Introduction
Prostate cancer is one of the most common types of
cancer worldwide, and acts as a leading cause of cancer-related
death in men (1). While early
prostate cancer can be cured, patients with advanced prostate
cancer often suffered from invasion and metastasis, which leads to
death of prostate cancer patients (2). Therefore, discovering novel molecular
biomarkers associated with prostate cancer progression, especially
those regulating the invasion and metastasis, may provide potential
molecular targets for the detection and therapy of prostate
cancer.
PAF is known as a phospholipid mediator of
inflammation, and further studies proved that PAF plays a pivotal
role in many diseases, including cancer (3,4). It
is reported that PAF promotes the migration and proliferation of
breast cancer cells (5), and
contributes to the invasiveness and motility of melanoma cells
(6). PAF exerts its biological
effects mainly via activating PAFR. As a member of G-protein
coupled receptor (GPCR) family, PAFR has been found to promote the
malignant development of esophageal squamous cell carcinoma via
PI3K/AKT pathway (7), and enhance
the growth of ovarian cancer cells through cooperating with EGFR
(8). However, it is reported that
elevated PAFR expression is significantly associated with smaller
tumor size, absence of lymph node and organ metastasis and low
tumor histopathological stage in gastric adenocarcinoma (9). In prostate cancer, the function of
PAFR remains elusive.
Given that PAFR may act as contrasting effect on the
progression of different cancers, the present study aimed to
investigate the effect of PAF and PAFR interaction in the
progression of prostate cancer cells. We unexpectedly found that
activation of PAFR by PAF stimulated the growth, invasion and
metastasis of prostate cancer cells via the ERK1/2 pathway.
Materials and methods
Cell lines and reagents
All cell lines were purchased from the American Type
Culture Collection (ATCC; Manassas, VA, USA). Human prostate cancer
cell lines LNCap, PC-3, PC-3M and DU-145 were maintained in
RPIM-1640 medium containing 10% fetal bovine serum (FBS) in 5%
CO2 atmosphere at 37°C. Normal prostate cell line RPWE-1
was maintained in K-SFM medium containing 10% FBS in 5%
CO2 atmosphere at 37°C. ERK1/2 special inhibitor U0126,
PAF and DMSO were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Antibodies against PAFR, E-cadherin, ERK1/2 and β-actin were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Antibody against phosho-ERK1/2 was obtained from Cell Signaling
Technology (Danvers, MA, USA).
Cell transfection
A specific siRNA targeting PAFR (siPAFR) was
purchased from GenePharma Co., Ltd., (Shanghai, China) with the
sequence of 5′-CUGGGCGUCAUCACUUAUA-3′ to transiently silence PAFR
expression. A scramble siRNA was used as control siRNA (siCtrl).
Cells were incubated with siPAFR or siCtrl for 36 h using the
Lipofectamine 2000 trans-fection reagent (Invitrogen), according to
the manufacturer's instruction. Knockdown efficiency was determined
by western blotting, and the cells were subjected to the
experiments described below.
Furthermore, to stably silence PAFR expression in
DU-145 cells, a PAFR shRNA (shPAFR) was obtained from GenePharma,
while a scrambled shRNA was used as control shRNA (shcontrol).
Cells were incubated with shPAFR or shControl for 36 h using the
Lipofectamine 2000 transfection reagent (Invitrogen), and the
stable clones were selected by G418. Knockdown efficiency was
detected by western blotting.
Western blotting
Total proteins from cell lines or transplanted tumor
tissues of mice were isolated using RIPA buffer with protease
inhibitor and phosphatase inhibitor. The concentration of protein
was determined by BCA assay (Applygen Technologies, Inc., Beijing,
Beijing, China). Then, equal amounts of protein were separated in
10% polyacrylamide gel and electrotransferred on PVDF membrane
(Millipore Corp., Billerica, MA, USA). The membrane was probed with
primary antibodies overnight, and then incubated with secondary
antibodies for 1 h. Next, the membrane was visualized by enhanced
chemiluminescence kit (Applygen Technologies). The density of each
band was analyzed with Quantity One software.
In vitro invasion assay and migration
assay
In vitro invasion assay and migration assay
were performed using Transwell chambers (Corning Costar, Corning,
NY, USA). Briefly, cells at the density of 1×106
cells/ml were plated in serum-free medium on upper chambers coated
with Matrigel in invasion assay, whereas 600 μl of 1640 medium
supplemented with 20% FBS was added into the lower chambers.
Sixteen hours later, cells passed through the membrane were stained
with crystal violet, and the numbers of cells were counted under a
light microscope in seven random fields. For in vitro
migration assay, cells at the density of 1×106 cells/ml
were plated in serum-free medium on upper chambers without
Matrigel, and cells were allowed to migrate for 16 h. The migrated
cells were stained with crystal violet, and the numbers of cells
were counted under a light microscope in seven random fields.
Real-time PCR
Total RNA was isolated from cells using TRIzol
reagent (Invitrogen), following the manufacturer's instruction.
Then, reverse transcription PCR was carried out using Omniscript
RT-PCR kit (Qiagen, Hilden, Germany). Next, real-time PCR was
performed using the primers of E-cadherin (forward,
5′-CGAGAGCTACACGTTCACGG-3′ and reverse,
5′-GGGTGTCGAGGGAAAAATAGG-3′; MMP-3 (forward,
5′-CTGGACTCCGACACTCTGGA-3′ and reverse,
5′-CAGGAAAGGTTCTGAAGTGACC-3′); or β-actin (forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGA-3′) under the following conditions: 10 min
at 94°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C.
β-actin served as an internal control. The relative mRNA expression
of MMP-3 and E-cadherin was determined by the 2−ΔΔCt
method.
ELISA assay
After stimulation with or without PAF, the
supernatants of DU-145 cells was collected and subjected to MMP-3
ELISA assay (Invitrogen), according to the manufacturer's
instruction. To detect MMP-3 level in transplanted tumor tissues of
mice, total proteins from transplanted tumor tissues of mice were
isolated in RIPA buffer with protease inhibitor, and then MMP-3
ELISA kit (Invitrogen) was used to determine MMP-3 protein
expression.
In vitro CCK-8 proliferation assay
Cell proliferation was determined by CCK-8 assay kit
(Jingmei Biotech Co., Ltd., Shanghai, China). Briefly, cells were
seeded at 800 cell/well in a 96-well plate, and incubated for 6 h.
Then, cells were stimulated with or without PAF for 72 h. Next, 15
μl of CCK-8 was added into the plate and cells were further
incubated for 2 h. Finally, optical density (OD) was observed using
a microplate reader (Bio-Rad Laboratories) at 490 nm.
