Introduction
As the most malignant type of brain tumor, gliomas
account for >70% of all brain tumors (1), and the most common type of glioma is
glioblastoma (GBM) (2). The
properties of GBM are generally thought to include high mortality
and recurrence rates (3),
uncontrollable invasiveness (4),
potent angiogenesis (5), a
widespread hypoxic region (6,7), and
upregulated autophagic processes (8,9).
Hypoxia is a common feature in solid tumors due to rapid
progression and a relatively inadequate blood supply, and tumor
hypoxia is an independent prognostic factor associated with poor
survival (6,10,11).
Numerous studies have suggested that hypoxia activates multiple
cellular processes in tumors, such as proliferation (12), angiogenesis (7), migration (13), invasion (12), and recently, autophagy (8,14).
The term autophagy means ‘self-eating’. Autophagy is
a process that involves the formation of double-membrane
autophagosomes containing damaged or old organelles (15,16).
Autophagy provides tumor cells with reclaimed essential elements
for survival through starvation, hypoxia, immune response and
chemoradiotherapy (17). Because
tumor cells are over-consumptive, these cells develop ubiquitous
autophagy to overcome the relatively infertile tumor
micro-environment (18–20). In recent years, accumulating
evidence has demonstrated that autophagy favors tumor development
(21). Nevertheless, the detailed
mechanisms by which hypoxia induces autophagy remain unclear.
CREBRF (CREB3 regulatory factor) is a novel cellular
protein and a specific negative regulator of CREB3 that can recruit
nuclear CREB3 out of nucleus and promote CREB3 protein degradation
(22). CREB3 (also called Luman)
is the primary member of the CREB (cyclic-AMP-responsive element
binding) family (CREB1, CREB2 and CREB3) (23). CREB family proteins bind to the
cAMP-responsive element (CRE) recognition sequence of
cAMP-sensitive genes to regulate transcription (23,24).
Furthermore, all CREB3 family sub-members appear to play a role in
the unfolded protein response (UPR) (22). Although CREB1 can upregulate
autophagy genes (25,26) and CREBRF is involved in inducing
cell apoptosis through the ER stress pathway (27), there is no evidence that the
CREBRF/CREB3 pathway is directly involved in regulating autophagy
in tumor cells.
In this study, we report a novel autophagy-promoting
mechanism in hypoxic tumor cells in which hypoxia downregulated
CREBRF expression and promotes tumor cell autophagy by upregulating
CREB3 in GBM cells through the ATG5-dependent pathway. We first
investigated the key autophagic regulating effect of CREBRF/CREB3
during the hypoxia process. We demonstrate that hypoxic
pretreatment decreases CREBRF expression in glioblastoma cells and
significantly increases CREB3 levels. Importantly, knocking down
endogenous CREB3 alleviates hypoxia-induced autophagy. To
understand the mechanisms of the autophagy induced by CREB3, we
screened most autophagy-related genes (ATGs) using quantitative
real-time PCR. Finally, we provide evidence that ATG5 has a central
role in the autophagic inducing effect of CREB3. Our results
suggest potential uses for anti-CREB therapeutic strategies in
adjuvant therapy for glioma patients.
Materials and methods
Tissue samples and cell lines
The human glioma cell lines T98G and U87 were
purchased from the Chinese Academy of Sciences Cell Bank and
identified by the STR site detection assay (Microread Genetics Co.,
Beijing, China). In addition, 76 human glioma tissue samples
including 32 low-grade gliomas (4 grade I tumors and 28 grade II
tumors), 44 high-grade gliomas (16 grade III tumors and 28 grade IV
tumors), and 2 normal brain tissues from decompression operation
were obtained from the Department of Neurosurgery of Qilu Hospital
of Shandong University from October 2013 to June 2015. The glioma
specimens were verified and classified according to the WHO
classification standard of tumors by two experienced clinical
pathologists. Our study was approved by the Institutional Review
Board of Shandong University. Written informed consent was obtained
from all the patients, and the hospital ethics committee approved
the experiments.
Cell culture and hypoxia treatment
The cells were cultured in DMEM (Invitrogen)
supplemented with 10% FBS (Gibco) and maintained at 37°C with 5%
CO2 in a humidified chamber. Hypoxic conditions were
induced by incubating the cells in a modular incubator chamber
flushed with a gas mixture containing 1% O2, 5%
CO2, and 94% N2 at 37°C.
Immunohistochemical staining
Human glioma tissue samples or solid tumors removed
from sacrificed mice were fixed with 4% formaldehyde.
Paraffin-embedded tumor tissues were sectioned to 5-μm thickness
and mounted on positively charged microscope slides, and 1 mM EDTA
(pH 8.0) for HIF-1α or citrate solution (pH 6.0) for other antigens
was used for antigen retrieval. Endogenous peroxidase activity was
quenched by incubating the slides in methanol containing 3%
hydrogen peroxide, followed by washing in PBS for 6 min. The
sections were incubated for 2 h at room temperature with normal
goat serum and subsequently incubated at 4°C overnight with primary
antibodies (Abcam, 1:200 HIF-1α, 1:400 LC3B, and 1:300 CREB3. Cell
Signaling Technology, 1:300 cleaved caspase 3). The sections were
then rinsed with PBS and incubated with horseradish
peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies,
followed by reaction with diaminobenzidine and counterstaining with
Mayer's hematoxylin. H&E staining was performed free of charge
by the pathology department of Qilu Hospital of Shandong
University. Evaluation of the staining reaction was performed in
accordance with the immunoreactive score (IRS): IRS = SI (staining
intensity) x PP (percentage of positive cells). An SI value of 0
was negative; 1, weak; 2, moderate; and 3, strong. A PP value of 0
was negative; 1, 10% positive cells; 2, 11–50% positive cells; 3,
51–80% positive cells; and 4, >80% positive cells. Five visual
fields from different areas of each tumor were used for the IRS
evaluation.
GFP-LC3 stable cell lines and
quantitative GFP-LC3 analyses
A T98G GFP-LC3 stable cell line was established by
transient transfection of the pSELECT-GFP-LC3 lentiviral vector
(Invivogen, USA). GFP-LC3 puncta formation under normoxic or
hypoxic conditions was determined by capturing images using a DP71
CCD digital camera microscope (Olympus). To quantify autophagic
cells after treatment, we counted the number of autophagic cells as
determined by the presence of GFP-LC3 puncta (≥20 puncta = a
positive cell) in 200 cells.
Western blot analysis
After the desired treatment, cells were washed twice
with cold PBS and harvested with a rubber scraper. Cell pellets
were lysed and kept on ice for ≥30 min in a buffer containing 50 mM
Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 1 mM
Na3VO4, 1 mM phenylmethylsulfonyl fluoride,
and 1 mM PMSF. The lysates were cleared by centrifugation, and the
supernatants were collected. Cell lysates were then separated by
SDS-PAGE and subjected to western blot analysis with primary
antibodies and horseradish peroxidase-conjugated secondary
antibodies. Antibodies against the following were used for western
blotting: P62 (Cell Signaling Technology, 5114S), LC3B (Abcam,
63817), GAPDH (Cell Signaling Technology, 3683S), caspase 3 (Cell
Signaling Technology, 9668T), cleaved caspase 3 (Cell Signaling
Technology, 9664S), CREBRF (Santa Cruz Biotechnology, sc-133747),
CREB3 (Abcam, ab42454), and ATG5 (Abcam, ab108327). GAPDH served as
the loading control in all experiments involving hypoxic treatment
due to the upregulation of β-actin under hypoxia.
