Introduction
Recent research has indicated that the
osteoclastogenesis induction factor, receptor activator of NF-κB
ligand (RANKL), is involved in all bone resorption diseases
regardless of inflammation or tumor induction (1). RANKL, which is expressed in cells
such as osteoblasts, promotes osteoclast differentiation through
signals from receptors expressed by osteoclast precursors (1,2). In
addition, RANKL induces osteoclast formation and bone resorption at
the disease site in rheumatoid arthritis through expression by
synovial fibroblasts (3,4), tumor cells themselves (5) and lymphocytes (6). On the basis of the results of studies
related to tooth germ development, cytokines and parathyroid
hormone-related protein produced by the odontogenic epithelium
promote osteoclast formation and protect the tooth germ from
developing bone tissue by inducing RANKL expression in osteoblasts
around the tooth germ and to be involved in the eruption of the
tooth germ by regulating bone resorption (7,8).
Transforming growth factor-β (TGF-β) is a
multifunctional cytokine that binds to its receptors on cell
membranes, causes TGF-β receptor phosphorylation, and activates
Smad2/3 to induce intracellular signal transmission (9). Interleukin-1α (IL-1α) is a
multifunctional inflammatory cytokine (10). Both cytokines are expressed by the
dental follicle (11).
Furthermore, TGF-β, IL-1α, and tumor necrosis factor-α promote
RANKL expression by the dental follicle, whereas the dental
follicle is indicated to possess a latent ability to promote
osteoclast formation (11–13). In addition, TGF-β directly promotes
osteoclast differentiation of osteoclast precursors (14), and IL-1α also promotes osteoclast
differentiation (15).
Typical neoplastic and cystic diseases of the
jawbone include keratocystic odontogenic tumors (KCOTs),
ameloblastomas, and follicular cysts; these diseases originate in
the tooth germ epithelial cells and enhance resorption of the
surrounding jawbone. These lesions, in the process of developing
within the jawbone, inevitably absorb or destroy the surrounding
jawbone. Although cyst epithelial cell proliferation (16,17)
and resorption resulting from intracystic fluid-induced compression
(18,19) have been widely accepted, the
molecular regulation of this biological event is still unclear.
With respect to the mechanism underlying this intracystic
fluid-induced pressure, the mechanism underlying bone resorption in
the primary lesion has not been explained, and there has been no
examination of the activation or involvement of osteoclasts in the
area surrounding the cysts and tumors. A previous study by Oka
et al (20) reported that
IL-1α produced by KCOTs promotes RANKL expression by stromal
fibroblasts, suggesting the RANKL-mediating mechanism in the
jawbones for tumors/cysts expansion.
We therefore hypothesized that there is a potential
cytokine network that induces both RANKL expression and bone
resorption in jawbone osteoclasts, thereby serving as a mechanism
for the growth of tumors and cysts.
Here we investigated the mechanism underlying RANKL
expression in stromal fibroblasts to determine whether other
cytokines produced by the odontogenic tumors and cell epithelium
also affect this osteoclast formation and to suggest another
mechanism underling tumors/cysts expansion in the jawbones.
Materials and methods
Cell culture and isolation of intracystic
fluid
Subjects were patients who underwent surgical
removal of jaw tumors/cysts at the First Department of Oral and
Maxillofacial Surgery at Osaka University Dental Hospital. After
the appropriate explanation of the use of specimens and samples for
research and obtaining consent, we harvested appropriately removed
odontogenic tumors and cysts (KCOTs, ameloblastomas, and follicular
cysts). We simultaneously performed fine-needle aspiration of
intracystic fluid. Stromal fibroblasts were isolated and cultured
with an explant method. For the experiment, we used stromal
fibroblasts that had undergone 2–9 subcultures.
Intracystic fluid was aspirated using a fine needle
and centrifuged at 740 × g at 4°C for 10 min; the supernatant was
then collected and centrifuged twice more at 13,400 × g at 4°C for
10 min. This supernatant was then collected, sterilized using a
filter with a pore size of 0.45 μm, and preserved at −80°C until
the experiment. For acid treatment of intracystic fluid, 12 M HCl
was added (final concentration 0.5 M HCl) to intracystic fluid,
which was then incubated at 4°C for 30 min. The acid was then
neutralized with 10 M NaOH, at which point intracystic fluid was
ready for use in the experiment.
Experiment reagents and antibodies
Recombinant human TGF-β1 (R&D Systems,
Minneapolis, MN, USA), recombinant human IL-1α (PeproTech, London,
UK), and prostaglandin E2 (PGE2; Cayman Chemical Co., Ann Arbor,
MI, USA) were used in the present experiment. The selective
inhibitors used were an IL-1 receptor antagonist (IL-1Ra;
PeproTech), TGF-β receptor inhibitor (SB-505124; Sigma-Aldrich),
and selective cyclooxygenase-2 (COX-2) inhibitor (CAY10404; Cayman
Chemical Co.). The antibodies used were mouse monoclonal
anti-TGF-β1,2,3 antibody (clone 1D11; R&D Systems), mouse
monoclonal anti-human RANKL antibody (clone 70525.11;
Sigma-Aldrich), goat anti-human COX-2 antibody (Cayman Chemical
Co.), and rabbit anti-phospho-Smad3 antibody (Rockland,
Gilbertsville, PA, USA).
Total RNA extraction and reverse
transcription polymerase chain reaction
Stromal fibroblasts were seeded into a 6-well
culture plate (Corning) and proliferated to confluency, at which
point the culture medium was changed. Stromal fibroblasts were
incubated with or without a sample or in the presence or absence of
a reagent, and total RNA was extracted using a RNeasy Mini kit
(Qiagen, Hilden, Germany). In some experiments, intracystic fluid
was reacted at room temperature for 1 h with mouse monoclonal
anti-TGF-β1,2,3 antibody or stromal fibroblasts were pretreated at
37°C for 1 h with 1 μM TGF-β receptor inhibitor, 50 ng/ml IL-1Ra,
or 1 μM COX-2 inhibitor; stromal fibroblasts were then incubated
with or without a sample or in the presence or absence of a
reagent, and total RNA was extracted. A total RNA template amount
of 0.8–1.5 μg was used. After incubating total RNA with ReverTra
Ace reverse transcriptase (Toyobo, Osaka, Japan), random hexamer
primer (Applied Biosystems, Foster City, CA, USA), and RNase
inhibitor (Promega, Madison, WI, USA) at 42°C for 30 min, tRNA was
then reacted at 99°C for 5 min and at 4°C for 5 min to synthesize
cDNA. The primers used were RANKL (sense,
5′-GGGTATGAGAACTTGGGATT-3′; and antisense,
5′-CACTATTAATGCCACCGAC-3′), osteoprotegerin (OPG) (sense,
5′-CCTGACCACTACTACACAGACA-3′; and antisense,
5′-GTTAGCAGGAGACCAAAGACACT GCA-3′), and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) (sense, 5′-CCATCACCATCTTCCAGGAG-3′; and
antisense, 5′-GCATGGACTGTGGTCATGAG-3′). PCR using the RANKL primer
consisted of 35 cycles of heat treatment at 94°C for 9 min followed
by thermal denaturation at 94°C for 1 min and annealing at 57°C for
1 min; elongation was then performed at 72°C for 10 min. PCR using
the OPG and GAPDH primers consisted of 30 cycles of annealing at
59°C for 1 min.
Total cellular protein extraction and
western blotting
Stromal fibroblasts were seeded into a 60-mm cell
culture dish (Corning) and proliferated to confluency. The culture
medium was then changed to serum-free α-MEM with 0.3% bovine serum
albumin, and stromal fibroblasts were cultured for 16 h. Culture
was subsequently performed in serum-free α-MEM with 0.3% BSA with
or without a sample or reagent. Stromal fibroblasts were placed on
ice for 20 min in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl,
1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.4; Santa
Cruz Biotechnology, Dallas, TX, USA) containing protease and
phosphatase inhibitor cocktails (both from Sigma-Aldrich) and
lysed. The cell lysate was then collected and centrifuged at 13,400
× g at 4°C for 20 min; the supernatant was boiled in 5X SDS sample
buffer and used in the following experiments. After performing
electrophoresis on 20 μg of total cellular protein in a 10% SDS
polyacrylamide gel, the gel was transferred to a PVDF membrane
(Bio-Rad, Hercules, CA, USA). As primary antibodies, rabbit
anti-phospho-Smad3 (diluted 1:2,000) and goat anti-human COX-2
antibodies were diluted with T-PBS and reacted at 4°C for 16 h. A
luminescence reaction was performed with an Amersham ECL Plus kit
(GE Healthcare, Uppsala, Sweden). Luminescent signals were detected
using a Kodak Gel Logic 2200 Imaging System (Carestream, Rochester,
NY, USA).
Immunohistochemical staining
Tissues from KCOTs and ameloblastomas were fixed in
10% formalin buffer solution and embedded in paraffin.
Four-micrometer-thick paraffin sections were created, and
deparaffinized sections were used in immunohistochemical staining.
Immunohistochemical staining was performed with a Vectastain ABC
kit (Vector Laboratories, Burlingame, CA, USA) as per
manufacturer's protocol. As primary antibodies, mouse monoclonal
anti-TGF-β1,2,3 (diluted 1:10), mouse monoclonal anti-human RANKL
(diluted 1:50), rabbit anti-phospho-Smad3 (diluted 1:500), and goat
anti-human COX-2 antibodies (diluted 1:500) were diluted with
blocking solution and reacted for 16 h at 4°C.
Measurements of concentrations of TGF-β1
and IL-1α
Concentrations of TGF-β1 and IL-1α in KCOT,
ameloblastoma, and follicular cyst intracellular fluid were
determined using a human TGF-β1 ELISA kit and IL-1α ELISA kit (both
from R&D Systems) while simultaneously measuring absorbance at
450 nm with a microplate reader (Model 680, Bio-Rad) in accordance
with the manufacturer's instructions. Experimental results are
presented as means ± standard deviation.
