Introduction
Prostate cancer is the most common diagnosed
non-cutaneous cancer and the second leading cause of cancer-related
death in American men (1). Similar
to many other cancers, once prostate cancer has metastasized, there
is no curative therapy. Currently, androgen ablation is the major
therapy for advanced prostate cancer. Most patients benefit
initially from androgen ablation therapy (2), but the tumor cells eventually develop
resistance to androgen ablation, becoming castration-resistant
prostate cancer (CRPC), with an invariably fatal outcome (3–6).
Currently, the discovery of a viable therapy for CRPC is a high
priority in the battle against prostate cancer. Two multi-center
clinical trials (7,8) have revealed that combinatorial
chemotherapy improved median survival in advanced prostate cancer
patients, leading to the FDA approval of such chemotherapy for CRPC
treatment and emphasizing the importance of chemotherapy in
CRPC.
Camptothecin, a plant alkaloid, was first isolated
from the Chinese tree Camptotheca acuminata by Wall and
colleagues (9). The elucidation of
the camptothecin antitumor mechanism by targeting intranuclear
enzyme topoisomerase 1 has made camptothecin a promising new class
of anticancer drugs, leading to the development of numerous
water-soluble derivatives with fewer side effects (10–12).
NSC606985 (NSC), a highly water-soluble camptothecin analog, has
recently been demonstrated to produce both caspase-dependent and
caspase-independent tumor cell death via differential mechanisms
(13,14). In a previous study, we demonstrated
the antitumor activity of NSC in prostate cancer cells through the
interaction with topoisomerase 1 and an activation of the
mitochondrial apoptotic pathway (15). However, the molecular pathway of
NSC activation of cell apoptosis in prostate cancer cells remains
to be further elucidated.
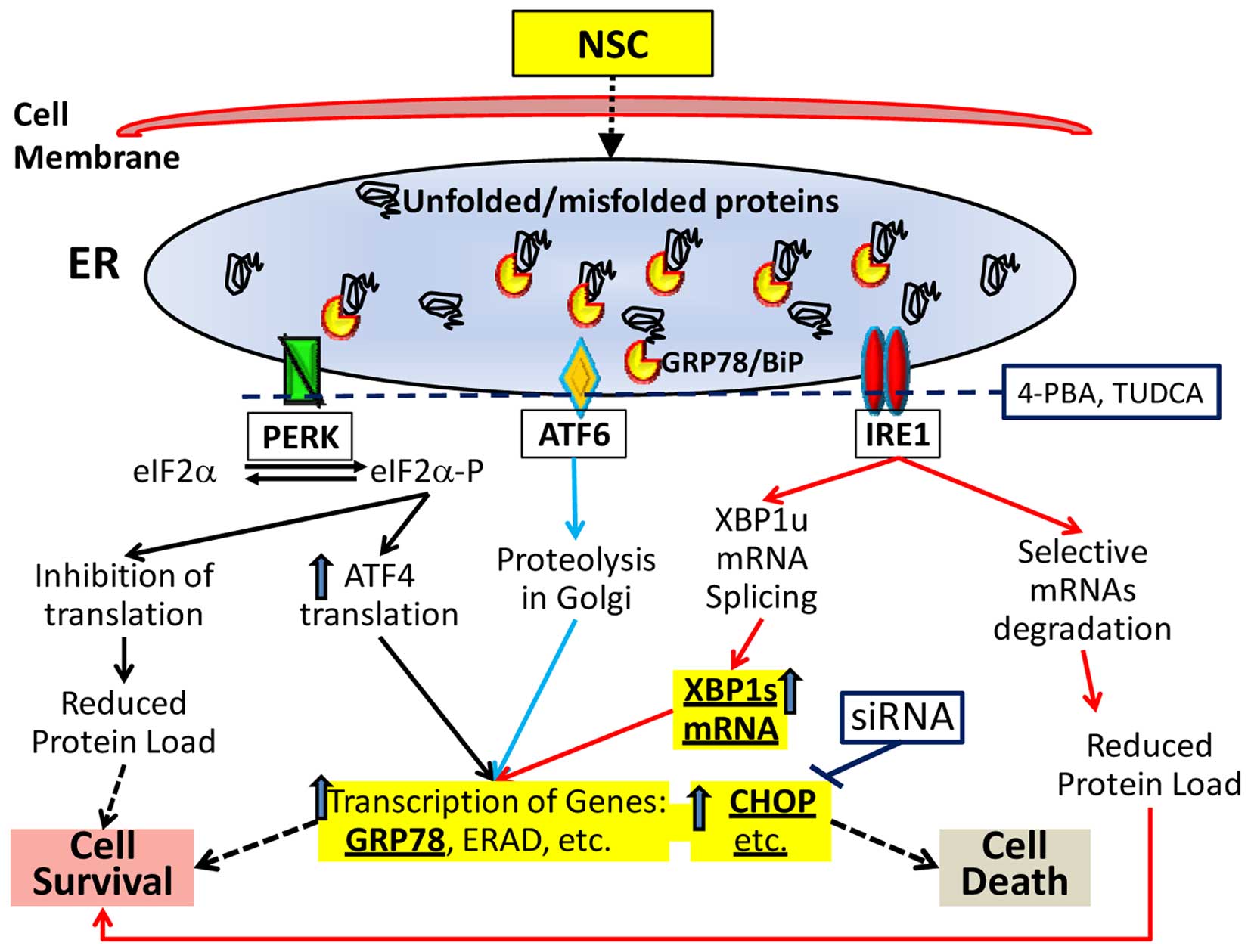
Endoplasmic reticulum (ER) plays an essential role
in cell function and survival. Accumulation of unfolded or
misfolded proteins in the lumen of the ER activates the unfold
protein response (UPR), resulting in ER-stress (16–18).
The complex cellular response of ER-stress is mediated through
three ER transmembrane systems: pancreatic ER kinase-like ER kinase
(PERK), activating transcription factor 6 (ATF6) and
inositol-requiring enzyme 1 (IRE1). With the accumulation of
unfolded or misfolded proteins, GRP78, a molecular chaperone,
dissociates from the three receptors, leading to an activation of
the three systems and triggering UPR. The activation of these three
systems leads to the alteration of multiple downstream events
including an alteration of X-box-binding protein-1 (XBP1) mRNA
splicing and an upregulation of C/EBP homologous protein (CHOP)
gene expression (Fig. 1). The UPR
is a pro-survival response to reduce accumulation of unfolded or
misfolded proteins and restore normal ER function (19). However, if ER-stress is prolonged,
or if the adaptive response fails, apoptosis ensues (19–21).
Cell death induced by ER-stress predominantly occurs via the
intrinsic mitochondrial apoptotic pathway mainly mediated through
CHOP that downregulates the anti-apoptotic mitochondrial protein
Bcl-2 (19). Release of cytochrome
c from the mitochondrial intermembrane space to cytosol is a
key event in intrinsic cell death and is associated with a loss of
mitochondrial transmembrane potential and an opening of the
mitochondrial permeability transition pore (22). Thus, an overwhelming ER-stress is
linked to cell apoptosis (Fig. 1).