Small interfering RNA transfection
CREB3 and negative control siRNAs were synthesized
by Rio-Bio (China). The sequences of CREB3 siRNAs were as follows:
CREB3-1299, 5′-GCA GUC AGA AGU GCC GAA ATT-3′ (sense) and 5′-TTC
GUC AGU CUU CAC GGC UUU-3′ (antisense). The sequences of negative
control were as follows: 5′-UUC UCC GAA CGU GUC ACG UTT-3′ (sense)
and 5′-ACG UGA CAC GUU CGG AGA ATT-3′ (antisense). The siRNAs were
transfected into T98G and U87 cells for 48 h using Lipofectamine
2000 according to the protocol of the manufacturer.
Cell viability assay
The CCK-8 assay (Dojindo, Japan) was used to test
cell viability. Tumor cells in medium containing 10% fetal bovine
serum were seeded into 96-well, flat-bottomed plates at
5×103 cells/well and incubated at 37°C overnight. After
the desired treatment, the cells were incubated for an additional 4
h with 100 μl of serum-free DMEM and 10 μl of CCK-8 at 37°C. The
absorbance at 450 nm was measured using a microplate reader.
Annexin V-fluorescein isothiocyanate
(FITC) assay
To assess the degree of apoptosis of different
groups of glioma cells, the extent of Annexin V-FITC/propidium
iodide (PI) staining was determined by flow cytometry using the
Annexin V/PI staining kit from Bender MedSystems (Vienna, Austria).
Samples were measured using an Epics XL-MCL flow cytometer (Beckman
Coulter, Brea, CA, USA) and analyzed with WinMDI 2.8 software.
TUNEL assay
Glioma cells were plated on glass slides in 24-well
culture plates at 2×105 cells/well for 24 h and
subsequently treated with drugs for an additional 48 h in
serum-free DMEM. Glass slides with glioma cells and
paraffin-embedded tumor sections were stained by the TUNEL
technique using a TACS®2 TdT-Fluor in situ
apoptosis detection kit (Trevigen, Inc.) according to the
instructions of the manufacturer. TUNEL-positive cells were counted
from ≥10 random fields under a fluorescence microscope.
RNA extraction and real-time quantitative
PCR
Total RNA was extracted using TRIzol according to
the manufacturer's protocol. Then, total RNA (50 ng) was
reverse-transcribed with miR stem-loop RT primers or with U6 RT
primers using a ReverTra Ace qPCR RT kit according to the
manufacturer's protocol to generate cDNA. Real-time PCR was
performed using a SYBR Premix Ex Taq™ kit with each primer. The
reactions were performed using a LightCycler 2.0 Instrument. U6
expression was used as the endogenous control. The absolute
expression levels were calculated as concentration ratios using a
Roche LightCycler® 2.0 system.
Statistical analysis
Data analyses were conducted with SPSS 16.0 (SPSS,
IL, USA) and GraphPad-Prism5 (GraphPad, CA, USA). Descriptive
statistics including means ± SD, Student's t-test, non-parametric
Kruskal-Wallis tests for multiple comparison, the Mann-Whitney U
test for two-group comparison, Kaplan-Meier plots, log-rank tests,
one-way ANOVAs, and Pearson's correlation test were used to analyze
significant difference; *P<0.05,
**P<0.01, and ***P<0.001 were
considered statistically significant.
Results
CREBRF/CREB3 levels correlate with human
glioma grade
To analyze CREBRF/CREB3 expression in clinical
samples, total RNA was extracted from surgically removed glioma
tissues of 20 patients (2 normal brain tissue samples, 9 patients
with WHO I–II and 9 patients with WHO III–IV gliomas), and RT-PCR
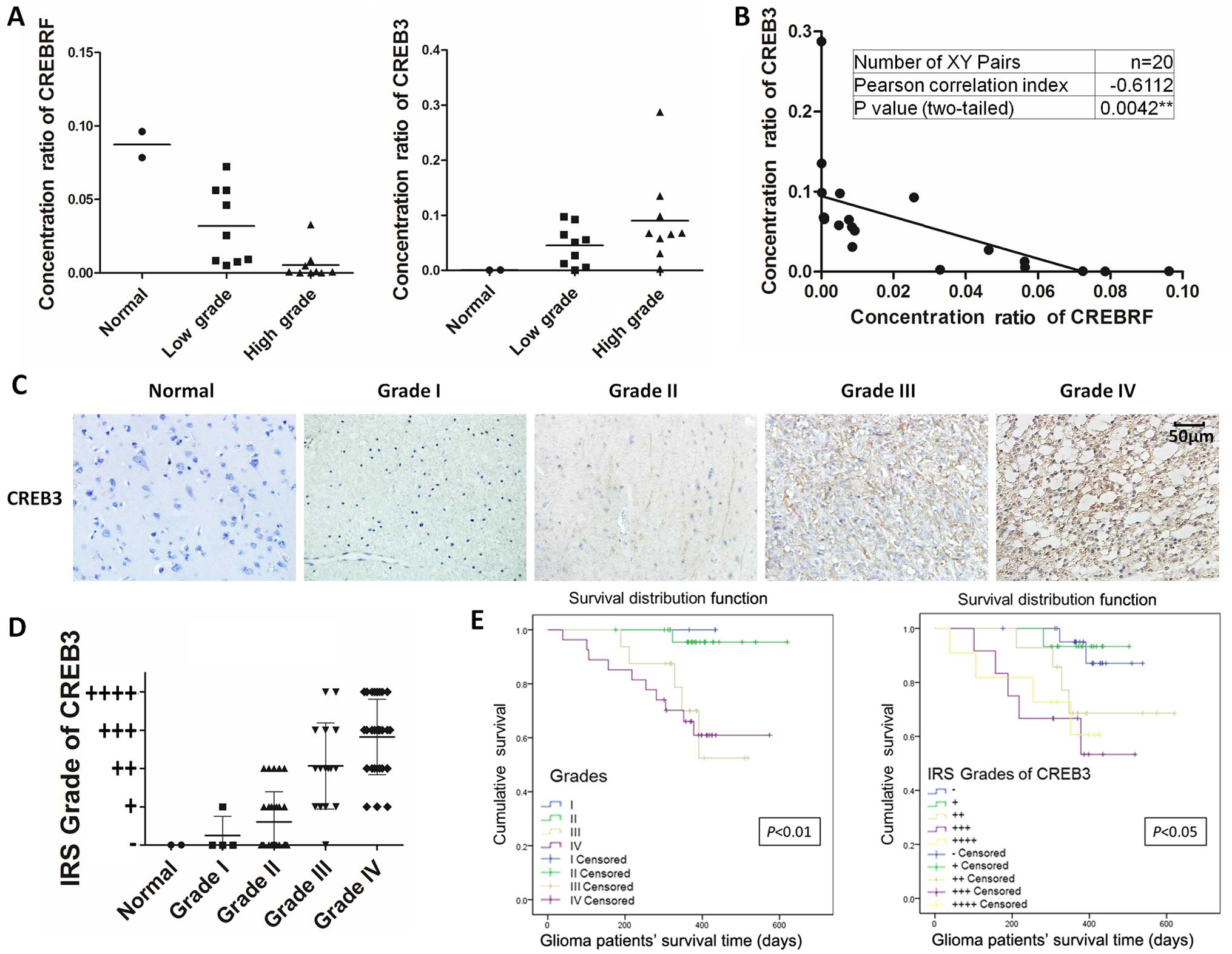
was performed. Interestingly, significant differences in CREBRF
expression were observed between low-grade (I–II) and high-grade
(III–IV) patients. CREBRF expression was significantly reduced in
high-grade glioma tissues compared with low-grade glioma tissues or
normal brain tissues (Fig. 1A,
left). Consistent with its negative regulating property, CREB3
expression was significantly reduced in low-grade glioma tissues
compared with high-grade glioma tissues (Fig. 1A, right) with a significant
negative correlation (Pearson correlation index, −0.6112;
P-value=0.0042) (Fig. 1B). To
further examine whether CREB3 expression correlates with the WHO
grades of human glioma, we performed immunohistochemical staining
to detect CREB3 expression in 76 human glioma specimens with
different grades and 2 normal brain tissues (Table I). As shown in Fig. 1C, CREB3 expression positively
correlated with the WHO grade of glioma (Fig. 1D). Furthermore, patients with
clinical survival information were analyzed by Kaplan-Meier
estimation. We found that glioma patients with high CREB3
expression exhibited significantly poor postoperative survival time
(Fig. 1E). The above findings
raised the intriguing possibility that CREBRF acts as a tumor
suppressor and downstream CREB3 acts as a prognostic biomarker of
glioma. The underlying mechanisms by which CREBRF suppresses the
malignant progression of glioma remain uncertain.