Measurement of concentration of
prostaglandin E2
Stromal fibroblasts were proliferated in a 24-well
culture plate (Corning) to confluency. After washing these stromal
fibroblasts twice with serum-free α-MEM culture medium containing
0.3% BSA, cells were cultured for 12 h in serum-free α-MEM culture
medium containing 0.3% BSA. The culture medium was then changed,
and cells were further incubated for 6 h with or without a sample
or reagent in serum-free α-MEM culture medium containing 0.3% BSA.
The culture supernatant was then collected and centrifuged at
13,400 × g at 4°C for 10 min. The resulting supernatant was then
collected and preserved at −80°C until used in experiments.
Concentrations of PGE2 in the culture supernatant were determined
using a PGE2 ELISA kit (Cayman Chemical Co.) while simultaneously
measuring absorbance at 420 nm with the microplate reader, in
accordance with the manual. Experimental results are expressed as
means ± standard deviation.
Statistical analyses
Statistical analyses were performed using Student's
t-test. P<0.05 was considered to be significant.
Results
Effects of KCOT fluid on RANKL and OPG
expression in KCOT stromal fibroblasts
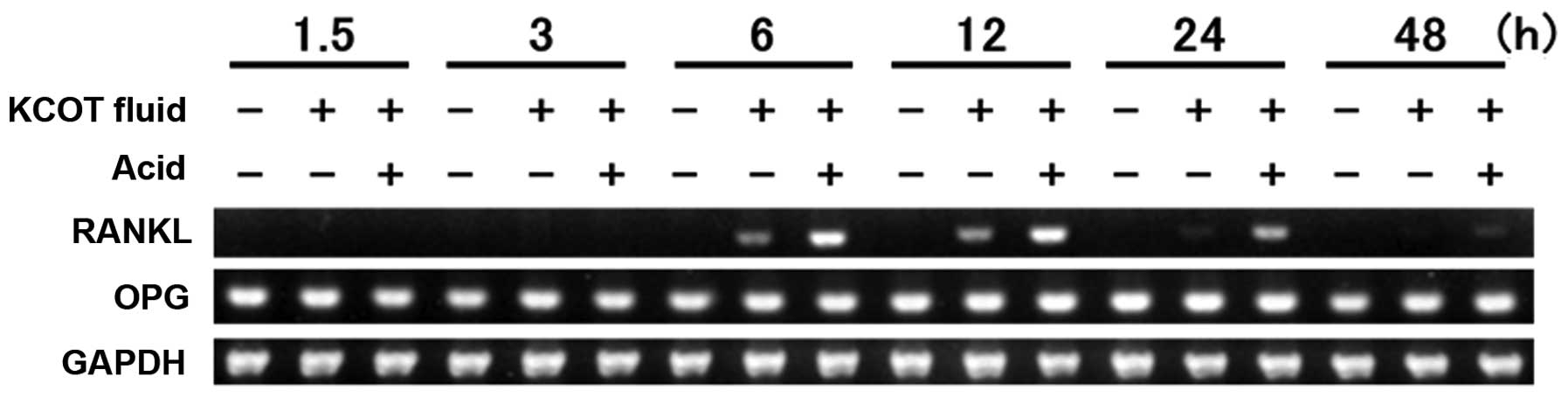
After KCOT stromal fibroblasts were cultured in the
presence of KCOT fluid, reverse transcription polymerase chain
reaction (RT-PCR) was used to examine RANKL and OPG expression. It
was found that RANKL expression was induced by KCOT fluid at 6 h
and the expression peaked at 12 h. At 24 h, RANKL expression
attenuated. There were no changes in OPG expression at any point in
time. Next, acid treatment was performed to determine the
physiochemical properties of intracystic fluid; it was found that
RANKL expression was enhanced at 6, 12, 24 and 48 h (Fig. 1).
Identical trends were observed in combinations of
stromal fibroblasts and ameloblastoma fluid in three cases of
ameloblastoma, stromal fibroblasts and follicular cyst fluid in two
cases of follicular cysts, and stromal fibroblasts and KCOT fluid
in three other cases of KCOT (data not shown). To ensure
reproducibility in subsequent experiments, a large volume of
intracystic fluid sample was collected, and the combination of KCOT
stromal fibroblasts and KCOT fluid was chosen because of strong
RANKL expression.
Effects of TGF-β1 and IL-1α on RANKL and
OPG expression in KCOT stromal fibroblasts
The enhancement of physiological activity resulting
from acid treatment indicated the possible involvement of key
cytokines, such as TGF-β, in this process, and we subsequently
addressed this point in detail. We also examined the effect of
IL-1α, another important factor present in odontogenic cysts that
is known to induce RANKL expression.
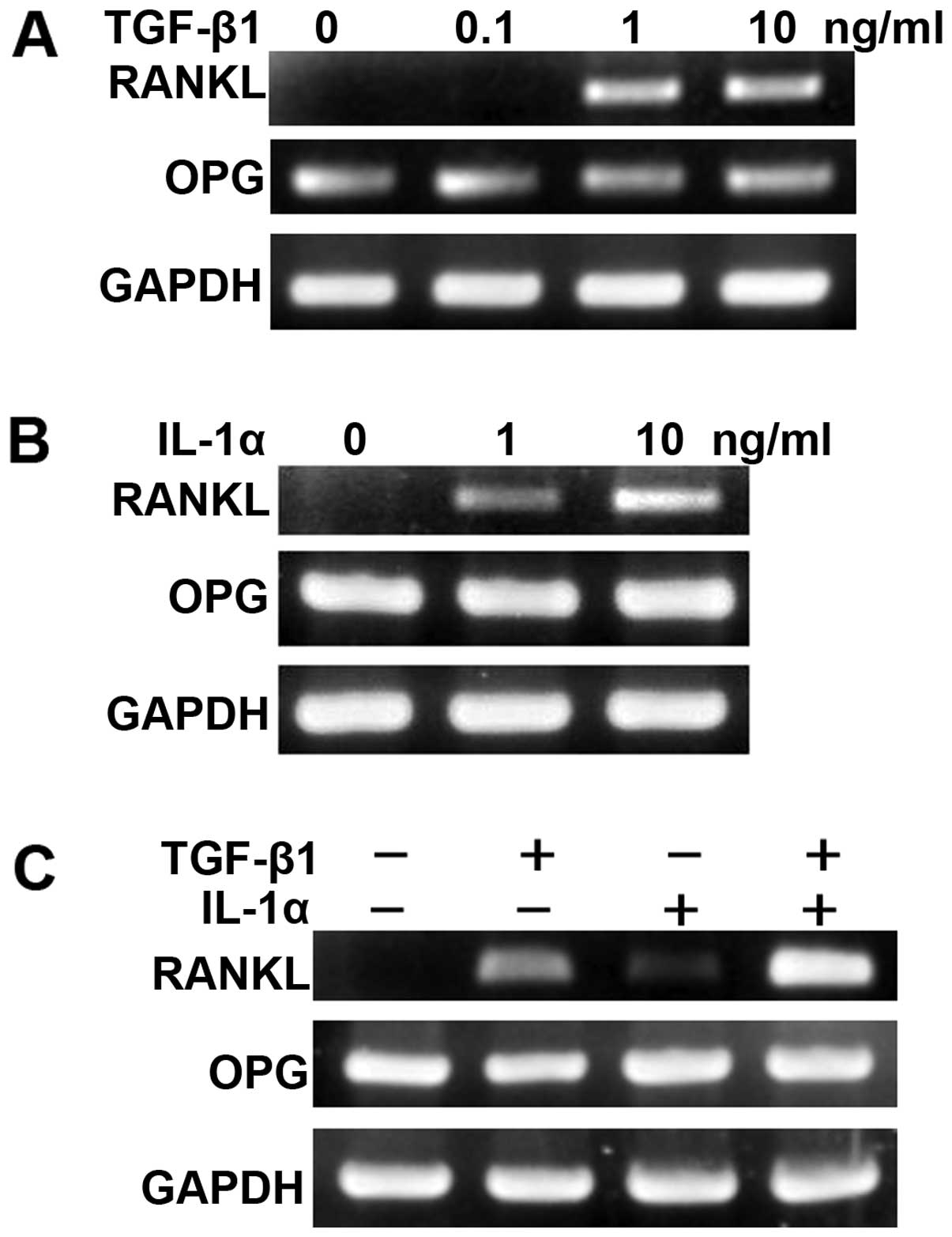
After KCOT stromal fibroblasts were cultured in the
presence or absence of TGF-β or IL-1α for 12 h, RT-PCR was used to
examine RANKL and OPG expression. It was thus found that both TGF-β
and IL-1α induced RANKL expression in a dose-dependent manner
(Fig. 2A and B). In addition, when
KCOT stromal fibroblasts were cultured in the combined presence of
TGF-β1 and IL-1α for 12 h, induction of RANKL expression was
further enhanced. OPG expression was not affected by culturing with
TGF-β alone, IL-1α alone, or a combination of the two (Fig. 2C).
