that leptin significantly increased tumor
volume, enhanced lung metastases, and increased expression of IL-8
and TAMs markers, which, nevertheless, were abolished by depletion
of macrophages by clophosome-clodronate liposomes. Taken together,
the assessment of the effect of TAMs on breast cancer suggests that
the tumor microenvironment consisting of these TAMs could dictate
the outcomes in breast cancer patients. Moreover, these findings
suggest that leptin may be a novel therapeutic target for breast
cancer treatment.
Our study showed that adipokine leptin triggered
macrophage-related cytokine IL-18 production, possibly contributing
to tumor progression. We anticipate the above results to have
important implication in the development of treatment strategies
for metastatic cancers, and their potential use in the
identification and screening of novel therapeutic targets.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (NSFC 81272544). The funding agencies
had no role in study design, collection, analysis, or
interpretation of data, writing of the manuscript, or the decision
to submit the manuscript for publication.
Abbreviations:
|
ATF-2
|
activating transcription factor-2
|
|
CD163
|
cluster of differentiation 163
|
|
CM
|
conditioned medium
|
|
i.p.
|
intraperitoneal
|
|
NF-κB
|
nuclear factor κB
|
|
TAMs
|
tumor associated macrophages
|
|
TME
|
tumor microenvrionment
|
References
|
1
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bhowmick NA, Neilson EG and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Robinson BD, Sica GL, Liu YF, Rohan TE,
Gertler FB, Condeelis JS and Jones JG: Tumor microenvironment of
metastasis in human breast carcinoma: A potential prognostic marker
linked to hematogenous dissemination. Clin Cancer Res.
15:2433–2441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tlsty TD and Coussens LM: Tumor stroma and
regulation of cancer development. Annu Rev Pathol. 1:119–150. 2006.
View Article : Google Scholar
|
|
5
|
Andò S and Catalano S: The multifactorial
role of leptin in driving the breast cancer microenvironment. Nat
Rev Endocrinol. 8:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo S, Liu M, Wang G, Torroella-Kouri M
and Gonzalez-Perez RR: Oncogenic role and therapeutic target of
leptin signaling in breast cancer and cancer stem cells. Biochim
Biophys Acta. 1825:207–222. 2012.PubMed/NCBI
|
|
7
|
Andò S, Barone I, Giordano C, Bonofiglio D
and Catalano S: The multifaceted mechanism of Leptin signaling
within tumor microenvironment in driving breast cancer growth and
progression. Front Oncol. 4:3402014.PubMed/NCBI
|
|
8
|
Procaccini C, Jirillo E and Matarese G:
Leptin as an immuno-modulator. Mol Aspects Med. 33:35–45. 2012.
View Article : Google Scholar
|
|
9
|
Oswal A and Yeo G: Leptin and the control
of body weight: A review of its diverse central targets, signaling
mechanisms, and role in the pathogenesis of obesity. Obesity
(Silver Spring). 18:221–229. 2010. View Article : Google Scholar
|
|
10
|
Gainsford T, Willson TA, Metcalf D,
Handman E, McFarlane C, Ng A, Nicola NA, Alexander WS and Hilton
DJ: Leptin can induce proliferation, differentiation, and
functional activation of hemopoietic cells. Proc Natl Acad Sci USA.
93:14564–14568. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zarkesh-Esfahani H, Pockley G, Metcalfe
RA, Bidlingmaier M, Wu Z, Ajami A, Weetman AP, Strasburger CJ and
Ross RJ: High-dose leptin activates human leukocytes via receptor
expression on monocytes. J Immunol. 167:4593–4599. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mattioli B, Straface E, Quaranta MG,
Giordani L and Viora M: Leptin promotes differentiation and
survival of human dendritic cells and licenses them for Th1
priming. J Immunol. 174:6820–6828. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chanmee T, Ontong P, Konno K and Itano N:
Tumor-associated macrophages as major players in the tumor
microenvironment. Cancers (Basel). 6:1670–1690. 2014. View Article : Google Scholar
|
|
14
|
Faggioni R, Fantuzzi G, Gabay C, Moser A,
Dinarello CA, Feingold KR and Grunfeld C: Leptin deficiency
enhances sensitivity to endotoxin-induced lethality. Am J Physiol.