 | Table IDemographic parameters of patients
participating in the study. |
Table I
Demographic parameters of patients
participating in the study.
| No. of
patients | N % |
|---|
| Assessable |
| Glioma | 76 | 97.44 |
| Normal brain
tissues | 2 | 2.56 |
| Gender |
| Male | 48 | 61.54 |
| Female | 30 | 38.46 |
| Age (years) |
| Median
(range) | 46.41 (6–75) | |
| Pathological
type |
| Astrocytoma | 22 | 28.21 |
| Anaplastic
astrocytoma | 12 | 15.38 |
| Pilocytic
astrocytoma | 3 | 3.85 |
|
Oligodendroglioma | 7 | 8.97 |
| Anaplastic
oligodendroglioma | 4 | 5.13 |
| Glioblastoma | 28 | 35.90 |
| Normal brain
tissues | 2 | 2.57 |
| WHO tumor grade at
diagnosis |
| I | 4 | 5.26 |
| II | 28 | 36.84 |
| III | 16 | 21.05 |
| IV | 28 | 36.84 |
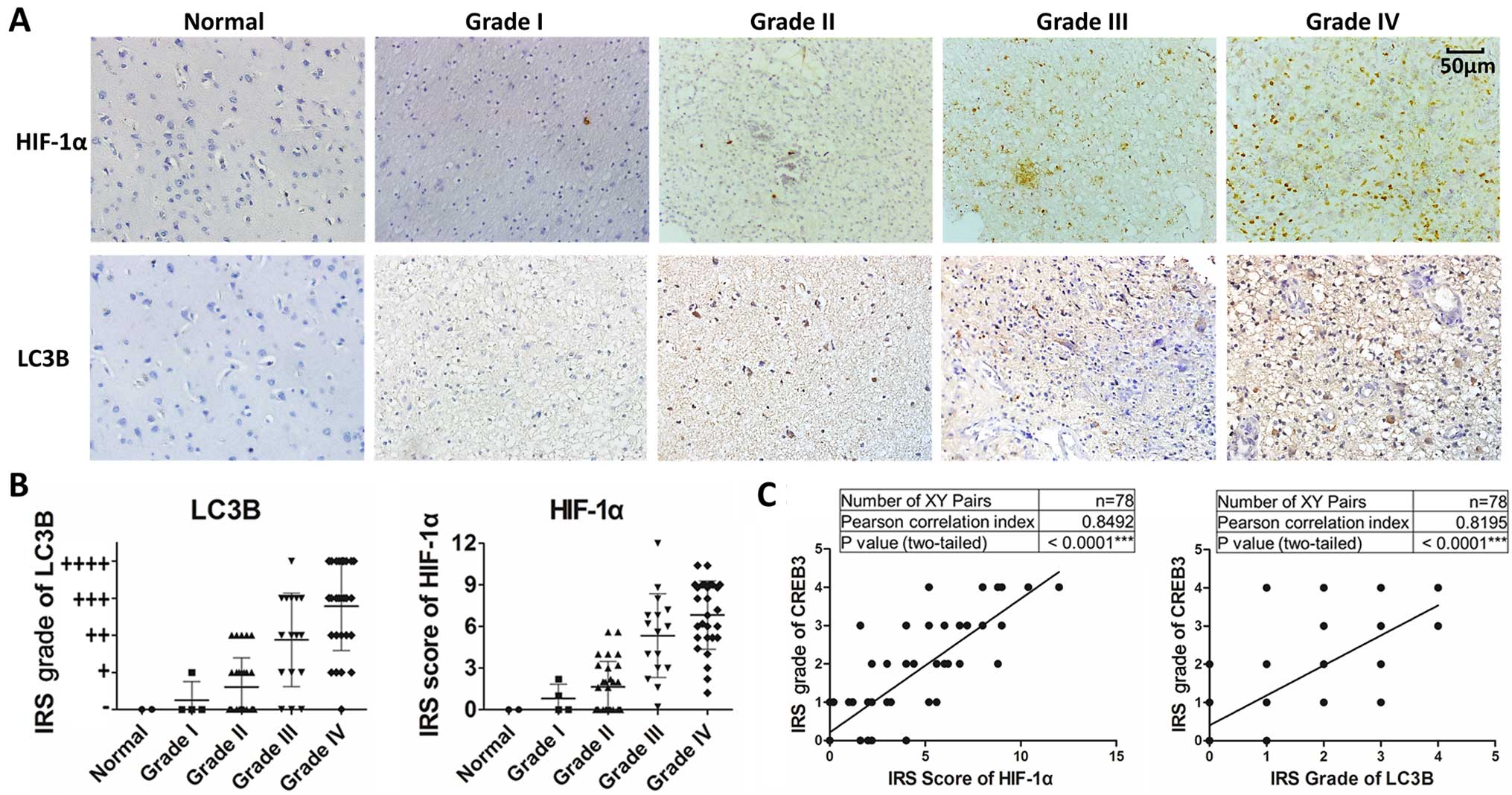
CREB3 levels correlate with the density
of autophagic cells and HIF-1α levels in human glioma tissues
Extensive autophagy in the hypoxic areas of tumors
has been described (14). However,
the association among CREB3 and LC3B levels and hypoxia levels in
gliomas has not been reported, and the CREB3-mediated upregulation
of autophagy in hypoxic areas is also unknown. To examine whether
CREB3 expression correlates with autophagy and hypoxia levels in
human glioma, we detected HIF-1α (a marker of hypoxia) and LC3B (a
marker of autophagy) expressions in 76 human glioma specimens,
which represent different grades and 2 normal brain tissue samples,
using immunochemical staining. As shown in Fig. 2A, HIF-1α and LC3B expression levels
strongly correlated with the WHO grade of glioma (Fig. 2B). In addition, HIF-1α, and LC3B
expression positively correlated with CREB3 levels in glioma
(Fig. 2C). Taken together, these
results demonstrate that CREB3 expression correlates with the
density of autophagic cells and HIF-1α levels in gliomas.