Involvement of TGF-β and IL-1 receptor
signals in KCOT fluid-induced RANKL expression
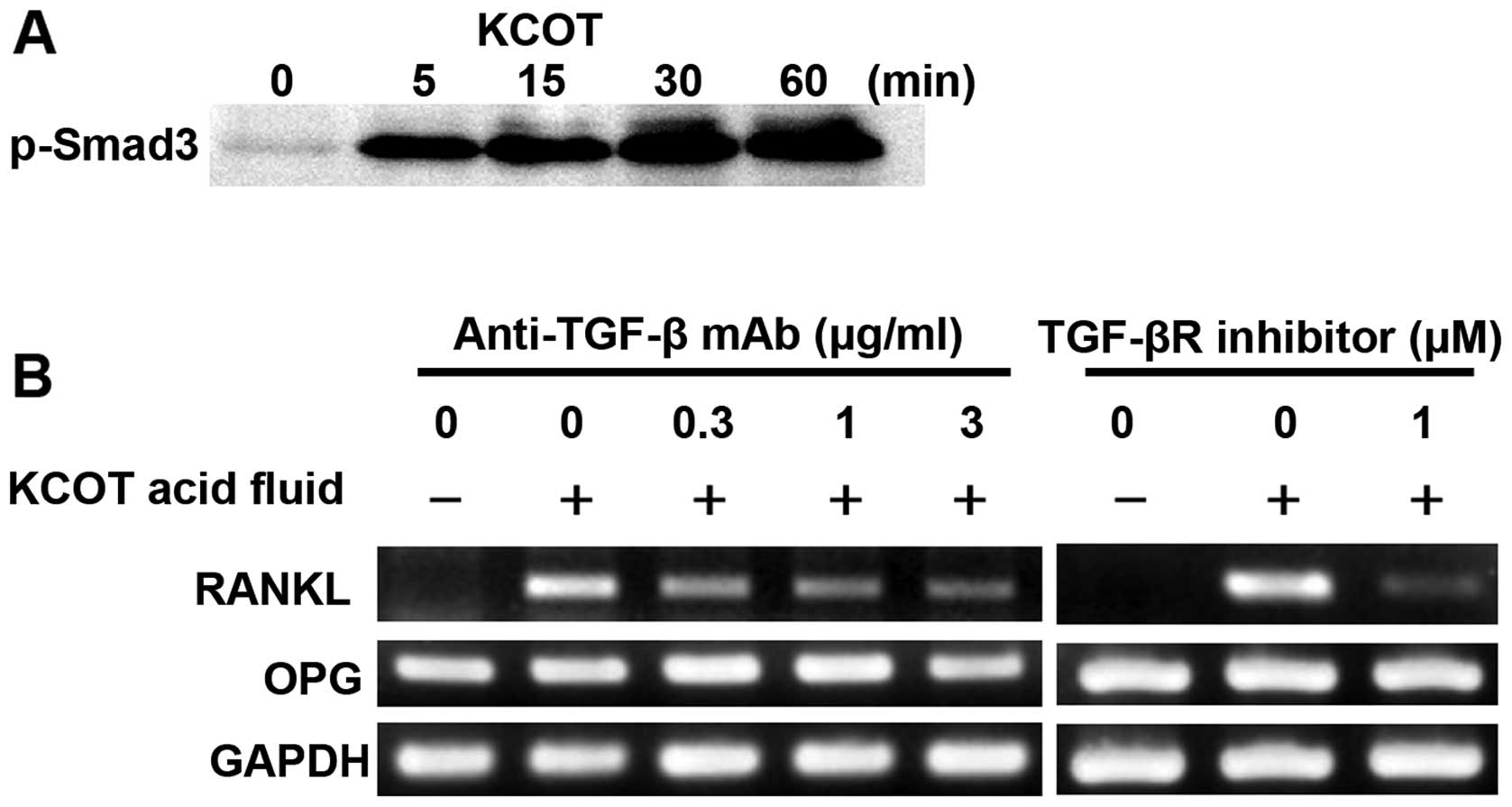
To investigate whether TGF-β is substantially
present in KCOT fluid, we examined Smad3 phosphorylation using
western blotting. Consequently, Smad3 phosphorylation was observed
5 min after KCOT fluid stimulation; this phosphorylation was
detected at 60 min as well (Fig.
3A). This result supports that TGF-β in KCOT fluid activated
TGF-β receptor signals in KCOT stromal fibroblasts. To investigate
whether TGF-β in KCOT fluid mediates RANKL expression in KCOT
fibroblasts, RT-PCR was used to examine RANKL and OPG expression in
KCOT stromal fibroblasts pretreated with anti-TGF-β antibody and
cultured with KCOT fluid for 12 h. It was found that KCOT
fluid-induced RANKL expression was inhibited by anti-TGF-β antibody
in a concentration-dependent manner (Fig. 3B). In addition, when KCOT stromal
fibroblasts were pretreated with TGF-β receptor inhibitor and
cultured for 12 h in the presence of KCOT fluid, RANKL expression
was partially inhibited by TGF-β receptor inhibitor (Fig. 3B). Incomplete blockade of
anti-TGF-β neutralizing antibody and TGF-β receptor inhibitor on
KCOT fluid-induced RANKL expression suggests that another factor in
intracystic fluid regulates RANKL expression; we therefore
investigated the involvement of IL-1α.
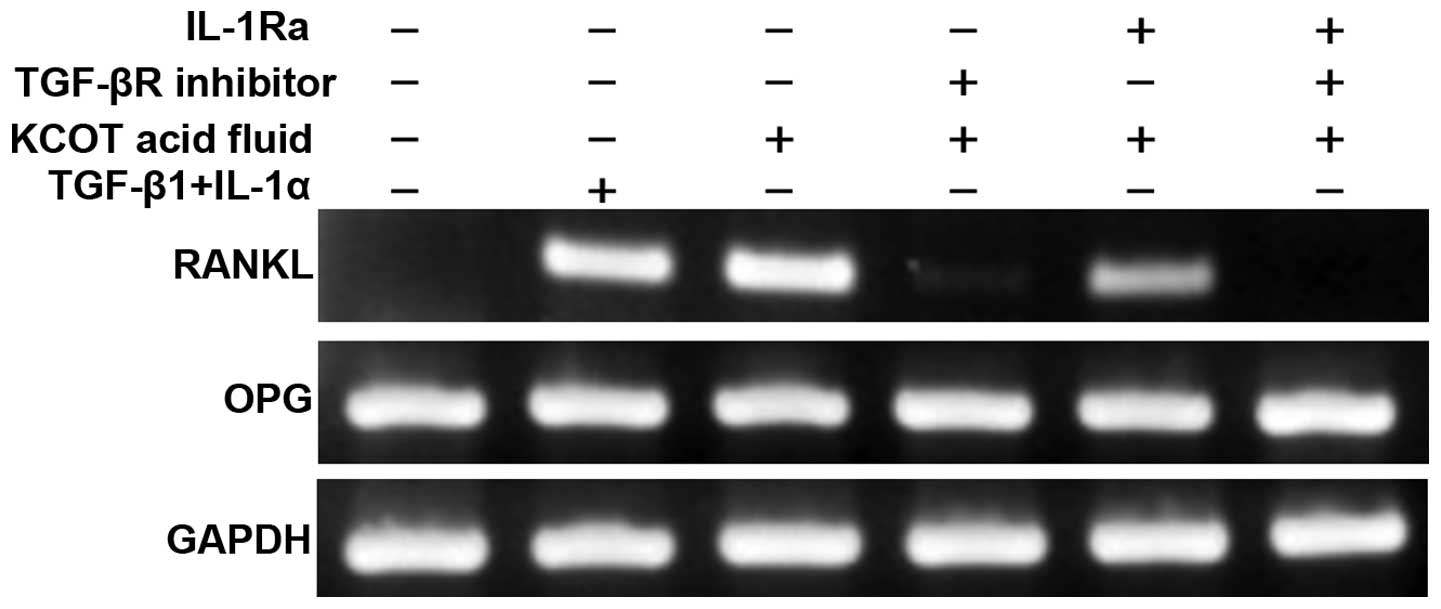
Following pretreatment with the TGF-β receptor
inhibitor, IL-1 receptor antagonist, or a combination of both, KCOT
stromal fibroblasts were cultured in the presence of KCOT fluid for
12 h. RT-PCR was then used to examine RANKL and OPG expression.
It was found that induction of RANKL expression when
stromal fibroblasts were cultured in the presence of TGF-β and
IL-1α was roughly equal to that when they were cultured with KCOT
fluid. RANKL expression induced by KCOT fluid was partially
inhibited by the IL-1α receptor antagonist, strongly inhibited by
TGF-β receptor inhibitor, and completely inhibited by a combination
of these two reagents (Fig. 4).
These results suggest that TGF-β and IL-1α in KCOT fluid cooperate
to induce RANKL expression in stromal fibroblasts.