In this study, we investigated the effect of NSC on ER-stress and
the association of NSC-induced ER-stress and cell death.
Materials and methods
Reagents
NSC606985 (NSC, glycine,
4-ethyl-3,4,12,14-tetrahydro-3,14-dioxo-1H-pyrano[3′,4′:6,7]
indolizino [1,2-b] quinolin-4-ylester, (S)-, monohydrochloride,
C22H19N3O5.ClH) was
kindly provided by the Drug Synthesis and Chemistry Branch,
Developmental Therapeutic Program, Division of Cancer Treatment and
Diagnosis, National Cancer Institute (Bethesda, MD, USA).
4-phenylbutyric acid (4-PBA), and tunicamycin were purchased from
Sigma (St. Louis, MO, USA). Tauroursodeoxycholic acid (TUDCA),
epoxomicin (EPO) and ALLN were from Calbiochem (San Diego, CA,
USA). Fetal bovine serum (FBS), L-glutamine, penicillin and
streptomycin were obtained from Gemini Bio-Products (Calabasas, CA,
USA). Reagents for real-time PCR such as dNTP Mix (10 mM), M-MLV
Reverse Transcriptase, RNase inhibitor, and M-MLV RT 5X buffer were
purchased from Promega (Madison WI, USA). Antibodies against GRP78
and CHOP were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA), β-actin antibody from Sigma, and cytochrome c
antibody from Pharmingen (San Diego, CA, USA). Fast-start universal
SYBR green master (Rox) and TriPure reagents were purchased from
Roche Diagnostic Inc. (Indianapolis, IN, USA). Lipofectamine 2000
and random primers were obtained from Invitrogen (Carlsbad, CA,
USA).
Cellcultures
LNCaP, PC3 and DU145 cells (ATCC, Rockville, MD,
USA) were respectively cultured in RPMI-1640 and Dulbecco's
modified Eagle's minimal essential medium (DMEM) supplemented with
10% fetal bovine serum (FBS), 2 mM L-glutamine, 50 U/ml of
penicillin and 50 μg/ml streptomycin as previously described
(23). Cells were maintained in a
5% CO2-95% air humidified atmosphere at 37°C and
cultured in phenol-red free medium with 5% stripped FBS (Gemini
Bio-Products) 24 h before experiments as described (23).
Cell proliferation assay
To measure viable cells, Pca DU145 cells were plated
in 96-well plates (5,000 cells/well) and treated with various drugs
as indicated at 24 h after plating. Viable cells were counted using
a CellTiter AQueous One Solution Cell Proliferation Assay kit
following the manufacturer's instructions (Promega, WI, USA).
Reverse transcription PCR (RT-PCR) and
quantitative RT-PCR
To measure gene expression, RT-PCR and real-time PCR
were performed as previously described (15,24).
Briefly, total cellular RNA was isolated using TriPure reagents,
and the level of specified RNAs were quantified using a
NanoDrop2000C. Reverse transcription was performed following the
protocol from Promega with 1 μg of total cellular RNA and real-time
PCR was carried out according to the protocol from Roche in a PCR
mixture containing 0.2 μM of each primer, 6 μl
Platinum®SYBR®Green qPCR SuperMix-UDG dNTPs
and 5 μl of diluted RT solution (1:50). Primers used for real-time
PCR quantification are listed in Table
I. Real-time PCR conditions were set according to the protocol
from Roche and performed in the Eco™ real-time PCR system
(Illumine). Glyceraldehydes-3-phosphate dehydrogenase (GAPDH) or
β-actin was used as an internal control. Differences between
control and treated samples were calculated using the ΔΔCt method
and presented as fold of control.
 | Table IPrimers for qRT-PCR. |
Table I
Primers for qRT-PCR.
| Gene | Primers |
|---|
| hGRP78 | F:
CGAGAACACGGTCTTTGACG
R: ACCACCTTGAACGGCAAGAACT |
| hCHOP | F:
5′-CCTGAGGAGAGAGTGTTCAAG-3′
R: 5′-CTCTTGCAGGTCCTCATACCA-3′ |
| hXBP-1 | F:
5′-TTACGAGAGAAAACTCATGGCC-3′
R: 5′-GGGTCCAAGTTGTCCAGAATGC-3′ |
| hXBP1s | F:
5′-TGAGAACCAGGAGTTAAGACAGCG-3′
R: 5′-CCTGCACCTGCTGCGGAC-3′ |
| hGAPDH | F:
5′-GAAGGTGAAGGTCGGAGTC-3′
R: 5′-GAAGATGGTGATGGGATTTC-3′ |
| hβ-actin |
F:5′-CTAGAAGCATTTGCGGTGGACGATG-3′
R:5′-TCATGAAGTGTGACGTGGACATCCG-3′ |
Western blotting
Western blotting was carried out following standard
methods with minor modifications (15). Briefly, cells were harvested and
total cellular proteins were extracted using a RIPA buffer (25 mM
Tris-HCl pH 7.6, 150 mM NaCI, 1% NP-40, 1% sodium deoxycholate,
0.1% SDS). Protein concentrations were measured with a Bio-Rad
protein assay following the manufacturer's instructions (Bio-Rad,
Hercules, CA, USA).Equal protein was fractionated on a Mini-PROTEAN
TGX™ Precast gel (Bio-Rad 456-8123) and transferred to PVDF
membrane using the Trans-blot Turbo system (Bio-Rad). Blots were
blocked with TBST buffer [500 mM NaCl, 20 mM Tris-HCl (pH 7.4), and
0.1% Tween 20] containing 5% non-fat dry milk overnight at 4°C and
then incubated with specified primary antibody in TBST buffer
containing 5% non-fat dry milk for 3 h at room temperature. After
secondary antibody incubation at room temperature for 1 h, the
signal was visualized with a SuperSignal West Pico Chemiluminescent
kit (Pierce Biotechnology Inc. Rockford, IL, USA), imaged and
quantified with a Bio-Rad ChemoDoc MP system. β-actin was used as
an internal control and detected via incubation of a specific
antibody against β-actin with the same membrane.
Small interference RNA (siRNA)
construction and transfection
Based on the human CHOP gene sequences (GenBank
accession no. XM_005357988.1), a custom silencer siRNA of CHOP with
a sense sequence of 5′-GAACGGCUCAAGCAGGAAAtt-3′ and an antisense
sequence of 5′-UUUCCUGCUUGAGCCGUUCat-3′, and a negative control
silencer siRNA (catalog no. 4390843) were obtained from Invitrogen.
