Introduction
Colorectal cancer (CRC) is the third most common
cancer in men and the second in women worldwide, accounting for
approximately 608,000 deaths worldwide (1). The most common cause of death from
CRC is hepatic metastasis. Approximately 50% of CRC patients are
diagnosed with hepatic metastases, either at the time of initial
presentation or as a result of disease recurrence (2). However, there have been no major
advances in the treatment of metastatic CRC during the last four
decades. Multiple new FDA-approved therapies were tried, the 5-year
survival remains extremely poor. Conventional chemotherapy
efficiently targets tumor bulk, but there exists a small
subpopulation of cells that contributes to resistance to
chemotherapy and tumor regrowth. These cells are termed cancer stem
cells (CSCs). Cumulative evidence has established that the majority
of tumors comprise a population of CSCs responsible for the
initiation and maintenance of tumors and resistance to cytotoxic
drugs (3).
Signal transducer and activator of transcription 3
(STAT3) is a latent cytoplasmic transcription factor that conveys
various cytokine and growth factor signals from the cell membrane
to the nucleus (4). It is involved
in many cellular processes including proliferation, survival, and
immune responses. The transient activation of STAT3 is tightly
regulated under normal conditions (5). In a variety of human malignancies,
constitutive activation of STAT3 is correlated with tumor
progression and poor prognosis (6). Recent reports showed that the STAT3
pathway preferentially regulates CSC self-renewal, tumor
initiation, and metastasis in many solid tumors (7–9). It
was also reported that the STAT3 pathway blockade causes a decrease
in CSCs and a significant reduction of tumor formation in mouse
xenograft models (10). Earlier
studies indicated that STAT3 can be an excellent cellular target
for anticancer agent development. However, STAT3 has generally been
considered in practice to be non-targetable, and the lag in
developing effective STAT3 inhibitors contributed to the current
lack of FDA-approved STAT3 inhibitors. Here, we investigated
whether targeting STAT3 with flavonoid morin, and targeting
telomerase with MST-312, can reduce the cancer stem cell
subpopulation in human colorectal and breast cancers.
Telomerase lengthens telomeres in DNA strands. A
number of clinical cases reveal that telomerase is specifically
activated in numerous human malignancies including colorectal
cancer (11). There is a report
that the prognosis of colorectal cancer patients with high
telomerase activity was significantly worse than that of patients
with moderate or low telomerase activity (P<0.01) (12). In the study, among the 87 patients
with surgically resectable and potentially curable tumors, the
disease-free survival rate of those with high telom-erase activity
was significantly poorer. These data suggest that inhibitors of
telomerase may prove efficacious in treating patients with advanced
disease. Recently, hTERT (human telomerase reverse transcriptase)
was shown to contribute to the epithelial-mesenchymal transition
and cancer stem cell traits in gastric cancer (13). Altogether, a growing body of
evidence suggests that telomerase can be a good candidate as a
cellular target for CRC therapy.
Morin (3,5,7,2′,4′-pentahydroxyflavone) is a
polyphenol compound originally isolated from members of the
Moraceae family such as mulberry figs and old fustic
(Chlorophora tinctoria). Earlier studies have shown that
morin suppresses the proliferation of a wide variety of tumor cells
including oral squamous cell carcinoma, leukemia, and COLO205
colorectal cancer cells in nude mice (14). Notably, the antitumor effect of
morin is mediated through the inhibition of NF-κB and STAT3
transcription factors and their regulated genes (15,16).
Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells
through activation of SHP1 protein tyrosine phosphatase. MST-312
(telomerase inhibitor IX) is a synthetic compound that acts as a
reversible telomerase inhibitor (17). MST-312 was also shown to have
strong anti-proliferative effects on lung cancer stem cells
(18).
We have demonstrated that the activated STAT3
transcriptionally upregulates hTERT (human telomerase reverse
transcriptase) expression, and consequently promotes CSC traits in
aggressive human breast cancers (19). This is in agreement with the recent
finding that telomerase acts as a transcriptional modulator of the
Wnt-β-catenin signaling pathway in stem cells and cancer cells
(20,21). The STAT3-telomerase signaling axis
is likely driving the CSC phenotype in human cancers. In this
study, we investigated whether combination treatment with morin and
MST-312, dually targeting STAT3 and telomerase, can reduce the CSC
populations. We also tested whether the morin/MST-312 combination
treatment could abolish tumorsphere formation and enhance
5-fluorouracil efficacy in human cancer cells originally resistant
to 5-FU treatments. Finally, we tried to determine the cell stress
and apoptosis gene signatures that were upregulated or
downregulated upon morin/MST-312 treatments. This study focused on
STAT3 and telomerase as potential therapeutic targets based on
their significant roles in the colorectal cancer growth and
maintenance.
Materials and methods
Cancer cell lines
HT-29, SW620 and MDA-MB-231 cancer cell lines were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). They were maintained in a monolayer culture in
DMEM/F12 (Dulbecco's modified Eagle's medium) with 10% fetal bovine
serum, 2.5% L-glutamine and 0.5% penicillin/streptomycin.
Reagents
Morin hydrate (Sigma-Aldrich, St. Louis, MO, USA;
catalog no. M4008) and MST-312 (Sigma-Aldrich; catalog no. M3949)
was purchased from Sigma-Aldrich Co. Morin was prepared in 50 mM
stock solution and MST-312 was in 10 mM stock solution. The working
concentration for morin was 50 mM whereas 10 mM for MST-312. Morin
and MST-312 were either used alone or in combination throughout
this study.
Tumorsphere formation assay
Matrigel (BD, Cambridge, MA, USA), 200 ml was spread
as a thick layer on wells of a 24-well plate and allowed to
polymerize at 37°C for 15 min. Cancer cells (2×104)
grown in monolayer were trypsinized to single cells and plated on
top of the pre-coated Matrigel. Plates were incubated at 37°C to
allow cells to fully settle down before media was replaced with
appropriate culture media containing 5% Matrigel. Cells were grown
for 15 days; fresh growth media with Matrigel was replenished every
two days. Images of representative fields were taken.
Cell invasion assay
Mouse fibroblasts (NIH-3T3) were used as a
chemoattractant, and grown in a 24-well plate in 2 ml of the
DMEM/F12 media. Boyden chambers were prepared with 25 μl of 1:6
diluted Matrigel and allowed to incubate for 2 h to solidify. Each
chamber received a different treatment: untreated, morin only,
MST-312 only and morin plus MST-312. After cell synchronization,
invasion was allowed to occur for 40 h. The cells were then fixed
with 0.5% glutaraldehyde and stained with 5% toluidine blue for
cell counting. Three different 40× microscope fields were used to
quantify the invasion statistics when counting cells.
Western blot analyses
Monolayer cultures of respective cell lines at
80–90% confluence were lysed using 100 μl of RIPA buffer (Thomas
Scientific Inc. Swedesboro, NJ, USA). Tris-glycine (Bio-Rad,
Irvine, CA, USA) gels were loaded with 50–100 μg of lysates. After
electrophoresis, the gel was transferred to a nitrocellulose
membrane for 2 h. The membrane was blocked for 1 h in 5% BSA or 5%
skim milk at 4°C. The membrane was then washed 3 times with 1X TTBS
and incubated overnight with the primary antibody at 4°C. Primary
antibodies of STAT3, pSTAT3 and β-actin were purchased from Cell
Signaling Technology (Danvers, MA, USA). After incubation with the
secondary antibodies conjugated with horseradish peroxidase (HRP),
the protein bands were developed with the chemiluminescent
reagents.
Telomerase activity assay
Cells were processed according to the manufacturer's
protocol for the TeloTAGGG Telomerase PCR ELISA kit (Roche, Orange,
CA, USA. catalog no. 11854666910). Briefly, cell pellets were
thawed in lysis reagent, incubated on ice for 30 min, and
centrifuged at 16,000 g for 20 min at 4°C. Telomerase activity was
immediately measured in the resultant supernatant using the
telomeric repeat amplification protocol in which telomerase, if
present in the cell lysate, adds telomeric repeats to the 3′ end of
a biotin-labeled synthetic P1-TS primer. Samples were amplified by
polymerase chain reaction (PCR), with P1-TS and P2 primers creating
an elongated telomere. The PCR product was denatured and hybridized
to a digoxigenin-labeled probe that detects telomeric repeats in a
subsequent enzyme-linked immunosorbent assay (ELISA). Samples were
considered positive for telomerase activity if the ELISA resulted
in a background-corrected absorbance of ≥0.2 U, resulting in binary
positive/negative data. Telomerase assays were performed three
times independently and P-values <0.05 were considered
statistically significant.
FACS profile analysis
Approximately 500,000 colorectal cancer cells were
washed with 1X PBS, trypsinized, and then transferred to a 15-ml
tube. Cell suspensions were centrifuged, re-suspended in 2 ml 1X
PBS, and then divided into two tubes of 0.5 ml each. One tube was
used as an unstained control and the other was stained with 5 μl
CD44 antibody (FITC Green; BD Biotech, San Jose, CA, USA) or CD133
antibody (PE Red; Miltenyl Biotec, San Diego, CA, USA). The tubes
were vortexed briefly and incubated at room temperature for 15 min
in the dark. Each tube was then washed with 3.5 ml 1X PBS and then
centrifuged for 6 min. The supernatant was removed by aspiration,
and the cells were re-suspended in 3 ml 1X PBS and subjected to
FACS profiling at the UCLA FACS Core Laboratory.