In vivo proliferation and metastasis
assay
Male BABL/c nude mice at four weeks old were
purchased and maintained in the pathogen-free conditions under the
approval of the Animal Care and Use Committee of Wenzhou Medical
University. Sixteen mice were randomly divided into two groups (n=8
each group). Then shControl cells or shPAFR cells (2×105
cells) were subcutaneously injected at the back of the mice. Tumors
were formed in one week. The length and width of tumors in mice
were measured every week, and tumor volumes were estimated with the
formula of 0.52 × length × width2. Seven weeks later,
the mice were sacrificed. Tumor tissues were lysed in RIPA buffer
to further detect MMP-3 and E-cadherin expression. The livers were
fixed in 4% paraformaldehyde, sectioned into slices and stained
with H&E. Then the number of micrometastasis in liver was
counted under a microscope.
Statistical analysis
Experiments were performed three times, and data are
presented as mean ± standard deviation (SD). Statistical analysis
was performed using SPSS version 17.0 software (SPSS, Inc.,
Chicago, IL, USA). The statistical significant difference was
determined by the Student's t-test between two groups, or by
non-parametric ANOVA among multiple groups. P<0.05 was
considered to be statistically significant.
Results
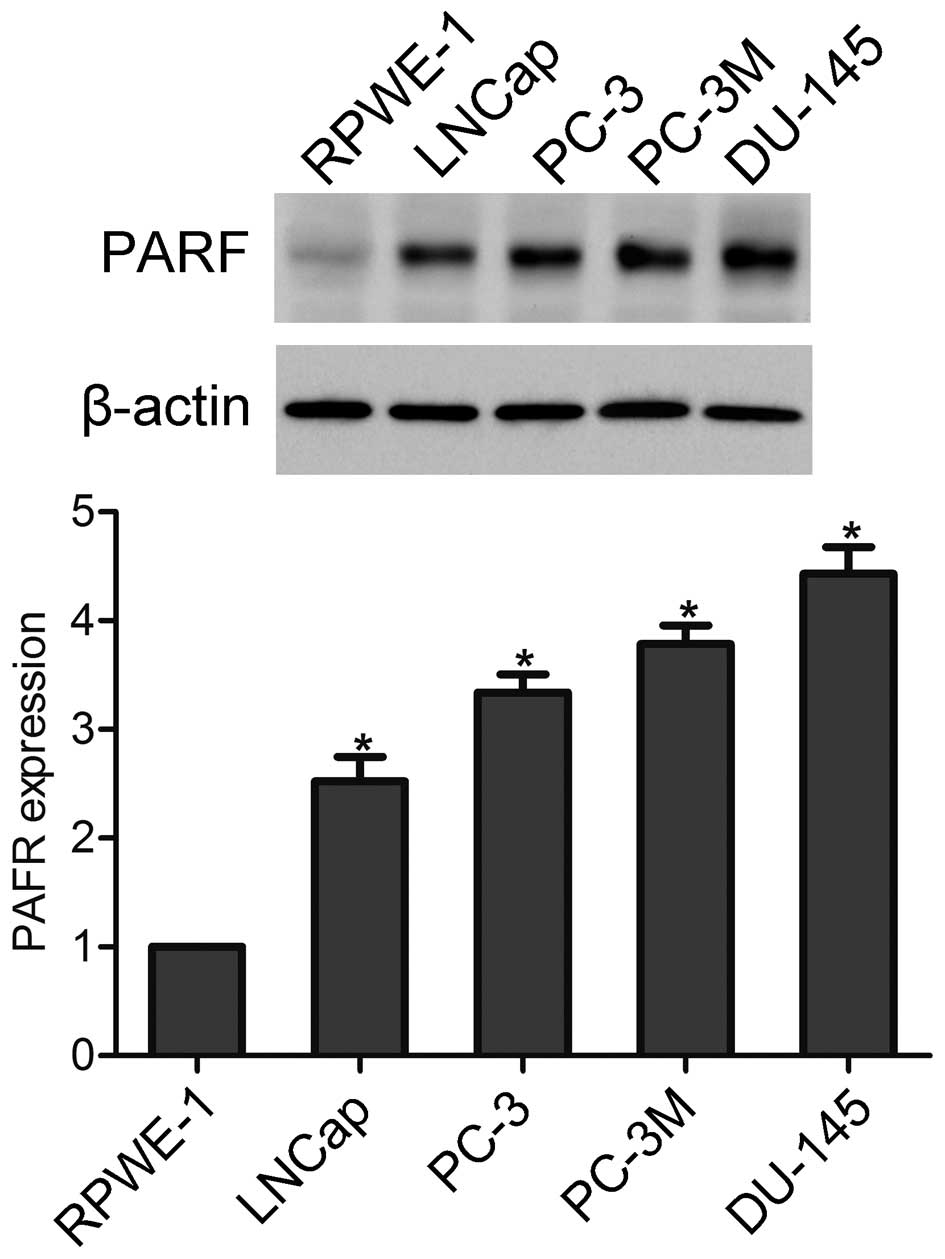
Overexpression of PAFR in prostate cancer
cells
In the present study we detected the protein level
of PAFR in human normal prostate RPWE-1 cells and prostate cancer
LNCap, PC-3, PC-3M and DU-145 cells by using western blotting. The
results showed that the PAFR expression level was significantly
higher in prostate cancer cells as compared to RPWE-1 cells
(Fig. 1).