For siRNA transfection, DU145 cells were seeded in a 96-well plate,
or 12-well plate or 60-mm dishes in phenol red-free DMEM containing
5% stripped FBS without antibiotics. Twenty-four hours later, the
cells were transfected with various concentrations of siRNA (10, 50
and 100 nM) using Lipofectamine 2000 according to the
manufacturer's instructions (Invitrogen) in OPTI-MEM medium as
previously described (24,25). Five hours after transfection,
transfection reagents were replaced with DMEM medium and cells were
treated with or without various dose NSC for different durations as
indicated in each experiment. At the end of the experiments, viable
cells were counted with a CellTiter AQueous One Solution Cell
Proliferation Assay kit. Knockdown of CHOP expression was verified
using quantitative RT-PCR and western blotting.
Analysis of cytochrome c release
Cytosolic cytochrome c was measured as
previously described with minor modifications (26). Briefly, cells were pelleted by
centrifugation and resuspended in 5X volumes of Buffer A (20 mM
HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM ethylene
glycol bis (β-aminoethylether)-N,N,N′,N′-tetra-acetic acid, 1 mM
DTT, 0.1 mM phenylmethylsulfonyl fluoride and complete protease
inhibitors) supplemented with 250 mM sucrose. After homogenization
with vortexing, the cytosolic fraction was obtained by sequential
centrifugations as described (26). Fractionated cytosolic proteins were
analyzed by western blotting on a Mini-PROTEAN TGX Precast gel as
described above.
Statistical analysis
Data are presented as means ± SEM. One-way analysis
of variance (ANOVA) followed by a post hoc Student-Newman-Keuls
test and ANOVA on Rank followed by a post hoc Dunn's test were used
to determine the difference among multiple groups for parametric
and non-parametric data, respectively. A p-value <0.05 was
considered statistically significant.
Results
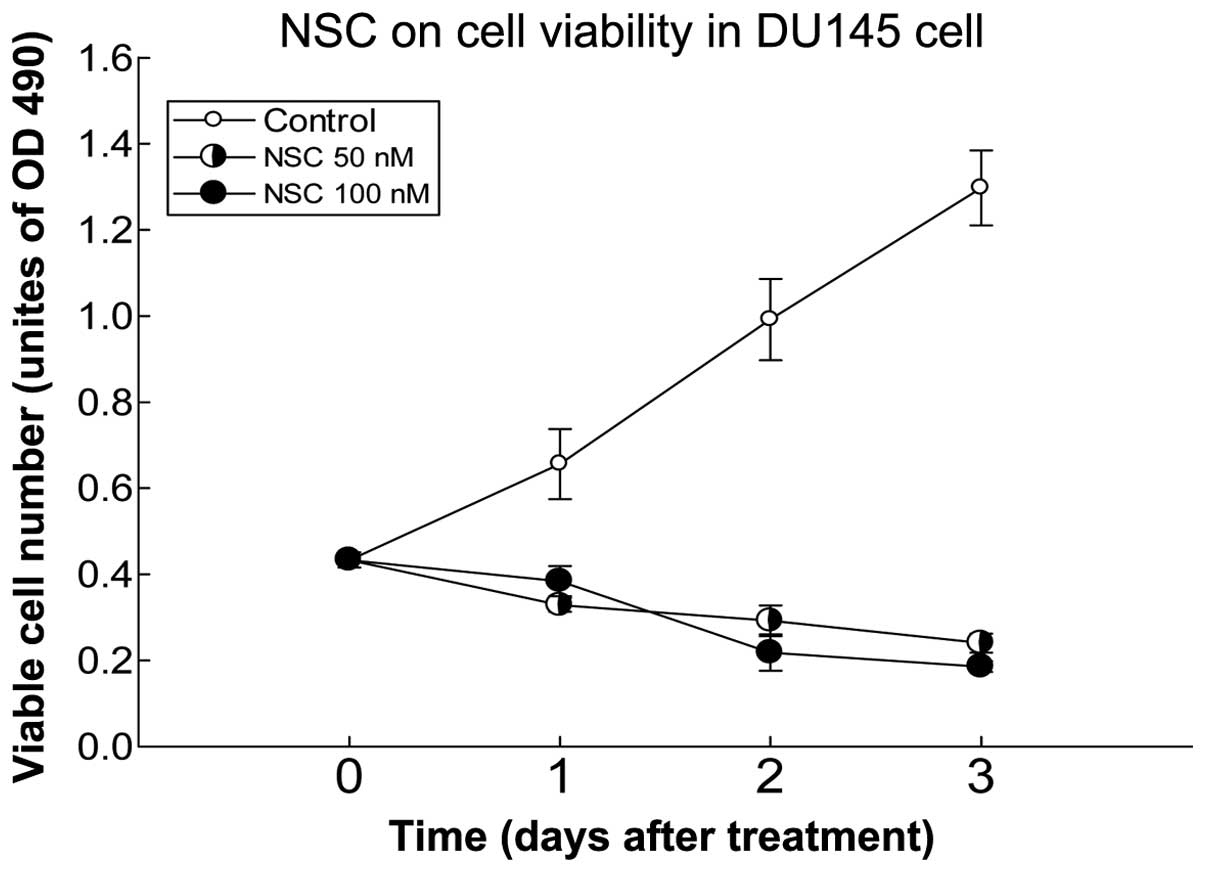
NSC decreases viable DU145 cells
We have previously reported that NSC produced a
time- and dose-dependent decrease of cell viability in Pca cells
(15). This effect was further
verified in this study using DU145 Pca cells (Fig. 2). At a dose of 100 nM, NSC produced
an ~60% reduction of viable cells at 72 h post-treatment compared
to pretreatment.