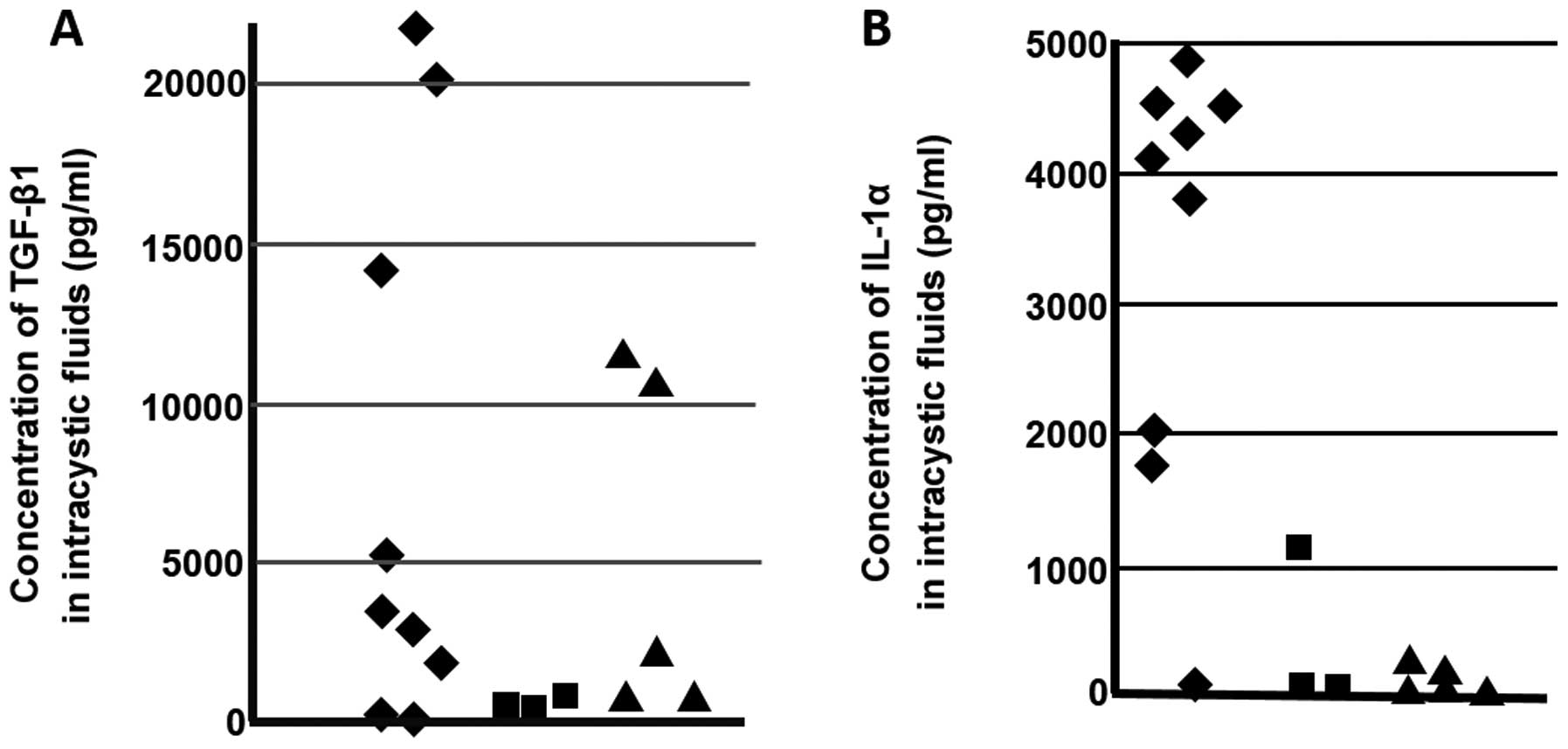
Measurements of concentrations of TGF-β1
and IL-1α in odontogenic tumor and cyst fluid
Concentrations of TGF-β1 and IL-1α in the
odontogenic tumor and cyst fluid were measured using ELISA. The
mean concentration of TGF-β1 in KCOT fluid in 10 cases was
7820.6±8625.2 pg/ml, which was markedly high, whereas that of IL-1α
was also markedly high at 3410.8±1592.8 pg/ml. The mean
concentrations of TGF-β1 and IL-1α in intracystic fluid in three
cases of ameloblastoma were 606.8±264.5 and 387.3±654.0 pg/ml,
respectively. Mean concentrations of TGF-β1 and IL-1α in
intracystic fluid in the five cases of follicular cyst were
5142.2±5338.1 and 73.5±102.7 pg/ml, respectively; both of these
concentrations were relatively low compared with those of KCOT
(Fig. 5).
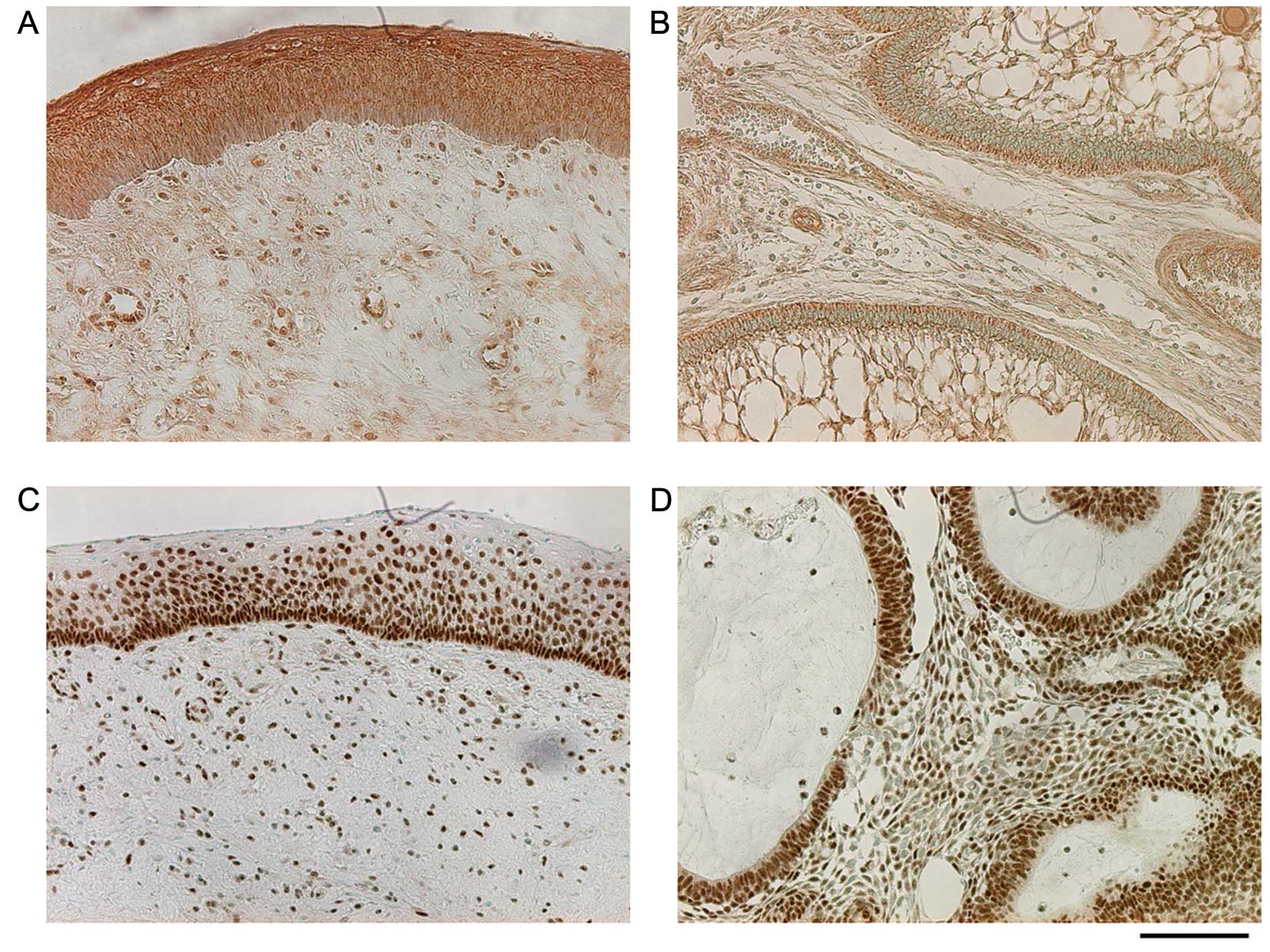
Investigation of localization of TGF-β,
phosphorylated Smad3, RANKL, and COX-2 in KCOT and ameloblastoma
tissues
Immunohistochemical staining was performed to
examine whether induction of RANKL expression in stromal
fibroblasts actually occurs within lesions via our model. Staining
for TGF-β in the KCOT tissue was strongly positive in the cyst
epithelium, positive in lymphocytes that had infiltrated the
interstitial tissue, positive in endothelial cells, and weakly
positive in stromal fibroblasts. Staining for TGF-β in
ameloblastoma was positive in tall columnar epithelial cells,
weakly positive in asteroid cells, positive in endothelial cells,
and weakly positive in stromal fibroblasts. Staining for
phosphorylated Smad3 in KCOT was strongly positive in all layers of
the cyst epithelium, particularly in basal cell nuclei, and was
positive in stromal fibroblast nuclei. Staining for phosphorylated
Smad3 in ameloblastoma was positive in tall columnar epithelial
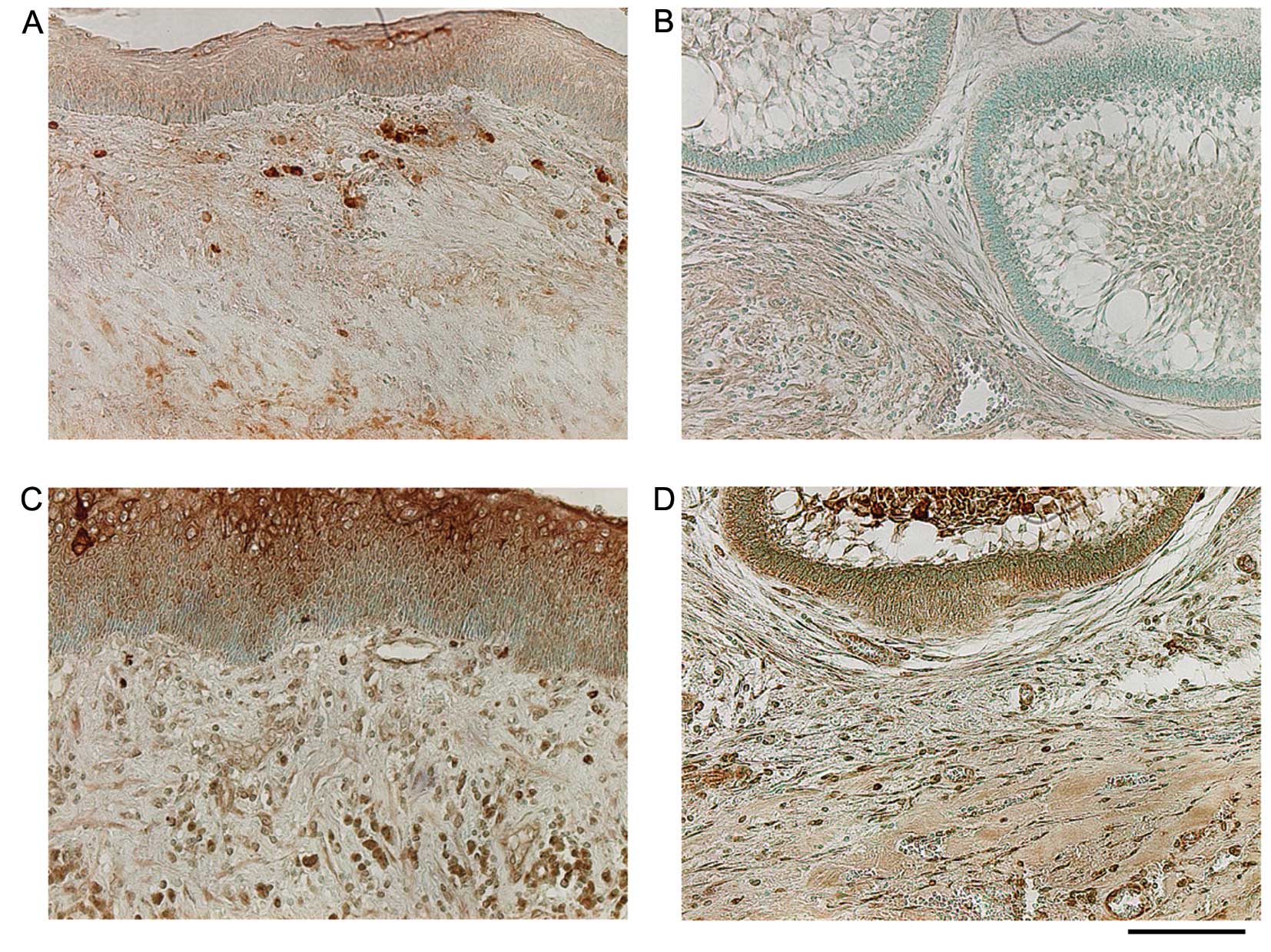
cells, endothelial cells, and stromal fibroblasts (Fig. 6). Staining for RANKL in the KCOT
tissue was strongly positive in infiltrative lymphocytes, positive
in the cyst epithelium, and positive in stromal fibroblasts.