Stress and apoptosis antibody array
The Stress and Apoptosis Signal Antibody Array kit
was purchased from Cell Signaling Technology (Cell Signaling
Technology, Beverly, MA, USA; catalog no. 12856). Each CRC cell
line had the following treatments in this order: untreated, morin
only, MST-312 only, and morin plus MST-312. Whole protein lysates
were prepared using the provided lysis buffer from the kit. One
hundred milliliters of each lysate were placed onto the membrane
window of the antibody slide. The treated slide was incubated
overnight at 4°C on an orbital shaker. The slide was then washed
with 100 ml 1X array wash buffer and incubated on an orbital shaker
for 5 min at room temperature. This procedure was repeated three
more times. 1X Detection Antibody Cocktail (75 μl) was added to
each of the 16-wells and the plate was covered with the provided
sealing tape. It was incubated for 1 h at room temperature on an
orbital shaker. Next, three wash cycles were performed and the
slide was incubated for 30 min with 75 μl 1X HRP-linked
streptavidin. The slide was washed and treated with Lumi Glo and
peroxide. The Bio-Rad Gel Documentation system was used to take
detailed pictures of the array using the Quantity One software
using the ChemiDoc XRS function. ImageJ software was used to
analyze the antibody array. All the array images were scanned and
saved as JPEG files. We utilized the ImageJ software to quantify
the expression levels of proteins. The quantified protein
expression levels were presented as histograms with statistic
significance.
Cell viability assay
The cell viability was evaluated by the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
uptake method. Briefly, the 5-FU chemo-resistant cell lines, HT-29
and SW620, were seeded in a 96-well plate (1,000 cells per well)
and exposed to different concentrations of 5-FU or 5-FU plus 5 μM
morin, or 5-FU plus 3 μM MST-312 in triplicate for 24 h.
Additionally, in another set of experiments, cells were co-treated
with 5 μM morin and 3 μM MST-312 with different concentrations of
5-FU (0, 0.1, 1, 2, 3 and 4 μM) for 24 h to obtain the optimum dose
for combination treatment. Cells were washed twice with PBS and
subsequently, MTT solution (5 mg/ml) was added to each well and the
plate was incubated for 4 h at 37°C. The 96-well plates were
wrapped with aluminum foil and gently swirled for 15 min at room
temperature. The absorbance of the cell suspension was measured at
570 nm. The data obtained were calculated and were represented as
hundredth of survival relative to controls. This experiment was
repeated 3 times independently, and statistical analysis was done
to obtain the final values.
Statistical analysis
Student's t-tests were used to evaluate the
significance of changes in all combination treatment assays
compared to controls. Differences were considered statistically
significant at P<0.05.
Results
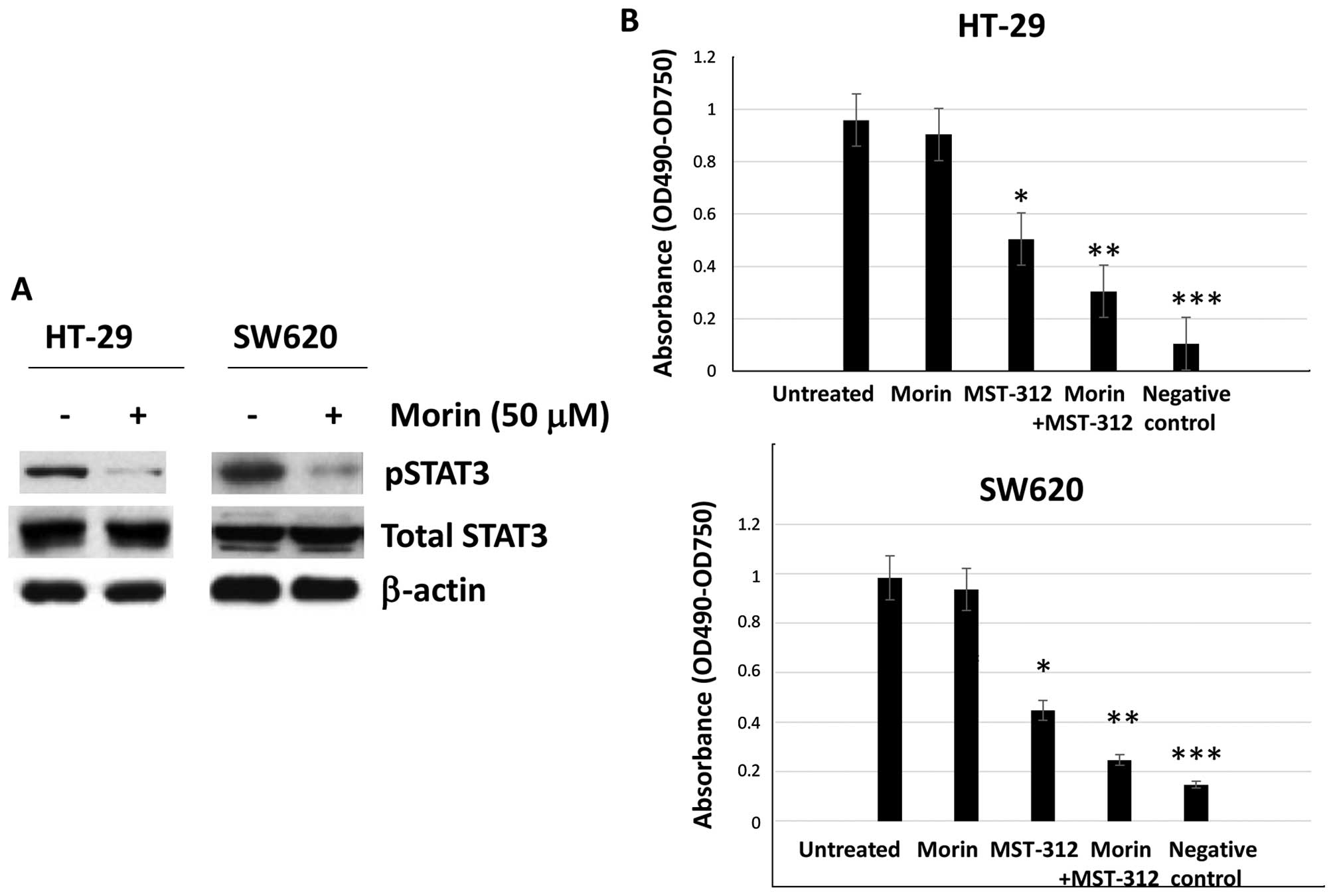
Morin inhibits STAT3 phosphorylation and
MST-312 inhibits telomerase activity in human colorectal cancer
cells
To confirm the molecular functions of morin and
MST-312, we tested two colorectal cancer cell lines which contain
the constitutively phosphorylated STAT3 (pSTAT3) and activated
telomerase, HT-29 and SW620. Morin inhibits STAT3 phosphorylation
in a dose-dependent and time-dependent manner (16). Initially, we treated HT-29 and
SW620 cells with morin at the concentration 50 μM for 24 h. After
the treatment, we ran a western blot analysis to examine STAT3
phosphorylation status. As shown in Fig. 1A, STAT3 phosphorylation was
inhibited in both HT-29 and SW620 cell lines whereas total STAT3
expression levels remained the same (Fig. 1A). Our data suggest that morin
specifically inhibited STAT3 phosphorylation step in colorectal
cancer cell lines.
Next we wished to determine the telomerase activity
in HT-29 and SW620 cell lines. MST-312 is a synthetic compound that
functions as a reversible telomerase inhibitor (17). To monitor telomerase activity,
TRAP-PCR-ELISA assay was performed as described in Materials and
methods. HT-29 and SW620 were treated with morin alone at a
concentration of 50 μM for 24 h, MST-312 alone at a concentration
of 10 μM for 24 h and morin and MST-312 combination for 24 h and
were applied to the telomere PCR-ELISA assays. As shown in Fig. 1B, MST-312 treatment inhibited
telomerase activity, average absorbance was clearly decreased from
0.98 to 0.47 (OD, 490–750) whereas morin slightly reduced the
absorbance from 0.95 to 0.90 in HT-29 (Fig. 1B). Morin and MST-312 combined
decreased the absorbance to 0.35. Similarly, MST-312 decreased the
absorbance from 0.99 to 0.41 (OD, 490–750) while morin reduced it
from 0.99 to 0.93 in SW620 cell lines. When morin and MST-312 were
combined, telomerase activity was decreased to 0.24. Our results
confirm that MST-312 inhibited telomerase activity in human
colorectal cancer cells and when combined with morin, the
inhibitory effects were further enhanced.