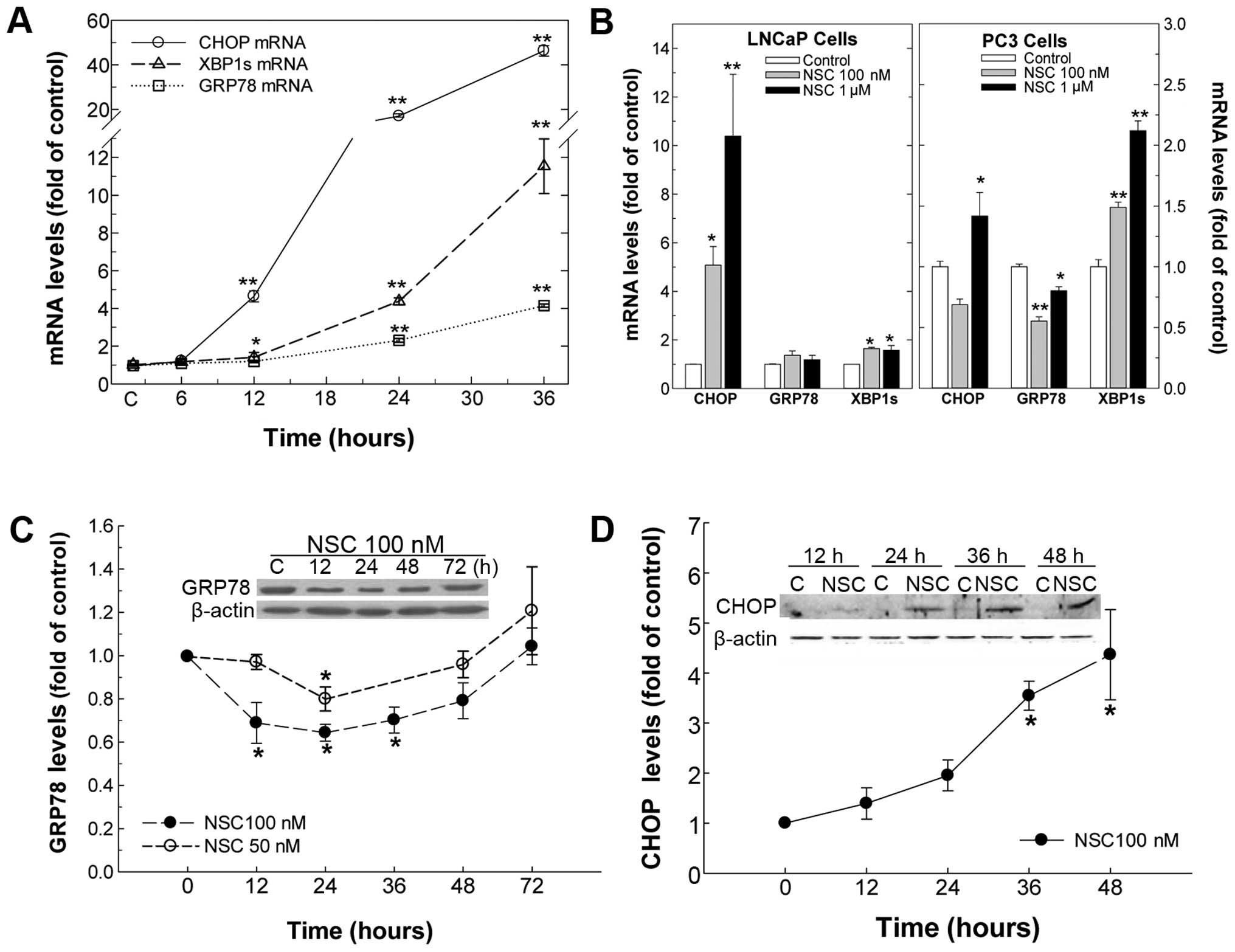
NSC induces ER-stress
To determine whether NSC induces ER-stress in Pca
cells, cells were treated with various doses of NSC for different
durations ranging from 6 to 72 h, and ER-stress biomarkers
including GRP78, CHOP and XBP1s mRNA were determined. In DU145 Pca
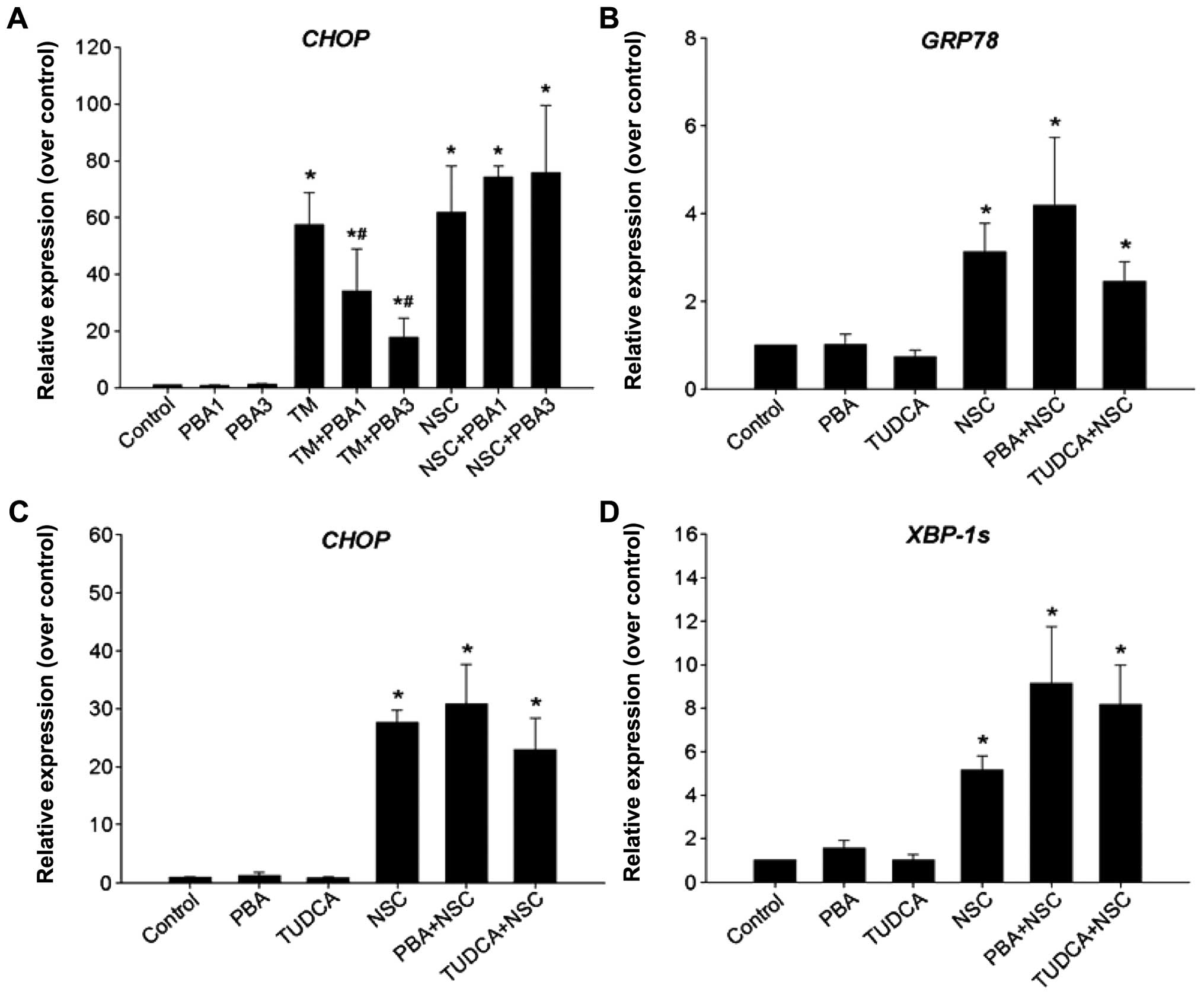
cells, NSC produced a time-dependent elevation of ER-stress
biomarkers, CHOP, XBP1s and GRP78, and their mRNA levels were
increased by >46-, 11- and 4-fold compared to vehicle control at
36 h of NSC (100 nM) treatment, respectively (Fig. 3A). Moreover, similar to the CHOP
mRNA change, the level of CHOP protein was elevated in a
time-dependent manner following NSC treatment and a >4-fold
increase in CHOP protein was observed at 48 h of NSC (100 nM)
treatment in DU145 cells (Fig.