Hypoxia induces autophagy activation and
CREB3 upregulation in glioblastoma cells
We further verified CREBRF and the CREB3 levels in
hypoxia T98G and U87 cells using quantitative real-time PCR, and
our results are consistent with results from human glioma tissue
samples. CREBRF expression significantly decreased in glioblastoma
cells after 24 h of hypoxia treatment, and CREB3 increased
accordingly (Fig. 3A). It is
generally accepted that hypoxia activates autophagy as an
evolutionarily conserved cellular catabolic process (8,28).
To verify this pivotal phenomenon, we performed a GFP-LC3B
puncta-formation assay and an LC3B conversion assay (29). Using GBM cells (T98G) that stably
express a GFP-LC3B fusion protein, the localization of GFP-LC3B was
examined by fluorescent microscopy. GFP-LC3B puncta that appear in
the cytoplasm reflect the recruitment of LC3B proteins to
autophagosomes. As shown in Fig.
3B, a significant increase in GFP-LC3B puncta was noted in
hypoxic cells, and this result was confirmed by the quantification
of GFP-LC3B dots per cell. Moreover, we detected the conversion of
LC3B-I to LC3B-II and P62 (autophagy marker) expression together
with CREBRF and CREB3 protein levels by western blot analysis under
different hypoxia treatment times. Consistent with the GFP-LC3B
puncta-formation assay, hypoxia led to a significant time-dependent
upregulation of LC3B-II and downregulation of P62 (Fig. 3C). Also consistent with the
quantitative real-time PCR results, hypoxia downregulated CREBRF
and upregulated CREB3 protein levels in a time-dependent manner
(Fig. 3C). Thus, both assays
suggested that hypoxia induces autophagosome accumulation and that
CREBRF/CREB3 may be involved in this process.
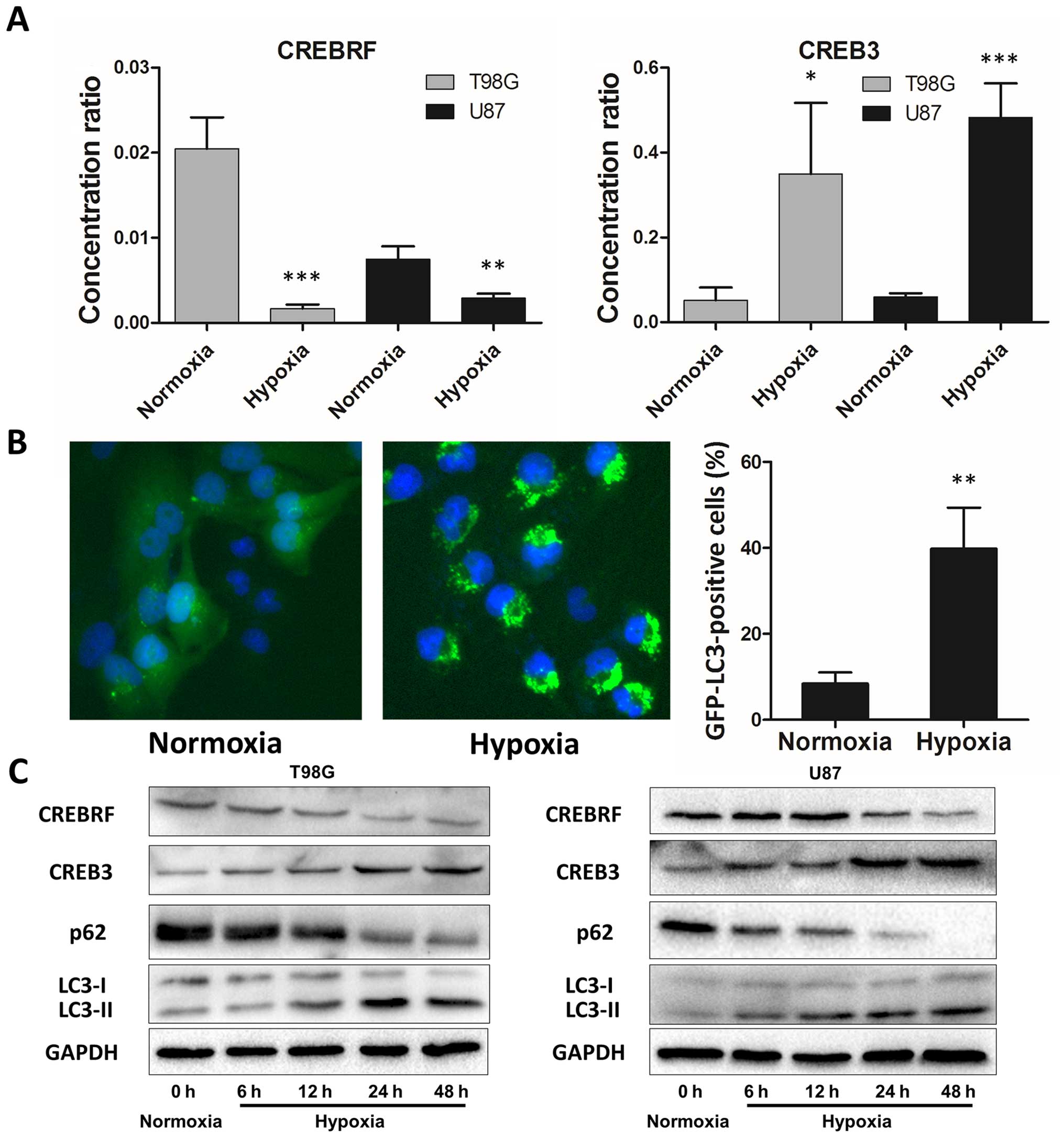 | Figure 3Hypoxia induces autophagy activation
and CREB3 upregulation in glioblastoma cells. (A) CREBRF and CREB3
expression levels in hypoxic T98G and U87 cells (hypoxia treatment
for 24 h) were assessed by quantitative real-time PCR. The data
presented are the mean ± SD of five independent experiments.
*P<0.05, **P<0.01,
***P<0.001, one-way ANOVA. (B) Hypoxia promotes
GFP-LC3B translocation. pSELECT-GFP-LC3B transfection revealed LC3B
puncta in T98G cells treated with hypoxia (1% O2, 5%
CO2, and 94% N2 at 37°C) for 24 h. Cells were
fixed and stained with DAPI for nuclear visualization.
Representative images are presented. Quantitative analysis of
GFP-LC3B puncta is presented in the right panel. At least 100 cells
were examined in each experimental group. The data presented are
the mean ± SD of three independent experiments.