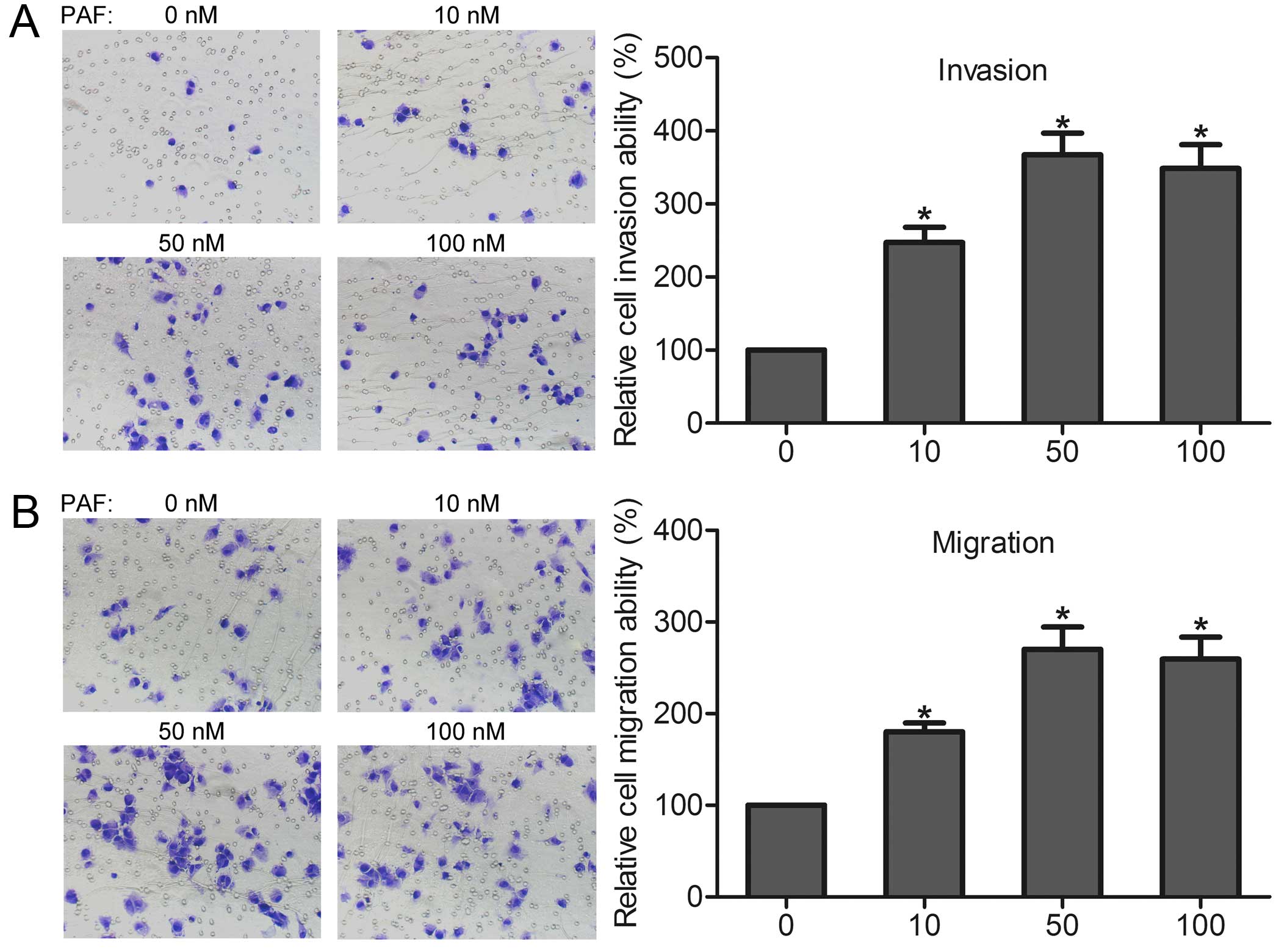
PAFR activation induces invasion and
migration of prostate cancer cells in vitro
To investigate the role of PAFR in prostate cancer
cells, DU-145 cells were selected and PAF was used to activate
PAFR. In vitro invasion and migration assays were performed
to determine the invasive and migration effect of PAFR activation
on prostate cancer cells. We found that cell invasion was
dose-dependently increased after incubation with different
concentration of PAF (Fig. 2A).
In vitro migration assay showed that PAF dose-dependently
induced the migration of DU-145 cells (Fig. 2B). These findings indicate that
PAFR activation is involved in prostate cancer cell invasion and
migration in vitro.
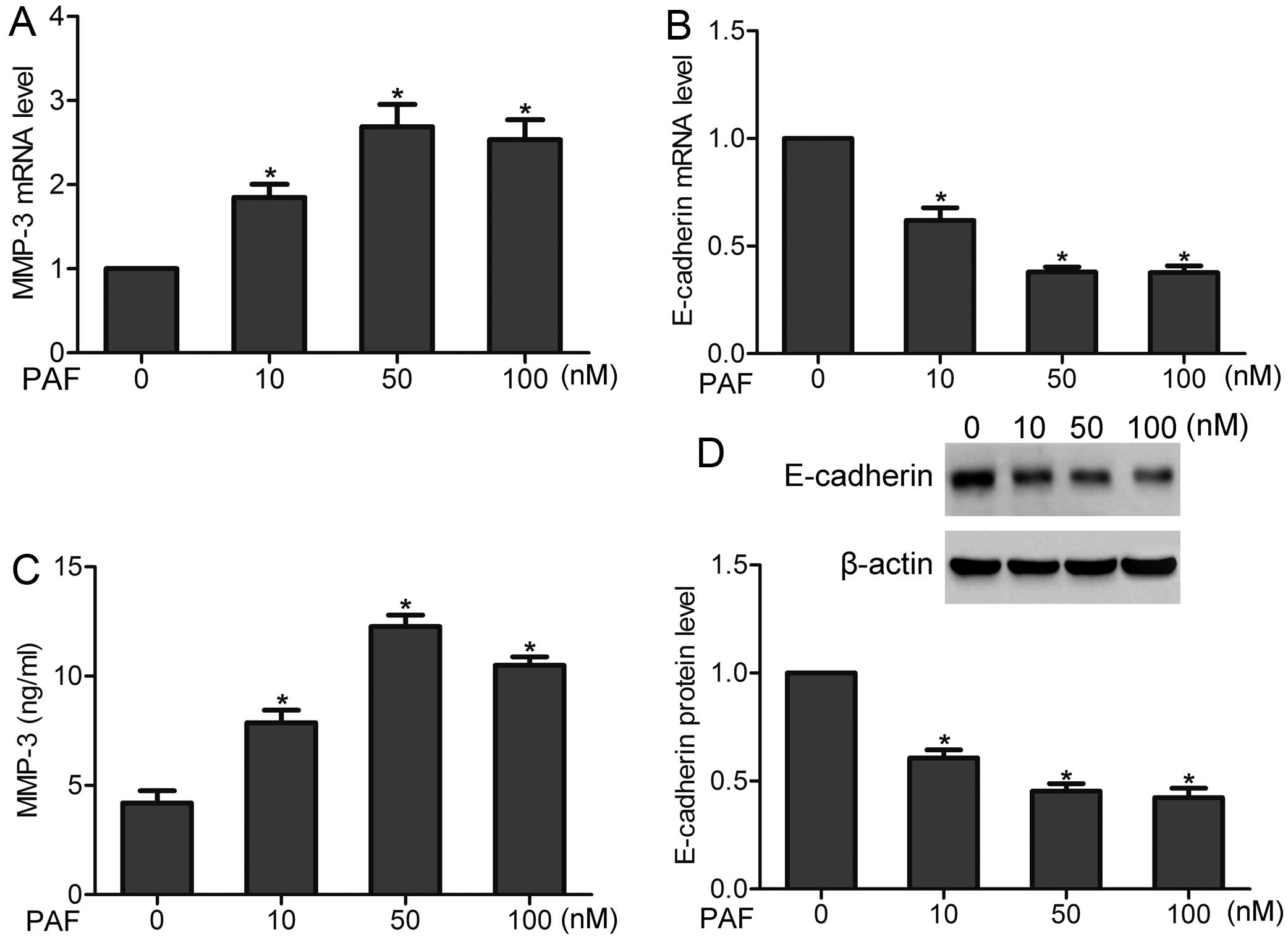
PAFR activation regulates the expressions
of MMP-3 and E-cadherin in vitro
After incubation of PAF with a concentration of 10,
50 and 100 nM, respectively, the mRNA and protein levels of MMP-3
were observed by real-time PCR and ELISA assay, respectively. The
results showed that the production of MMP-3 was increased after PAF
stimulation, in a dose-dependent manner (Fig. 3A and C). We further found that PAF
stimulation decreased the expression of E-cadherin at mRNA and
protein levels (Fig. 3B and D).