3D). Surprisingly, unlike the change in GRP78 mRNA, NSC caused
a dose- and time-dependent U-shaped change in GRP78 protein. From
12 to 36 h, NSC treatment resulted in a dose- and time-dependent
decrease in GRP78 protein, which was gradually recovered by 48–72 h
following NSC treatment (Fig. 3C).
Similarly, NSC induced ER-stress in LNCaP and PC3 Pca cells as
evident by an increase in ER-stress biomarkers, CHOP and XBP1s mRNA
(Fig. 3B).
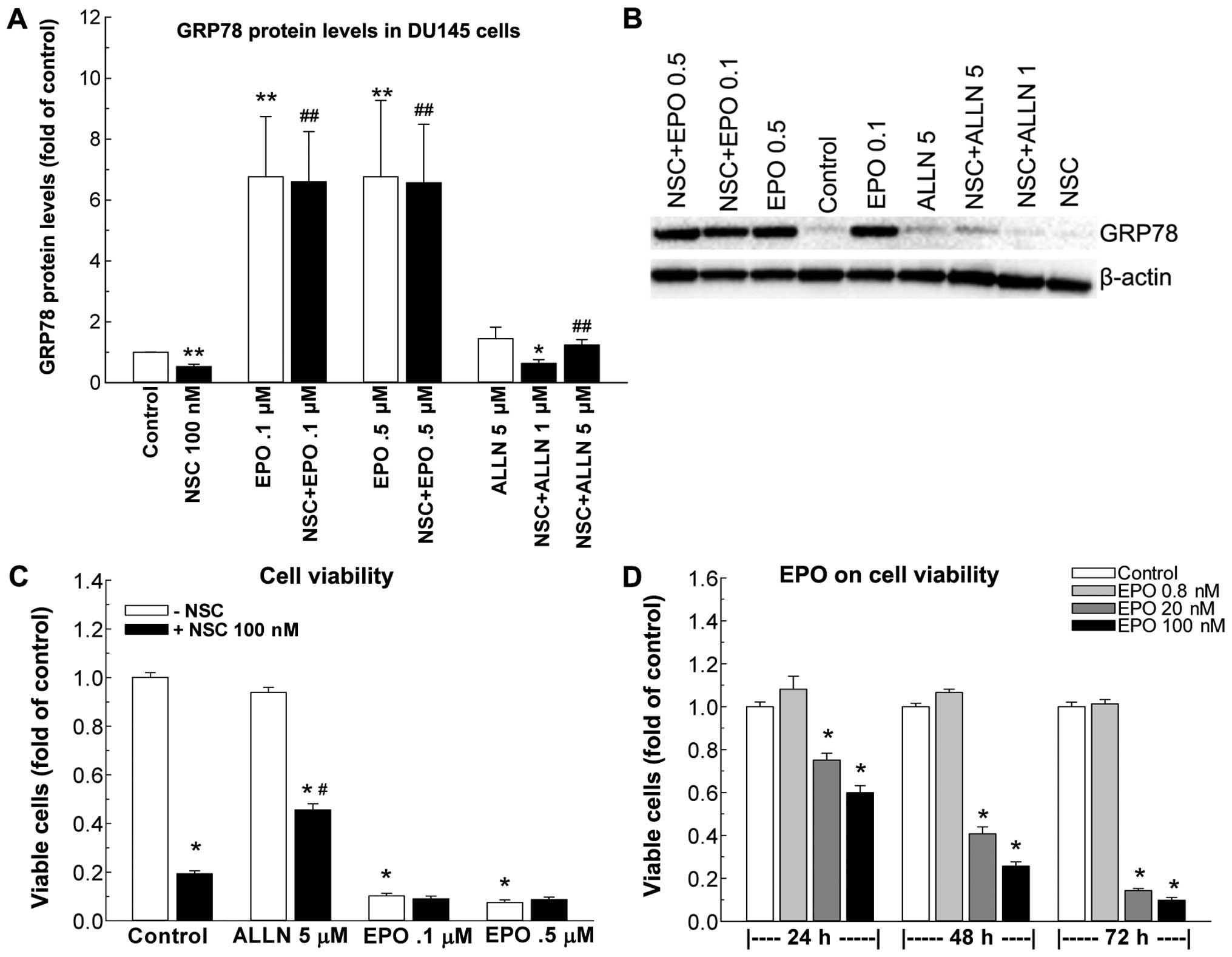
NSC-induced GRP78 protein reduction and
cell death are blocked by protease inhibitors in DU145 cells
To determine if the NSC-induced decrease in GRP78
protein is related to acceleration of protein degradation, two
protease inhibitors were employed, EPO that inhibits protein
degradation in the ubiquitin-proteasome pathway (27), and ALLN that inhibits protein
degradation in the autophagy-lysosomal pathway (28). Treatment of DU145 cells with ALLN
at 1 and 5 μM doses produced a dose-dependent blockade of
NSC-induced GRP78 reduction (Fig. 4A
and B) and partially rescued NSC-induced cell death (Fig. 4C). On the contrary, EPO at both 0.1
and 0.5 μM concentration inhibited NSC-induced decreases in GRP78
protein levels (Fig. 4A and B)
without any effect on NSC-induced cell death (Fig. 4C). Moreover, EPO alone, but not
ALLN, markedly increased GRP78 protein levels (Fig. 4A and B), while producing a dose-
and time-dependent reduction of viable cell numbers (Fig. 4D). At 72 h post treatment with 100
nM EPO, the viable cell number was reduced by 90% (Fig. 4D).
