Introduction
Pancreatic cancer is the fourth or fifth most common
cause of cancer-related death in most western industrialized
countries. The disease has a poor prognosis with a 5-year survival
rate of <5%. In unresectable cases, chemo- or radiotherapy is
used for palliation of symptoms and improvement of survival
(1,2). Gemcitabine is currently the standard
systemic treatment for unresectable and advanced pancreatic cancer,
which can extend patient survival by 5–6 months (3). To date, a more satisfactory outcome
has not been reported when gemcitabine is used in combination with
other cytotoxic drugs (3–5).
The molecular basis for the resistance of pancreatic
cancers to chemotherapy is not fully understood. It appears to
result from various mechanisms that can each influence tumor
resistance to gemcitabine. One form of drug resistance, termed
multidrug resistance (MDR), is linked to the expression of proteins
that act as drug efflux pumps. MDR is characterized by the
development of broad cross-resistance to functionally and
structurally unrelated drugs. This enhanced efflux of drugs is
mediated by a super family of membrane glycoproteins, the
ATP-binding cassette (ABC) transporters. The ABCB superfamily
includes a 170-kDa transporter protein (ABCB1), also termed
P-glycoprotein (P-gp). P-gp is comprised of the protein product of
the human MDR1 gene in complex with the multidrug
resistance-associated protein (MRP) (6). The expression of P-gp correlates with
both the decreased accumulation of drugs, and the degree of
chemoresistance in many different human cancer cells (7–9).
Generally, 40–50% of patient tumors exhibit overexpression of P-gp
(10). Although gemcitabine is a
substrate for the ATP-dependent efflux pump (11), it is predominantly transported into
the cell via facilitated diffusion mediated by the equilibrative
nucleoside transporter (ENT), and sodium-dependent transporters
(concentrative nucleoside transporter, CNT) (12,13).
Pancreatic tumor cells can express high levels of ENT1, whereas
members of the CNT family are present at only negligible levels
(14).
Emerging evidence suggests that cancer stem cells
(CSCs), a small subset of undifferentiated cells found within
tumors, play a key role in tumorigenicity and malignancy (15). SP cells represent a small
subpopulation of tumor cells that have properties associated with
CSC in that they can rapidly efflux lipophilic fluorescent dyes
producing a characteristic profile in fluorescence-activated flow
cytometric analysis (16). SP
cells are generally defined by their ability to efflux the
fluorescent DNA binding dye Hoechst 33342 (H33342) and the
differential emission spectra of this dye when it binds chromatin
(16). The pumps responsible for
dye efflux are attributed to the ABC superfamily transporters,
including the MDR1/P-glycoprotein, ABCG2, MRP1 and ABCA2 (17–20).
These pumps also transport chemotoxic drugs out of the cell,
including vinblastine, doxorubicin, daunorubicin and paclitaxel
(21). SP cells exhibit increased
chemoresistance following in vitro exposure to gemcitabine
(22,23). The FACS-based assay used to detect
the presence of side populations is also currently under evaluation
as a general method to identify and isolate CSCs subpopulations
within tumor samples.
Verapamil is a calcium channel blocker that is
utilized clinically to treat cardiac arrhythmias (24). It is also a first generation
inhibitor of P-gp (25). When
combined with chemotherapeutic agents, verapamil can help to
promote intra-cellular drug accumulation (26). This has been demonstrated in
non-small cell lung cancer, colorectal carcinoma, leukemia, and
neuroblastoma cell lines (27–30).
Based on this ability of verapamil to inhibit P-gp transport
activity, it can also be used as an ‘SP’ blocker in the Hoechst
33342 assay as it will substantially reduce SP cells as visualized
by flow cytometry analysis. Based on these observations, we
hypothesized that verapamil treatment may directly exert anti-SP
effects and therefore enhanced gemcitabine sensitivity in
pancreatic cancer.
In this study, the biological characteristics of
CSCs in pancreatic cancer SP cells including their self-renewal
ability, resistance to gemcitabine, and general tumorigenicity were
investigated in the context of verapamil treatment.
Materials and methods
Human pancreatic cancer cells and culture
conditions
Human pancreatic adenocarcinoma cell lines L3.6pl
(31) and AsPC-1 (American Tissue
Culture Collection) were maintained in Dulbecco's minimal essential
medium (D-MEM; Invitrogen GmbH, Karlsruhe, Germany), supplemented
with 10% fetal bovine serum (Biochrom AG, Berlin, Germany), 2% MEM
vitamin mixture (PAN Biotech GmbH, Aidenbach, Germany), 2% MEM NEAA
(PAN Biotech GmbH), 1% penicillin streptomycin (PAN Biotech GmbH,
Aidenbach, Germany) and 2% glutamax (Invitrogen GmbH). Cells were
incubated in a humidified incubator (37°C, 5% CO2),
grown in cell culture flasks, and passaged on reaching 70–80%
confluence.
A gemcitabine-resistant pancreatic cancer cell line,
termed L3.6plGres, was developed from the parental
L3.6pl cell line by gradually increasing the concentration of
gemcitabine (Gemzar; Lilly Deutschland GmbH, Giessen, Germany) in
the cultured cells. Gemcitabine was first added at a concentration
of 0.5 ng/ml (based on the IC50 value of L3.6pl). When
the cells reached exponential growth, they were subcultured for two
additional passages with 0.5 ng/ml gemcitabine or until the cells
grew stably. The concentration of gemcitabine was then increased to
100 ng/ml and the cells were passaged until a stable
gemcitabine-resistant pancreatic cancer cell line
(L3.6plGres) was established.
Isolation of SP- and non-SP-cell
fractions from L3.6plGres and AsPC-1 cell lines
SP- and non-SP-cell fractions were identified and
isolated using a modified protocol described by Goodell et
al (16). Briefly,
1×106/ml cells were re-suspended in D-MEM containing 2%
fetal bovine serum and labeled with H33342 (Sigma-Aldrich GmbH,
Steinheim, Germany) at a concentration of 2.5 μg/ml for 60 min in
37°C water bath, either alone or with 225 μM verapamil
hydrochloride (Sigma-Aldrich GmbH). After 60 min the cells were
centrifuged (300 g, 4°C) for 5 min, and then resuspended in
ice-cold PBS containing 2% fetal bovine serum. The cells were
passed through a 40-μm mesh filter and maintained at 4°C in the
dark until flow cytometry analysis or sorting. Cells were
counter-stained with 10 μg/ml propidium iodide to label dead cells,
and the entire preparation was then analyzed using a BD-LSRII flow
cytometer (BD Biosciences, Heidelberg, Germany) and FlowJo software
(Treestar Inc., Ashland, OR, USA), or sorted using a MoFlo cell
sorter with the Summit 4.3 software (Beckmann Coulter GmbH,
Krefeld, Germany). Hoechst dye was excited at 355 nm (32), and fluorescence was measured at two
wavelengths using a 450/50-nm (blue) band-pass filter and a
670/30-nm (33) long-pass edge
filter. Following isolation the SP and non-SP cell fractions were
used for in vitro and in vivo assays.
Cell viability and proliferation
assay
Trypan blue (Sigma-Aldrich) staining was used to
test for cell viability. The dye stains dead cells, and livings are
distinguished by their ability to exclude the dye performing phase
contrast microscopy. Cell viability was calculated using the
following formula: Cell viability = unstained cells/unstained +
trypan blue stained cells × 100%. Cell proliferation was measured
using the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto,
Japan) according to the manufacturer's instructions. In this assay
5,000–8,000 cells/well were plated in a 96-well plate and grown
overnight, and then treated for 24 h with gemcitabine or verapamil.
Cell proliferation was then determined using a VersaMax tunable
microplate reader and Softmaxpro 5.2 software for data analysis
(Molecular Devices, Sunnyvale, CA, USA).
Apoptosis assay
Cell apoptosis was analyzed using an Annexin V-FITC
assay (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) according
to the manufacturer's instructions. After determination of cell
numbers, 1×106 cells were washed with binding buffer and
centrifuged. The cell pellet was resuspended in binding buffer, and
10 μl of Annexin V-FITC per 106 cells was added, mixed,
and the preparation was incubated for 15 min in the dark at room
temperature. The cells were then washed, and the cell pellet
resuspended in binding buffer. The PI solution was added
immediately prior to analysis using a BD-LSRII flow cytometer (BD
Biosciences, Heidelberg, Germany) and FlowJo software version 7.6
(Treestar Inc.). Experiments were repeated at least three
times.
Colony formation assay
To determine the ability of cells to form colonies,
500 cells in 2 ml of D-MEM medium were seeded into each well of a
6-well plate. The medium was changed two times per week and the
assay was stopped when colonies were clearly visible without using
a microscope (i.e., each single colony comprised approximately ≥50
cells). The resulting colonies were stained with 0.1% crystal
violet and counted.
Flow cytometry analysis
For this staining procedure, the cells were
incubated in the dark and on ice. Firstly they were blocked with
PBS containing 0.5% albumin bovine (BSA) and 0.02% NaN3
with FCR blocking reagent (Miltenyi Biotec) for 15 min. Cells were
then stained with the P-glycoprotein mouse mAb primary antibody
(clone C219, Calbiochem, Darmstadt, Germany) for 45 min on ice.
Afterwards they were incubated with the fluorescence-conjugated
secondary antibody, goat anti-mouse IgG FITC (Abcam, Cambridge, UK)
for 45 min. FITC isotype mouse immunoglobulins were used as
negative controls. FITC was excited using a 488-nm laser and
detected by a 530/30 band-pass filters. Dead cells were excluded by
gating on forward and side scatter and eliminating PI-positive
population cells. All the data were analyzed using FlowJo software
version 7.6 (Treestar Inc.).