Staining for RANKL in ameloblastoma was weakly positive in
epithelial cells and stromal fibroblasts. Staining for COX-2 in the
KCOT tissue was strongly positive in the cornified layer of the
cyst epithelium and positive in stromal fibroblasts, infiltrative
lymphocytes, and endothelial cells. Staining for COX-2 in
ameloblastoma was positive in epithelial cells, stromal
fibroblasts, and endothelial cells (Fig. 7).
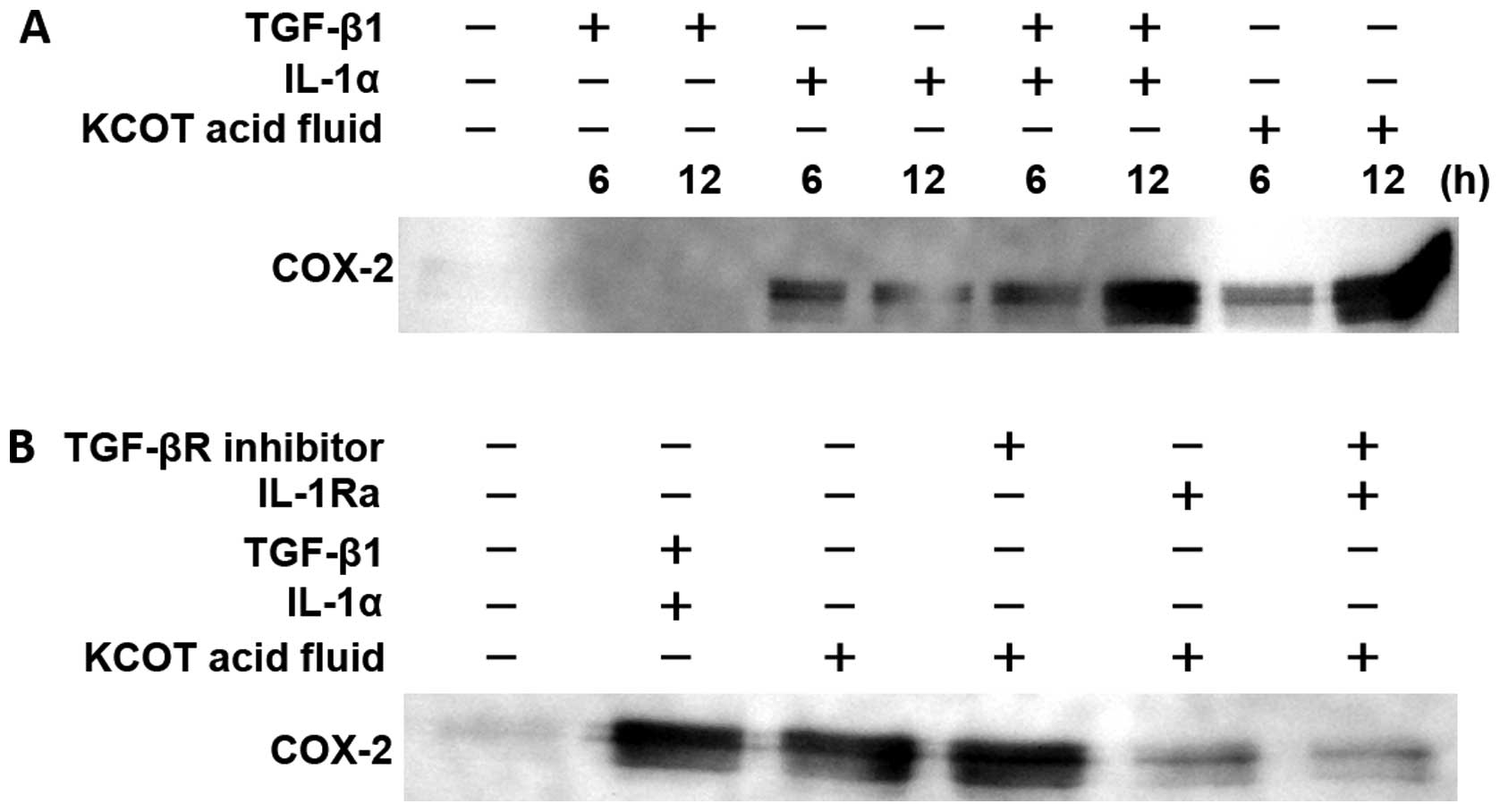
Mechanisms of COX-2 expression, PGE2
synthesis, and RANKL expression in KCOT stromal fibroblasts
Both TGF-β and IL-1α in intracystic fluid were shown
to induce RANKL expression in stromal fibroblasts, whereas a
combination of TGF-β and IL-1α was shown to strongly induce RANKL
expression. The mechanism by which IL-1α induces RANKL expression
is mediated by COX-2 and PGE2 synthesis (20,21).
Therefore, we investigated whether TGF-β is also involved in these
processes and whether this PGE synthesis promotes the induction of
RANKL expression in stromal fibroblasts.
We examined the concentration of COX-2 protein
resulting from culturing stromal fibroblasts for 6 and 12 h in the
presence of KCOT fluid, TGF-β1, IL-1α, and a combination of TGF-β1
and IL-1α. It was found that although TGF-β1 did not increase COX-2
protein expression at 6 or 12 h, IL-1α did increase its expression.
In addition, the combination of TGF-β1 and IL-1α synergistically
increased COX-2 protein expression. Furthermore, KCOT fluid yielded
an increase in COX-2 protein expression roughly equivalent to that
yielded by a combination of TGF-β1 and IL-1α (Fig. 8A).
Next, we cultured KCOT stromal fibroblasts for 12 h
in the presence of KCOT fluid following pretreatment with the TGF-β
receptor inhibitor, IL-1 receptor antagonist, or a combination of
both and then examined subsequent COX-2 expression. It was found
that KCOT fluid-induced COX-2 protein expression was not inhibited
by TGF-β receptor inhibitor alone, but was nearly completely
inhibited by the IL-1 receptor antagonist. The combination of TGF-β
receptor inhibitor and IL-1α receptor antagonist did not inhibit
COX-2 protein expression more than that by the IL-1α receptor
antagonist alone (Fig. 8B).