These findings suggest that PAFR activation can regulate the levels
of MMP-3 and E-cadherin in prostate cancer cells.
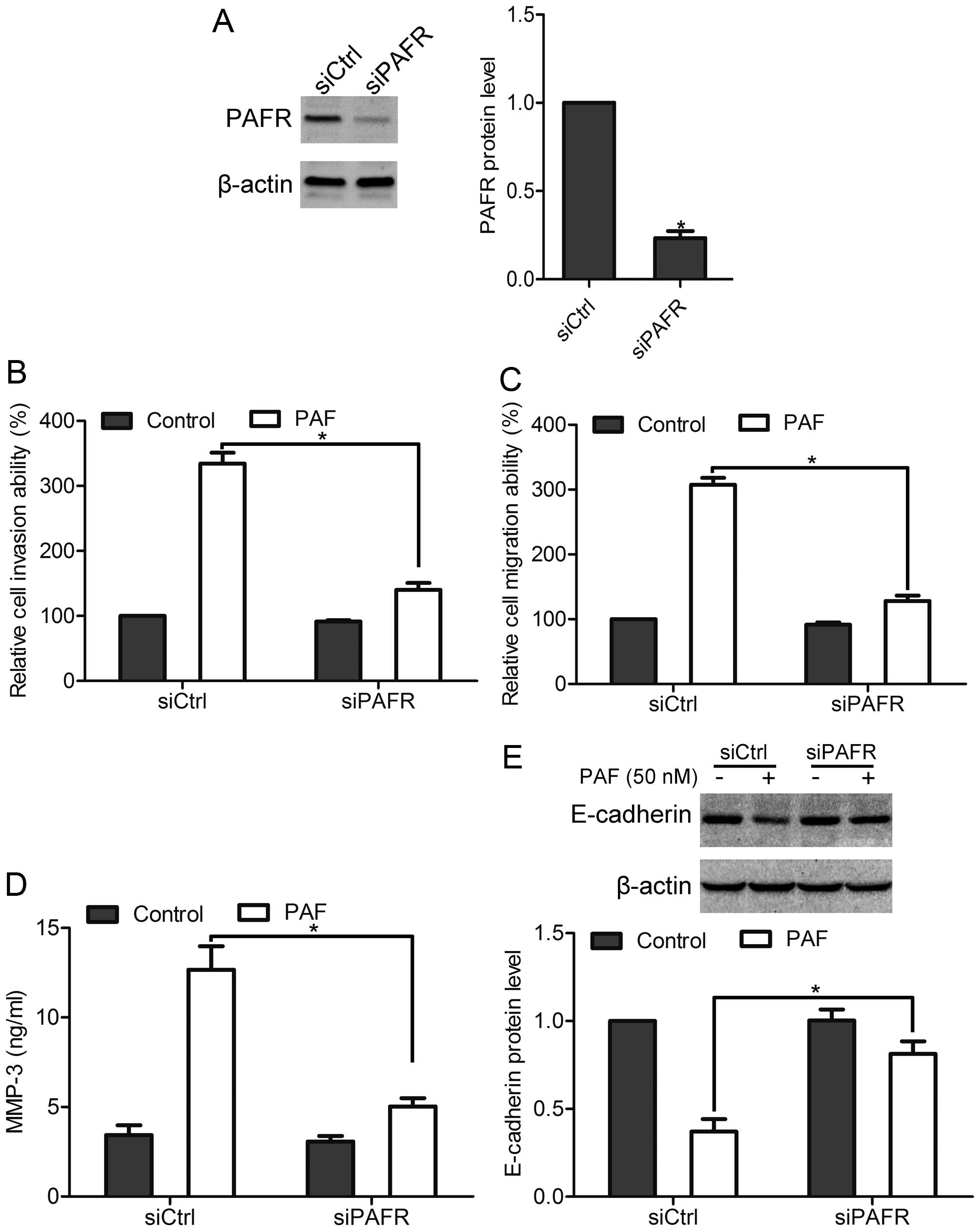
Knockdown of PAFR inhibits prostate
cancer cell invasion and migration in vitro
To confirm the effect of PAFR on prostate cancer
cells, DU-145 cells were transfected with siPAFR to reduce PAFR
expression. Using western blotting, we found that siPAFR
significantly reduced PAFR expression by >70% compared with the
siCtrl transfection (Fig. 4A).
Using in vitro invasion assay and migration assay, we found
that PAF stimulation promoted the invasion and migration in siCtrl
cells, whereas knockdown of PAFR attenuated the invasion and
migration of DU-145 cells induced by PAF (Fig. 4B and C). Furthermore, after
knockdown of PAFR, PAF-mediated increase of MMP-3 expression, as
well as decrease of E-cadherin was suppressed (Fig. 4D and E). These data confirm that
PAFR contributes to prostate cancer cell invasion and
migration.