NSC-induced ER-stress is not affected by
4-PBA and TUDCA in DU145 cells
TUDCA and 4-PBA have been previously reported to
function as chemical chaperons to reduce ER-stress induced by
multiple stimuli (29–31). To determine whether NSC-induced
ER-stress is inhibited by 4-PBA and TUDCA, DU145 cells were
pretreated with different dose 4-PBA or TUDCA for 1 h and then
treated with NSC (100 nM) for 36 h. The doses of 4-PBA and TUDCA
were based on previous reports (32,33)
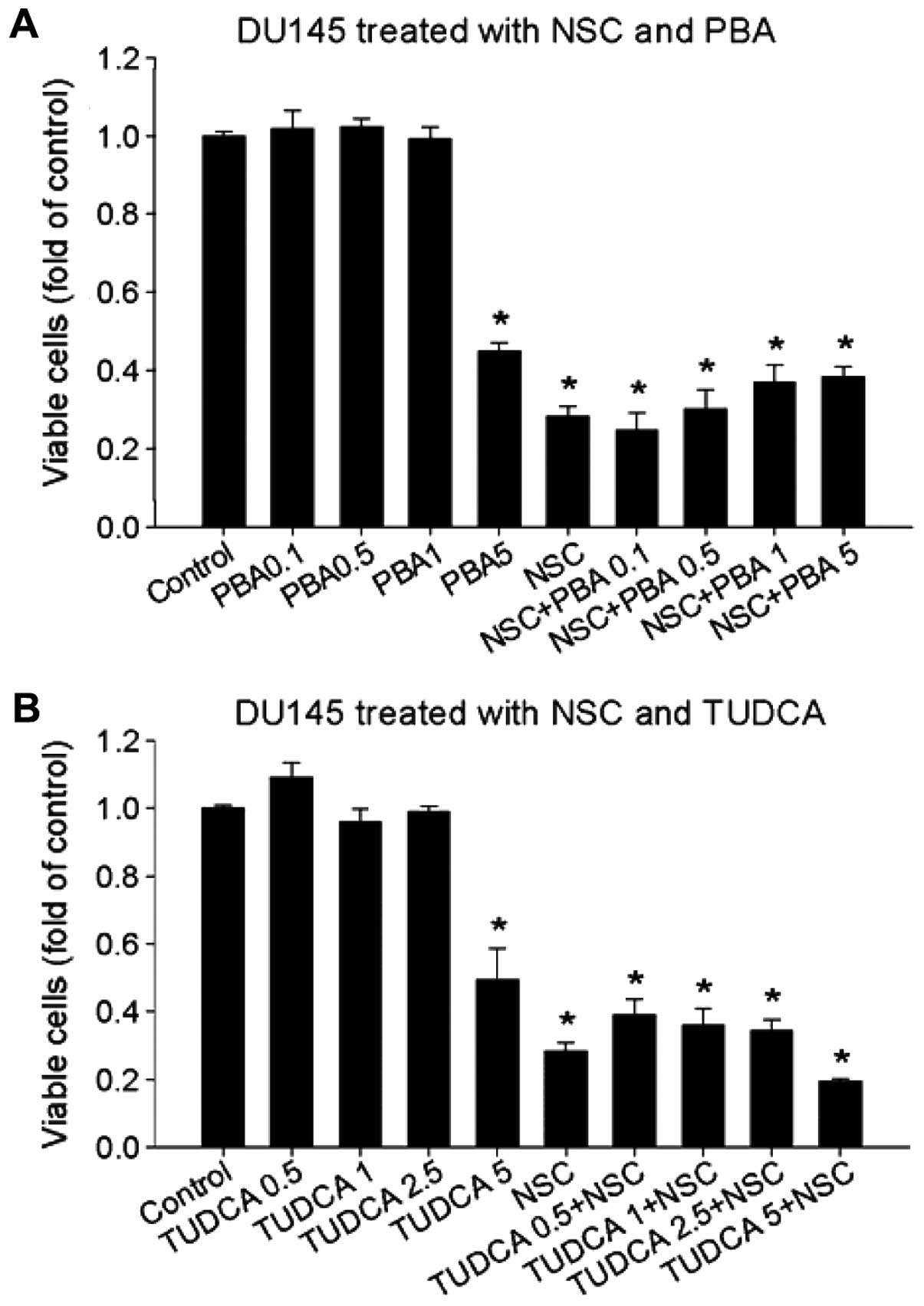
and our results in viable cells (Fig.
6). Pretreatment of DU145 cells with 4-PBA (1–3 μM) or TUDCA
(2.5–5 μM) significantly blocked CHOP mRNA expression induced by
tunicamycin (TM), a positive inducer of ER-stress by disrupting ER
Ca2+ homeostasis (34)
(Fig. 5A and data not shown).
However, neither 4-PBA nor TUDCA inhibited NSC-induced GRP78
(Fig. 5B), CHOP (Fig. 5A and C) and XBP1s mRNA expression
(Fig. 5D) in DU145 cells. Thus,
NSC-induced ER-stress is differentiated from TM-induced ER-stress
because chemical chaperons, 4-PBA and TUDCA failed to block it.
To determine the effect of chemical chaperone 4-PBA
and TUDCA on NSC-induced cell death, DU145 cells were pretreated
with various concentrations of 4-PBA and TUDCA 1 h prior to NSC
(100 nM) treatment for 72 h. Fig.
6 shows that neither 4-PBA nor TUDCA altered NSC-induced cell
death, in agreement with changes in ER-stress biomarkers (Fig. 5). Thus, chemical chaperones, 4-PBA
and TUDCA, failed to modify NSC-induced ER-stress and cell death in
DU145 cells.
Knockdown of NSC-induced CHOP expression
failed to block NSC-induced cytochrome c release and cell
death
Because CHOP is a major mediator of
ER-stress-induced apoptosis (35,36),
we investigated the role of CHOP in NSC-induced cytochrome c
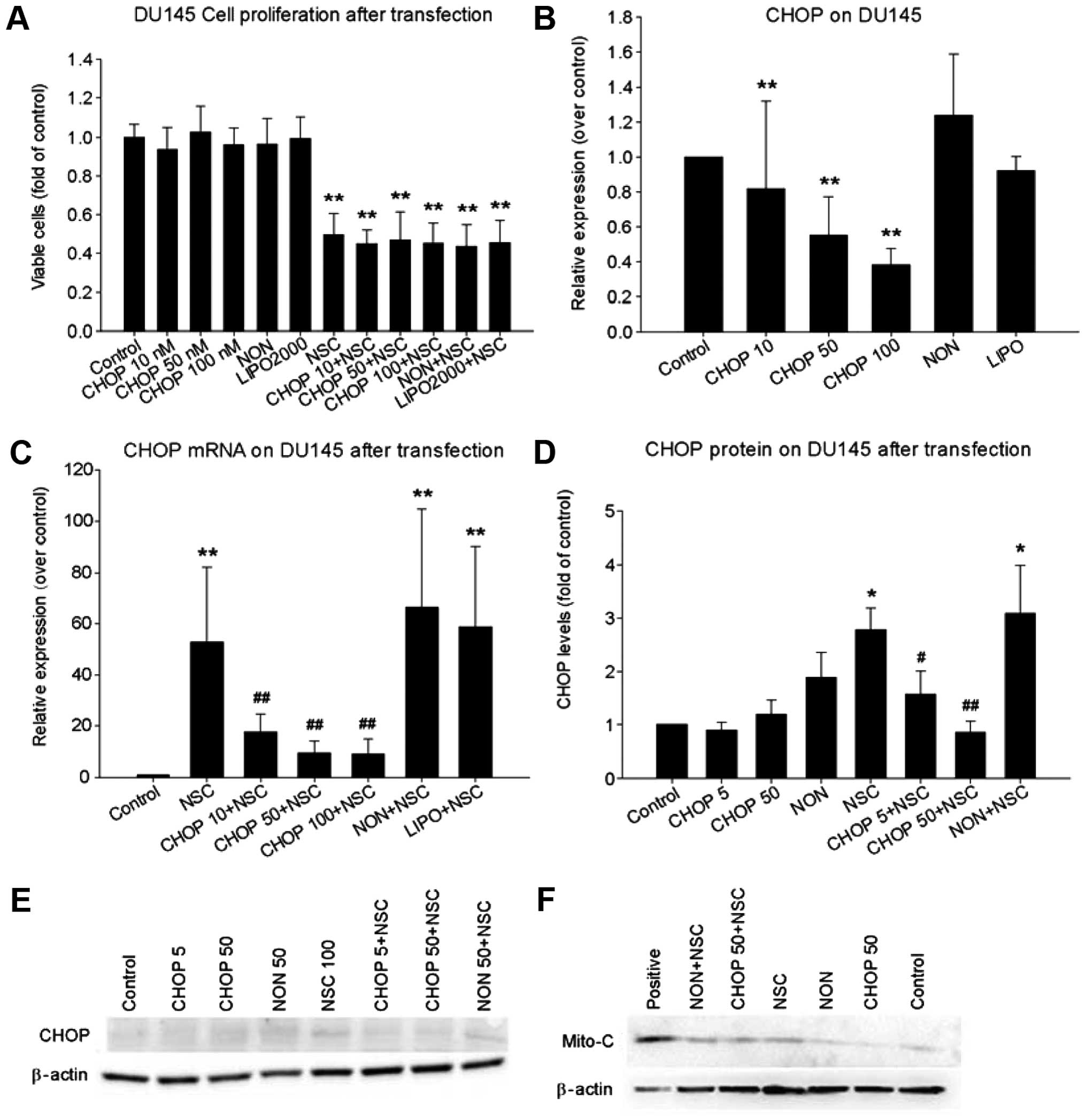
release and cell death. Although transfection of a specific CHOP
siRNA markedly reduced basal (Fig.