Western blot assay
The proteins ENT1 and P-gp were assayed by
immunoblotting. Cells were resuspended in ice-cold RIPA buffer
supplemented with a cocktail of protease/phosphatase inhibitors
(Roche, Mannheim, Germany). Cells were further incubated on ice for
10 min and centrifuged at 14,000 g at 4°C for 10 min. After
determination of the protein concentration using the BCA protein
assay (Thermo Fisher Scientific, Rockford, IL, USA), an equal
amount of protein was run on polyacrylamide gels and transferred
semidry to polyvinylidene difluoride membranes (Amersham,
Braunschweig, Germany). The corresponding primary antibody was
incubated at 4°C overnight. After blocking for 2 h, the secondary
antibody was added and incubated for 2 h at room temperature,
following by washing. An enhanced chemiluminescense
system(Amersham) was used for detection. Afterwards the membranes
were reused for β-actin staining (Sigma-Aldrich GmbH) to ensure
equal protein levels. The polyclonal goat anti-mouse/rabbit
immunoglobulin HRP (Dako, Glostrup, Denmark) was used according to
the manufacturer's instructions. For detection of the ENT1 protein,
the anti-ENT1 antibody (Abcam) was used at 1:500 in 5% BSA-TBST
buffer. For P-gp protein detection, the monoclonal mouse C219
antibody (Calbiochem) was used at 1:100 dilutions in 7.5% skim
dried milk-PBST buffer. This antibody recognizes the two P-gp
isoforms (34).
Orthotopic pancreatic cancer mouse
model
Approval by the animal rights commission of the
state of Bavaria, Germany was obtained for all animal experiments.
Male athymic Balb/c nu/nu mice were purchased from Charles River,
Inc. (Sulzfeld, Germany). Mice aged 6–8 weeks with an average
weight of 20 g were maintained under a 12:12-h light-dark cycle and
were used for the orthotopic pancreatic cancer mouse model. They
were anesthetized using ketamine [100 mg/kg body weight (BW),
xylazine (5 mg/kg BW] and atropine. The operation was carried out
in a sterile manner. A 1-cm left abdominal flank incision was made,
the spleen was exteriorized and 1×105 isolated SP- and
non-SP L3.6plGres cells were injected into the
subcapsular region of the pancreas. Before injection, cell
viability was assessed by trypan blue staining and only cell
preparations that showed ≥95% viability were used for injection.
Mice were divided into an SP control group, non-SP control group
and different drug treatment SP-groups. Four weeks after orthotopic
implantation, therapy was initiated. One treatment group obtained
daily (every workday) intraperitoneal injection of low
concentration of verapamil (200 μM, 0.5 mg/Kg BW), the second
treatment group received a higher concentration of verapamil (10
mM, 25 mg/Kg BW). Nine weeks after the injection of tumor cells,
all the mice were sacrificed and examined for orthotopic tumor
growth and development of metastases. The pancreatic tumors as well
as other organs were isolated, weighed, and then used for H&E
and immunohistochemical staining.
Immunohistochemistry
Formaldehyde-fixed and paraffin-embedded tissues
were serially sectioned at 3 μm and allowed to dry overnight.
Sections were deparaffinized in xylene followed by a graded series
of ethanol (100, 95 and 80%) and rehydrated in phosphate-buffered
solution, pH 7.5. Paraffin-embedded tissues were used for the
Ki67 proliferation index assay, microvascular density
analysis, and the TUNEL assay. TUNEL assay was carried out using an
in situ cell death detection kit, Fluorescein (Roche
Diagnostics GmbH, Mannheim, Germany), according to the
manufacturer's instructions. The antibodies used for
paraffin-embedded tissues were monoclonal rabbit anti-Ki67
antibody (ab 16667, Abcam) and polyclonal rabbit anti-CD31 (ab
28364, Abcam). The primary antibodies were diluted in PBS
containing 3% bovine serum albumin (BSA). The sample slides were
treated for 20 min with blocking solution (8% goat serum or rabbit
serum in PBS with 3% BSA) before the primary antibody was applied.
Endogenous peroxidase was blocked by incubation with 3% hydrogen
peroxide (H2O2). Endogenous avidin and biotin
were blocked using the Avidin/Biotin Blocking kit (Vector,
USA).
Overnight incubation with the primary antibodies was
followed by incubation with the respective biotinylated secondary
antibodies (goat anti-rabbit, BA-1000, Vector), followed by the ABC
reagent for signal amplification (Vectastain ABC-Peroxidase kits,
PK-4000, Vector). Between the incubation steps, the slides were
washed in TBS. 3,3′-diaminobenzidine (DAB, Dako, USA) was used to
develop the color. Slides were counter-stained with hematoxylin,
mounted in Kaisers Glycerinegelatine (Merck, Germany), and
covered.
For quantification of the staining intensity, each
index (Ki67, microvascular density CD31, and TUNEL) was
evaluated in a blinded manner. Slides were observed under a
high/low magnification scope (×200/×100), each one was evaluated
within 3 fields, and the data were analyzed as mean positive signal
(Ki67-positive cells, amount of microvessels, cells with
strong FITC-fluorescence) of 3 fields.
Statistical analysis
Statistical evaluation was performed using the
paired Student's t-test or ANOVA test (Microcal Origin) with
p<0.05 considered to be statistically significant [p<0.05
(&); p<0.01 (#) p<0.001
(*); p<0.0001 (**)]. GraphPad
Prism® 5.0 or Microsoft excel 2010 software were used to
generate graphs and tables.
Results
Side population cells are enriched in
L3.6plGres cells and show higher colony formation
The human pancreatic adenocarcinoma cell line L3.6pl
was continuously cultured with gemcitabine, starting at 0.5 ng/ml,
which was then gradually increased to 100 ng/ml over multiple cell
passages to eventually develop a gemcitabine-resistant version of
the cell line (L3.6plGres). The 24 h IC50
increased from 6.1±0.9 ng/ml in the parental L3.6pl cells, to
498.8±3.2 ng/ml in the L3.6plGres cells (L3.6pl vs.
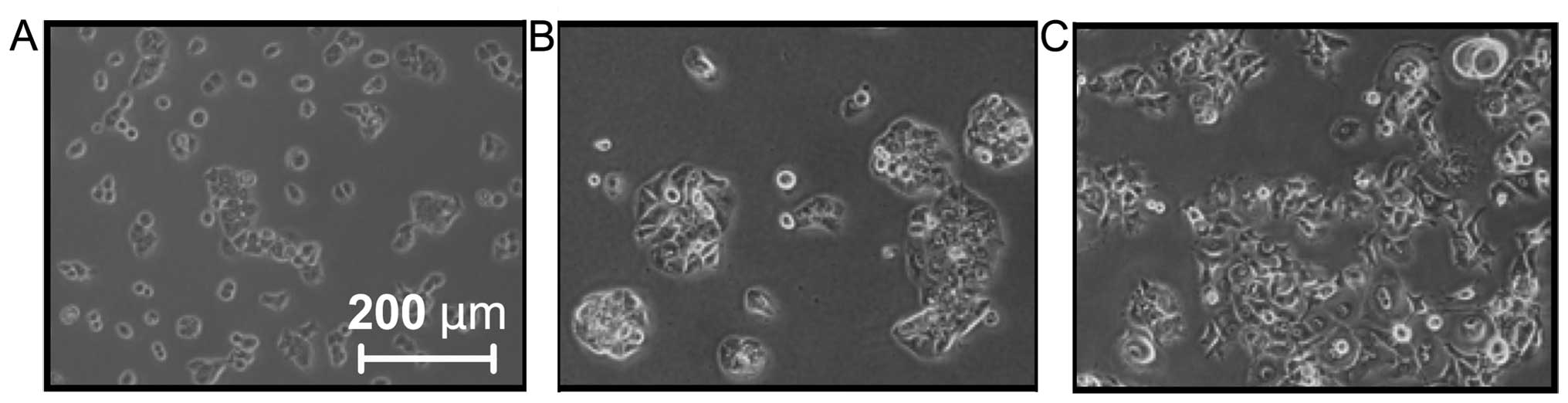
L3.6plGres p<1E−9). The general morphology
of L3.6plGres cells was also changed with gemcitabine
resistance into large fibroblastoid-like tumor cells (Fig. 1), while the parental L3.6pl cell
line maintained its original round shape. By contrast, the AsPC-1
line, which was largely resistant to gemcitabine and presented with
a spindle-like shape, did not show a change in morphology under
gemcitabine selection (data not shown).