After culturing stromal fibroblasts for 6 h in the
presence of KCOT fluid, TGF-β1, IL-1α, and a combination of TGF-β1
and IL-1α, we measured the concentration of PGE2 in the resulting
supernatant. The mean concentration of PGE2 in untreated culture
medium was 313.0±142.38 pg/ml. The mean concentrations of PGE2 in
culture in the presence of TGF-β1, IL-1α, both TGF-β1 and IL-1α,
KCOT fluid, ameloblastoma fluid, and follicular cyst fluid were
403.3±83.28, 2160.4±65.04, 16046.2±264.87, 10970.8±405.21,
11683.2±602.28 and 2393.7±43.71 pg/ml, respectively (~1.3, 6.9,
51.3, 35, 37.3 and 7.6 times, respectively, higher than that
yielded by untreated culture medium). While PGE2 concentrations in
cultured samples of other intracystic fluids demonstrated widely
varying increases from 1.4 to 537.6 times, these fluids displayed
greatly increased concentrations of PGE2 (data not shown). In a
mechanism consistent with the changes in COX-2 protein shown in
Fig. 8A, TGF-β1 did not promote
PGE2 synthesis, whereas IL-1α promoted PGE2 synthesis, and a
combination of the two promoted PGE2 synthesis synergistically. In
addition, PGE2 synthesis was strongly promoted by jawbone tumor and
cyst fluids (Fig. 9). Finally, we
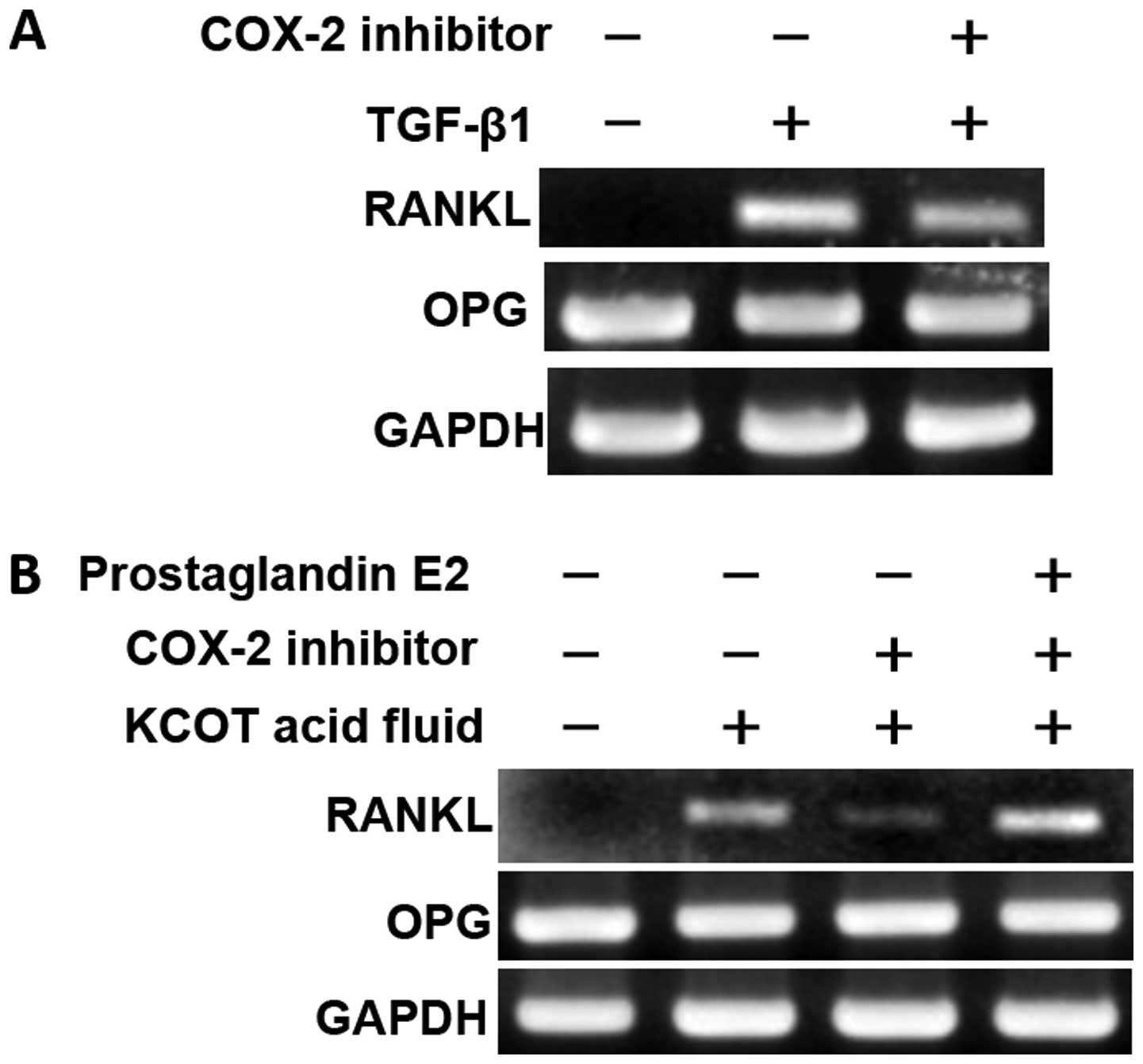
examined COX-2-mediated RANKL expression in stromal fibroblasts.
Following pretreatment with a selective COX-2 inhibitor, KCOT
stromal fibroblasts were cultured for 12 h in the presence of
TGF-β1 or KCOT fluid; RT-PCR was then used to examine RANKL
expression. It was found that RANKL expression induced by TGF-β1
was only weakly inhibited by the selective COX-2 inhibitor
(Fig. 10A). Moreover, RANKL
expression induced by KCOT fluid was strongly inhibited by the
selective COX-2 inhibitor (Fig.
10B). In addition, recovery from this inhibition was achieved
by culture in the presence of PGE2, a downstream target of COX-2
(Fig. 10B).
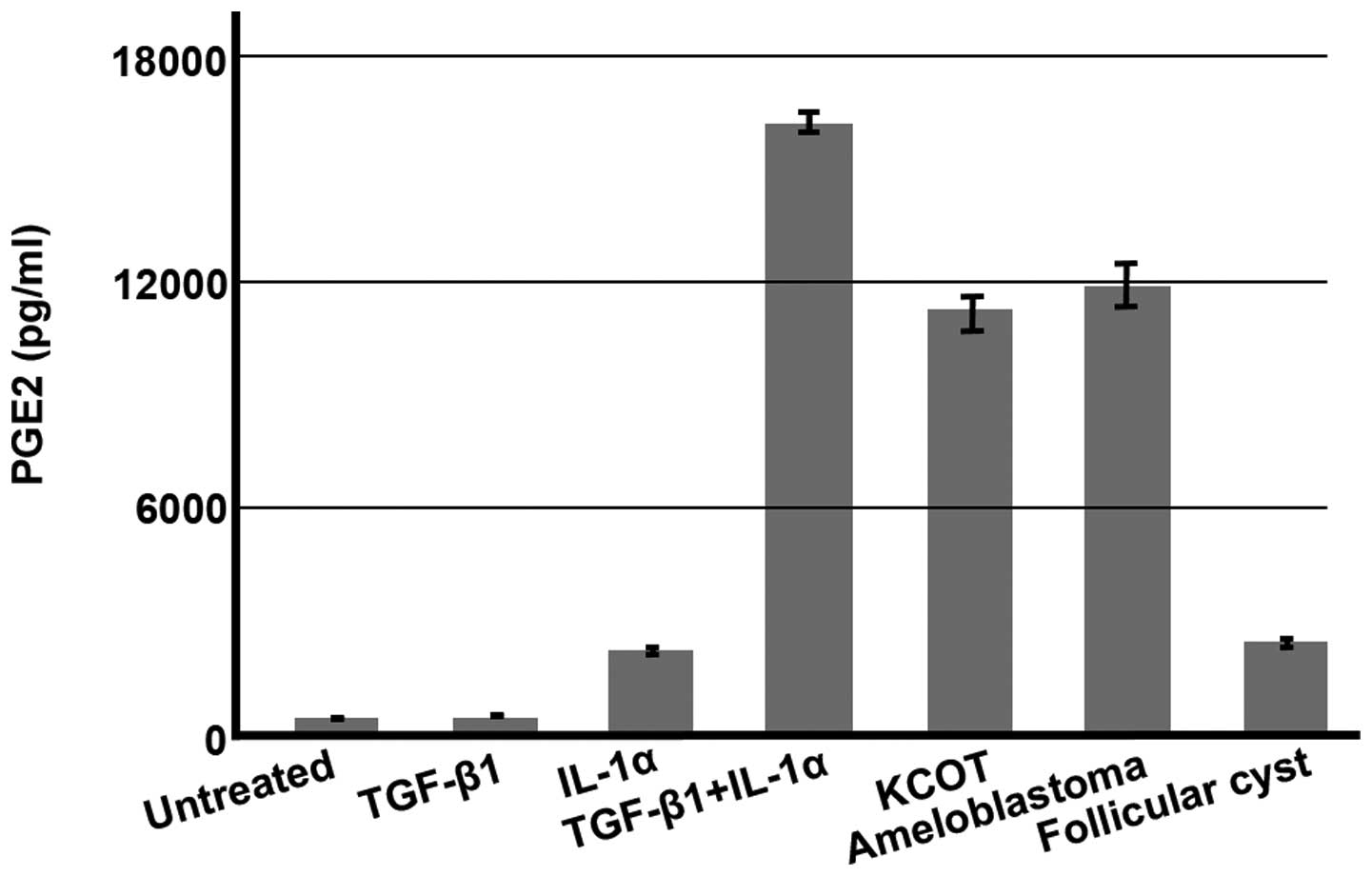 | Figure 9Concentrations of PGE2 in KCOT
stromal fibroblast culture supernatant. Stromal fibroblasts were
cultured for 6 h in the presence of TGF-β (1 ng/ml), IL-1α (1
ng/ml), and intracystic fluids; the concentration of PGE2 in the
resulting culture supernatant was then measured. Representative
results are shown. The mean concentrations of PGE2 in culture
medium were 313.0±142.38 (untreated), 403.3±83.28 (TGF-β1-treated),
2,160.4±65.04 (IL-1α-treated), 16,046.2±264.87 (TGF-β1 + IL-1α),
10,970.8±405.21 (1% KCOT-treated), 1,1683.2±602.28 (2%
ameloblastoma fluid-treated), and 2,393.7±43.71 pg/ml (1%
follicular cyst fluid-treated). |
Discussion
Recent research has suggested that RANKL is involved
in all bone resorption diseases regardless of inflammation or tumor
induction (1). However, although
cyst epithelial cell proliferation (16,17)
and resorption resulting from intracystic fluid-induced compression
(18,19) have long been proposed as mechanisms
for the enlargement of maxillary tumors and cysts and have garnered
widespread acceptance, no action has been taken on these proposals.
Therefore, we determined whether the cytokines and stromal
fibroblasts produced by odontogenic tumors and cysts are involved
in osteoclast formation and to determine the molecular mechanism
underlying this involvement. To address these points, we focused on
the role of TGF-β and IL-1α, and we analyzed the mechanism
underlying osteoclast formation. Direct isolation of cytokines
produced by odontogenic epithelial cells is technically difficult,
and cytokines produced extracellularly were thought to be present
in intracystic fluid. Therefore, by the stimulation of intracyctic
cytokines, we investigated the regulation of RANKL expression, an
osteoclast formation-inducing factor, in stromal fibroblasts.