7B) and NSC-induced CHOP mRNA and protein expression (Fig. 7C–E), it failed to block NSC-induced
cytochrome c release from mitochondria to cytosol (Fig. 7F) and NSC-induced cell death
(Fig. 7A), indicating that CHOP is
not a major mediator of NSC-induced apoptosis and cell death.
Discussion
In a previous study, we demonstrated that NSC, a
water-soluble camptothecin analog, produced a dose- and
time-dependent inhibition of cell growth and promotion of cell
apoptosis in multiple prostate cancer cell lines (15). In DU145 cells, NSC, through
interaction with topoisomerase 1, activated the mitochondrial
apoptotic pathway, leading to cytochrome c release from
mitochondria into the cytosol and a caspase-dependent cell
apoptosis (15). To further
elucidate the effect and mechanism of NSC in prostate cancer cells,
we studied NSC-induced ER-stress and its association with
NSC-induced cell apoptosis in this study. The results indicate that
NSC produced atypical ER-stress that was dissociated from
NSC-induced cell death.
The ER is an essential cellular organelle
responsible for protein synthesis, maturation, folding,
transportation, calcium storage, biosynthesis of lipids and sterols
(19). ER-stress is the
consequence of disequilibrium between the ER folding capacity and
the accumulation of unfolded or misfolded proteins in the ER
(37). To cope with ER-stress, the
cell triggers UPR, initiated via the dissociation of GRP78 from the
three ER-stress sensors: PERK, IRE1α and ATF6 (Fig. 1) (18,38).
When PERK is activated, it phosphorylates eukaryotic
translation-initiation factor 2α (eIF2α), reducing mRNA translation
and biosynthetic protein-folding load in the ER. Once IRE1α is
activated, it functions as an endoribonuclease to remove a
26-nucleotide region from unspliced X-box binding protein 1 (XBP1u)
mRNA, generating a spliced XBP1 (XBP1s) mRNA that yields an active
transcription factor (XBP1s) to regulate the expression of various
subsets of genes involved in protein folding (18,38).
On the other hand, the activated IRE1α causes a degradation of
selective mRNAs, resulting in an overall decrease in protein
biosynthesis. When ATF6 is activated, it can decrease ER protein
load via signaling proteolysis in Golgi. These responses
collectively result in an upregulation of genes involved in protein
folding, maturation and transport such as GRP78, and genes in
ER-associated protein degradation (ERAD), leading to a greater
chance of cell survival (Fig. 1).
However, chronic or overwhelming ER-stress elicits apoptotic
signals such as CHOP expression followed by an activation of
intrinsic mitochondrial apoptotic pathway (18,37).
In this study, we observed that NSC produced a dose- and
time-dependent elevation of GRP78, XBP1s and CHOP mRNAs (Fig. 3), three ER-stress biomarkers,
suggesting that NSC induces ER-stress in Pca cells. However, unlike
the classic ER-stress in which GRP78 protein is elevated, NSC
produced a time- and dose-dependent U-shaped change in GRP78
protein (Fig. 3C), suggesting that
NSC-induced ER-stress in DU145 cells is distinguished from the
conventional ER-stress induced by other stimuli.
GRP78, an ER protein chaperone, has greater affinity
to unfolded or misfolded proteins and therefore, when these
proteins are accumulated in the ER due to various stimuli, GRP78
dissociates from its receptors, triggering the UPR and the
activation of three signal pathways (Fig. 1) (18,39).
In return, GRP78 as an endpoint marker of the UPR, is regulated by
all the three branches of the UPR activated by various stimuli
(40–45). GRP78 expression is generally
expected to be elevated during ER-stress although the observable
increase in protein accumulation usually occurs more slowly than
the accumulation of mRNA due in part to the long half-life of GRP78
protein (37,46). In agreement with this concept, the
expression of GRP78 mRNA was elevated in a time-dependent manner
upon NSC treatment. In contrast, GRP78 protein level significantly
decreased between 12 and 36 h, before recovering back to control
levels around 72 h post-NSC treatment, forming a time-dependent
U-shaped change (Fig. 3). Although
a decrease in GRP78 protein levels is unusual, such ER-stress
associated GRP78 reduction has been reported by other investigators
in different cells (47,48). It is obvious that the decrease in
GRP78 protein is not due to a decrease in GRP78 mRNA since NSC
induced a time-dependent increase in GRP78 mRNA levels. We
therefore investigated the hypothesis that NSC induces an
acceleration of GRP78 protein degradation in DU145 cells. ER
associated degradation (ERAD) (Fig.
7), a process which directs unfolded or misfolded proteins for
destruction by the cytoplasmic proteasome pathways, is enhanced
when cells are subjected to ER-stress (18,37,49).