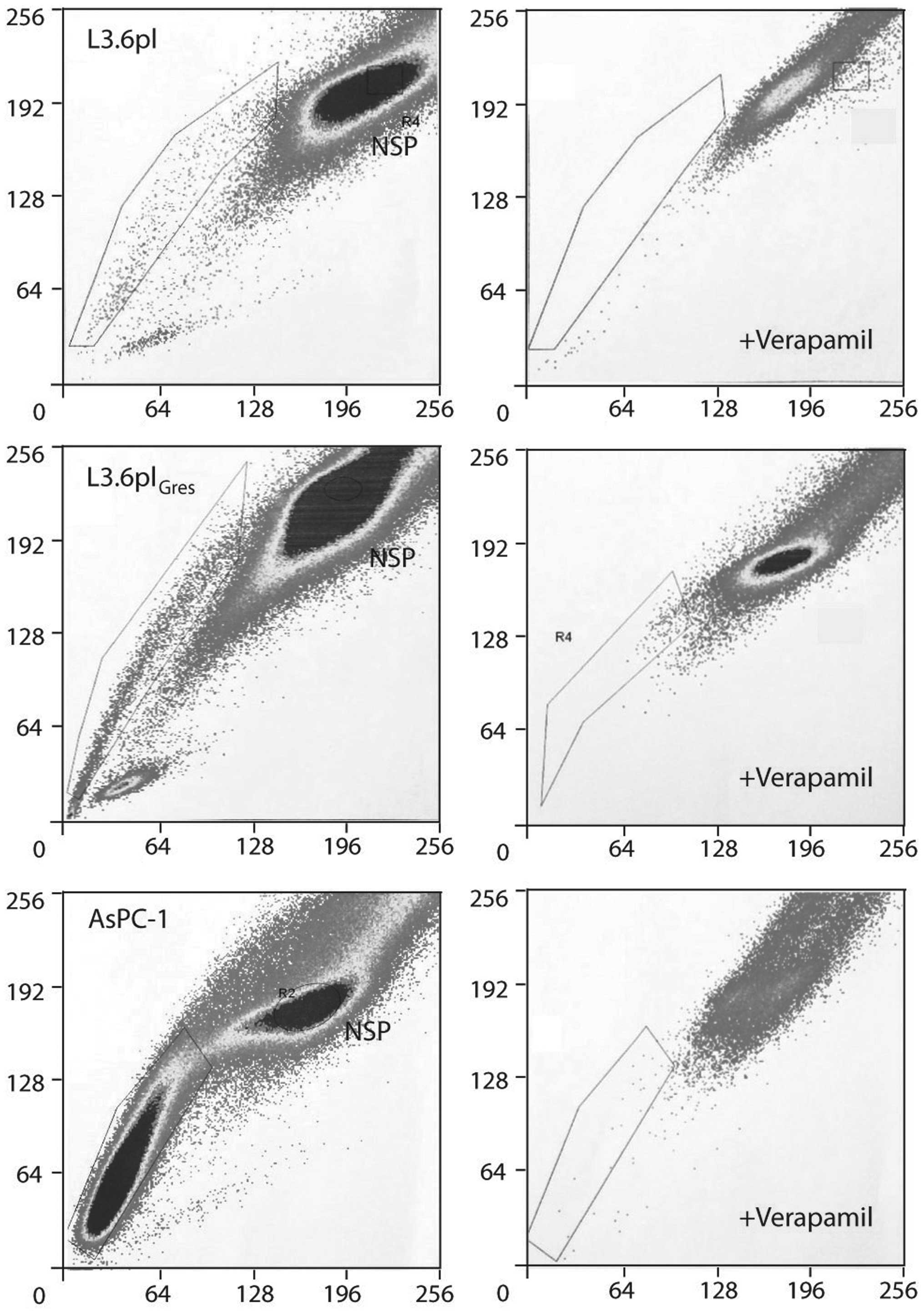
The L3.6pl and AsPC-1 cell lines were assayed for
the presence of SP-cells. Verapamil hydrochloride, which blocks
transporters of the ABC family and abrogates the ability of cells
to efflux the dye, served as an SP cell control in the H33342 assay
(SP cells will disappear or decrease in the presence of ABCG2
inhibitors owing to inhibition of Hoechst efflux from the cells).
Both cell lines were found to contain a distinct proportion of SP
cells. The percentage of SP cells increased from 0.9±0.22% in
L3.6pl, to 5.38±0.99% in the L3.6plGres cells in
response to continuous gemcitabine treatment. This proportion of SP
cells could be diminished by treatment with verapamil
hydrochloride. In the AsPC-1 cell line, 21.35±3.48% SP cells were
identified, which was found to diminish to 3.56±0.87% in response
to verapamil blockade (p<5E−10) (Fig. 2).
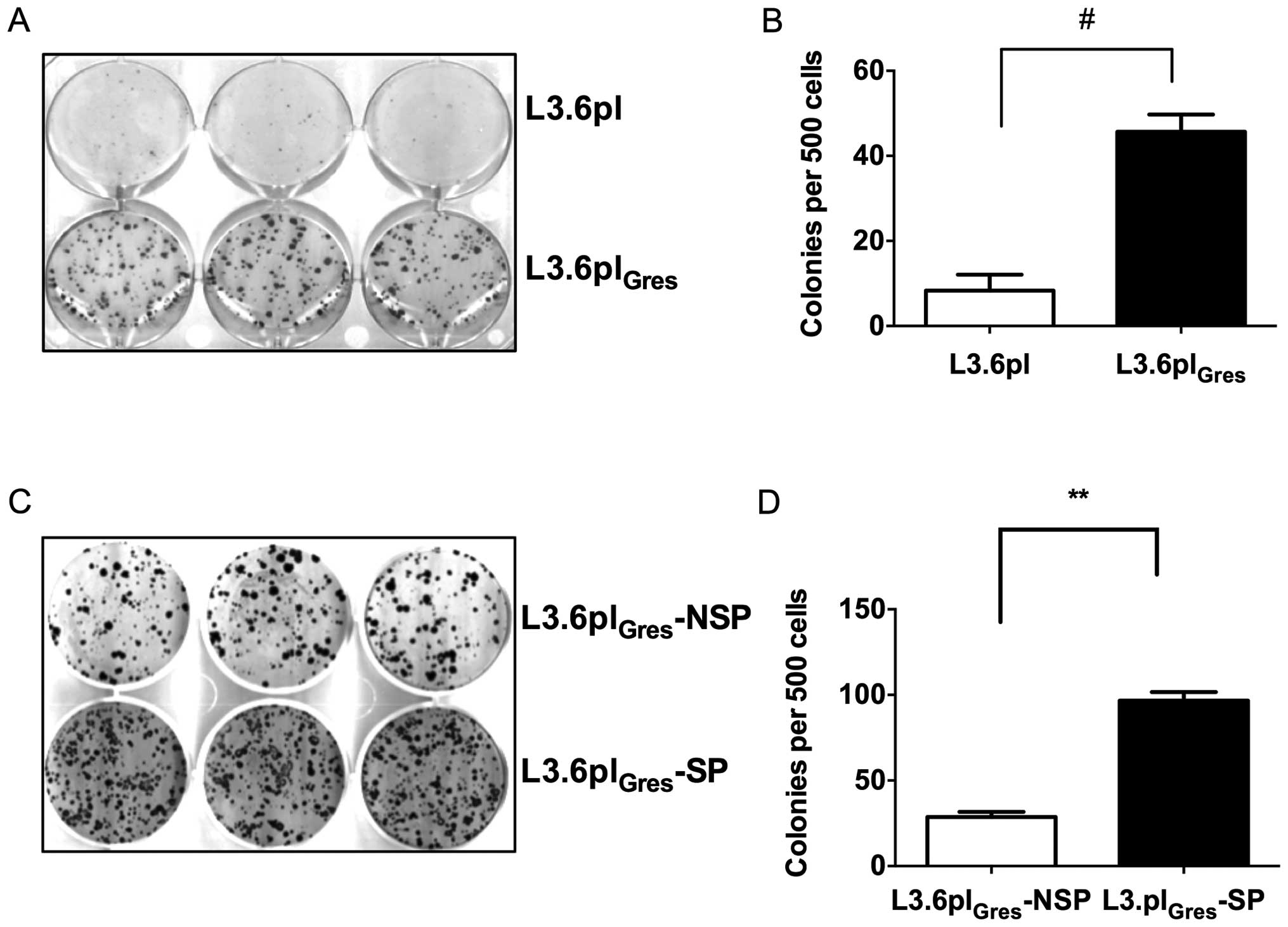
We then compared the colony formation ability of
L3.6plGres, L3.6pl, as well as L3.6plGres-SP
and NSP cells. The assay showed that L3.6plGres cells
and L3.6plGres-SP cells have significantly higher colony
formation ability, and thus enhanced CSC-like characteristics when
compared to the L3.6pl or L3.6plGres-NSP cells (Fig. 3).
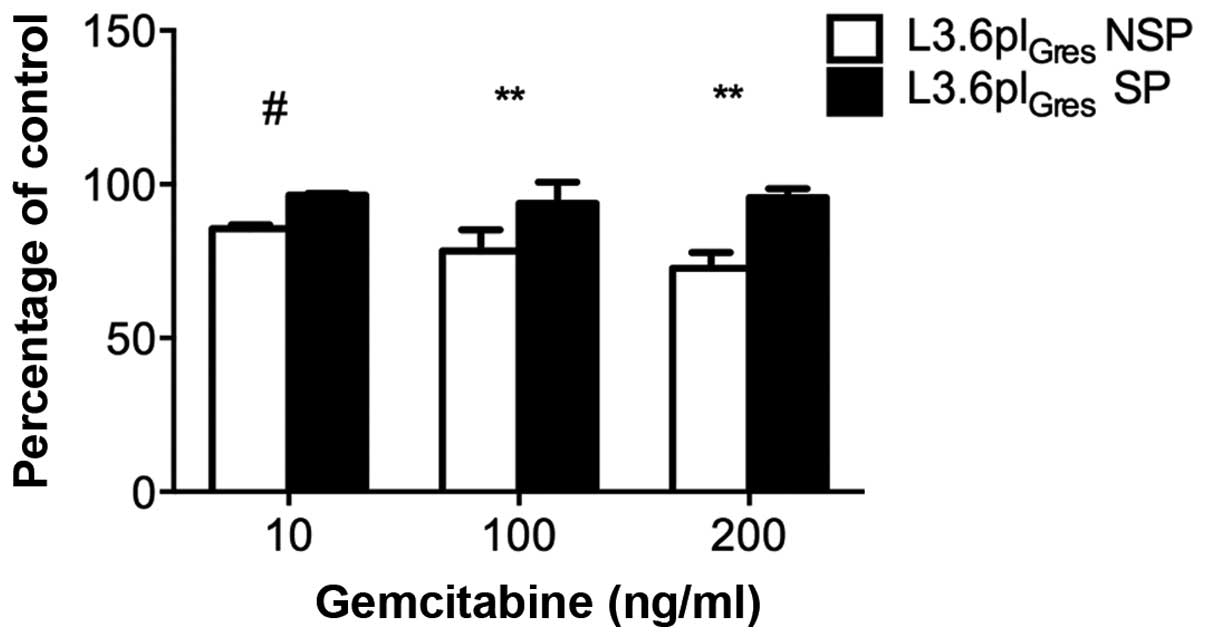
The gemcitabine resistance of L3.6plGres
cells was further validated by cell viability assays. The results
show that L3.6plGres-SP cells have enhanced resistance
to gemcitabine relative to respective NSP cells (Fig. 4).