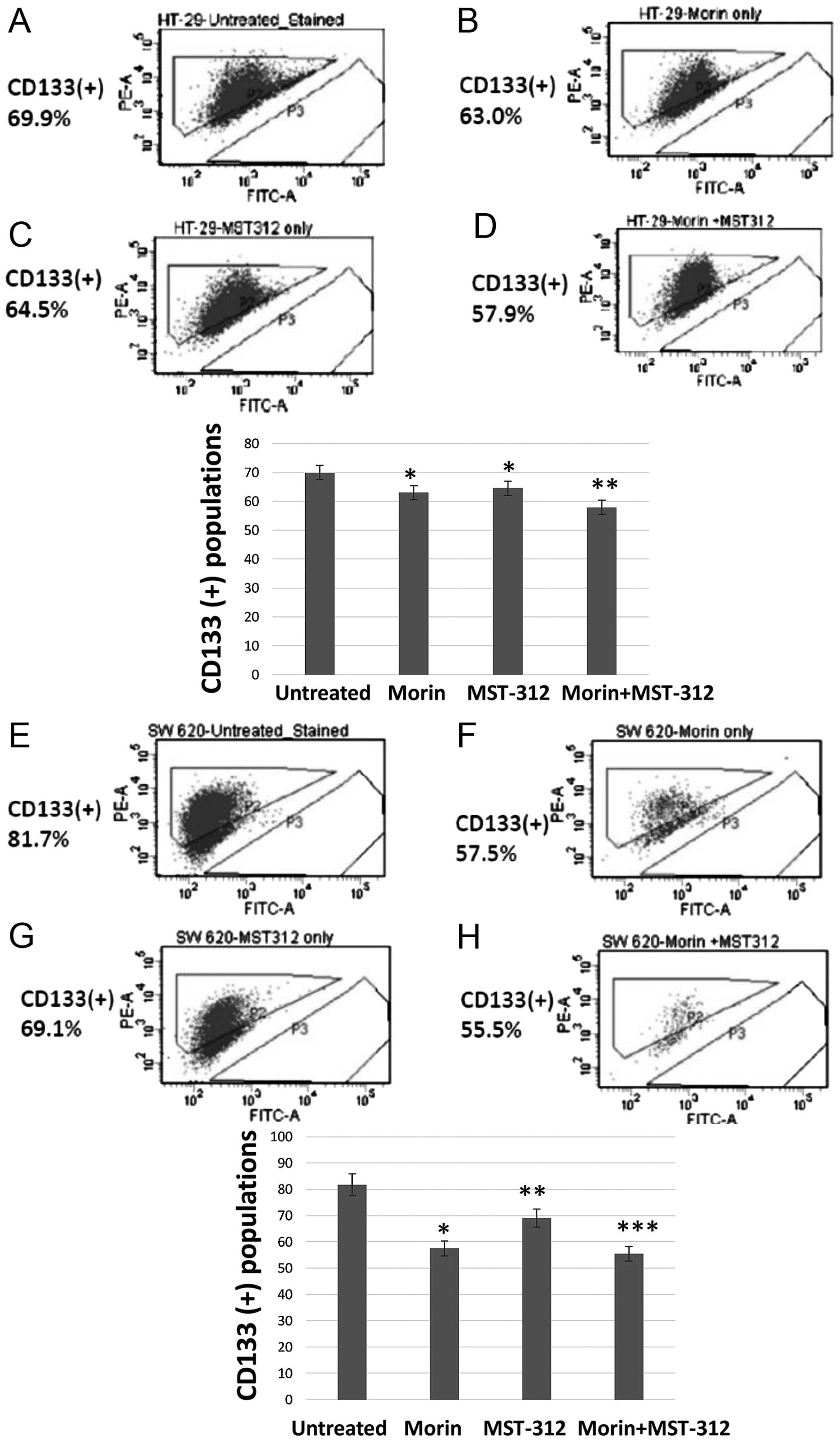
Combined treatment of morin and MST-312
show reduction of the CD133 (+) subpopulation from HT-29 and SW620
cells
To quantify the cancer stem cell population, we
chose CD133 as a biomarker. FACS profiling revealed a clear
reduction of the CD133+ population in the cancer cells
treated with morin and MST-312. Untreated HT-29 showed that 69.9%
of cells were CD133 (+) (Fig. 2A).
However, when we treated HT-29 with morin (50 μM for 24 h), the
CD133-positive subpopulation was reduced to 63% (Fig. 2B). Similarly, MST-312 treatment (10
μM for 24 h) reduced the CD133 (+) population to 64.5% (Fig. 2C). Moreover, the CD133 (+)
population was decreased to 57.9% after combined treatment with
morin and MST-312 in HT-29 cells (Fig.
2D). Representative histograms are presented with the
statistical significance.
We also observed the same CD133 (+) pattern from the
metastatic colorectal cancer cell line SW620. In the untreated
control SW620, 81.7% of cells were CD133 (+) (Fig. 2E). After treatment with morin (50
μM, 24 h), the CD133 (+) population was reduced to 57.5% (Fig. 2F). With MST-312 treatment, CD133
(+) was decreased to 69.1% (Fig.
2G). Notably, the combined treatment of morin and MST-312
showed enhanced reduction of CD133 (+) to 55.5% (Fig. 2H). Our results demonstrate that
combined treatments with morin and MST-312 indeed reduced the CD133
(+) subpopulations in both primary and metastatic human colorectal
cancer cell lines. Histograms of the CD133 (+) populations are
presented with statistical significance.
Morin and MST-312 treatments abolish the
tumorsphere formation and suppress cell invasiveness in human
colorectal cancer cell lines
As combined treatment of morin and MST-312 reduced
CD133 positivity, we next examined the cell-level invasiveness of
the colorectal cancer cells. To this end, we employed two cell
invasion assays, tumorsphere formation assay and boyden chamber
assays. In tumorsphere culture condition, we created a
three-dimensional microenvironment by adding 5% Matrigel to the
24-well plates. As shown in Fig.
3, both HT-29 and SW620 cells formed tumorspheres in 7 days.
However, when we treated cells with morin and MST-312, the
tumorsphere forming capacity was significantly reduced. Untreated
control HT-29 formed ~32 tumorspheres per wells (Fig. 3A). However, when cells were treated
with morin and MST-312, the formation was reduced to 15 and 21
tumorspheres, respectively. The combined treatment of morin and
MST-312 abolished tumorsphere formation from HT-29. We observed a
similar pattern in the SW620. Untreated control cells formed 45
tumor-spheres per wells (Fig. 3B).
Morin treatment resulted in a reduction to 32 tumorspheres and
MST-312 treatment a reduction to 21 tumorspheres per well. When we
treated SW620 with morin and MST-312 together, tumorsphere
formation capacity was abolished.
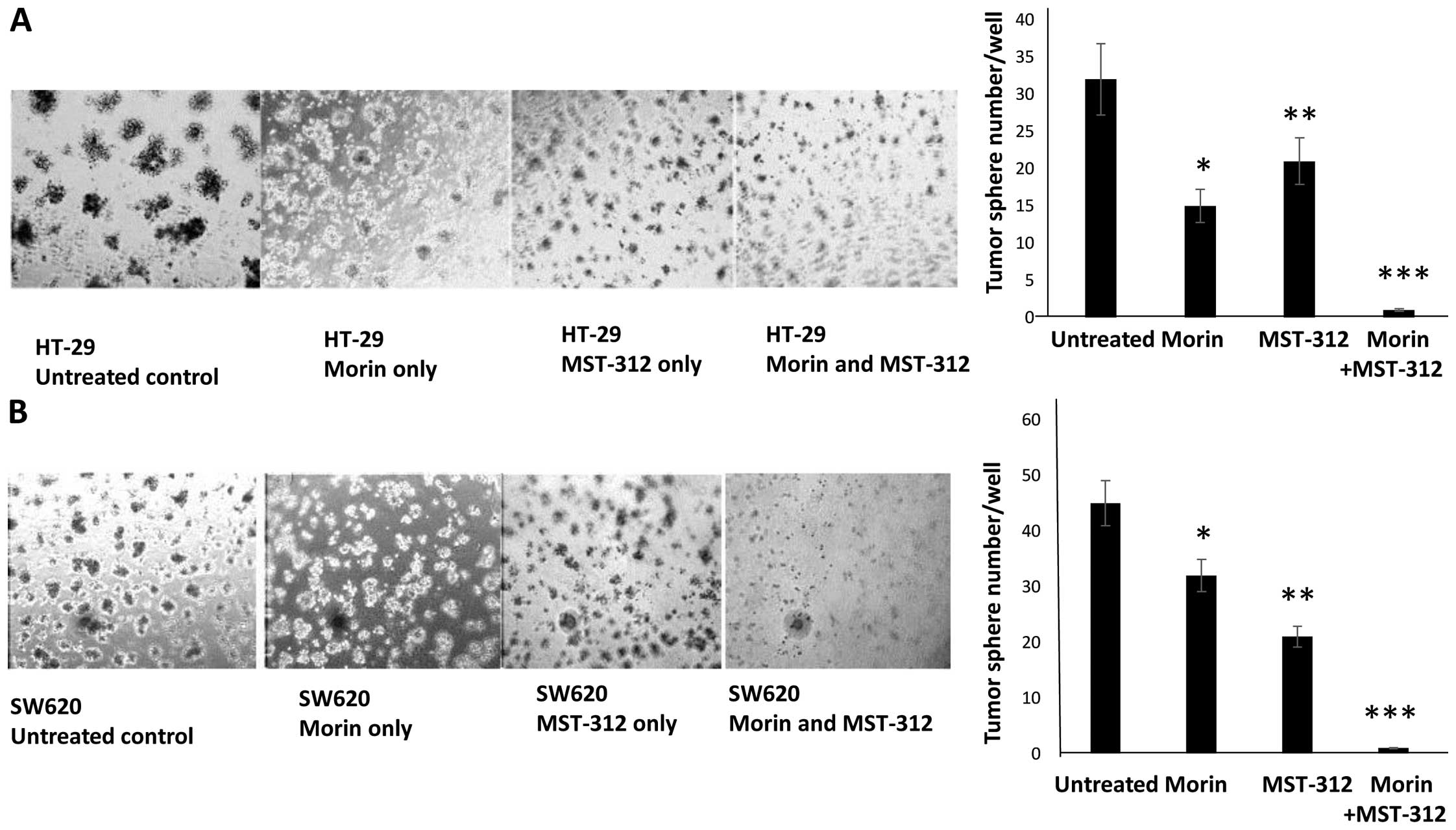 | Figure 3Inhibition of tumorsphere formation
with morin and MST-312 treatment in HT-29 and SW620. Combined
treatments of morin and MST-312 reduced tumorsphere formation
capability from colorectal cancer cells. (A) HT-29 untreated
control, pre-treated with morin alone at 50 μM for 24 h,
pre-treated with MST-312 10 μM for 24 h and pre-treated with morin
and MST-312 combination at concentrations of 50 and 10 μM for 24 h,
respectively. Cells were cultured for 7 days. On the right side,
graphic presentation of HT-29 tumor-sphere formation results. Data
are presented as mean ± SD (n=3 in each group).