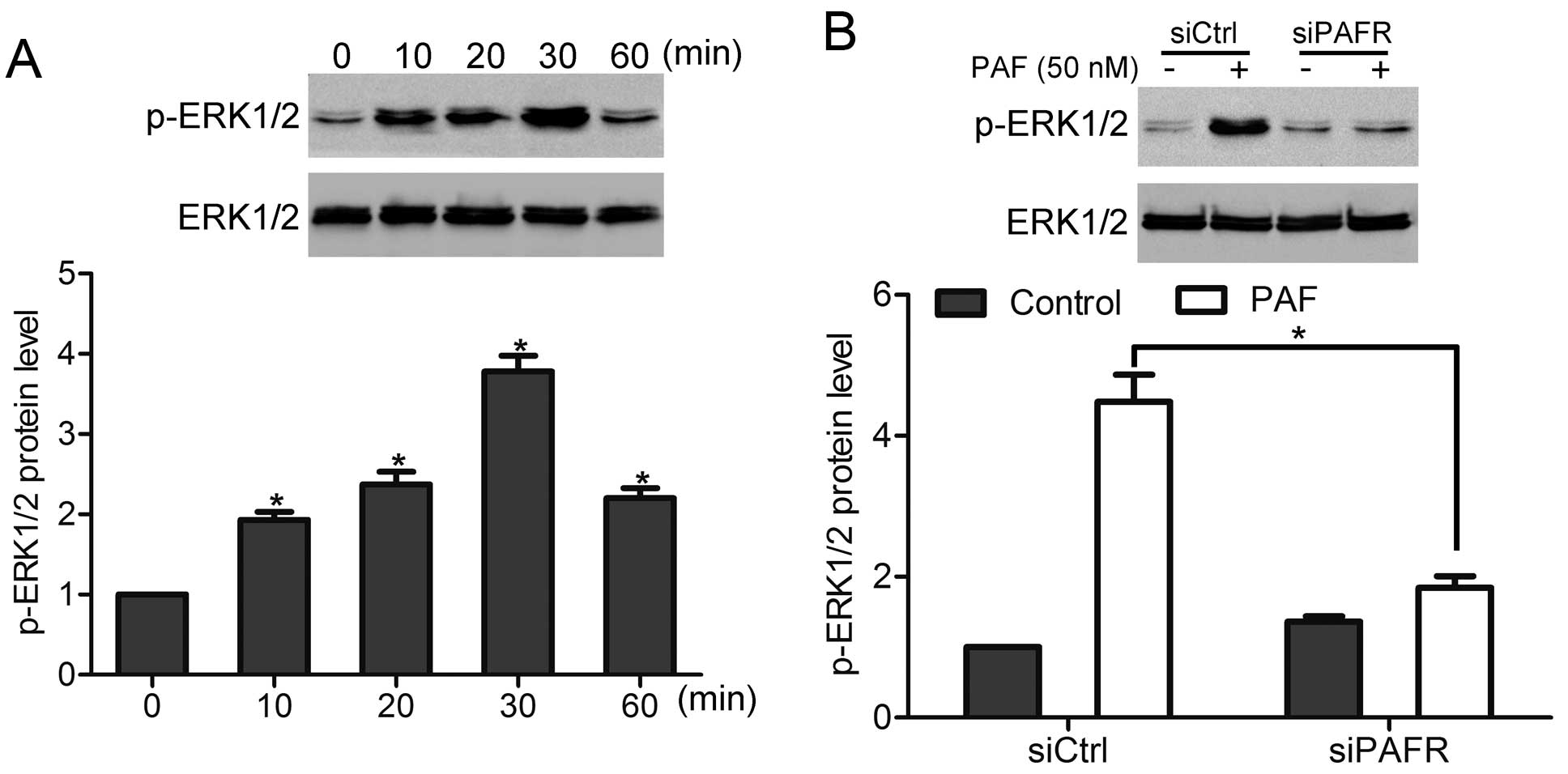
PAFR activation induces activation of
ERK1/2 in vitro
Next, we found that PAF stimulation induced
activation of ERK1/2 in DU-145 cells, in a time-dependent manner,
and peak activation was observed at 30 min (Fig. 5A). However, knockdown of PAFR
inhibited PAF-induced activation of ERK1/2 (Fig. 5B). These findings suggest that PAF
can mediate ERK1/2 activity via PAFR.
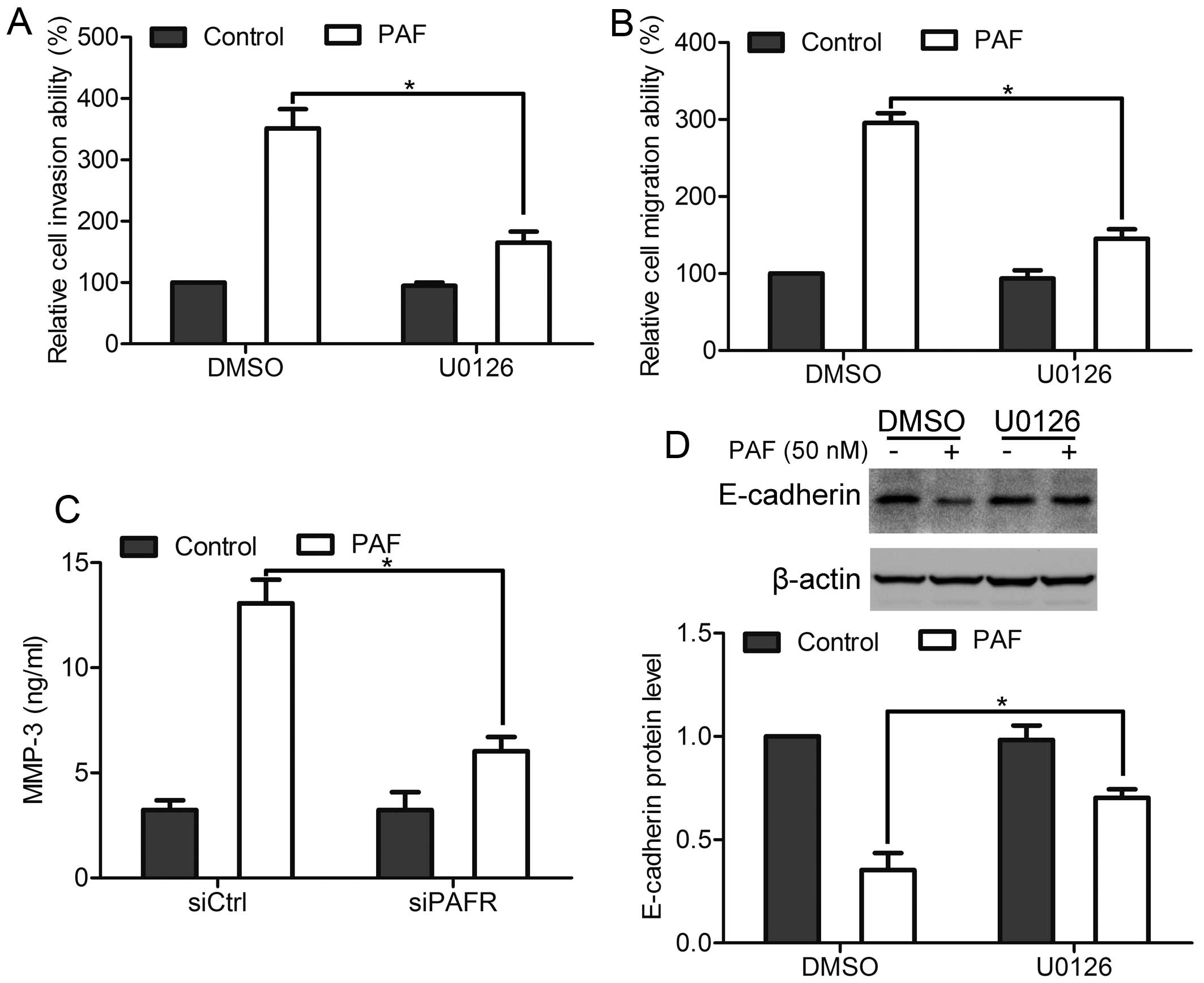
ERK1/2 pathway is required for
PAFR-mediated prostate cancer cell invasion and migration
To investigate the function of ERK1/2 pathway in
PAFR-mediated prostate cancer cell invasion and migration, cells
were incubated with U0126 for 1 h before PAF stimulation to inhibit
ERK1/2 activation. In vitro invasion assay and migration
assay showed that inhibition of ERK1/2 activation suppressed
PAF-mediated cell invasion and migration (Fig. 6A and B). Moreover, inhibition of
ERK1/2 activation inhibited PAF-mediated increase of MMP-3
expression and decrease of E-cadherin expression (Fig. 6C and D). Together, these data
suggest that ERK1/2 pathway is essential for PAFR-mediated prostate
cancer cell invasion and migration.