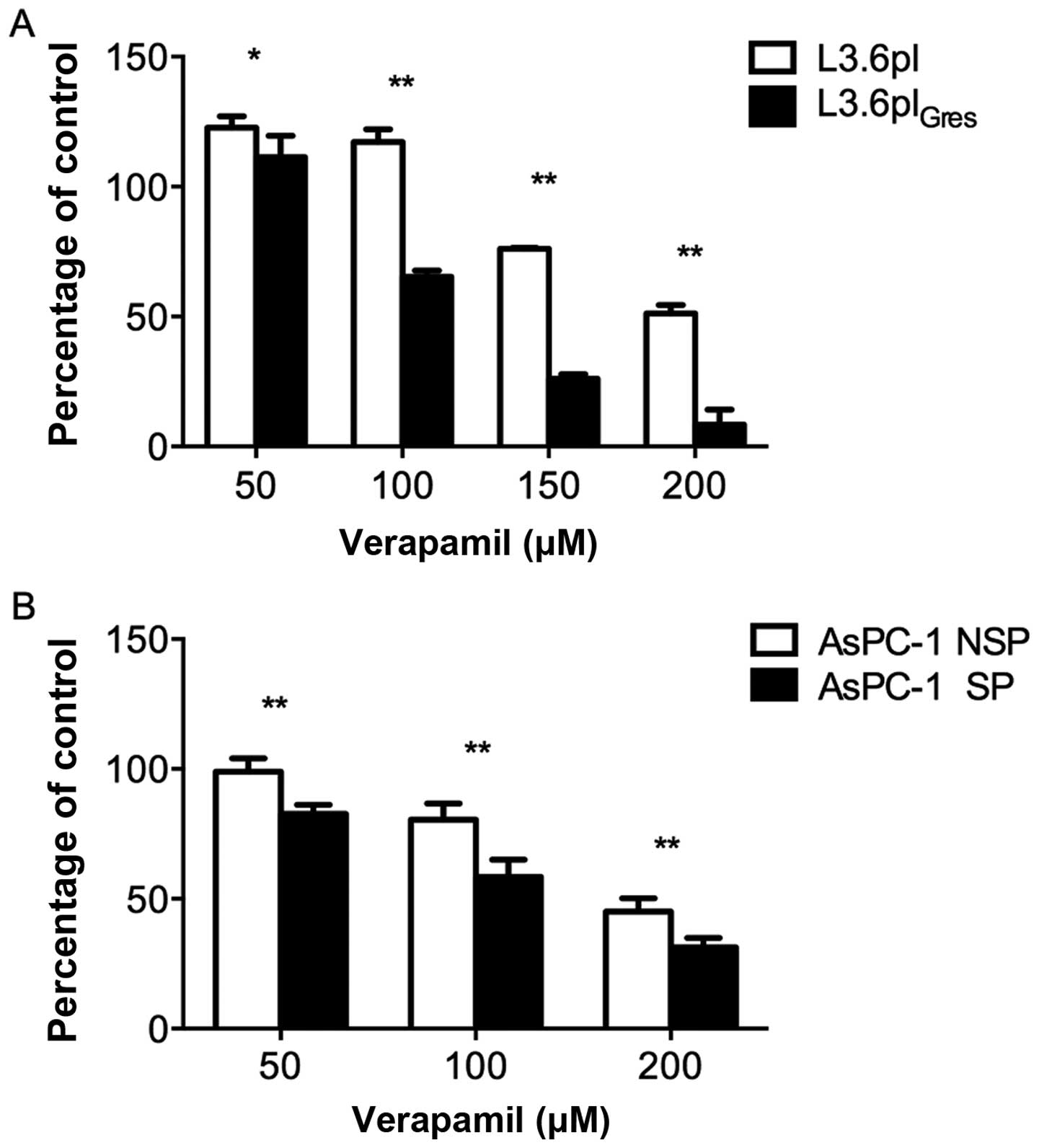
Verapamil can effectively inhibit
L3.6plGres and AsPC-1 SP cell proliferation
Since the percentage of SP cells in the parental
L3.6pl cell line was relatively low, it was difficult to harvest
sufficient cell numbers by FACS sorting for cell proliferation
assays. Therefore, the proliferation capacity of the
L3.6plGres cell line (enriched SP cells) was compared to
control L3.6pl cells. In parallel, the SP and Non-SP cells from the
AsPC-1 cell line were directly compared.
To investigate whether verapamil alone is effective
against SP cells, we analyzed its effect on L3.6pl- and
L3.6plGres-, AsPC-1-SP and -NSP cell proliferation. All
of the cell lines tested showed a concentration-dependent reduction
in cell proliferation. In response to 50, 100, 150 and 200 μM of
verapamil, the L3.6plGres- and AsPC-1-SP cells were more
sensitive to verapamil effects than L3.6pl- and AsPC-1-NSP cells
(Fig. 5).
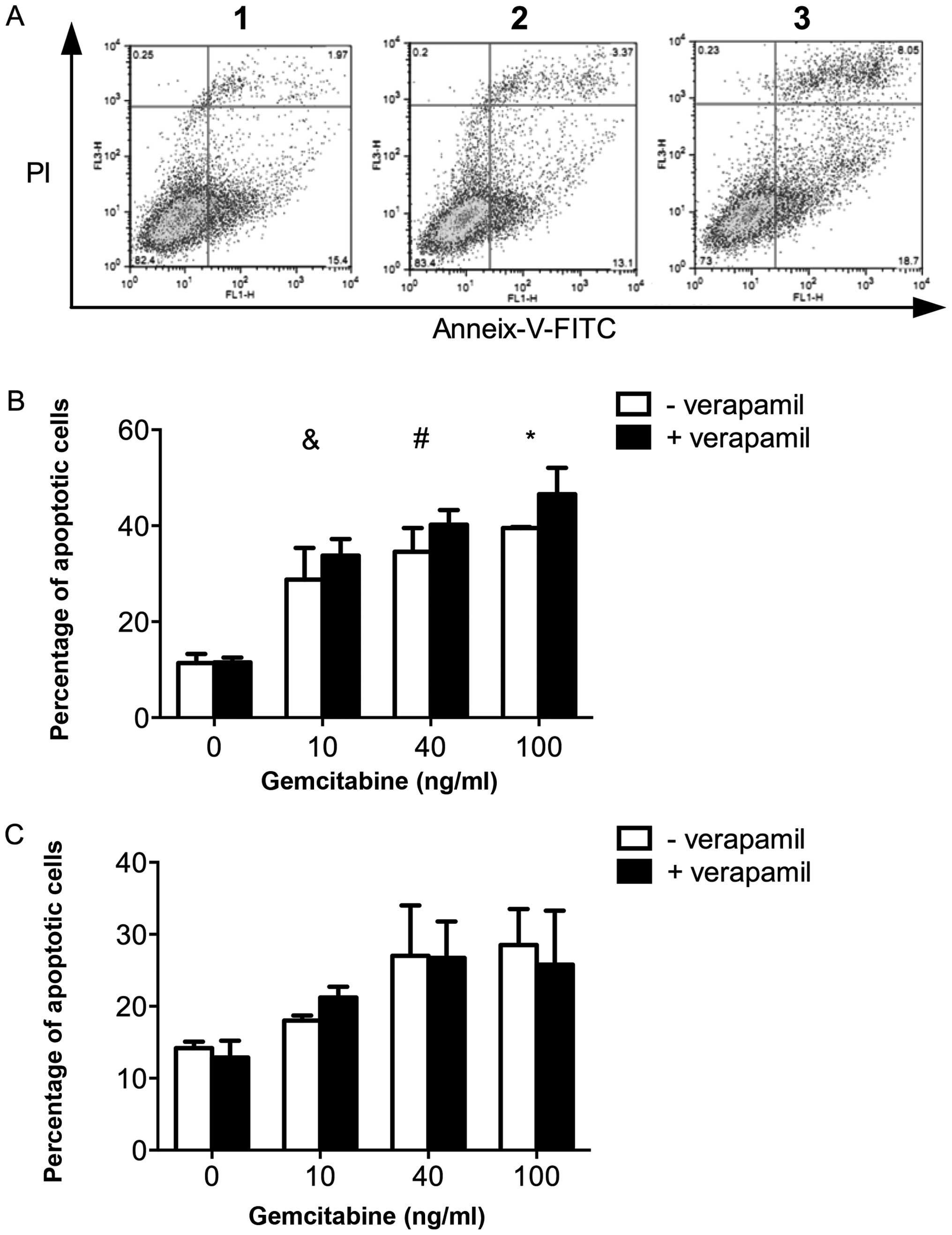
Pro-apoptotic effect of verapamil in
combination with gemcitabine in L3.6pl and L3.6plGres
cells
The potential additive effect of verapamil in
combination with gemcitabine was then investigated.
L3.6plGres-SP cells were treated separately with
verapamil (50 μM), gemcitabine (10 ng/ml), and in combination.
Using FACS analysis, the level of apoptotic cells after combined
treatment (at 24 h) was found to increase relative to treatment
with verapamil or gemcitabine alone (Fig. 6A). In dose response experiments,
the apoptosis of L3.6pl and L3.6plGres cells in response
to increasing concentrations of gemcitabine was determined, while
maintaining the concentration of verapamil at 50 μM. A
dose-dependent increase in apoptosis was observed in L3.6pl cells,
but not in the L3.6plGres cells (Fig. 6B and C).
The expression level of drug transporter
proteins in L3.6pl and L3.6plGres
The expression of the drug transporter proteins P-gp
and ENT1 were then analyzed by western blotting. The results
revealed that the expression of P-gp was increased in
L3.6plGres cells as compared to L3.6pl cells, while
expression of ENT1 in L3.6plGres cells was found to be
substantially lower than in L3.6pl cells. The expression of P-gp in
both cell lines was found to decrease after 24-hour treatment with
verapamil (50 μM), and in particular, showed a marked reduction in
the L3.6plGres cells. The expression of ENT1 remained
unchanged after verapamil treatment (Fig. 7).