*P<0.05, **P<0.01,
***P<0.001 vs. untreated control. (B) SW620 untreated
control, pre-treated with morin alone at 50 μM for 24 h,
pre-treated with MST-312 alone at 10 μM for 24 h and pre-treated
with morin and MST-312 combination at concentrations of 50 and 10
μM for 24 h, respectively. Cells were seeded onto 3-D cultures and
tumorsphere formation was observed. On the right side, quantitative
graph of SW620 tumorsphere formation results were presented. Data
are presented as mean ± SD (n=3 in each group).
*P<0.05, **P<0.01,
***P<0.001 vs. untreated control. |
In agreement with the tumorsphere formation, cell
invasion assays also revealed that combined treatment inhibited
cell invasiveness. On average, the number of cells invading from
the boyden chamber decreased from 55 to 2 when compared to
untreated control HT-29 and morin/MST-312-treated cells (Fig. 4A). In SW620, the average invaded
cell numbers decreased from 75 of untreated control to 12 of
morin/MST-312-treated cells (Fig.
4B). Our results suggest that morin/MST-312 combined treatment
can efficiently reduce the cancer stem cell subpopulations from
human colorectal cancer cells.
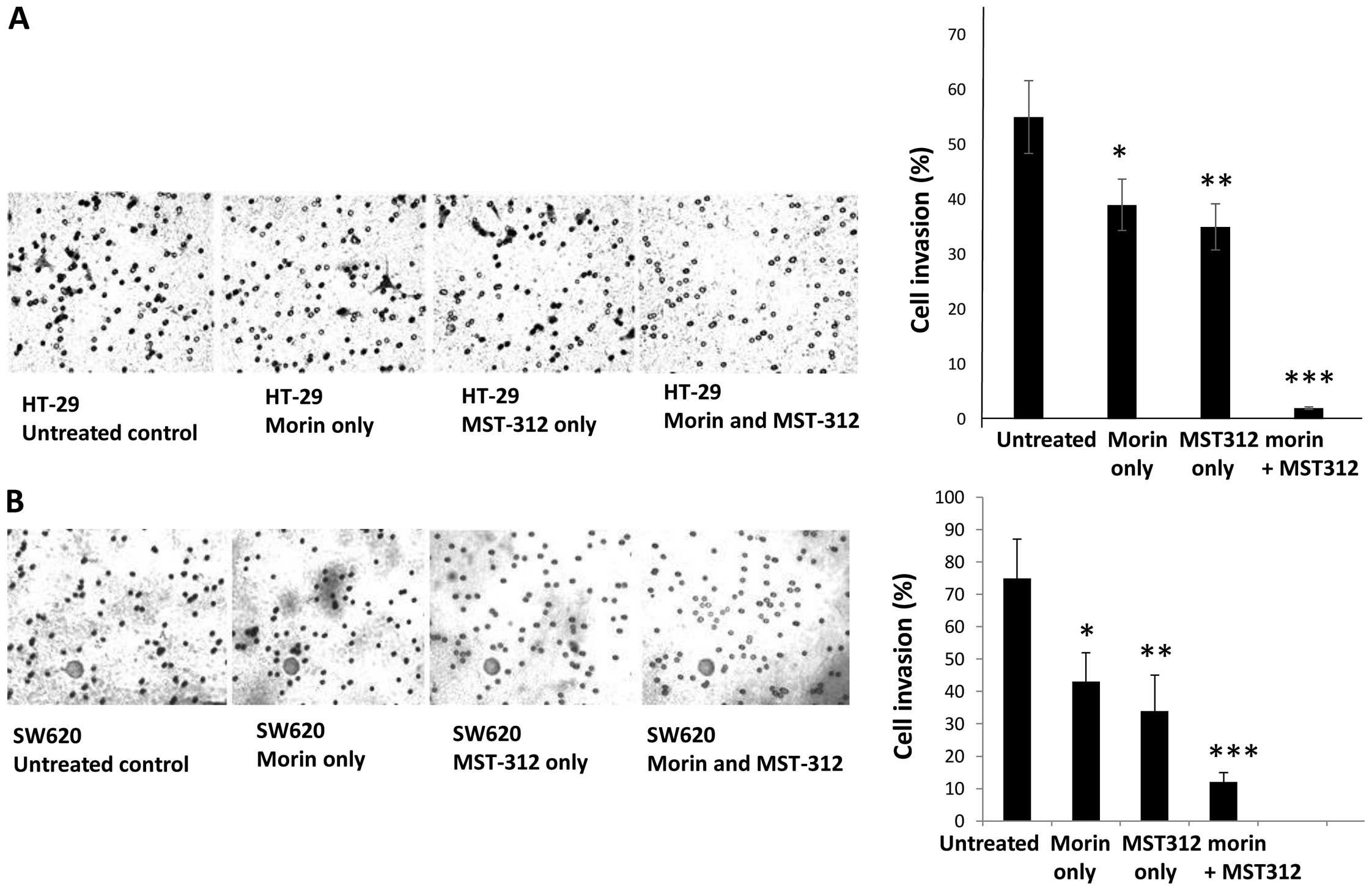 | Figure 4Decreased cell invasiveness with
morin and MST-312 treatment in HT-29 and SW620. Cell invasiveness
was examined by employing Boyden chamber assays. (A) HT-29
untreated control, pre-treated with morin alone, MST-312 alone and
morin and MST-312 combination. On the right side, the cell invasion
assay was quantitatively measured in graphic representation. Data
are presented as mean ± SD (n=3 in each group).
*P<0.05, **P<0.01,
***P<0.001 vs. untreated control. (B) SW620 untreated
control, pre-treated with morin alone, MST-312 alone and morin and
MST-312 combination. Cells were placed on the Boyden chamber and
cell invasion was measured. On the right side, the cell invasion
assay was quantitatively measured in graphic representation. Data
are presented as mean ± SD (n=3 in each group).