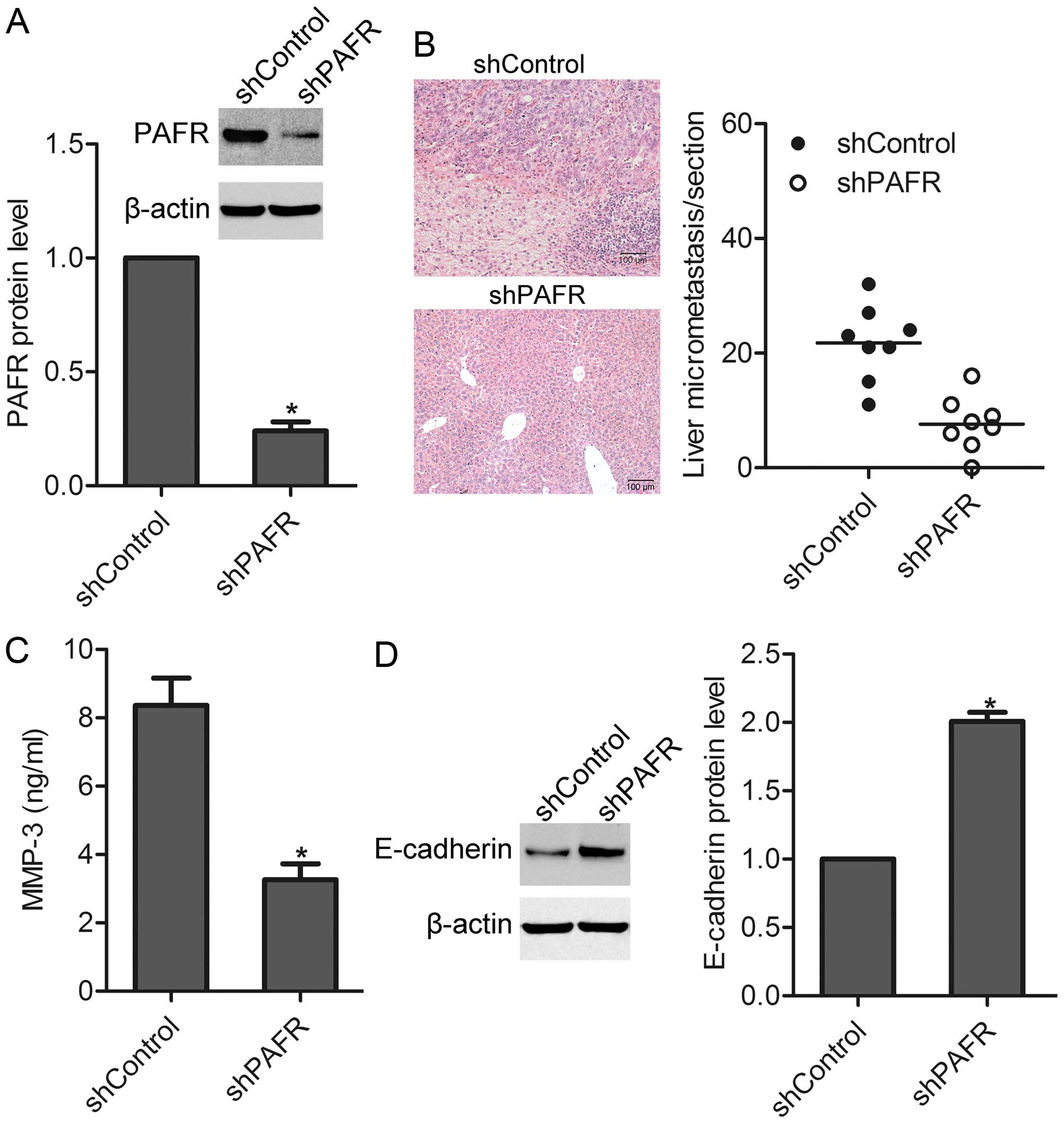
PAFR activation promotes the metastasis
of prostate cancer cells in vivo
To detect the effect of PAFR on the metastasis of
prostate cancer cells in vivo, cells were transfected with
pcDNA3.1-shPAFR to stably decrease PAFR expression (Fig. 7A). Then, the mice were injected
subcutaneously with shControl and shPAFR cells into the back,
respectively. Eight weeks later, the mice were sacrificed, and
liver micrometastasis was observed under a microscope. The results
showed that knockdown of PAFR greatly decreased the number of
micrometastasis in the liver (Fig.
7B). We found that after knockdown of PAFR, E-cadherin
expression was increased while MMP-3 expression was decreased in
tumor tissues (Fig. 7C and D).
These results suggest that PAFR participates in the metastasis of
prostate cancer cells in vivo.
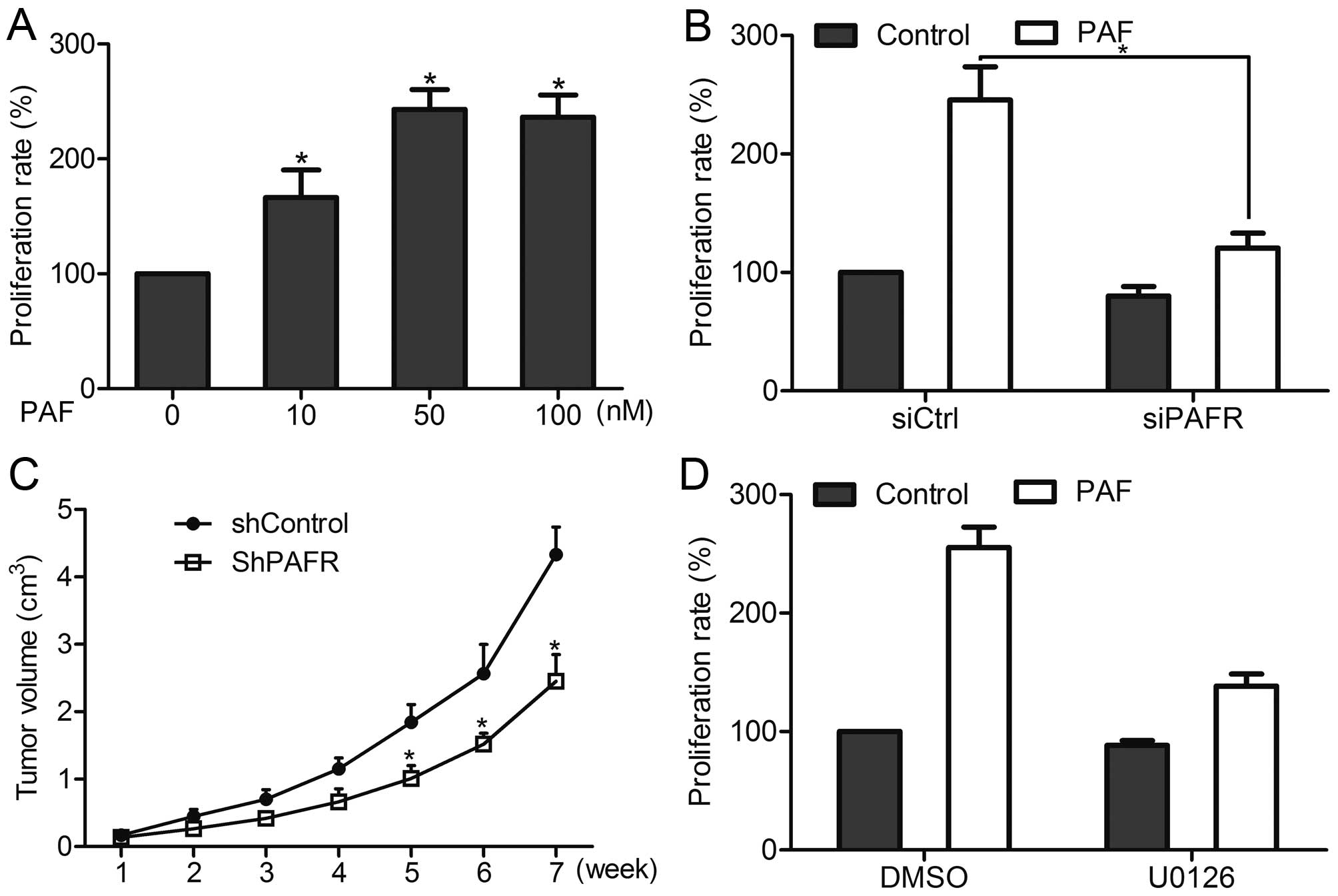
Effect of PAFR on prostate cancer cell
proliferation in vitro and in vivo
We examined the effect of PAFR on cell proliferation
of prostate cancer cells. In vitro CCK-8 proliferation assay
showed that PAF stimulation dose-dependently promoted the
proliferation of DU-145 cells in vitro, whereas knockdown of
PAFR suppressed the effect of PAF on cell proliferation (Fig. 8A and B). Similarly, in vivo
growth assay showed that knockdown of PAFR inhibited the
proliferation of DU-145 cells in mice (Fig. 8C). These results suggest that PAFR
is involved in prostate cancer cell proliferation. Moreover,
inhibition of ERK1/2 pathway attenuated PAF-mediated cell growth of
DU-145 cells in vitro (Fig.
8D), indicating PAFR regulated prostate cancer cell growth via
the ERK1/2 pathway.
Discussion
In the present study, we found that PAFR induced
activation of the ERK1/2 pathway, leading to the upregulation of
MMP-3 and downregulation of E-cadherin expression, ultimately
inducing the invasion and metastasis of prostate cancer cells in
vitro and in vivo. We also found that PAFR contributed
to prostate cancer cell proliferation via ERK1/2 pathway. These
findings suggest that PAFR may be an essential mediator in prostate
cancer progression.
PAF plays an important role in many cellular
processes. It is reported that PAF can induce activation of matrix
metallo-proteinase-2 activity and vascular endothelial cell
invasion and migration (10). In
cancer, Melnikova et al (11) have reported that PAF contributs to
the metastasis of melanoma. PAF binds the PAFR, which belongs to
GPCR family and is involved in the tumorigenesis and progression of
many cancers. It is reported that PAFR is essential for the
malignant potential in BRCA1 dysfunctional at-risk ovarian
epithelium (12), and PAFR
activation promotes the growth of ovarian cancer cells (13). In addition, studies have found that
crosstalk between protease-activated receptor 1 and PAFR regulates
melanoma metastasis (14) and PAFR