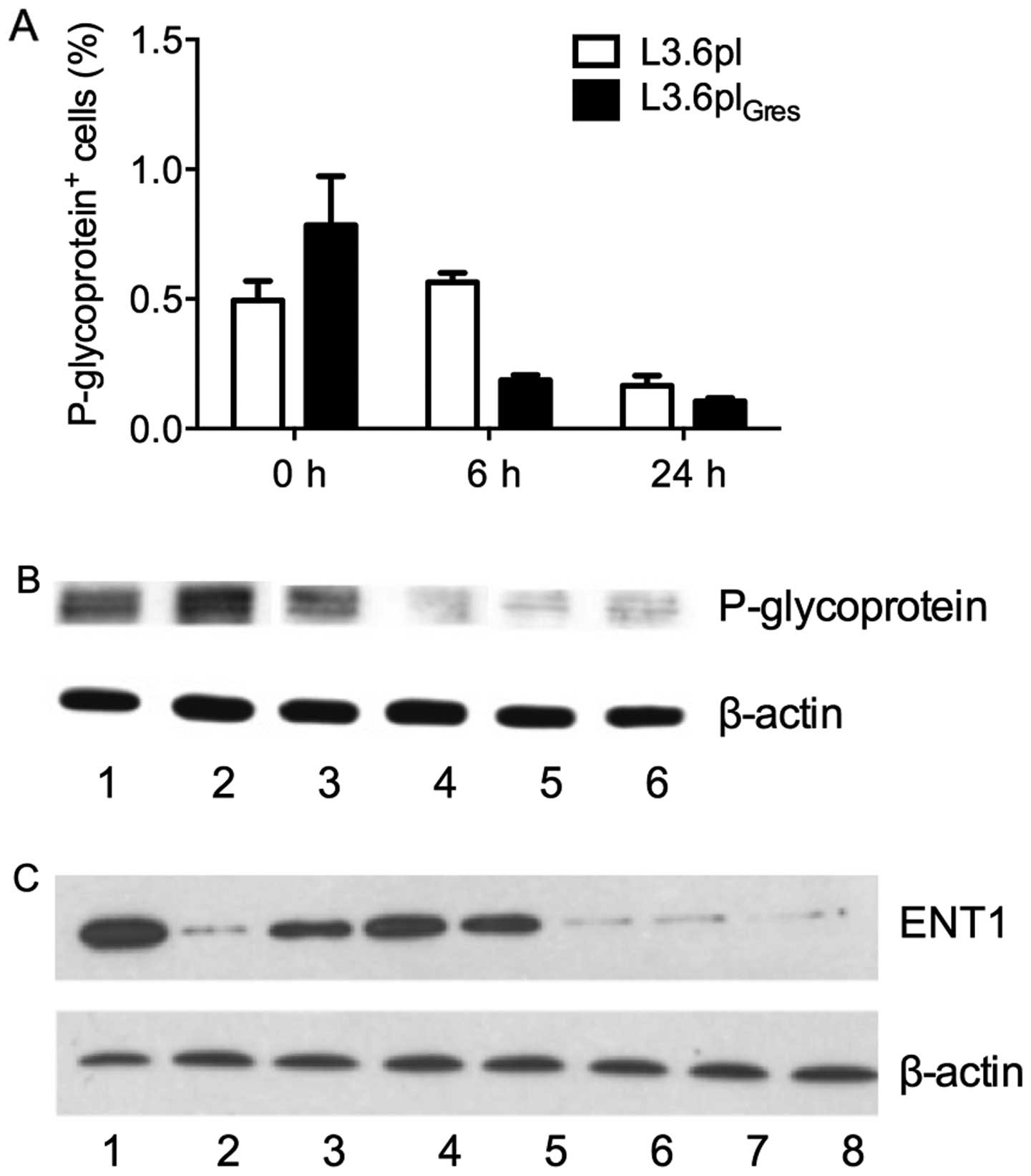 | Figure 7Expression of drug transporter
proteins in L3.6pl and L3.6plGres. (A) After treatment
of L3.6pl and L3.6plGres cells with verapamil (50 μM)
for 0, 6 and 24 h, all cells were analyzed by FACS. The proportion
of P-glycoprotein positive cells decreased after treatment in both
cell lines, however, significantly more in L3.6plGres
(p<0.01). (B) The protein lysates from: 1, L3.6pl; 2,
L3.6plGres; 3, L3.6pl treated with verapamil (50 μM) for
6 h; 4, L3.6pl treated with verapamil (50 μM) for 24 h; 5,
L3.6plGres treated with verapamil (50 μM) for 6 h; 6,
L3.6plGres treated with verapamil (50 μM) for 24 h. The
expression of P-glycoprotein was significantly higher in
L3.6plGres, than L3.6pl cells. Under treatment of
verapamil for 24 h, the expression level of P-glycoprotein
decreased in both cell lines, and showed a marked reduction in the
L3.6plGres cells. (C) The expression level of ENT1
before/after verapamil treatment in L3.6pl and
L3.6plGres cells. The protein lysates from: 1, L3.6pl;
2, L3.6plGres; 3, L3.6pl treated with verapamil (50 μM)
for 24 h; 4, L3.6pl treated with verapamil (100 μM) for 24 h; 5,
L3.6pl treated with verapamil (200 μM) for 24 h; 6,
L3.6plGres treated with verapamil (50 μM) for 24 h; 7,
L3.6plGres treated with verapamil (100 μM) for 24 h; 8,
L3.6plGres treated with verapamil (200 μM) for 24 h. The
results indicate that the expression of ENT1 was lower in
L3.6plGres cells than in L3.6pl cells, and were
unchanged under different concentrations (50, 100 and 200 μM) of
verapamil. |
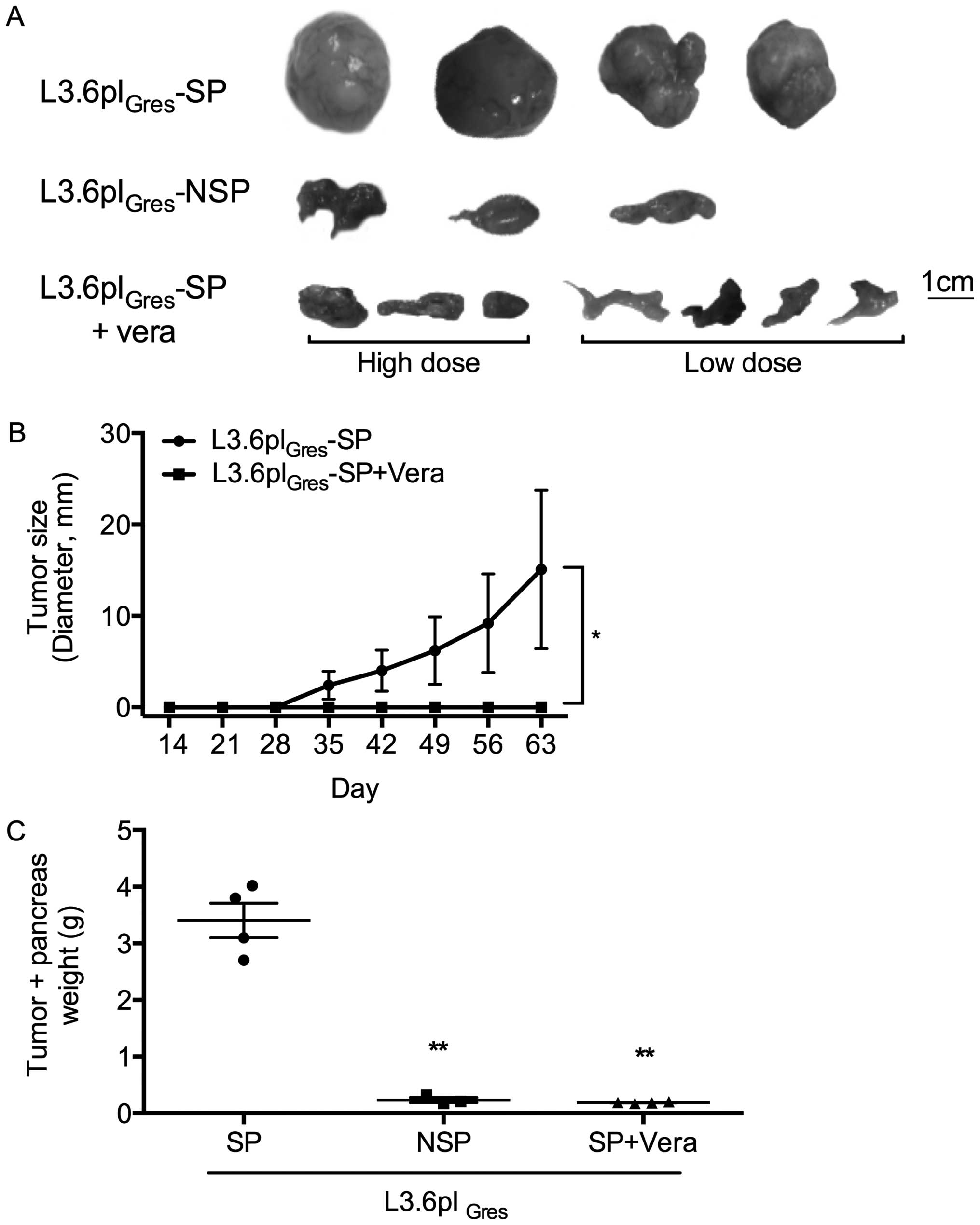
Verapamil can effectively inhibit
L3.6plGres -SP tumor growth in vivo
L3.6plGres-SP and NSP cells were injected
into the pancreas of athymic Balb/c nu/nu mice. The following
experimental groups were compared: group 1, L3.6plGres
-SP control group (5 mice); group 2, L3.6plGres-NSP cell
control group (3 mice); group 3, L3.6plGres-SP treatment
group with high concentration of verapamil (25 mg/kg BW, 10 mM, 3
mice); and group 4, L3.6plGres-SP treatment group with
low concentration of verapamil (0.5 mg/kg BW, 200 μM, 4 mice). In
all groups, verapamil treatment was initiated four weeks after
orthotopic tumor cell injection. Nine weeks later all the mice were
sacrificed. Tumors and other organs were then harvested for
analysis.