**P<0.01, two-tailed t-test. (C) Hypoxia induced LC3B
conversion and SQSTM1 degradation in T98G and U87 cells (hypoxia
treatment for 0, 6, 12, 24 and 48 h). CREBRF, CREB3, LC3B and p62
levels were examined by western blot analysis in glioblastoma cells
after hypoxia treatment (1% O2, 5% CO2, and
94% N2 at 37°C). GAPDH served as the loading
control. |
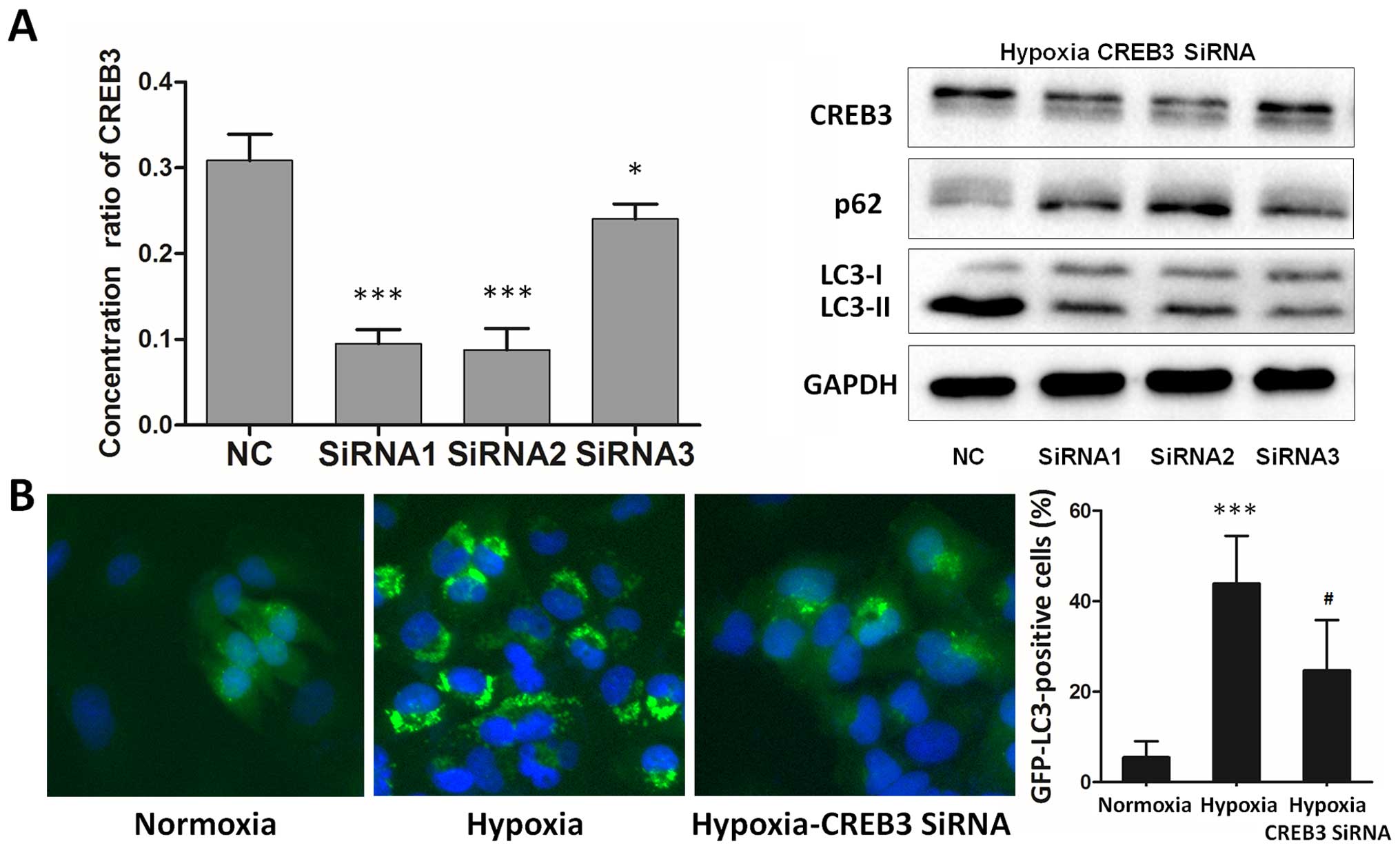
Inhibition of endogenous CREB3 represses
hypoxia-induced autophagy in glioblastoma cells
To further support the fact that endogenous CREB3 is
involved in hypoxia-induced autophagy, we examined LC3B-II and P62
protein levels after attenuating CREB3 activity in hypoxic
glioblastoma cells using RNA interference (RNAi) (Fig. 4A, left). Western blot analysis
clearly indicated that CREB3 knockdown inhibited the
hypoxia-induced LC3B-II increase and P62 degradation in
glioblastoma cells (Fig. 4A,
right). Using T98G cells transiently transfected with GFP-LC3B, our
study revealed that the inhibition of endogenous CREB3 inhibited
autophagy under hypoxic conditions (Fig. 4B). The data indicate that a
blockade of endogenous CREB3 repressed autophagy in hypoxic
glioblastoma cells.