*P<0.05, **P<0.01,
***P<0.001 vs. untreated control. |
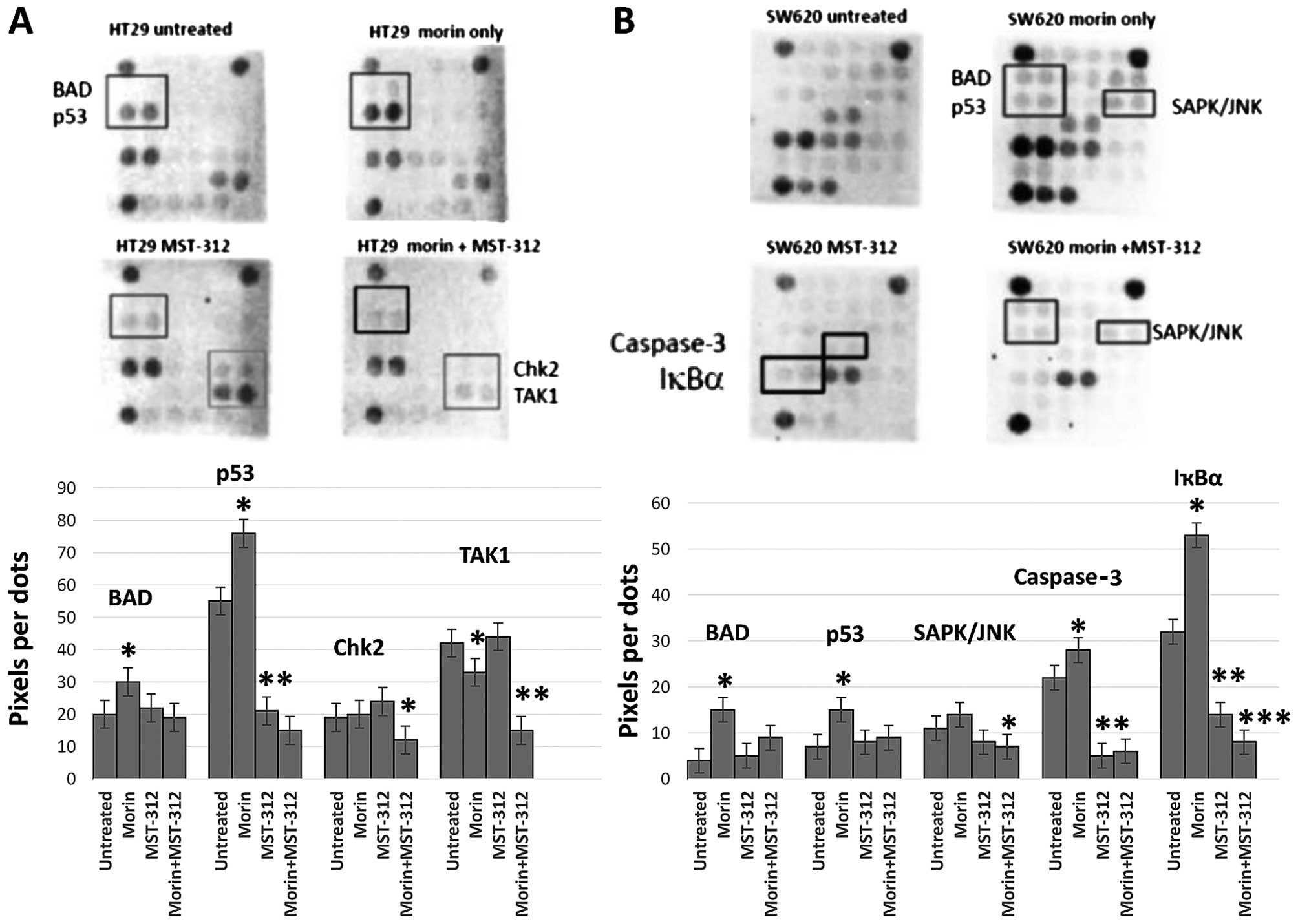
Distinct stress and apoptosis-related
gene expression patterns responding to morin and MST-312
treatments
The findings that morin and MST-312 treatment
inhibited CSC phenotype suggest that it may directly trigger
certain signaling pathways. Therefore, we wished to determine how
morin and MST-312 treatments elicit cellular stress and apoptosis
responses from colorectal cancer cells. To assess the net effects
of the treatments, we decided to monitor the changes in key
signaling components related to cellular stress and apoptosis. To
this end, we utilized the stress and apoptosis signaling antibody
array kit (Cell Signaling Technology). By using this array, we were
able to simultaneously interrogate 19 signaling molecules that are
involved in the regulation of stress response and apoptosis.
Target-specific capture antibodies were spotted in duplicate onto
nitrocellulose-coated glass slides. Cancer cell lysates were
incubated on the slide followed by a biotinylated antibody. The
expression differences from the antibody arrays were presented as
histograms with statistical significance of the differential
expression proteins in HT-29 and SW620. As shown in Fig. 5, morin treatment on HT-29 enhanced
the levels of BAD and p53 phosphorylation (Fig. 5A). BAD is a member of the Bcl-2
family and a regulator of the programmed cell death pathway
(22). Tumor suppressor p53 plays
an important role in cellular responses to DNA damage (23). Thus, morin treatment activated BAD
and p53 and concurrently induced apoptosis in HT-29 cells. MST-312
treatment inhibited p53 phosphorylation on the contrary, indicating
that MST-312 inactivates p53, possibly independent of morin
pathway. When morin and MST-312 were treated in combination, p53,
Chk2 and TAK1 phosphorylation were inhibited in HT-29. Chk2 kinase
acts downstream of ATM/ATR and plays an important role in DNA
damage check point control (24).
TAK1 is a kinase that can be activated by TFG-β, bone morphogenetic
proteins and other cytokines (25). Morin treatment attenuated p53, Chk2
and TAK1 phosphorylation which are important for DNA damage control
and cell survival. Increased apoptotic effects of morin and MST-312
treatments might be through the impaired DNA damage repair system
and suppressed cell survival signaling.
Morin treatment on SW620 enhanced the
phosphorylation of BAD, p53 and SAPK (Fig. 5B). SAPK kinase is activated through
a dual phosphorylation of Thr202 and Tyr204 in response to
pro-inflammatory cytokines and genotoxic stress (26). BAD and p53 activation with morin
treatment is similar to HT-29. MST-312 treatment inhibited
caspase-3 cleavage and downregulated IκBα expression level in
SW620. Caspase-3 protease exerts a pro-apoptotic function through
cleavage of multiple targets (27). Caspase-3 is activated at Asp175.
IκBα is targeted to the proteasome via phosphorylation at Ser32 and
Ser36 (28). Morin and MST-312
combined treatment activated BAD, p53 and SAPK phosphorylation
whereas inhibited caspase-3 cleavage and IκBα phosphorylation in
SW620. The enhanced apoptotic effects may be result from the
inhibited cytokine signaling required for cancer cell survival.
This distinct subset of gene inhibition in caspase-3 cleavage and
IκBα is possibly due to the cell line specific differences between
HT-29 and SW620. Taken together, our data revealed the existence of
distinct expression patterns from the stress and apoptosis genes
responding to morin and MST-312 treatments in the colorectal cancer
cell lines.
Morin and MST-312 co-treatments
chemosensitized 5-FU resistant human colorectal cancer cells
Human colorectal cancer cell lines were sub-grouped
based on their growth inhibition (GI50) values against
5-FU treatments in a previous study (29). Three subgroups were chosen,
consisting of 5-FU sensitive, intermediate and resistant colorectal
cancer cell lines. Two 5-FU chemo-resistant cell lines, HT-29 and
SW620, were used in our study to investigate the effects of
morin/MST-312 and/or 5-FU on cell viability. The GI50
values of HT-29 and SW620 were 14.90 and 17.97 μM, respectively.
The cytotoxic effects of 5-FU or morin plus MST-312 on two colon
cancer cell lines were determined using the MTT assay. The cancer
cells were treated with different concentrations of 5-FU (0, 1, 5,
10, 20 and 50 μM) alone or combined with 5 μM morin and 3 μM
MST-312. We observed that 5-FU blocked the proliferation of the
cell lines HT-29 and SW620 in a dose-dependent manner with 5 μM
morin and 3 μM MST-312 co-treatments (Fig. 6A). 5-FU efficacy was enhanced to
the extent that the IC50 level was reduced to 0.5 μM for
HT-29 and 1 μM for SW620 (Fig.
6B). Both 5-FU chemo-resistant cell lines became equally
sensitive to 5-FU with the co-treatment of 5 μM morin and 3 μM
MST-312. Our data suggest that the morin/MST-312 combination
treatment as an approach for the better treatment of human colon
tumors with the potentially enhanced chemo-sensitivity to 5-FU.