In this study, KCOT fluid induced RANKL expression
in stromal fibroblasts. This effect was enhanced on treating KCOT
fluid with an acid (Fig. 1).
Because TGF-β is known to be activated by acid treatment, we
believed that TGF-β was present in KCOT fluid and was involved in
RANKL expression. TGF-β1 induced RANKL expression in KCOT stromal
fibroblasts in a concentration-dependent manner (Fig. 2A). RANKL expression was induced in
a time-dependent manner from 3 to 12 h of culture and was
attenuated beyond 24 h (data not shown). In TGF-β signal
transduction, Smad2/3 is characteristically phosphorylated
(9). Although KCOT fluid
stimulation resulted in phosphorylation of Smad3 in KCOT stromal
fibroblasts (Fig. 3A),
phosphorylation of Smad2 was not detected with TGF-β stimulation
(data not shown). Thus, the results in KCOT stromal fibroblasts
suggested that TGF-β1 activates Smad3 and exerts a biochemical
effect. Treatment of KCOT fluid and KCOT stromal fibroblasts with
anti-TGF-β antibody and TGF-β receptor inhibitor, respectively,
inhibited the induction of RANKL expression (Fig. 3B). These results suggested that
TGF-β is present in KCOT fluid and that TGF-β receptors activate
Smad3 and induce RANKL expression in KCOT stromal fibroblasts.
The effects of TGF-β in relation to the regulation
of RANKL and OPG expression in osteoblasts, as reported in previous
studies, differ from the results obtained in stromal fibroblasts in
this study as well as that obtained in a report on the effects of
TGF-β in tooth germ fibroblasts (11). In particular, these past reports
stated that TGF-β inhibited RANKL expression and promoted OPG
expression (22,23). However, TGF-β increases RANKL
expression in human osteoblasts isolated from clinical samples
(24). Thus, it is possible that
the mechanism by which TGF-β regulates RANKL expression differs
according to the type of cell and stage of cell differentiation. It
is also possible that RANKL expression in odontogenic tumor and
cyst stromal and tooth germ fibroblasts (11) is promoted by TGF-β.
KCOT fluid-induced RANKL expression was not
completely inhibited by TGF-β antibody or TGF-β receptor inhibitor
(Fig. 3B), but was completely
inhibited by the IL-1 receptor antagonist (Fig. 4). This finding demonstrated that
TGF-β1 and IL-1α in intracystic fluid induce RANKL expression in
stromal fibroblasts. IL-1α promotes RANKL expression in KCOT
stromal fibroblasts (20). In this
study, IL-1α consistently induced RANKL expression in KCOT stromal
fibroblasts (Fig. 2B), a result
which is consistent with that observed in previous studies.
Both TGF-β1 and IL-1α were present in KCOT fluid in
high concentrations. In addition, the concentrations of TGF-β1 and
IL-1α in ameloblastoma fluid, though lower than that in KCOT fluid,
were nonetheless high. The concentration of TGF-β1 in follicular
cyst fluid, though lower than that in KCOT fluid, was still high,
and the concentration of IL-1α was low. The concentration of IL-1α
in KCOT fluid is high, whereas the concentration of IL-6 in
ameloblastoma fluid is high and shows disease specificity (25). In the results of this study as well
KCOT fluid showed high concentrations of both TGF-β1 and IL-1α;
this characteristic distinguishes KCOT from follicular cysts in
which concentrations of IL-1α are low and suggests that TGF-β1 and
IL-1α are disease specific and concentration of IL-1α could be used
as a clinical marker to distinguish between these two lesions.
In immunohistochemical staining, KCOT and
ameloblastoma epithelial cells were positive for TGF-β, which is
consistent with the results of previous studies (26,27).
Furthermore, immunohistochemical results for TGF-β and
phosphorylated Smad3 demonstrated that TGF-β is produced by
odontogenic epithelial cells and Smad3 is activated in the
epithelial tissue and stromal fibroblasts, thus triggering
intracellular signal transduction. In addition, RANKL expression
was confirmed in the KCOT and ameloblastoma interstitial tissue
(Fig. 7A and B). Epithelial cells
were also positive for RANKL. RANKL is expressed in vitro in
ameloblastoma-derived epithelial cells (28) and that ameloblastoma epithelial
cells are positive for RANKL staining (29,30);
however, the function of RANKL in ameloblastoma remains unknown and
thus must be further investigated.
PGE2, which is synthesized by COX in the arachidonic
acid cascade, is a biologically active substance that strongly
promotes RANKL expression (31).
In addition, COX exists both as COX-1 and COX-2. Although COX-1 is
constantly expressed in cells and involved in the production of
PGE2, which is necessary for physiological functions, COX-2 is
expressed only when induced by stimuli, such as cytokines and
growth factors, and is known to temporarily increase production of
tissue-specific PGE2 (32).
Therefore, regulation of inducible COX-2 expression is important
for synthesis of PGE2 by cellular stimulation. IL-1α promotes RANKL
expression via synthesis of COX-2 and PGE2 (20,21).
This study determined that TGF-β and IL-1α induce
RANKL expression in KCOT stromal fibroblasts. Based on this finding
and on the fact that a combination of the above two factors
enhanced induction of RANKL expression (Fig. 2C), we investigated the involvement
of COX-2 and PGE2 in TGF-β-induced RANKL expression. We determined
that although TGF-β1 did not promote synthesis of COX-2 protein or
PGE2 in stromal fibroblasts, it did synergistically promote
IL-1α-induced synthesis of COX-2 protein and PGE2 (Figs. 8 and 9). Also, although TGF-β1-induced RANKL
expression was only weakly inhibited by a selective COX-2
inhibitor, intracystic fluid-induced RANKL expression was strongly
inhibited by the selective COX-2 inhibitor (Fig. 10). IL-1α-induced RANKL expression,
which is mediated by COX-2/PGE2 synthesis, may have been inhibited
and thus TGF-β1-induced RANKL expression was observed, which is not
mediated by COX-2/PGE2 synthesis. The above findings demonstrate
that induction of RANKL expression in stromal fibroblasts occurs by
two pathways: a TGF-β pathway, which is not mediated by COX-2/PGE2
synthesis, and an IL-1α pathway. It was also demonstrated that
TGF-β signals synergistically promote IL-1α signal-induced
COX-2/PGE2 synthesis, thus promoting RANKL expression.
In addition, TGF-β and IL-1α act directly on
osteoclast precursors and promote osteoclast differentiation
(14,15). TGF-β promoted RANKL-dependent
osteoclast differentiation of human peripheral blood mononuclear
cells and murine macrophage-like cell line RAW264, which are
osteoclast precursors, and IL-1α also promoted osteoclast
differentiation of the murine macrophage-like cell line RAW264
(data not shown).
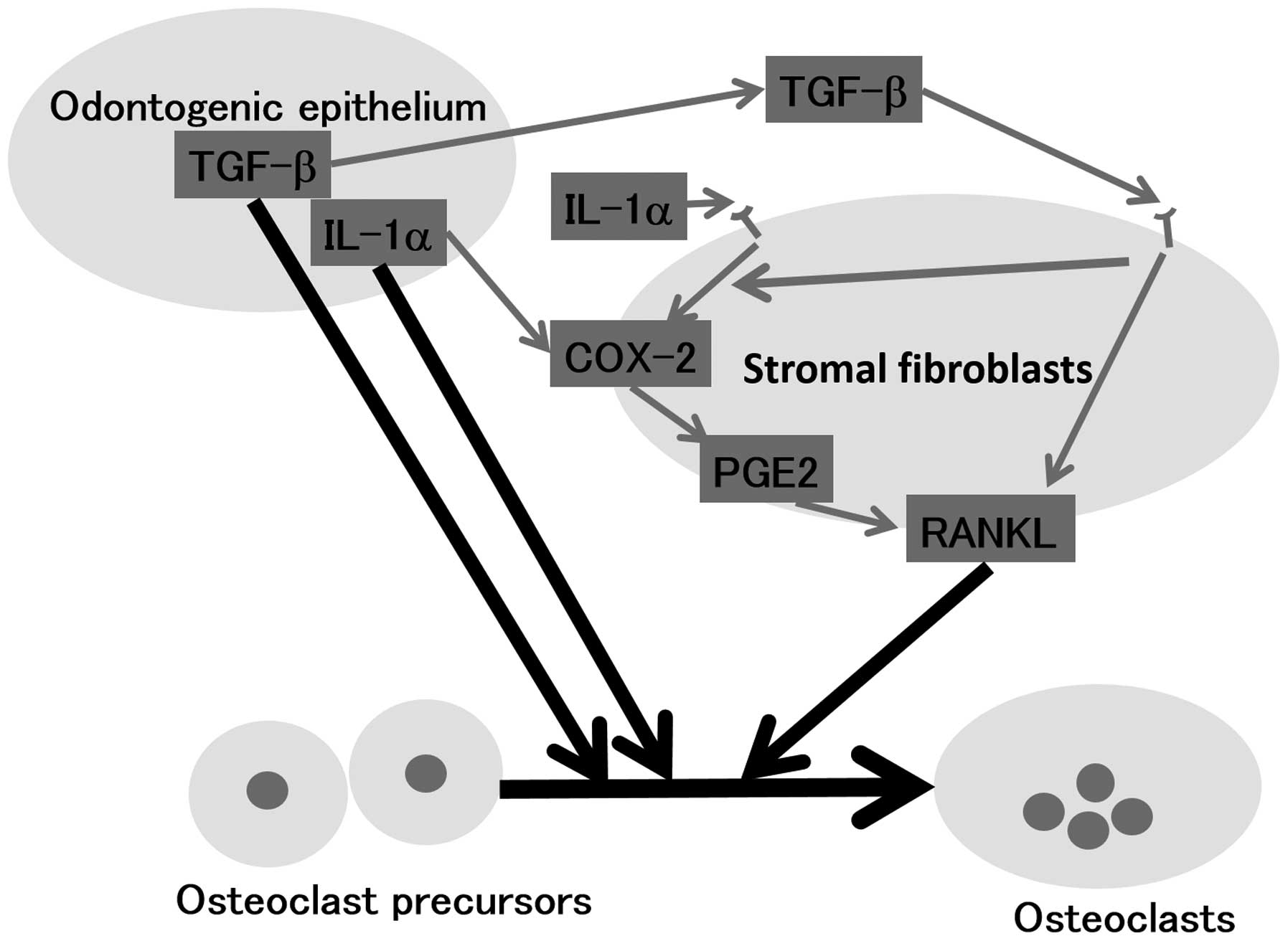
The experiments described above demonstrated that
TGF-β and IL-1α, cytokines which are produced by KCOT,
ameloblastoma, and follicular cyst epithelial cells, possess the
latent capacity to directly promote osteoclast formation from
osteoclast precursors and may be involved in jawbone resorption by
inducing RANKL expression in stromal fibroblasts and promoting
osteoclast formation (Fig. 11).