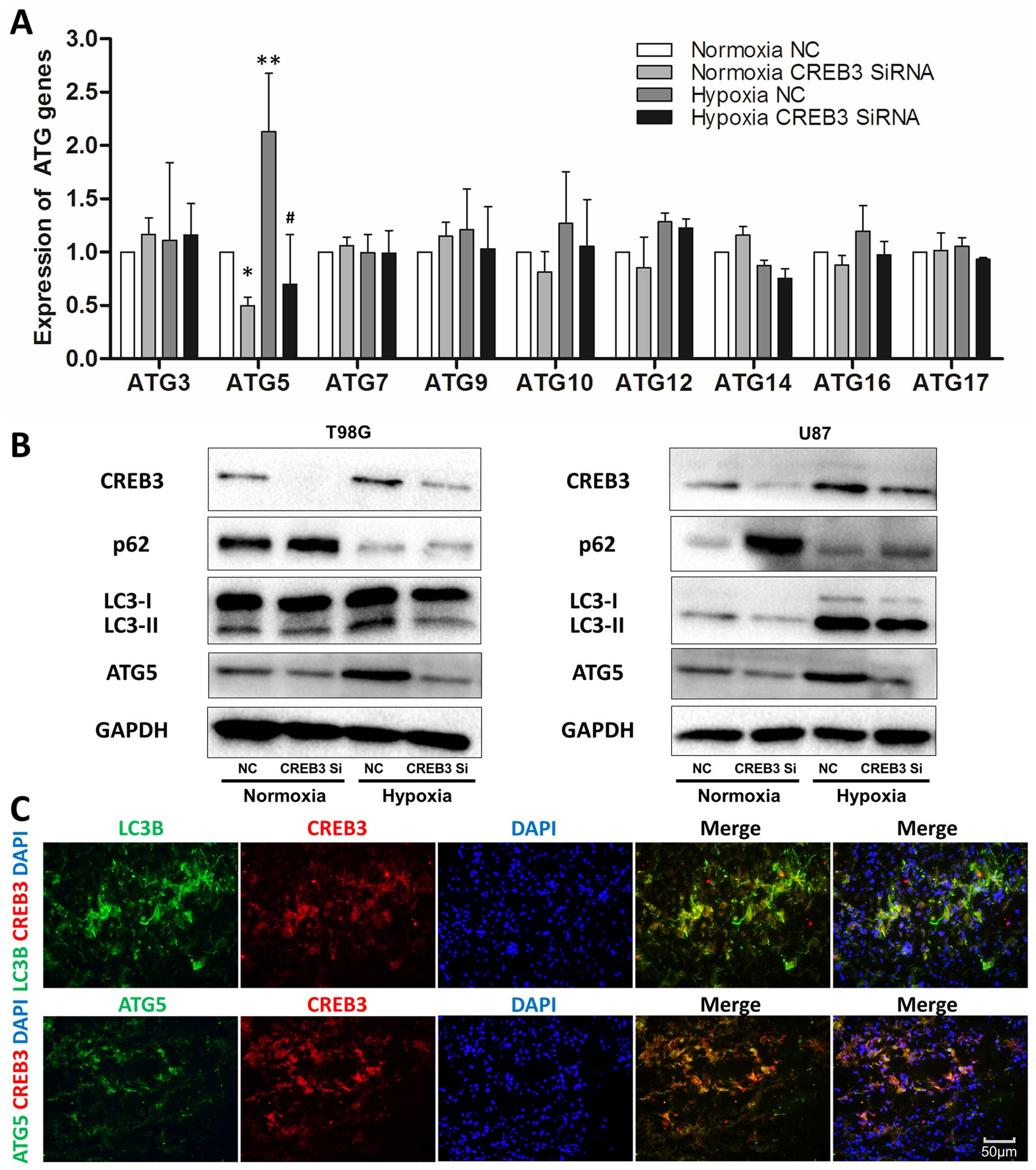
CREB3 enhances hypoxia-induced autophagy
by targeting ATG5 in human glioblastoma cells
To elucidate the mechanisms of CREB3-induced
autophagy, several autophagy-related genes (ATGs) were examined by
quantitative real-time PCR in T98G cells, revealing a significant
decrease in ATG5 after CREB3 knockdown (Fig. 5A) that was confirmed by western
blot analysis in both T98G and U87 cell lines (Fig. 5B). To further investigate the
relationship between CREB3 and ATG5 in hypoxic glioblastoma cells,
we collected 3 fresh, surgically removed WHO grade IV glioma tissue
samples for fast-frozen sectioning, and staining revealed the
co-localization of CREB3, ATG5, and LC3B proteins in high-grade
glioma tissues (Fig. 5C).
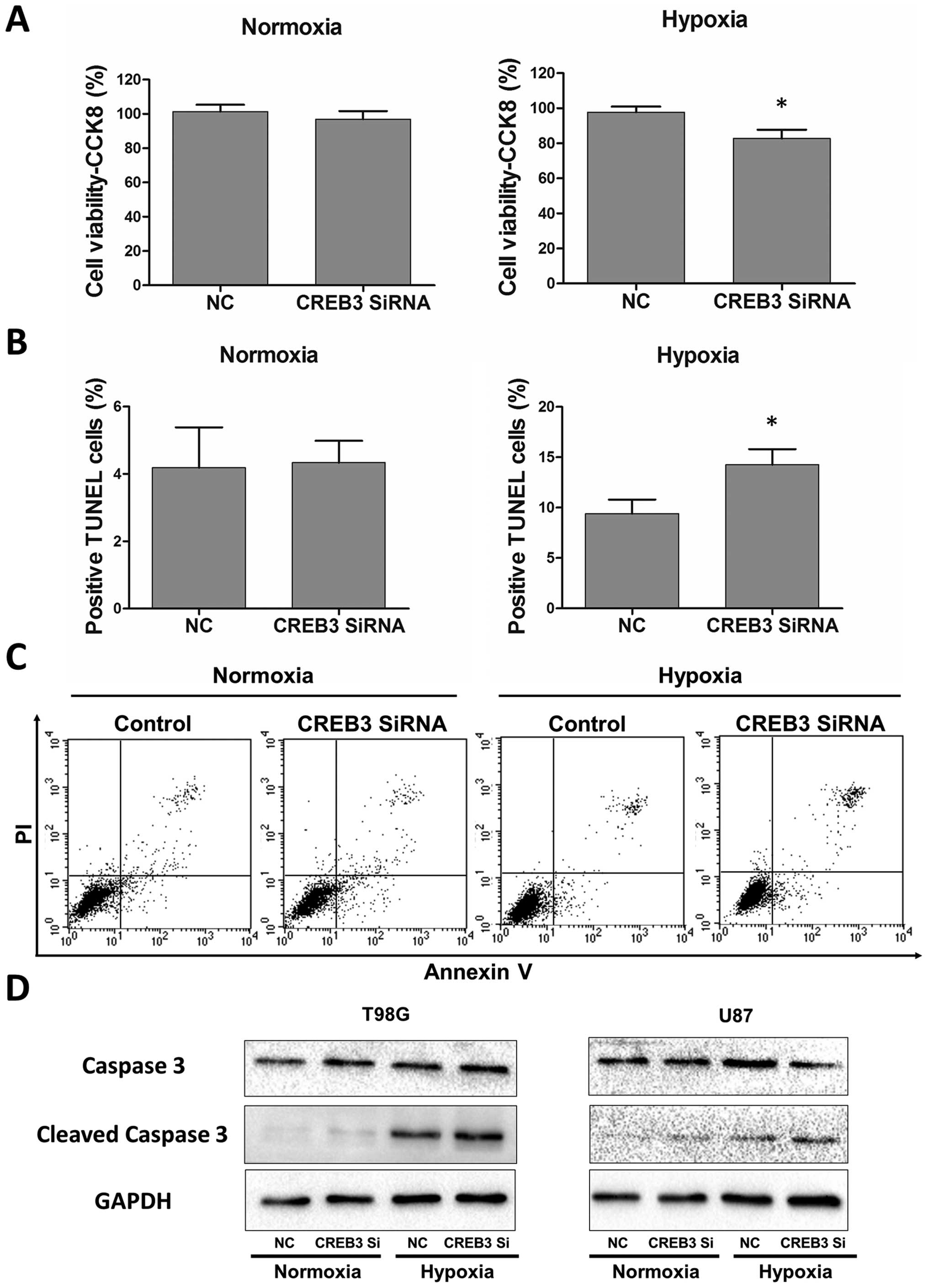
CREB3 knockdown induces
autophagy-enhanced apoptosis in GBM cells
In recent years, accumulating evidence has
demonstrated that autophagy may serve as a protective mechanism in
tumor cells and that therapy-induced apoptosis can be potentiated
by autophagy inhibition (30,31).
To determine the biological significance of CREB3-induced autophagy
on apoptotic cell death, CREB3 interfering RNA was utilized in
glioblastoma cells. A CCK-8 assay was utilized to determine cell
viability. TUNEL staining assays and Annexin V-FITC/ PI were
performed to examine the level of apoptosis in glioblastoma cells.