276:R136–R142. 1999.PubMed/NCBI
|
|
15
|
Lee FY, Li Y, Yang EK, Yang SQ, Lin HZ,
Trush MA, Dannenberg AJ and Diehl AM: Phenotypic abnormalities in
macrophages from leptin-deficient, obese mice. Am J Physiol.
276:C386–C394. 1999.PubMed/NCBI
|
|
16
|
Gordon S: The macrophage: Past, present
and future. Eur J Immunol. 37(Suppl 1): S9–S17. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ojalvo LS, King W, Cox D and Pollard JW:
High-density gene expression analysis of tumor-associated
macrophages from mouse mammary tumors. Am J Pathol. 174:1048–1064.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Woldbaek PR, Tønnessen T, Henriksen UL,
Florholmen G, Lunde PK, Lyberg T and Christensen G: Increased
cardiac IL-18 mRNA, pro-IL-18 and plasma IL-18 after myocardial
infarction in the mouse; a potential role in cardiac dysfunction.
Cardiovasc Res. 59:122–131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Colston JT, Boylston WH, Feldman MD,
Jenkinson CP, de la Rosa SD, Barton A, Trevino RJ, Freeman GL and
Chandrasekar B: Interleukin-18 knockout mice display maladaptive
cardiac hypertrophy in response to pressure overload. Biochem
Biophys Res Commun. 354:552–558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dinarello CA: Interleukin-18. Methods.
19:121–132. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Y, Hahm E, Kim Y, Kang J, Lee W, Han
I, Myung P, Kang H, Park H and Cho D: Regulation of IL-18
expression by CRH in mouse microglial cells. Immunol Lett.
98:291–296. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vidal-Vanaclocha F, Mendoza L, Telleria N,
Salado C, Valcárcel M, Gallot N, Carrascal T, Egilegor E,
Beaskoetxea J and Dinarello CA: Clinical and experimental
approaches to the pathophysiology of interleukin-18 in cancer
progression. Cancer Metastasis Rev. 25:417–434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim KE, Song H, Kim TS, Yoon D, Kim CW,
Bang SI, Hur DY, Park H and Cho DH: Interleukin-18 is a critical
factor for vascular endothelial growth factor-enhanced migration in
human gastric cancer cell lines. Oncogene. 26:1468–1476. 2007.
View Article : Google Scholar
|
|
26
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Daigneault M, Preston JA, Marriott HM,
Whyte MK and Dockrell DH: The identification of markers of
macrophage differentiation in PMA-stimulated THP-1 cells and
monocyte-derived macrophages. PLoS One. 5:e86682010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qualls JE, Kaplan AM, van Rooijen N and
Cohen DA: Suppression of experimental colitis by intestinal
mononuclear phagocytes. J Leukoc Biol. 80:802–815. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van Rooijen N and Sanders A: Liposome
mediated depletion of macrophages: Mechanism of action, preparation
of liposomes and applications. J Immunol Methods. 174:83–93. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tang X: Tumor-associated macrophages as
potential diagnostic and prognostic biomarkers in breast cancer.
Cancer Lett. 332:3–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Newman G and Gonzalez-Perez RR:
Leptin-cytokine crosstalk in breast cancer. Mol Cell Endocrinol.
382:570–582. 2014. View Article : Google Scholar
|
|
32
|
Yang Y, Cheon S, Jung MK, Song SB, Kim D,
Kim HJ, Park H, Bang SI and Cho D: Interleukin-18 enhances breast
cancer cell migration via down-regulation of claudin-12 and
induction of the p38 MAPK pathway. Biochem Biophys Res Commun.
459:379–386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lewis C and Murdoch C: Macrophage
responses to hypoxia: Implications for tumor progression and
anti-cancer therapies. Am J Pathol. 167:627–635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun B, Nishihira J, Yoshiki T, Kondo M,
Sato Y, Sasaki F and Todo S: Macrophage migration inhibitory factor
promotes tumor invasion and metastasis via the Rho-dependent
pathway. Clin Cancer Res. 11:1050–1058. 2005.PubMed/NCBI
|
|
35
|
De Palma M and Lewis CE: Macrophage
regulation of tumor responses to anticancer therapies. Cancer Cell.
23:277–286. 2013. View Article : Google Scholar : PubMed/NCBI
|