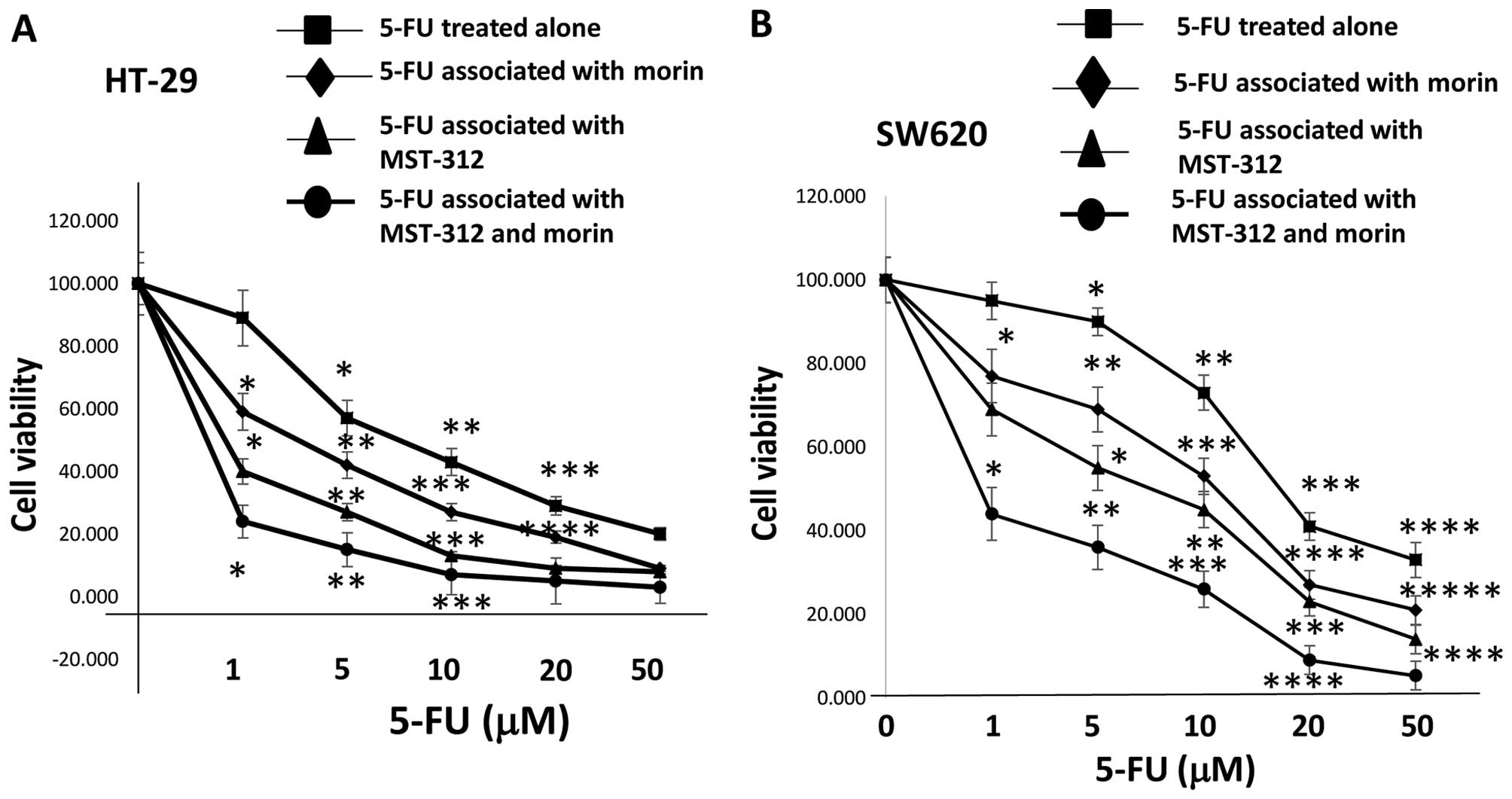 | Figure 6Cell viability is reduced by 5-FU,
morin, MST-312 and the combination treatment in 5-FU-resistant cell
lines HT-29 and SW620. Co-treatment with 5 μM morin or morin and 3
μM MST-312 combination chemosensitized drug-resistant colorectal
cancer cells to 5-fluorouracil. (A) HT-29 was treated with
different concentrations of 5-FU (0, 1, 5, 10, 20 and 50 μM) alone
and associated with 5 μM morin or 3 μM MST-312 or morin and 3 μM
MST-312 combination. Cell viability was measured with the MTT
method. The results are provided as mean values with standard
deviations from at least three independent experiments. Data are
presented as mean ± SD (n=3 in each group). *P<0.05,
**P<0.01, ***P<0.001 vs. untreated
control. (B) SW620 cell line was treated with different
concentrations of 5-FU (0, 1, 5, 10, 20 and 50 μM) alone or
associated with 5 μM morin or 3 μM MST-312 or morin and 3 μM
MST-312 combination. Data are presented as mean ± SD (n=3 in each
group). *P<0.05, **P<0.01,
***P<0.001 vs. untreated control. |
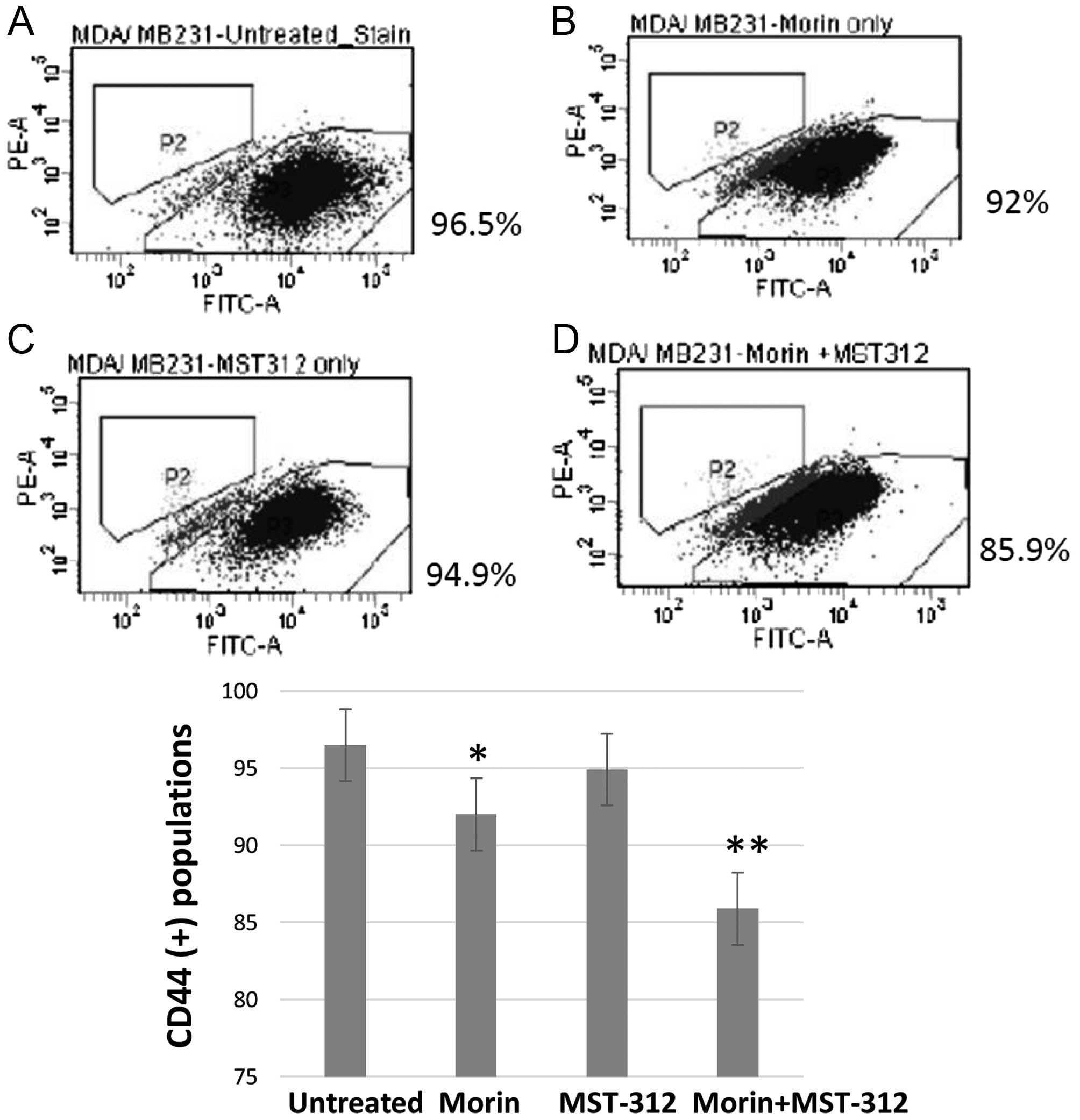
Morin and MST-312 combination treatment
reduced the CD44 (+) subpopulation and inhibited wound healing from
human breast cancer cells
Morin and MST-312 treatment inhibited the CSC
phenotype in human colorectal cancer cells. Next we wished to
determine whether this effect holds true in other human cancers. To
test this, we chose the human triple-negative breast cancer cell
line, MDA-MB-231. It also contains constitutively activated STAT3
phosphorylated by JAK2 kinase at the site of Tyr705 (30) and activated telomerase. Morin (10
μM for 24 h) and MST-312 (10 μM for 24 h) were used alone or in
combination. Untreated control and treated cells were subsequently
applied to FACS analysis for CD44 (+) profiling (Fig. 7A). CD44 is a well-established
biomarker for breast CSC population. When treatment was used, the
CD44 (+) subpopulation was reduced slightly from 96.5%
(CD44+ of the untreated control) to 92% (Fig. 7B). Similarly, MST-312 treatment
reduced the CD44 (+) population to 94.9% (Fig. 7C). The combined treatment with
morin and MST-312 reduced the CD44 (+) to 85.9% (Fig. 7D). These data suggest that the
synergism of morin and MST-312 may be conserved in multiple human
cancers.