activation augments the growth and metastasis of lung cancer
(15). However, some studies have
found that lower expression of PAFR correlats with poor
differentiation and a poor prognosis in patients with
hepatocellular carcinoma after hepatectomy (16), and PAFR expression is negative
associated with histopathological stage and grade and patient
survival in gastric adenocarcinoma (9). In prostate cancer cells, Jan and Chao
(17) found that a specific PAFR
antagonist inhibits prostate cancer cell growth, but the function
of PAFR in the progression of prostate cancer cells and the
underlying molecular mechanisms are still not very clear. In the
present study, we found that PAFR expression was upregulated in
prostate cancer cells. Activation of PAFR by PAF dose-dependently
stimulated the growth, invasion and migration of prostate cancer
cells in vitro. Knockdown of PAFR inhibited PAF-mediated
cell growth, invasion and migration. Furthermore, knockdown of PAFR
suppressed the growth and metastasis of prostate cancer cells in
vivo. These data suggest that PAFR may act as a vital mediator
in prostate cancer cells.
We further found that PAFR activation decreased
E-cadherin expression and increased MMP-3 expression of prostate
cancer cells in vitro, whereas knockdown of PAFR attenuated
the effect of PAF on E-cadherin and MMP-3 expression. Moreover,
knockdown of PAFR increased E-cadherin expression and decreased
MMP-3 expression of prostate cancer cells in vivo. MMP-3 is
an important member of the matrix metalloproteinase family, which
is required for the dissolution of stromal collagen during tumor
dissemination (18). It is
considered that MMP-3 is involved in the invasion and metastasis of
many cancers including prostate cancer (19–21).
E-cadherin is a cell-cell adhesion protein and a well-documented
tumor suppressor (22). Many
studies have proved that downregulation of E-cadherin participates
in the occurrence and development of prostate cancer (23,24).
In the present study, we showed that PAFR induced prostate cancer
cell invasion and metastasis in vitro and in vivo,
and decreased E-cadherin expression and increased MMP-3 expression,
suggesting that PAFR may induce the invasion and metastasis of
prostate cancer cells via regulation of MMP-3 and E-cadherin
expression.
The MAPK ERK1/2 pathway is involved in a wide
variety of cellular processes in cancer development (25,26).
Studies have shown that ERK1/2 activation participates in cell
growth, invasion, malignant transformation and drug resistance of
prostate cancer (27,28). The ERK1/2 pathway can be activated
by many growth factors and cytokines that are important in the
progression of cancer. In the present study, we found that PAF
induced activation of ERK1/2 via PAFR. Moreover, using an ERK1/2
specific inhibitor, we found that ERK1/2 pathway was required for
PAFR-mediated cell growth, invasion and metastasis of prostate
cancer cells.