Previously reported mechanisms of growth of jawbone tumors, growth
of cysts in the jawbone, and bone resorption include proliferation
of cyst epithelial cells (16,17)
and intracystic fluid-induced compressive resorption (18,19).
The results of this study demonstrate the involvement of another
mechanism in which TGF-β1 and IL-1α produced by the jawbone tumor
and cyst epithelial cells induce RANKL expression in stromal
fibroblasts and promote osteoclast formation. We believe that these
findings will help in elucidating the mechanism underlying bone
resorption in primary lesions.
Acknowledgements
This study was supported by a Grant-in-Aid for
Scientific Research from Japan Society for the Promotion of Science
(no. 24592986 and no. 16K11682 to T.A., no. 21592523 to S.I. and
T.A., and no. 25893125 to C.Y.).
Abbreviations:
|
TGF-β
|
transforming growth factor-β
|
|
RANKL
|
receptor activator of NF-κB ligand
|
|
COX-2
|
cyclooxygenase-2
|
|
PGE2
|
prostaglandin E2
|
|
IL-1α
|
interleukin-1α
|
|
KCOT
|
keratocystic odontogenic tumors
|
|
OPG
|
osteoprotegerin
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
|
RT-PCR
|
reverse transcription polymerase chain
reaction
|
References
|
1
|
Kong YY, Feige U, Sarosi I, Bolon B,
Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, et al:
Activated T cells regulate bone loss and joint destruction in
adjuvant arthritis through osteoprotegerin ligand. Nature.
402:304–309. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakashima T and Takayanagi H: Osteoclasts
and the immune system. J Bone Miner Metab. 27:519–529. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takayanagi H, Oda H, Yamamoto S, Kawaguchi
H, Tanaka S, Nishikawa T and Koshihara Y: A new mechanism of bone
destruction in rheumatoid arthritis: Synovial fibroblasts induce
osteoclastogenesis. Biochem Biophys Res Commun. 240:279–286. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takayanagi H, Iizuka H, Juji T, Nakagawa
T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K and
Tanaka S: Involvement of receptor activator of nuclear factor
kappaB ligand/osteoclast differentiation factor in
osteoclastogenesis from synoviocytes in rheumatoid arthritis.
Arthritis Rheum. 43:259–269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagai M, Kyakumoto S and Sato N: Cancer
cells responsible for humoral hypercalcemia express mRNA encoding a
secreted form of ODF/TRANCE that induces osteoclast formation.
Biochem Biophys Res Commun. 269:532–536. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kawai T, Matsuyama T, Hosokawa Y, Makihira
S, Seki M, Karimbux NY, Goncalves RB, Valverde P, Dibart S, Li YP,
et al: B and T lymphocytes are the primary sources of RANKL in the
bone resorptive lesion of periodontal disease. Am J Pathol.
169:987–998. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Philbrick WM, Dreyer BE, Nakchbandi IA and
Karaplis AC: Parathyroid hormone-related protein is required for
tooth eruption. Proc Natl Acad Sci USA. 95:11846–11851. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kitahara Y, Suda N, Kuroda T, Beck F,
Hammond VE and Takano Y: Disturbed tooth development in parathyroid
hormone-related protein (PTHrP)-gene knockout mice. Bone. 30:48–56.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyazono K, ten Dijke P and Heldin CH:
TGF-beta signaling by Smad proteins. Adv Immunol. 75:115–157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Allan SM, Tyrrell PJ and Rothwell NJ:
Interleukin-1 and neuronal injury. Nat Rev Immunol. 5:629–640.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wise GE, Frazier-Bowers S and D'Souza RN:
Cellular, molecular, and genetic determinants of tooth eruption.
Crit Rev Oral Biol Med. 13:323–334. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao S, Ring S, Henk WG and Wise GE: In
vivo expression of RANKL in the rat dental follicle as determined
by laser capture microdissection. Arch Oral Biol. 49:451–456. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu D, Yao S, Pan F and Wise GE:
Chronology and regulation of gene expression of RANKL in the rat
dental follicle. Eur J Oral Sci. 113:404–409. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fox SW and Lovibond AC: Current insights
into the role of transforming growth factor-beta in bone
resorption. Mol Cell Endocrinol. 243:19–26. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim JH, Jin HM, Kim K, Song I, Youn BU,
Matsuo K and Kim N: The mechanism of osteoclast differentiation
induced by IL-1. J Immunol. 183:1862–1870. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thosaporn W, Iamaroon A, Pongsiriwet S and
Ng KH: A comparative study of epithelial cell proliferation between
the odontogenic keratocyst, orthokeratinized odontogenic cyst,
dentigerous cyst, and ameloblastoma. Oral Dis. 10:22–26. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
el Murtadi A, Grehan D, Toner M and
McCartan BE: Proliferating cell nuclear antigen staining in
syndrome and nonsyndrome odontogenic keratocysts. Oral Surg Oral
Med Oral Pathol Oral Radiol Endod. 81:217–220. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubota Y, Yamashiro T, Oka S, Ninomiya T,
Ogata S and Shirasuna K: Relation between size of odontogenic jaw
cysts and the pressure of fluid within. Br J Oral Maxillofac Surg.
42:391–395. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Marker P, Brøndum N, Clausen PP and
Bastian HL: Treatment of large odontogenic keratocysts by
decompression and later cystectomy: A long-term follow-up and a
histologic study of 23 cases. Oral Surg Oral Med Oral Pathol Oral
Radiol Endod. 82:122–131. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oka S, Kubota Y, Yamashiro T, Ogata S,
Ninomiya T, Ito S and Shirasuna K: Effects of positive pressure in
odontogenic keratocysts. J Dent Res. 84:913–918. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ogata S, Kubota Y, Yamashiro T, Takeuchi
H, Ninomiya T, Suyama Y and Shirasuna K: Signaling pathways
regulating IL-1alpha-induced COX-2 expression. J Dent Res.
86:186–191. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takai H, Kanematsu M, Yano K, Tsuda E,
Higashio K, Ikeda K, Watanabe K and Yamada Y: Transforming growth
factor-beta stimulates the production of
osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow
stromal cells. J Biol Chem. 273:27091–27096. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Quinn JM, Itoh K, Udagawa N, Hausler K,
Yasuda H, Shima N, Mizuno A, Higashio K, Takahashi N, Suda T, et
al: Transforming growth factor beta affects osteoclast
differentiation via direct and indirect actions. J Bone Miner Res.
16:1787–1794. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jurado S, Garcia-Giralt N, Díez-Pérez A,
Esbrit P, Yoskovitz G, Agueda L, Urreizti R, Pérez-Edo L, Saló G,
Mellibovsky L, et al: Effect of IL-1beta, PGE(2), and TGF-beta1 on
the expression of OPG and RANKL in normal and osteoporotic primary
human osteoblasts. J Cell Biochem. 110:304–310. 2010.PubMed/NCBI
|
|
25
|
Kubota Y, Nitta S, Oka S, Nakagawa S,
Ninomiya T and Shirasuna K: Discrimination of ameloblastomas from
odontogenic keratocysts by cytokine levels and gelatinase species
of the intracystic fluids. J Oral Pathol Med. 30:421–427. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piattelli A, Rubini C, Fioroni M, Favero L
and Strocchi R: Expression of transforming growth factor-beta 1
(TGF-beta 1) in odontogenic cysts. Int Endod J. 37:7–11. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kumamoto H, Yoshida M and Ooya K:
Immunohistochemical detection of hepatocyte growth factor,
transforming growth factor-beta and their receptors in epithelial
odontogenic tumors. J Oral Pathol Med. 31:539–548. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sandra F, Hendarmin L, Kukita T, Nakao Y,
Nakamura N and Nakamura S: Ameloblastoma induces
osteoclastogenesis: A possible role of ameloblastoma in expanding
in the bone. Oral Oncol. 41:637–644. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tay JY, Bay BH, Yeo JF, Harris M, Meghji S
and Dheen ST: Identification of RANKL in osteolytic lesions of the
facial skeleton. J Dent Res. 83:349–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
da Silva TA, Batista AC, Mendonça EF,
Leles CR, Fukada S and Cunha FQ: Comparative expression of RANK,
RANKL, and OPG in keratocystic odontogenic tumors, ameloblastomas,
and dentigerous cysts. Oral Surg Oral Med Oral Pathol Oral Radiol
Endod. 105:333–341. 2008. View Article : Google Scholar
|
|
31
|
Blackwell KA, Raisz LG and Pilbeam CC:
Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol Metab.
21:294–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vane J: Towards a better aspirin. Nature.
367:215–216. 1994. View Article : Google Scholar : PubMed/NCBI
|