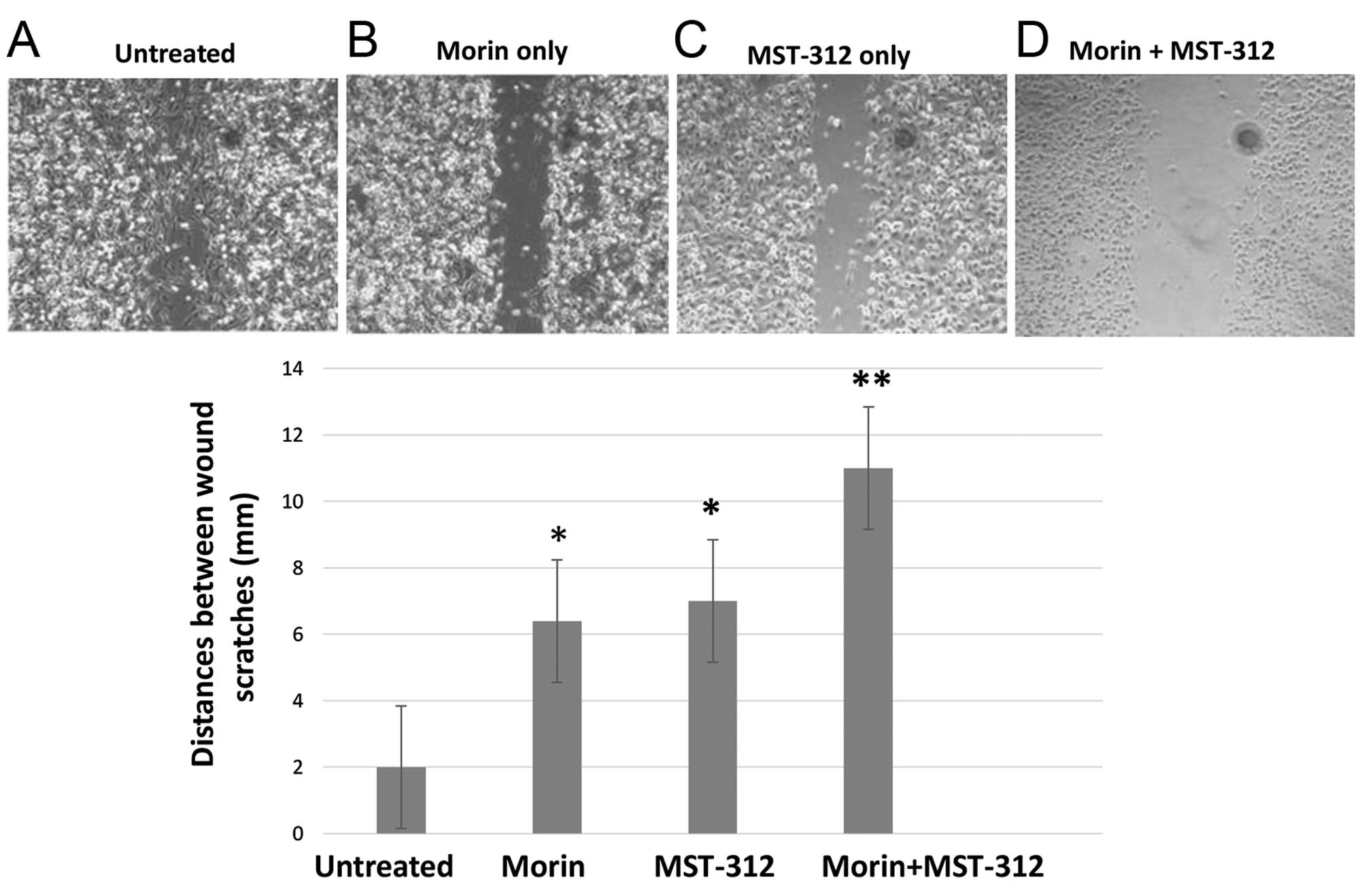
In agreement with FACS analyses, wound healing
studies also showed synergistic effects of morin and MST-312
treatments. MDA-MB-231 cells were pre-treated with either
morin/MST-312 alone or in combination for 24 h at the same
concentration (morin; 50 μM and MST-312; 10 μM). Afterwards, the
cells were seeded onto 24-well plates and strait scratches were
performed. We observed wound healing up to 48 h. As shown in
Fig. 8, untreated control cells
rapidly healed the wounds (Fig.
8A). However, morin and MST-312 treatments clearly inhibited
the wound healing (Fig. 8B and C).
The inhibition effect was enhanced upon morin and MST-312
combination treatment (Fig. 8D).
Together, our results indicate that morin and MST-312 combination
treatment can downregulate breast cancer stem cell phenotype.
Discussion
Cancer stem cells (CSCs) are considered to be
responsible for the maintenance of the whole cancer cell population
and tumor re-initiation after therapy. Considering the CSC traits,
it is important to understand the signal pathways specifically
activated in CSCs so that we can devise strategies to target them.
This study is built on our previous findings that constitutively
activated STAT3 upregulates hTERT expression and promotes cancer
stem cell traits in human breast cancer. Subsequently, hTERT
upregulated expression of the breast CSC marker, CD44. The
STAT3-telomerase axis was selectively activated in cancer stem cell
subpopulations.
In this study, we demonstrate that combined
treatments of morin and MST-312 inhibit the cancer stem cell
phenotype. We tested our hypothesis that flavonoid morin and
telomerase inhibitor MST-312 co-treatments might result in an
enhanced inhibition of cancer stem cell traits through dual
targeting of STAT3 and telomerase. The colorectal CSC marker CD133
(+) subpopulation was reduced by the combination treatments. In
accordance, tumor-sphere formation and cell invasiveness were
decreased in the colorectal cancer cell lines.
To identify gene signatures responding to
morin/MST-312 treatments, we studied the cellular stress and
apoptosis antibody arrays. We identified specific subsets of genes
that are upregulated and downregulated upon the combination
treatments. Combined treatment of morin/MST-312 inhibited p53, Chk2
and TAK1 phosphorylation and enhanced apoptosis of the colorectal
cancer cells. Since p53 and Chk2 both play roles in DNA damage
check point control, the impaired DNA damage of the cancer cells
likely led to cell death. TAK1 kinase is activated in TFG-β and
other cytokines. The cytokine signaling important for cancer cell
survival was possibly suppressed by the morin and MST-312 in HT-29.
There was caspase-3 and IκBα inhibited cell death in SW620.
Malfunctioning protein cleavage from caspase-3 inhibition and
disturbed targeting to the proteasome from IκBα might result in
cell death in SW620 cells. The distinct subset of apoptosis genes
exist in difference cancer cell lines, implicating differences in
cell line characteristics. More colorectal cell lines and other
cancer cell studies are warranted for the mechanistic work of morin
and MST-312 in apoptosis and cell stress mechanism.
Morin and MST-312 combination treatments
chemosensitized 5-FU-resistant human colorectal cancer cells. Since
the STAT3-telomerase axis driven CSC phenotype is conserved in
breast cancer, morin/MST-312 treatment also showed the same
inhibitory effects against breast cancer stem cells.
Constitutively activated STAT3 has been reported in
numerous human malignancies. Active STAT3 has been reported to
participate in tumorigenesis by regulating the expression of genes
involved in tumor cell proliferation, survival, invasion and
metastasis (31). STAT3 activation
also has been linked to chemo-resistance and radio-resistance
(9,32). We noted that constitutively
activated STAT3 proteins are enriched in the CSC subpopulations.
Mounting evidence suggests that STAT3 is an attractive therapeutic
target for the development of anticancer stem cell agents.
Morin was originally isolated from members of the
Moraceae family such as mulberry figs and old fustic.
Earlier studies demonstrate that morin inhibits STAT3
phosphorylation at the Tyr705 site. We used morin at 50 μM dosage
because we observed that morin clearly suppressed constitutive
pSTAT3 at that concentration in a gradient of 0, 5, 10, 25 and 50
μM with human colorectal cancer cells (data not shown). Other
groups have shown that morin reduces the incidence of
lipopolysaccharide-induced septic shock (33) and suppresses the phorbol
ester-induced transformation of hepatocytes (34). Morin has also been found to exert
chemopreventive effects in a model of dimethylhydrazine-induced
colon carcinogenesis (35). Here,
we tested morin's anti-CSC effects based on the selective
activation of STAT3 in the cancer stem cell population. Morin
indeed reduced the cancer stem cell phenotype in human colorectal
and breast cancers.
Telomeres function to protect DNA integrity, but
unfortunately fragile sites and DNA damage can result at telomeric
sites following disruption of telomere-telomerase homeostasis.
MST-312 is a reversible telomerase inhibitor as it reduced
telomerase activity and induced telomere dysfunction. We have
observed that MST-312 clearly inhibited telomerase activity at 10
μM in a gradient of 0, 1, 5 and 10 μM concentrations with human
colorectal cancer cells (data not shown). It was recently reported
that MST-312 exposure to breast cancer cells elevated level of
double strand breaks (DSBs) based on the presence of the γ-H2AX
proteins (36). This acute
induction of DSBs resulted in growth arrest and was more evident in
the metastatic breast cancer cell type MDA-MB-231 than MCF-7. We
chose MST-312 because it inhibits telomerase and induce growth
arrest selectively in aggressive tumor cells. MST-312 does not
inhibit normal cell growth but inhibits effectively metastatic
cancer cells (36). This makes it
an attractive anticancer, anti-metastatic compound. Moreover,
MST-312 is chemically more stable and more potent than its analog,
green tea epigallocatechin gallate (EGCG) (17). MST-312 induced DNA damage at
telomeres and elevated the level of DSBs leading to growth arrest.
So, even after the MST-312 is removed, the inhibitory effects on
telomere dynamics and telomerase will likely remain for certain
time. In addition, MST-312 chemosensitized 5-FU in colorectal
cancer cells and when combined with morin, showed well enhanced
antitumor effects.
We reasoned that if we targeted STAT3 and telomerase
together, we could synergistically inhibit cancer stem cell traits
since STAT3 regulates hTERT and telomerase activity is required for
CSC growth. As morin inhibits STAT3 phosphorylation, it
downregulates STAT3 target gene expression resulting in inhibition
of CSC growth. Similarly, MST-312 inhibits telomerase and reduces
the cancer stem cell population. One step further, we tested
whether morin/MST-312 co-treatment augment 5-FU efficacy on the
chemo-resistant colorectal cancer cells. In agreement with CSC
trait reduction data, the co-treatment chemosensitized the
5-FU-resistant cancer cell lines. Taken together, this study
suggests that novel targeted-therapy may be implemented using
combination treatment for inhibiting STAT3 and telomerase. The
in vivo animal study is underway to validate the reduction
of tumor formation and metastasis with the morin/MST-312
combination treatment.