As shown in Fig. 6A, CREB3
knock-down significantly suppressed glioblastoma cells exclusively
under hypoxic conditions. Consistent with this observation,
hypoxia-induced apoptotic cell death was augmented in the presence
of CREB3 interfering RNA, which was demonstrated by the TUNEL assay
(Fig. 6B) and the Annexin
V-FITC/PI assay (Fig. 6C). In the
process of apoptosis, caspase 3, as the final effector molecule,
can be cleaved and activated as the most common characteristic of
apoptosis. Subsequently, we examined cleaved caspase 3 protein
levels by western blot analysis, and the data further indicated
that CREB3 knock-down led to the activation of caspase 3 apoptotic
signaling pathways (Fig. 6D). This
type of crosstalk between autophagy and apoptosis provides a
possible avenue for interference with hypoxia-induced autophagy of
glioblastoma cells through anti-CREB3 adjuvant therapy to treat
glioma patients.
Discussion
Autophagy benefits glioblastoma cell survival under
hypoxic metabolic stress and mediates resistance to radiotherapy
and chemotherapy (32,33), and these strategies trigger
cellular autophagy (14).
Recently, accumulating evidence has demonstrated that the
inhibition of autophagy triggers apoptosis and kills more tumor
cells (30). Our study also
revealed a novel anti-autophagy therapeutic target by regulating
hypoxia-dependent CREBRF/CREB3 in glioblastoma cells. Given that
upregulated autophagy inhibits tumor apoptosis and promotes
proliferation (34), we inhibited
the CREB3-dependent pathway using specific siRNAs that
significantly suppressed glioblastoma cell viability by converting
autophagy into apoptosis. This work is significant because it
suggests a potential anti-glioma effect of the anti-CREB3 strategy
in vitro for the first time and characterizes the pivotal
role of the CREBRF/ CREB3/ATG5 pathway in hypoxia-induced autophagy
in glioblastoma cells.
The tumor microenvironment plays a critical role in
tumor progression. As the tumor rapidly outgrows its blood supply,
malignant proliferative tumor cells are deprived of oxygen
(35). Tumor hypoxia can
powerfully induce cells to develop an aggressive and
treatment-resistant phenotype that leads to rapid progression and
poor prognosis (6,13), this development is even the case
for immune cells (36–39). HIF-1α is a key transcription factor
in the adaptation to hypoxic stress and plays an important role in
hypoxia-induced autophagy (40),
which activates several autophagy-inducing molecules, such as BNIP3
(41) and IGFBP3 (42). In addition, HIF-1α-independent AMPK
activates more severe hypoxia, and AMPK could contribute to
autophagy via the mTOR inhibition mechanism (43,44).
Nevertheless, in this study, we found that CREB3 also plays a role
in hypoxia-induced autophagy. We also assumed that CREBRF might act
as a tumor suppressor by inhibiting CREB3-induced autophagy. In
addition, CREBRF and CREB3 levels in patient glioma samples might
also be used as prognostic factors.
CREBRF is a negative regulator of CREB3 that can
recruit nuclear CREB3 to discrete foci in the nucleus, promote
CREB3 protein degradation and repress CREB3-mediated activation of
unfolded protein response element (UPRE)-containing promoters
(22). CREB3 (also called Luman or
LZIP) is the primary member of the CREB3 family. All CREB3 family
members appear to play a role in the unfolded protein response
(UPR) (45), during which
endoplasmic reticulum (ER)-resident molecular chaperones and
foldases are induced, attenuating translation to reduce the load on
the ER (46). Unfolded proteins
can also be targeted for proteasomal degradation via ubiquitination
(47). CREB (cyclic-AMP-responsive
element binding) family proteins, including CREB1, CREB2 and CREB3,
bind to the cAMP-responsive element (CRE) recognition sequence of
cAMP-sensitive genes to regulate transcription (23). CREB1 upregulates autophagy genes
ATG7, Ulk1, and Tfeb (25) and
CREBRF is involved in inducing cell apoptosis through the ER stress
pathway (27). Our study is the
first to confirm that CREB3 is also involved in regulating
autophagy in tumor cells.
The cAMP signal transduction pathway is activated
through ligand binding to G-protein coupled receptors and
culminates with the phosphorylation of the CREB family protein,
thus allowing it to bind the cAMP responsive element (CRE)
recognition sequence of cAMP-sensitive genes and to regulate
transcription (23). Details
regarding the positive correlation between CREB3 and ATG5 in our
study remain unclear. The CRE sequence in ATG5 must be verified
first. Alternatively, other intermediate regulator molecules might
play a vital role if the regulation of CREB3 is indirect.
In conclusion, this study demonstrated that the
hypoxia-dependent CREBRF/CREB3 pathway is a potent autophagy
regulator in glioma by regulating ATG5 and that CREB3-induced
autophagy protects glioblastoma cells from apoptotic death. We also
suggested the potential uses for anti-CREB3 therapeutic strategies
in adjuvant therapy for glioma patients and the prognosis
predicting effect of CREBRF in tissues of glioma patients. Future
in vitro studies should be directed toward the
identification of the regulation mechanism of CREB3 and ATG5, and
future in vivo research should be investigated.
Acknowledgements
We thank Professor Xun Qu for helpful comments and
advice on this study, and D. Nicole, Senior Editor at American
Journal Experts, for language advice. This study was supported by
grants from the National Natural Science Foundation of China (nos.
81101594, 81372719, 81172403, 81300510, 81402077, 81571284 and
91542115) and Taishan Scholars of Shandong Province of China (no.
ts201511093).
References
|
1
|
Ohgaki H: Epidemiology of brain tumors.
Methods Mol Biol. 472:323–342. 2009. View Article : Google Scholar
|
|
2
|
Ostrom QT, Gittleman H, Stetson L, Virk SM
and Barnholtz-Sloan JS: Epidemiology of gliomas. Cancer Treat Res.
163:1–14. 2015. View Article : Google Scholar
|
|
3
|
Davis ME and Stoiber AM: Glioblastoma
multiforme: Enhancing survival and quality of life. Clin J Oncol
Nurs. 15:291–297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sayegh ET, Kaur G, Bloch O and Parsa AT:
Systematic review of protein biomarkers of invasive behavior in
glioblastoma. Mol Neurobiol. 49:1212–1244. 2014. View Article : Google Scholar
|
|
5
|
McNamara MG and Mason WP: Antiangiogenic
therapies in glioblastoma multiforme. Expert Rev Anticancer Ther.
12:643–654. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaupel P: Hypoxia and aggressive tumor
phenotype: Implications for therapy and prognosis. Oncologist.
13(Suppl 3): 21–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mongiardi MP: Angiogenesis and hypoxia in
glioblastoma: A focus on cancer stem cells. CNS Neurol Disord Drug
Targets. 11:878–883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: Cell death or cell survival? Curr Opin
Cell Biol. 22:177–180. 2010. View Article : Google Scholar
|
|
9
|
Yuan G, Yan SF, Xue H, Zhang P, Sun JT and
Li G: Cucurbitacin I induces protective autophagy in glioblastoma
in vitro and in vivo. J Biol Chem. 289:10607–10619. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Evans SM, Judy KD, Dunphy I, Jenkins WT,
Hwang WT, Nelson PT, Lustig RA, Jenkins K, Magarelli DP, Hahn SM,
et al: Hypoxia is important in the biology and aggression of human
glial brain tumors. Clin Cancer Res. 10:8177–8184. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brahimi-Horn C and Pouysségur J: The role
of the hypoxia-inducible factor in tumor metabolism growth and
invasion. Bull Cancer. 93:E73–E80. 2006.PubMed/NCBI
|
|
13
|
Ji RC: Hypoxia and lymphangiogenesis in
tumor microenvironment and metastasis. Cancer Lett. 346:6–16. 2014.