Macroscopically, the L3.6plGres-SP tumors
showed a more aggressive phenotype compared to the
L3.6plGres-NSP tumors. Verapamil treatment (low/high
concentrations) substantially inhibited tumor growth and metastasis
of the L3.6plGres-SP cells (Fig. 8). No changes in body weight, or
general behavior were observed in the verapamil treatment groups
(groups 3 and 4).
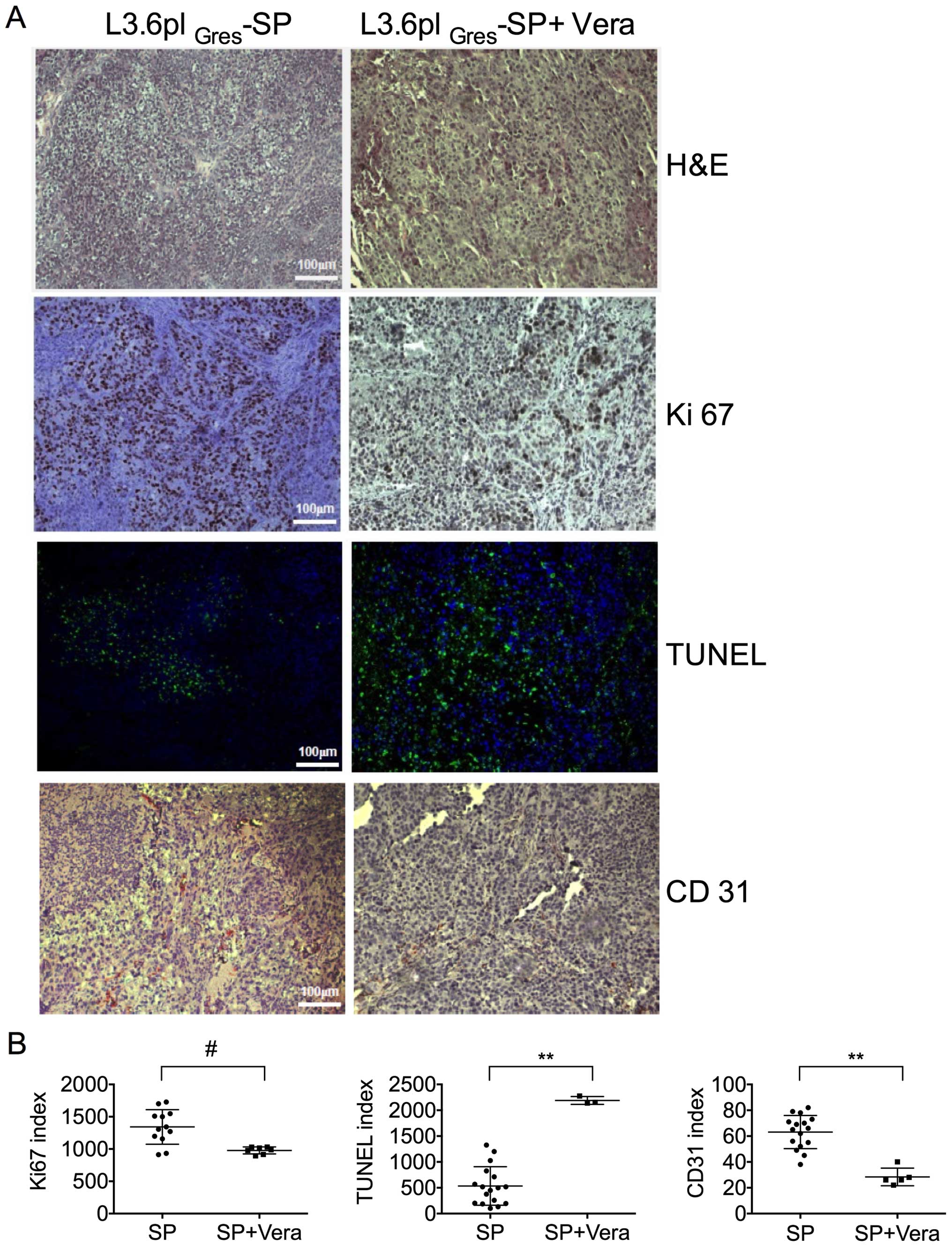
Immunohistochemical staining was used to establish a
proliferation index, microvascular density, and apoptosis level in
the experimental pancreatic tumors derived from the
L3.6plGres-SP control and verapamil treatment groups
(Fig. 9). The median percentage of
Ki67+ cells within the tumors of the verapamil
treatment group differed slightly from that seen in the
L3.6plGres-SP group. The apoptotic signal was increased
in the verapamil treatment group as compared to the
L3.6plGres-SP control group. The microvascular density
measured by CD31 staining revealed significantly reduced
angiogenesis in tumors following verapamil treatment, as compared
to the L3.6plGres-SP tumors in the control group.
H&E staining showed large, pleomorphic cells with
hyperchromatic nuclei in the L3.6plGres-SP control
group.
Discussion
The general concept of cancer stem cells (CSCs) is a
topic of increasing clinical interest. CSCs are proposed to show
stem-like self-renewal and tumor initiation characteristics that
are thought to help foster tumor recurrence and resistance to
chemotherapy. CSCs can be enriched by several methods including by
growth in serum-free defined media to induce sphere formation. They
can also be directly isolated based on their expression of specific
surface marker combinations, i.e.,
CD44+/CD24−/lin- for human breast cancers
(35),
EpCAMhigh/CD44+/CD166+ for
colorectal cancer (36),
CD34+/CD38− for acute myeloid leukemia
(37), and
Stro1+/CD105+/CD44+, a surface
marker for bone sarcoma (38). CSC
subpopulations can also be isolated by FACS sorting of side
population (SP) cells based on their ability to efflux fluorescent
dyes or chemotherapy reagents (39). SP cells have been widely linked to
CSCs in various tumors and cancer cell lines, e.g., acute myeloid
leukemia (40), neuroblastoma
(19), melanoma (41), ovarian cancer (42), glioma cell lines (43), various human gastrointestinal
cancer cell lines (44), and
pancreatic cancer cell lines, including; MIAPaCa2, PANC-1, Capan-2,
KP-1 NL and SW1990 (23,45–47).
In this study, we isolated and characterized SP
cells from a highly metastatic pancreatic adenocarcinoma cell line
L3.6pl, a chemotherapy-resistant variant of this line
L3.6plGres, and from the AsPC-1 human pancreatic cell
line (Fig. 2).
The SP-enriched L3.6plGres cells, which
were shown in a previous study to have significantly increased
ABCG2+, and CD24+ cells, showed higher colony
formation ability in vitro than did the parental L3.6pl
cells. L3.6plGres-SP cells also showed increased colony
formation ability relative to L3.6plGres-NSP cells
(Fig. 3) and exhibited increased
gemcitabine chemotherapy resistance relative to the
L3.6plGres-NSP cells suggesting enhanced stem cell-like
properties (Fig. 4). SP and non-SP
of AsPC-1 did not work as a good model for stem cell like SP study
by our previous data (data not shown). SP proportion is highly
abound in AsPC-1 and did not show statistic significance in
tumorigenicity in vivo. Therefore, in this study, we focused
on SP cells from L3.6pl and its corresponding resistant
variant.
These observations were then validated in
vivo. Following orthotopic implantation, significantly larger
primary pancreatic tumors, and a higher incidence of liver
metastases, were found in the L3.6plGres-SP cells, as
compared to L3.6plGres-NSP cells, supporting the
hypothesis that they show enhanced stem cell-like characteristics
(Fig. 9).
Morphologic changes were associated with the
establishment of gemcitabine-resistant L3.6pl cells. A
fibroblastoid phenotype with loss of polarity, increased
intercellular separation and pseudopodia was detected in the
L3.6plGres cells (Fig.
1). This observation is consistent with the phenomenon of
epithelial-to-mesenchymal transition (EMT). Molecular and
phenotypic associations have been previously linked to the
establishment of chemoresistance including the acquisition of an
EMT-like cancer cell phenotype (48). SP cells have been previously
associated with chemotherapy resistance and enhanced EMT (49,50).
Goodell et al (16) have shown that the molecular pumps
responsible for H33342 efflux (used to differentiate SP from NSP
cells) are generally members of the ABC superfamily including
MDR1/P-gp, ABCG2, and MRP (7–9,51).
Verapamil, the first generation P-gp inhibitor, has been reported
to block dye efflux activity and potentially reverse the drug
resistance caused by expression of P-gp (26). We show that verapamil effectively
reduces Hoechst 33342 staining of FACS-associated SP cells in
L3.6pl, L3.6plGres, and AsPC-1 cell lines (Fig. 2). Verapamil appears to target
important characteristics associated with CRC/SP cells and may
therefore show therapeutic efficacy for treating pancreatic cancer
stem cells. We found that verapamil could also directly inhibit
cell proliferation in a dose-dependent manner in vitro, and
was able to inhibit SP cells more efficiently than NSP cells in
both pancreatic cell lines tested (L3.6plGres and
AsPC-1) (Fig. 5). Verapamil was
further found to effectively prevent L3.6plGres-SP tumor
growth in orthotopic in vivo tumor models (Fig. 8 and Table I). For a potential side effect, we
checked the mice by bodyweight regular detection and animal
behaviour, and speculated that no serious side effects existed. It
is a limitation in this study that we did not have more biochemical
parameters for a better evaluation of side effect.
 | Table IThe incidence of primary pancreatic
tumors as well as metastatic spread in L3.6plGres-SP,
L3.6plGres-NSP and the therapy groups is
indicated.a |
Table I
The incidence of primary pancreatic
tumors as well as metastatic spread in L3.6plGres-SP,
L3.6plGres-NSP and the therapy groups is
indicated.a
| |
L3.6plGres |
|---|
| |
|
|---|
| | | SP + verapamil |
|---|
| | |
|
|---|
| Group | SP | NSP | Low dose | High dose |
|---|
| Primary tumor | 4/5 | 0/4 | 1/3 | 0/3 |
| Liver
metastasis | 2/5 | 2/4 | 0/3 | 0/3 |
The direct effects of verapamil on pancreatic cancer
cells seen are supported by earlier reports that showed verapamil
inhibition of proliferation and induced differentiation of human
promyelocytic HL-60 cells (52), a
growth inhibitory effect on human colonic tumor cells (54), and antiproliferative effects on
brain tumor cells in vitro (55). A reversible, antiproliferative
action of verapamil on human medulloblastoma, pinealoblastoma,
glioma, and neuroblastoma tumor lines has also been reported
(55).