Acknowledgements
This study was supported by the National Institutes
of Health (NIH, NCI, NIMHD, NCATS) grants to J.V. Vadgama: U54
CA143931, U54MD007598, UL1TR000124. S. Steven Chung is a scholar
supported by the Clinical Research Education and Career Development
by the NIMHD R25 MD 007610, pilot project award from U54 MD 007598
and Emerging scientist award from the Life Science Institute-CDU
S21 MD 000103. We thank the division of cancer research and
training members for their helpful suggestions. We also thank Dr
Robert Gelfand for careful reading and proofreading of the
manuscript.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luu C, Arrington AK, Schoellhammer HF,
Singh G and Kim J: Targeted therapies in colorectal cancer:
surgical considerations. J Gastrointest Oncol. 4:328–336.
2013.PubMed/NCBI
|
|
3
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells - perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhong Z, Wen Z and Darnell JE Jr: Stat3: A
STAT family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirano T, Ishihara K and Hibi M: Roles of
STAT3 in mediating the cell growth, differentiation and survival
signals relayed through the IL-6 family of cytokine receptors.
Oncogene. 19:2548–2556. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Turkson J and Jove R: STAT proteins: Novel
molecular targets for cancer drug discovery. Oncogene.
19:6613–6626. 2000. View Article : Google Scholar
|
|
7
|
Azare J, Doane A, Leslie K, Chang Q,
Berishaj M, Nnoli J, Mark K, Al-Ahmadie H, Gerald W, Hassimi M, et
al: Stat3 mediates expression of autotaxin in breast cancer. PLoS
One. 6:e278512011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dauer DJ, Ferraro B, Song L, Yu B, Mora L,
Buettner R, Enkemann S, Jove R and Haura EB: Stat3 regulates genes
common to both wound healing and cancer. Oncogene. 24:3397–3408.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tseng LM, Huang PI, Chen YR, Chen YC, Chou
YC, Chen YW, Chang YL, Hsu HS, Lan YT, Chen KH, et al: Targeting
signal transducer and activator of transcription 3 pathway by
cucurbit-acin I diminishes self-renewing and radiochemoresistant
abilities in thyroid cancer-derived CD133+ cells. J
Pharmacol Exp Ther. 341:410–423. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan KS, Sano S, Kiguchi K, Anders J,
Komazawa N, Takeda J and DiGiovanni J: Disruption of Stat3 reveals
a critical role in both the initiation and the promotion stages of
epithelial carcinogenesis. J Clin Invest. 114:720–728. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bertorelle R, Rampazzo E, Pucciarelli S,
Nitti D and De Rossi A: Telomeres, telomerase and colorectal
cancer. World J Gastroenterol. 20:1940–1950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tatsumoto N, Hiyama E, Murakami Y, Imamura
Y, Shay JW, Matsuura Y and Yokoyama T: High telomerase activity is
an independent prognostic indicator of poor outcome in colorectal
cancer. Clin Cancer Res. 6:2696–2701. 2000.PubMed/NCBI
|
|
13
|
Liu Z, Li Q, Li K, Chen L, Li W, Hou M,
Liu T, Yang J, Lindvall C, Björkholm M, et al: Telomerase reverse
transcriptase promotes epithelial-mesenchymal transition and stem
cell-like traits in cancer cells. Oncogene. 32:4203–4213. 2013.
View Article : Google Scholar
|
|
14
|
Brown J, O'Prey J and Harrison PR:
Enhanced sensitivity of human oral tumours to the flavonol, morin,
during cancer progression: Involvement of the Akt and stress kinase
pathways. Carcinogenesis. 24:171–177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Manna SK, Aggarwal RS, Sethi G, Aggarwal
BB and Ramesh GT: Morin (3,5,7,2′,4′-Pentahydroxyflavone) abolishes
nuclear factor-kappaB activation induced by various carcinogens and
inflammatory stimuli, leading to suppression of nuclear
factor-kappaB-regulated gene expression and up-regulation of
apoptosis. Clin Cancer Res. 13:2290–2297. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gupta SC, Phromnoi K and Aggarwal BB:
Morin inhibits STAT3 tyrosine 705 phosphorylation in tumor cells
through activation of protein tyrosine phosphatase SHP1. Biochem
Pharmacol. 85:898–912. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seimiya H, Oh-hara T, Suzuki T, Naasani I,
Shimazaki T, Tsuchiya K and Tsuruo T: Telomere shortening and
growth inhibition of human cancer cells by novel synthetic
telomerase inhibitors MST-312, MST-295, and MST-1991. Mol Cancer
Ther. 1:657–665. 2002.PubMed/NCBI
|
|
18
|
Serrano D, Bleau AM, Fernandez-Garcia I,
Fernandez-Marcelo T, Iniesta P, Ortiz-de-Solorzano C and Calvo A:
Inhibition of telomerase activity preferentially targets aldehyde
dehydrogenase-positive cancer stem-like cells in lung cancer. Mol
Cancer. 10:962011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chung SS, Aroh C and Vadgama JV:
Constitutive activation of STAT3 signaling regulates hTERT and
promotes stem cell-like traits in human breast cancer cells. PLoS
One. 8:e839712013. View Article : Google Scholar
|
|
20
|
Park JI, Venteicher AS, Hong JY, Choi J,
Jun S, Shkreli M, Chang W, Meng Z, Cheung P, Ji H, et al:
Telomerase modulates Wnt signalling by association with target gene
chromatin. Nature. 460:66–72. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hoffmeyer K, Raggioli A, Rudloff S, Anton
R, Hierholzer A, Del Valle I, Hein K, Vogt R and Kemler R:
Wnt/β-catenin signaling regulates telomerase in stem cells and
cancer cells. Science. 336:1549–1554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Korsmeyer SJ: BCL-2 gene family and the
regulation of programmed cell death. Cancer Res. 59(Suppl):
S1693–S1700. 1999.
|
|
23
|
Kern SE, Kinzler KW, Bruskin A, Jarosz D,
Friedman P, Prives C and Vogelstein B: Identification of p53 as a
sequence-specific DNA-binding protein. Science. 252:1708–1711.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamaguchi K, Shirakabe K, Shibuya H, Irie
K, Oishi I, Ueno N, Taniguchi T, Nishida E and Matsumoto K:
Identification of a member of the MAPKKK family as a potential
mediator of TGF-beta signal transduction. Science. 270:2008–2011.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tibbles LA and Woodgett JR: The
stress-activated protein kinase pathways. Cell Mol Life Sci.
55:1230–1254. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alnemri ES, Livingston DJ, Nicholson DW,
Salvesen G, Thornberry NA, Wong WW and Yuan J: Human ICE/CED-3
protease nomenclature. Cell. 87:1711996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jacobs MD and Harrison SC: Structure of an
IkappaBalpha/NF-kappaB complex. Cell. 95:749–758. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bracht K, Nicholls AM, Liu Y and Bodmer
WF: 5-Fluorouracil response in a large panel of colorectal cancer
cell lines is associated with mismatch repair deficiency. Br J
Cancer. 103:340–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garcia R, Bowman TL, Niu G, Yu H, Minton
S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, et
al: Constitutive activation of Stat3 by the Src and JAK tyrosine
kinases participates in growth regulation of human breast carcinoma
cells. Oncogene. 20:2499–2513. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takemoto S, Ushijima K, Kawano K,
Yamaguchi T, Terada A, Fujiyoshi N, Nishio S, Tsuda N, Ijichi M,
Kakuma T, et al: Expression of activated signal transducer and
activator of transcription-3 predicts poor prognosis in cervical
squamous-cell carcinoma. Br J Cancer. 101:967–972. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Wang G, Zhao Y, Liu X, Ding Q, Shi
J, Ding Y and Wang S: STAT3 mediates resistance of
CD44(+)CD24(−/low) breast cancer stem cells to tamoxifen in vitro.
J Biomed Res. 26:325–335. 2012. View Article : Google Scholar
|
|
33
|
Fang SH, Hou YC, Chang WC, Hsiu SL, Chao
PD and Chiang BL: Morin sulfates/glucuronides exert
anti-inflammatory activity on activated macrophages and decreased
the incidence of septic shock. Life Sci. 74:743–756. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hsiang CY, Wu SL and Ho TY: Morin inhibits
12-O-tetradecanoyl-phorbol-13-acetate-induced hepatocellular
transformation via activator protein 1 signaling pathway and cell
cycle progression. Biochem Pharmacol. 69:1603–1611. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sreedharan V, Venkatachalam KK and
Namasivayam N: Effect of morin on tissue lipid peroxidation and
antioxidant status in 1, 2-dimethylhydrazine induced experimental
colon carcinogenesis. Invest New Drugs. 27:21–30. 2009. View Article : Google Scholar
|
|
36
|
Gurung RL, Lim SN, Low GK and Hande MP:
MST-312 alters telomere dynamics, gene expression profiles and
growth in human breast cancer cells. J Nutrigenet Nutrigenomics.
7:283–298. 2014. View Article : Google Scholar
|