View Article : Google Scholar
|
|
14
|
Hu YL, Jahangiri A, De Lay M and Aghi MK:
Hypoxia-induced tumor cell autophagy mediates resistance to
anti-angiogenic therapy. Autophagy. 8:979–981. 2012. View Article : Google Scholar :
|
|
15
|
Meijer AJ and Codogno P: Autophagy:
Regulation and role in disease. Crit Rev Clin Lab Sci. 46:210–240.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen N and Karantza-Wadsworth V: Role and
regulation of autophagy in cancer. Biochim Biophys Acta.
1793:1516–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: A double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar
|
|
20
|
Guo JY, Xia B and White E:
Autophagy-mediated tumor promotion. Cell. 155:1216–1219. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aredia F, Guamán Ortiz LM, Giansanti V and
Scovassi AI: Autophagy and cancer. Cells. 1:520–534. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Audas TE, Li Y, Liang G and Lu R: A novel
protein, Luman/ CREB3 recruitment factor, inhibits Luman activation
of the unfolded protein response. Mol Cell Biol. 28:3952–3966.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mamdani F, Alda M, Grof P, Young LT,
Rouleau G and Turecki G: Lithium response and genetic variation in
the CREB family of genes. Am J Med Genet B Neuropsychiatr Genet.
147B:500–504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cao G, Ni X, Jiang M, Ma Y, Cheng H, Guo
L, Ji C, Gu S, Xie Y and Mao Y: Molecular cloning and
characterization of a novel human cAMP response element-binding
(CREB) gene (CREB4). J Hum Genet. 47:373–376. 2002. View Article : Google Scholar
|
|
25
|
Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar
S, Sun X, Yoon G, Kang Y, Zhong W, et al: Transcriptional
regulation of autophagy by an FXR-CREB axis. Nature. 516:108–111.
2014.PubMed/NCBI
|
|
26
|
Liu YL, Lai F, Wilmott JS, Yan XG, Liu XY,
Luan Q, Guo ST, Jiang CC, Tseng HY, Scolyer RA, et al: Noxa
upregulation by oncogenic activation of MEK/ERK through CREB
promotes autophagy in human melanoma cells. Oncotarget.
5:11237–11251. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang Y, Lin P, Chen F, Wang A, Lan X, Song
Y and Jin Y: Luman recruiting factor regulates endoplasmic
reticulum stress in mouse ovarian granulosa cell apoptosis.
Theriogenology. 79:633–639. e1–3. 2013. View Article : Google Scholar
|
|
28
|
Mucaj V, Shay JE and Simon MC: Effects of
hypoxia and HIFs on cancer metabolism. Int J Hematol. 95:464–470.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boya P, González-Polo RA, Casares N,
Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D,
Souquere S, Yoshimori T, et al: Inhibition of macroautophagy
triggers apoptosis. Mol Cell Biol. 25:1025–1040. 2005. View Article : Google Scholar :
|
|
31
|
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A,
Wu Q, Zhang J and Hong Y: Caspases: A molecular switch node in the
crosstalk between autophagy and apoptosis. Int J Biol Sci.
10:1072–1083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang Z and Klionsky DJ: Eaten alive: A
history of macroautophagy. Nat Cell Biol. 12:814–822. 2010.
View Article : Google Scholar
|
|
33
|
Marino ML, Pellegrini P, Di Lernia G,
Djavaheri-Mergny M, Brnjic S, Zhang X, Hägg M, Linder S, Fais S,
Codogno P, et al: Autophagy is a protective mechanism for human
melanoma cells under acidic stress. J Biol Chem. 287:30664–30676.
2012. View Article : Google Scholar :
|
|
34
|
Mukhopadhyay S, Panda PK, Sinha N, Das DN
and Bhutia SK: Autophagy and apoptosis: Where do they meet?
Apoptosis. 19:555–566. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Eng JW, Kokolus KM, Reed CB, Hylander BL,
Ma WW and Repasky EA: A nervous tumor microenvironment: The impact
of adrenergic stress on cancer cells, immunosuppression, and
immunotherapeutic response. Cancer Immunol Immunother.
63:1115–1128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang M, Ma C, Liu S, Shao Q, Gao W, Song
B, Sun J, Xie Q, Zhang Y, Feng A, et al: HIF-dependent induction of
adenosine receptor A2b skews human dendritic cells to a
Th2-stimulating phenotype under hypoxia. Immunol Cell Biol.
88:165–171. 2010. View Article : Google Scholar
|
|
37
|
Seo YJ, Koh SH, Kang HJ, Shin HY, Jeong G
and Ahn HS: Hypoxia inhibits the SDF-1-dependent migration of human
leukemic cell line HL-60 via blocking of Akt activation. Biochem
Biophys Res Commun. 364:388–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qu X, Yang MX, Kong BH, Qi L, Lam QL, Yan
S, Li P, Zhang M and Lu L: Hypoxia inhibits the migratory capacity
of human monocyte-derived dendritic cells. Immunol Cell Biol.
83:668–673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao W, Darmanin S, Fu Q, Chen J, Cui H,
Wang J, Okada F, Hamada J, Hattori Y, Kondo T, et al: Hypoxia
suppresses the production of matrix metalloproteinases and the
migration of human monocyte-derived dendritic cells. Eur J Immunol.
35:3468–3477. 2005. View Article : Google Scholar
|
|
40
|
Unwith S, Zhao H, Hennah L and Ma D: The
potential role of HIF on tumour progression and dissemination. Int
J Cancer. 136:2491–2503. 2015. View Article : Google Scholar
|
|
41
|
Hu YL, DeLay M, Jahangiri A, Molinaro AM,
Rose SD, Carbonell WS and Aghi MK: Hypoxia-induced autophagy
promotes tumor cell survival and adaptation to antiangiogenic
treatment in glioblastoma. Cancer Res. 72:1773–1783. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hsieh DJ, Kuo WW, Lai YP, Shibu MA, Shen
CY, Pai P, Yeh YL, Lin JY, Viswanadha VP and Huang CY:
17β-estradiol and/ or estrogen receptor β attenuate the autophagic
and apoptotic effects induced by prolonged hypoxia through
HIF-1α-mediated BNIP3 and IGFBP-3 signaling blockage. Cell Physiol
Biochem. 36:274–284. 2015. View Article : Google Scholar
|
|
43
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lu R, Yang P, O'Hare P and Misra V: Luman,
a new member of the CREB/ATF family, binds to herpes simplex virus
VP16-associated host cellular factor. Mol Cell Biol. 17:5117–5126.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mori K: Tripartite management of unfolded
proteins in the endoplasmic reticulum. Cell. 101:451–454. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sifers RN: Cell biology. protein
degradation unlocked. Science. 299:1330–1331. 2003. View Article : Google Scholar : PubMed/NCBI
|