SP cells isolated from L3.6plGres and
AsPC-1 cell lines were more sensitive to verapamil than NSP cells.
SP-enriched L3.6plGres cells expressed higher levels of
P-gp than NSP L3.6pl cells. Verapamil treatment of
L3.6plGres cells led to a significant reduction in P-gp
expression as compared to L3.6pl cells (Fig. 7), which may explain in part the
targeted effect of verapamil detected in SP cells.
Pro-apoptotic effects of verapamil were demonstrated
by in vitro apoptosis assays (Fig. 6) and by ex vivo
immunohistochemistry analysis of the tumor samples (Fig. 9). This enhanced apoptosis by
verapamil may be explained by its actions as a calcium channel
antagonist (52,54). Calcium ions (Ca2+) are
cellular messengers that control cell and tissue physiology, and
potentially ‘death’ signals (55,56).
Calcium antagonists disrupt intracellular and extracellular
Ca2+ equilibrium (57).
Ca2+ is toxic at high concentrations, thus verapamil may
help to foster apoptosis though disruption of the Ca2+
balance.
Other ABC superfamily members are also thought to
contribute to SP chemotherapy resistance. Recently it was shown
that verapamil and its derivative-NMeOHI(2) can trigger apoptosis though
glutathione (GSH) extrusion mediated by MRP1 (58). The loss of GSH from cells can
result in increased oxidative stress, a well-recognized trigger for
apoptosis (59).
Although verapamil can enhance cytotoxicity when
used in combination with chemotherapy agents, such as
anthracyclines (28), paclitaxel
(60), epipodophyllotoxins
(61) and melphalan (62), little is known about the potential
effects of verapamil when used alone or in combination with
gemcitabine, the standard chemotherapy reagent for pancreatic
cancer. We identified combined pro-apoptotic effects of verapamil
and gemcitabine on L3.6pl but not on L3.6plGres cells.
This may be explained in part by the expression level of ENT1, a
major transporter for gemcitabine that was substantially lower in
L3.6plGres cells compared to L3.6pl cells. The absence
of ENT1 is also associated with a reduced survival in patients with
gemcitabine treated pancreatic adenocarcinoma (63).
In conclusion, this study revealed that verapamil
may act on pancreatic cancer cells in vitro by inhibiting
P-gp transporters and inducing apoptosis of stem-like SP cells in
L3.6pl, L3.6plGres and AsPC-1 cells. Verapamil can
significantly inhibit pancreatic cancer tumor growth in vivo
potentially by targeting stem-like side population cells.
Acknowledgements
The authors thank Anneli Tischmacher for her
technical assistance. This study was supported by the German
Research Society (DFG) grant BR 1614/7-1 and the DKTK/DKFZ 2013
(‘Stem cells in Oncology’) to C.J.B. L.Z., Y.Z. and Q.B. were
supported by LMU-CSC (The China Scholarship Council)
scholarship.
Abbreviations:
|
ABC
|
ATP-binding cassette
|
|
CSCs
|
cancer stem cells
|
|
CNT
|
concentrative nucleoside
transporter
|
|
ENT
|
equilibrative nucleoside
transporter
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
GSH
|
glutathione
|
|
H33342
|
Hoechst 33342
|
|
HSC
|
hematopoietic stem cells
|
|
MDR
|
multidrug resistance
|
|
MRP
|
multidrug resistance-associated
protein
|
|
PKC
|
protein kinase C
|
|
P-gp
|
P-glycoprotein
|
|
SP
|
side population
|
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernandez E, La Vecchia C, Porta M, Negri
E, Lucchini F and Levi F: Trends in pancreatic cancer mortality in
Europe, 1955–1989. Int J Cancer. 57:786–792. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen M, Xue X, Wang F, An Y, Tang D, Xu Y,
Wang H, Yuan Z, Gao W, Wei J, et al: Expression and promoter
methylation analysis of ATP-binding cassette genes in pancreatic
cancer. Oncol Rep. 27:265–269. 2012.
|
|
4
|
Arbuck SG: Overview of chemotherapy for
pancreatic cancer. Int J Pancreatol. 7:209–222. 1990.PubMed/NCBI
|
|
5
|
Suwa H, Ohshio G, Arao S, Imamura T,
Yamaki K, Manabe T, Imamura M, Hiai H and Fukumoto M:
Immunohistochemical localization of P-glycoprotein and expression
of the multidrug resistance-1 gene in human pancreatic cancer:
Relevance to indicator of better prognosis. Jpn J Cancer Res.
87:641–649. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grant CE, Valdimarsson G, Hipfner DR,
Almquist KC, Cole SP and Deeley RG: Overexpression of multidrug
resistance-associated protein (MRP) increases resistance to natural
product drugs. Cancer Res. 54:357–361. 1994.PubMed/NCBI
|
|
7
|
Germann UA, Chambers TC, Ambudkar SV,
Licht T, Cardarelli CO, Pastan I and Gottesman MM: Characterization
of phosphorylation-defective mutants of human P-glycoprotein
expressed in mammalian cells. J Biol Chem. 271:1708–1716. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Childs S and Ling V: The MDR superfamily
of genes and its biological implications. Important Adv Oncol.
21–36. 1994.PubMed/NCBI
|
|
9
|
Chin KV, Pastan I and Gottesman MM:
Function and regulation of the human multidrug resistance gene. Adv
Cancer Res. 60:157–180. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thomas H and Coley HM: Overcoming
multidrug resistance in cancer: an update on the clinical strategy
of inhibiting P-glycoprotein. Cancer Control. 10:159–165.
2003.PubMed/NCBI
|
|
11
|
Hagmann W, Jesnowski R and Löhr JM:
Interdependence of gemcitabine treatment, transporter expression,
and resistance in human pancreatic carcinoma cells. Neoplasia.
12:740–747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mackey JR, Mani RS, Selner M, Mowles D,
Young JD, Belt JA, Crawford CR and Cass CE: Functional nucleoside
transporters are required for gemcitabine influx and manifestation
of toxicity in cancer cell lines. Cancer Res. 58:4349–4357.
1998.PubMed/NCBI
|
|
13
|
Ritzel MW, Ng AM, Yao SY, Graham K, Loewen
SK, Smith KM, Ritzel RG, Mowles DA, Carpenter P, Chen XZ, et al:
Molecular identification and characterization of novel human and
mouse concentrative Na+-nucleoside cotransporter
proteins (hCNT3 and mCNT3) broadly selective for purine and
pyrimidine nucleosides (system cib). J Biol Chem. 276:2914–2927.
2001. View Article : Google Scholar
|
|
14
|
Garcia-Manteiga J, Molina-Arcas M, Casado
FJ, Mazo A and Pastor-Anglada M: Nucleoside transporter profiles in
human pancreatic cancer cells: Role of hCNT1 in
2′,2′-difluorodeoxy-cytidine- induced cytotoxicity. Clin Cancer
Res. 9:5000–5008. 2003.
|
|
15
|
Al-Hajj M and Clarke MF: Self-renewal and
solid tumor stem cells. Oncogene. 23:7274–7282. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goodell MA, Brose K, Paradis G, Conner AS
and Mulligan RC: Isolation and functional properties of murine
hematopoietic stem cells that are replicating in vivo. J Exp Med.
183:1797–1806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bunting KD, Zhou S, Lu T and Sorrentino
BP: Enforced P-glycoprotein pump function in murine bone marrow
cells results in expansion of side population stem cells in vitro
and repopulating cells in vivo. Blood. 96:902–909. 2000.PubMed/NCBI
|
|
18
|
Zhou S, Schuetz JD, Bunting KD, Colapietro
AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M,
Nakauchi H, et al: The ABC transporter Bcrp1/ABCG2 is expressed in
a wide variety of stem cells and is a molecular determinant of the
side-population phenotype. Nat Med. 7:1028–1034. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hirschmann-Jax C, Foster AE, Wulf GG,
Nuchtern JG, Jax TW, Gobel U, Goodell MA and Brenner MK: A distinct
‘side population’ of cells with high drug efflux capacity in human
tumor cells. Proc Natl Acad Sci USA. 101:14228–14233. 2004.
View Article : Google Scholar
|
|
20
|
Benchaouir R, Rameau P, Decraene C,
Dreyfus P, Israeli D, Piétu G, Danos O and Garcia L: Evidence for a
resident subset of cells with SP phenotype in the C2C12 myogenic
line: A tool to explore muscle stem cell biology. Exp Cell Res.
294:254–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao J, Cai HH, Wei JS, An Y, Ji ZL, Lu ZP,
Wu JL, Chen P, Jiang KR, Dai CC, et al: Side population in the
pancreatic cancer cell lines SW1990 and CFPAC-1 is enriched with
cancer stem-like cells. Oncol Rep. 23:1375–1382. 2010.PubMed/NCBI
|
|
23
|
Zhang SN, Huang FT, Huang YJ, Zhong W and
Yu Z: Characterization of a cancer stem cell-like side population
derived from human pancreatic adenocarcinoma cells. Tumori.
96:985–992. 2010.
|
|
24
|
Vohra J: Verapamil in cardiac arrhythmias:
An overview. Clin Exp Pharmacol Physiol Suppl. 6:129–134.