In conclusion, our data demonstrate that PAFR is
over-expressed in prostate cancer cells. PAFR promotes prostate
cancer cell invasion and metastasis of prostate cancer cells in
vitro and in vivo, possibly via activation of ERK1/2
pathway and regulation of E-cadherin and MMP-3 expression. We also
showed that PAFR stimulates the growth of prostate cancer cells via
ERK1/2 pathway. The present study provides evidence showing that
PAFR may have a potential value in early detection and therapy for
prostate cancer.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheville JC, Tindall D, Boelter C, Jenkins
R, Lohse CM, Pankratz VS, Sebo TJ, Davis B and Blute ML: Metastatic
prostate carcinoma to bone: Clinical and pathologic features
associated with cancer-specific survival. Cancer. 95:1028–1036.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zimmerman GA, McIntyre TM, Prescott SM and
Stafforini DM: The platelet-activating factor signaling system and
its regulators in syndromes of inflammation and thrombosis. Crit
Care Med. 30(Suppl): S294–S301. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Melnikova V and Bar-Eli M: Inflammation
and melanoma growth and metastasis: The role of platelet-activating
factor (PAF) and its receptor. Cancer Metastasis Rev. 26:359–371.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bussolati B, Biancone L, Cassoni P, Russo
S, Rola-Pleszczynski M, Montrucchio G and Camussi G: PAF produced
by human breast cancer cells promotes migration and proliferation
of tumor cells and neo-angiogenesis. Am J Pathol. 157:1713–1725.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fallani A, Calorini L, Mannini A,
Gabellieri S, Mugnai G and Ruggieri S: Platelet-activating factor
(PAF) is the effector of IFN gamma-stimulated invasiveness and
motility in a B16 melanoma line. Prostaglandins Other Lipid Mediat.
81:171–177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen J, Lan T, Zhang W, Dong L, Kang N,
Zhang S, Fu M, Liu B, Liu K, Zhang C, et al: Platelet-activating
factor receptor-mediated PI3K/AKT activation contributes to the
malignant development of esophageal squamous cell carcinoma.
Oncogene. 34:5114–5127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Y, Zhang M, Zhang X, Cai Q, Hong S,
Jiang W and Xu C: Synergistic effects of combined
platelet-activating factor receptor and epidermal growth factor
receptor targeting in ovarian cancer cells. J Hematol Oncol.
7:392014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giaginis C, Kourou E, Giagini A, Goutas N,
Patsouris E, Kouraklis G and Theocharis S: Platelet-activating
factor (PAF) receptor expression is associated with
histopathological stage and grade and patients' survival in gastric
adenocarcinoma. Neoplasma. 61:309–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Axelrad TW, Deo DD, Ottino P, Van Kirk J,
Bazan NG, Bazan HE and Hunt JD: Platelet-activating factor (PAF)
induces activation of matrix metalloproteinase 2 activity and
vascular endothelial cell invasion and migration. FASEB J.
18:568–570. 2004.PubMed/NCBI
|
|
11
|
Melnikova VO, Mourad-Zeidan AA, Lev DC and
Bar-Eli M: Platelet-activating factor mediates MMP-2 expression and
activation via phosphorylation of cAMP-response element-binding
protein and contributes to melanoma metastasis. J Biol Chem.
281:2911–2922. 2006. View Article : Google Scholar
|
|
12
|
Zhang L, Wang D, Jiang W, Edwards D, Qiu
W, Barroilhet LM, Rho JH, Jin L, Seethappan V, Vitonis A, et al:
Activated networking of platelet activating factor receptor and
FAK/STAT1 induces malignant potential in BRCA1-mutant at-risk
ovarian epithelium. Reprod Biol Endocrinol. 8:742010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Y, Zhang X, Hong S, Zhang M, Cai Q,
Zhang M, Jiang W and Xu C: The expression of platelet-activating
factor receptor modulates the cisplatin sensitivity of ovarian
cancer cells: A novel target for combination therapy. Br J Cancer.
111:515–524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Melnikova VO, Balasubramanian K, Villares
GJ, Dobroff AS, Zigler M, Wang H, Petersson F, Price JE, Schroit A,
Prieto VG, et al: Crosstalk between protease-activated receptor 1
and platelet-activating factor receptor regulates melanoma cell
adhesion molecule (MCAM/MUC18) expression and melanoma metastasis.
J Biol Chem. 284:28845–28855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hackler PC, Reuss S, Konger RL, Travers JB
and Sahu RP: Systemic platelet-activating factor receptor
activation augments experimental lung tumor growth and metastasis.
Cancer Growth Metastasis. 7:27–32. 2014.PubMed/NCBI
|
|
16
|
Kitagawa D, Taketomi A, Kayashima H,
Kuroda Y, Itoh S, Yamashita Y and Maehara Y: Expression of
platelet-activating factor receptor: A novel prognosticator in
patients with hepatocellular carcinoma following hepatectomy.
Oncology. 72:381–387. 2007. View Article : Google Scholar
|
|
17
|
Jan CR and Chao YY: Novel effect of
Y-24180, a presumed specific platelet activation factor receptor
antagonist, on Ca2+ levels and growth of human prostate
cancer cells. Cell Signal. 16:959–965. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Littlepage LE, Sternlicht MD, Rougier N,
Phillips J, Gallo E, Yu Y, Williams K, Brenot A, Gordon JI and Werb
Z: Matrix metalloproteinases contribute distinct roles in
neuroendocrine prostate carcinogenesis, metastasis, and
angiogenesis progression. Cancer Res. 70:2224–2234. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Slavin S, Yeh CR, Da J, Yu S, Miyamoto H,
Messing EM, Guancial E and Yeh S: Estrogen receptor α in
cancer-associated fibroblasts suppresses prostate cancer invasion
via modulation of thrombospondin 2 and matrix metalloproteinase 3.
Carcinogenesis. 35:1301–1309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu F, Liu P, Li J and Zhang Y: Eotaxin-1
promotes prostate cancer cell invasion via activation of the
CCR3-ERK pathway and upregulation of MMP-3 expression. Oncol Rep.
31:2049–2054. 2014.PubMed/NCBI
|
|
21
|
Yamashita CM, Radisky DC, Aschner Y and
Downey GP: The importance of matrix metalloproteinase-3 in
respiratory disorders. Expert Rev Respir Med. 8:411–421. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Canel M, Serrels A, Frame MC and Brunton
VG: E-cadherin-integrin crosstalk in cancer invasion and
metastasis. J Cell Sci. 126:393–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Davies G, Jiang WG and Mason MD:
E-cadherin and associated molecules in the invasion and progression
of prostate cancer. Oncol Rep. 5:1567–1576. 1998.PubMed/NCBI
|
|
24
|
Khamis ZI, Iczkowski KA and Sang QX:
Metastasis suppressors in human benign prostate, intraepithelial
neoplasia, and invasive cancer: Their prospects as therapeutic
agents. Med Res Rev. 32:1026–1077. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
De Luca A, Maiello MR, D'Alessio A,
Pergameno M and Normanno N: The RAS/RAF/MEK/ERK and the PI3K/AKT
signalling pathways: Role in cancer pathogenesis and implications
for therapeutic approaches. Expert Opin Ther Targets. 16(Suppl 2):
S17–S27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
28
|
Sosa MS, Avivar-Valderas A, Bragado P, Wen
HC and Aguirre-Ghiso JA: ERK1/2 and p38α/β signaling in tumor cell
quiescence: opportunities to control dormant residual disease. Clin
Cancer Res. 17:5850–5857. 2011. View Article : Google Scholar : PubMed/NCBI
|