1982.PubMed/NCBI
|
|
25
|
Bellamy WT: P-glycoproteins and multidrug
resistance. Annu Rev Pharmacol Toxicol. 36:161–183. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Broxterman HJ, Lankelma J and Pinedo HM:
How to probe clinical tumour samples for P-glycoprotein and
multidrug resistance-associated protein. Eur J Cancer.
32A:1024–1033. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ince P, Appleton DR, Finney KJ, Sunter JP
and Watson AJ: Verapamil increases the sensitivity of primary human
colorectal carcinoma tissue to vincristine. Br J Cancer.
53:137–139. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Merry S, Fetherston CA, Kaye SB, Freshney
RI and Plumb JA: Resistance of human glioma to adriamycin in vitro:
The role of membrane transport and its circumvention with
verapamil. Br J Cancer. 53:129–135. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morrow M, Wait RB, Rosenthal RA and
Gamelli RL: Verapamil enhances antitumor activity without
increasing myeloid toxicity. Surgery. 101:63–68. 1987.PubMed/NCBI
|
|
30
|
Tsuruo T, Iida H, Tsukagoshi S and Sakurai
Y: Overcoming of vincristine resistance in P388 leukemia in vivo
and in vitro through enhanced cytotoxicity of vincristine and
vinblastine by verapamil. Cancer Res. 41:1967–1972. 1981.PubMed/NCBI
|
|
31
|
Bruns CJ, Harbison MT, Kuniyasu H, Eue I
and Fidler IJ: In vivo selection and characterization of metastatic
variants from human pancreatic adenocarcinoma by using orthotopic
implantation in nude mice. Neoplasia. 1:50–62. 1999. View Article : Google Scholar
|
|
32
|
Guillermet-Guibert J, Davenne L,
Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C,
Delisle MB, Cuvillier O, Susini C and Bousquet C: Targeting the
sphingolipid metabolism to defeat pancreatic cancer cell resistance
to the chemotherapeutic gemcitabine drug. Mol Cancer Ther.
8:809–820. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reddy KL, Zullo JM, Bertolino E and Singh
H: Transcriptional repression mediated by repositioning of genes to
the nuclear lamina. Nature. 452:243–247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Georges E, Bradley G, Gariepy J and Ling
V: Detection of P-glycoprotein isoforms by gene-specific monoclonal
antibodies. Proc Natl Acad Sci USA. 87:152–156. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Al-Hajj M, Wicha MS, Benito-Hernandez A,
Morrison SJ and Clarke MF: Prospective identification of
tumorigenic breast cancer cells. Proc Natl Acad Sci USA.
100:3983–3988. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dalerba P, Dylla SJ, Park IK, Liu R, Wang
X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al:
Phenotypic characterization of human colorectal cancer stem cells.
Proc Natl Acad Sci USA. 104:10158–10163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Clayton S and Mousa SA: Therapeutics
formulated to target cancer stem cells: Is it in our future? Cancer
Cell Int. 11:72011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gibbs CP, Kukekov VG, Reith JD,
Tchigrinova O, Suslov ON, Scott EW, Ghivizzani SC, Ignatova TN and
Steindler DA: Stem-like cells in bone sarcomas: Implications for
tumorigenesis. Neoplasia. 7:967–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Challen GA and Little MH: A side order of
stem cells: The SP phenotype. Stem Cells. 24:3–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wulf GG, Wang RY, Kuehnle I, Weidner D,
Marini F, Brenner MK, Andreeff M and Goodell MA: A leukemic stem
cell with intrinsic drug efflux capacity in acute myeloid leukemia.
Blood. 98:1166–1173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Grichnik JM, Burch JA, Schulteis RD, Shan
S, Liu J, Darrow TL, Vervaert CE and Seigler HF: Melanoma, a tumor
based on a mutant stem cell? J Invest Dermatol. 126:142–153. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szotek PP, Pieretti-Vanmarcke R, Masiakos
PT, Dinulescu DM, Connolly D, Foster R, Dombkowski D, Preffer F,
Maclaughlin DT and Donahoe PK: Ovarian cancer side population
defines cells with stem cell-like characteristics and Mullerian
inhibiting substance responsiveness. Proc Natl Acad Sci USA.
103:11154–11159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kondo T, Setoguchi T and Taga T:
Persistence of a small subpopulation of cancer stem-like cells in
the C6 glioma cell line. Proc Natl Acad Sci USA. 101:781–786. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haraguchi N, Utsunomiya T, Inoue H, Tanaka
F, Mimori K, Barnard GF and Mori M: Characterization of a side
population of cancer cells from human gastrointestinal system. Stem
Cells. 24:506–513. 2006. View Article : Google Scholar
|
|
45
|
Asuthkar S, Stepanova V, Lebedeva T,
Holterman AL, Estes N, Cines DB, Rao JS and Gondi CS:
Multifunctional roles of urokinase plasminogen activator (uPA) in
cancer stemness and chemoresistance of pancreatic cancer. Mol Biol
Cell. 24:2620–2632. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou J, Wang CY, Liu T, Wu B, Zhou F,
Xiong JX, Wu HS, Tao J, Zhao G, Yang M, et al: Persistence of side
population cells with high drug efflux capacity in pancreatic
cancer. World J Gastroenterol. 14:925–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kabashima A, Higuchi H, Takaishi H,
Matsuzaki Y, Suzuki S, Izumiya M, Iizuka H, Sakai G, Hozawa S,
Azuma T, et al: Side population of pancreatic cancer cells
predominates in TGF-beta-mediated epithelial to mesenchymal
transition and invasion. Int J Cancer. 124:2771–2779. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang AD, Fan F, Camp ER, van Buren G, Liu
W, Somcio R, Gray MJ, Cheng H, Hoff PM and Ellis LM: Chronic
oxaliplatin resistance induces epithelial-to-mesenchymal transition
in colorectal cancer cell lines. Clin Cancer Res. 12:4147–4153.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Okano M, Konno M, Kano Y, Kim H, Kawamoto
K, Ohkuma M, Haraguchi N, Yokobori T, Mimori K, Yamamoto H, et al:
Human colorectal CD24+ cancer stem cells are susceptible
to epithelial-mesenchymal transition. Int J Oncol. 45:575–580.
2014.PubMed/NCBI
|
|
50
|
Zhao Y, Bao Q, Schwarz B, Zhao L,
Mysliwietz J, Ellwart J, Renner A, Hirner H, Niess H, Camaj P, et
al: Stem cell-like side populations in esophageal cancer: A source
of chemotherapy resistance and metastases. Stem Cells Dev.
23:180–192. 2014. View Article : Google Scholar
|
|
51
|
Doyle L and Ross DD: Multidrug resistance
mediated by the breast cancer resistance protein BCRP (ABCG2).
Oncogene. 22:7340–7358. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jensen RL, Lee YS, Guijrati M, Origitano
TC, Wurster RD and Reichman OH: Inhibition of in vitro meningioma
proliferation after growth factor stimulation by calcium channel
antagonists: Part II - Additional growth factors, growth factor
receptor immunohistochemistry, and intracellular calcium
measurements. Neurosurgery. 37:937–946; discussion 946-937. 1995.
View Article : Google Scholar
|
|
53
|
Zhao Y, Zhao L, Ischenko I, Bao Q, Schwarz
B, Nieß H, Wang Y, Renner A, Mysliwietz J, Jauch KW, et al:
Antisense inhibition of microRNA-21 and microRNA-221 in
tumor-initiating stem-like cells modulates tumorigenesis,
metastasis, and chemotherapy resistance in pancreatic cancer.
Target Oncol. 10:535–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jensen RL, Petr M and Wurster RD: Calcium
channel antagonist effect on in vitro meningioma signal
transduction pathways after growth factor stimulation.
Neurosurgery. 46:692–702; discussion 702-693. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Berridge MJ, Lipp P and Bootman MD: Signal
transduction. The calcium entry pas de deux. Science.
287:1604–1605. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hajnóczky G, Csordás G, Madesh M and
Pacher P: Control of apoptosis by IP(3) and ryanodine receptor
driven calcium signals. Cell Calcium. 28:349–363. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao QZ, Niu G and Tan HR: In vitro growth
inhibition of human colonic tumor cells by Verapamil. World J
Gastroenterol. 11:2255–2259. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Trompier D, Chang XB, Barattin R, du
Moulinet D'Hardemare A, Di Pietro A and Baubichon-Cortay H:
Verapamil and its derivative trigger apoptosis through glutathione
extrusion by multidrug resistance protein MRP1. Cancer Res.
64:4950–4956. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ghibelli L, Coppola S, Rotilio G, Lafavia
E, Maresca V and Ciriolo MR: Non-oxidative loss of glutathione in
apoptosis via GSH extrusion. Biochem Biophys Res Commun.
216:313–320. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wang F, Zhang D, Zhang Q, Chen Y, Zheng D,
Hao L, Duan C, Jia L, Liu G and Liu Y: Synergistic effect of
folate-mediated targeting and verapamil-mediated P-gp inhibition
with paclitaxel-polymer micelles to overcome multi-drug resistance.
Biomaterials. 32:9444–9456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yalowich JC and Ross WE: Verapamil-induced
augmentation of etoposide accumulation in L1210 cells in vitro.
Cancer Res. 45:1651–1656. 1985.PubMed/NCBI
|
|
62
|
Robinson BA, Clutterbuck RD, Millar JL and
McElwain TJ: Verapamil potentiation of melphalan cytotoxicity and
cellular uptake in murine fibrosarcoma and bone marrow. Br J
Cancer. 52:813–822. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Spratlin J, Sangha R, Glubrecht D, Dabbagh
L, Young JD, Dumontet C, Cass C, Lai R and Mackey JR: The absence
of human equilibrative nucleoside transporter 1 is associated with
reduced survival in patients with gemcitabine-treated pancreas
adenocarcinoma. Clin Cancer Res. 10:6956–6961. 2004. View Article : Google Scholar : PubMed/NCBI
|