ERAD involves the chaperone of unfolded or misfolded proteins by
GRP78 to the cytosol and then degradation of these proteins through
either ubiquitin-proteasome or autophagy-lysosome pathway. To
determine the role of ubiquitin-proteasome pathway in NSC-induced
GRP78 reduction, EPO, a specific ubiquitin-proteasome inhibitor
(50), was co-administered with
NSC in DU145 cells and it prevented to some extent the NSC-induced
decrease in GRP78 protein levels (Fig.
4). However, since EPO alone markedly increased GRP78 protein
levels, the results do not allow us to conclude that GRP78
degradation via ubiquitin-proteasome pathway alone can explain the
NSC-induced reduction in GRP78 protein levels. We then explored a
second intracellular pathway of protein degradation, the
autophagy-lysosome pathway, a process involving multiple proteases
including cysteine protease calpain 1, cathepsin B and cathepsin L
(28,51,52).
Using a specific cysteine protease inhibitor, ALLN, we observed
that ALLN blocked NSC-induced reduction of GRP78 protein in a
dose-dependent manner while ALLN alone did not alter GRP78 protein
levels (Fig. 4). Taken together,
the data suggest that NSC-induced decreases in the GRP78 protein
are mediated through protein degradation, mainly the
autophagy-lysosome pathway.
It is well documented that ER-stress-triggered UPR
activates simultaneously both adaptive and pro-apoptotic pathways
(Fig. 1). During chronic or
overwhelming ER-stress, the cells enter apoptotic death when the
adaptive responses are unable to resolve ER-stress. There are
multiple pathways involved in ER-stress-induced cell apoptosis or
cell death and the major and best-characterized pathway is through
the production of CHOP (37,49).
CHOP initiated pro-apoptotic programs involve the downregulation of
Bcl2 which leads to outer mitochondrial membrane permeabilization
(53), induction of oxidative
stress (54), upregulation of
death receptor 5 and activation of caspase-8 (55). CHOP expression in most cells is low
under non-stressed conditions, and increased in response to
ER-stress through IRE1-, PERK-, and ATF6-dependent transcriptional
induction (Fig. 1) (18,20,37).
CHOP has been reported to mediate ER-stress-induced cell apoptosis
in prostate cancer cells (56).
Overexpression of CHOP induces cell cycle arrest and cell apoptosis
in several cell lines, whereas CHOP-deficient cells are protected
from ER-stress-induced apoptosis (20,34,37).
Taken together, the data suggest that CHOP is a key mediator of
ER-stress-related cell apoptosis.
Consistent with the concept of overwhelming
ER-stress, CHOP expression at both the mRNA and protein levels was
greatly increased following NSC treatment in this study (Fig. 3). To determine whether NSC-induced
ER-stress-CHOP activation is involved in NSC-induced cell apoptotic
death through an activation of the intrinsic mitochondrial
apoptotic pathway (15), two
approaches were applied: pretreatment with chemical chaperons and
knockdown of CHOP. Chemical chaperones are known to reduce
ER-stress by facilitating protein folding and accelerating ER
protein trafficking (57).
Previous studies have shown that 4-PBA and TUDCA, two chemical
chaperons, were able to inhibit ER-stress associated gene
expression including CHOP and cell apoptotic death initiated by
various stimuli (29,58–61).
We therefore pretreated cells with either 4-PBA or TUDA at various
doses to test if they inhibit NSC-induced ER-stress and cell death.
Unexpectedly, both chemical chaperons failed to inhibit NSC-induced
GRP78, XBP1s and CHOP mRNA expression (Fig. 5), and NSC-induced cell apoptotic
death (Fig. 6) although they
significantly inhibited CHOP mRNA expression stimulated by TM
(Fig. 5A and data not shown), a
typical ER-stress inducer (34).
Furthermore, to define the role of CHOP protein in NSC-induced cell
death, a specific CHOP siRNA was used to knockdown CHOP expression.
Again, a complete knockdown of NSC-induced CHOP expression did not
block NSC-induced cytochrome c release (Fig. 7F) and cell apoptotic death
(Fig. 7A). The data collectively
suggest that the ER-stress-CHOP pathway is not a major player in
modulating NSC-induced cell apoptotic death. The reasons why
chemical chaperons, 4-PBA and TUDCA, and CHOP knockdown failed to
alleviate NSC-induced changes in DU145 cells are currently unknown.
It is possible that NSC induces an atypical ER-stress that is not
modified by these chemical chaperons. Alternatively, TUDCA and
4-PBA may display other biological actions in addition to their
role as inhibitors of the ER-stress response, which may cause
complicated unpredictable effects. Finally, the action pathway(s)
for NSC induction of this gene expression and cell apoptotic death
may completely deviate from the ER stress-CHOP pathway although we
cannot exclude the possibility that low levels of CHOP is
sufficient to induce cell apoptosis.
It is interesting to note that blockade of
NSC-induced GRP78 protein reduction by ALLN resulted in a partial
recovery of NSC-induced cell death (Fig. 4C), suggesting that a decrease in
GRP78 protein and/or an activation of the autophagy pathway may
involve NSC-induced cell death. Previous studies have indicated
that GRP78 was an anti-apoptotic factor (46), and GRP78 overexpression was able to
protect against camptothecin-induced cell apoptotic death (62). However, an increase in GRP78
protein alone is not sufficient to protect NSC-induced cell death
since EPO blocked NSC-induced reduction of GRP78 protein and
markedly elevated GRP78 protein levels, but failed to inhibit
NSC-induced cell death (Fig. 4C).
Actually, EPO alone produced a marked time- and dose-dependent
induction of cell death in DU145 cells (Fig. 4D), consistent with previous reports
in other systems (63). Taken
together, these results support the hypothesis that a partial
blockade of NSC-induced cell death by ALLN involves the autophagy
pathway. The relationship between NSC-induced ER-stress, intrinsic
mitochondrial apoptosis and autophagy is currently under
investigation in this laboratory.
In conclusion, this study in Pca cells demonstrated
that NSC produced an atypical ER-stress characterized by an
elevation of XBP1s and CHOP expression, an opposite change in GRP78
mRNA and protein levels and a failure of alleviation by chemical
chaperons. The NSC-induced decrease in GRP78 protein levels is most
likely mediated through protein degradation, mainly the
autophagy-lysosome pathway. Furthermore, both chemical chaperons
and CHOP knockdown failed to inhibit NSC-induced cell apoptosis
while inhibition of cysteine proteases partially restored
NSC-induced cell death. The data collectively suggest that even
though ER-stress is associated with NSC-induced alterations in
DU145 cells, it is dissociated from NSC-induced cell apoptotic
death. As a result, elevation of GRP78 mRNA levels may be a more
reliable marker of ER-stress than protein levels. This study also
provides a clue that NSC may activate cell autophagy, an innovative
hypothesis that is under investigation.
Acknowledgements
This study was supported in part by a Cohen Research
Fund from the Division of Endocrinology, Weill Cornell Medicine,
and a Youth-Talent Fund (N-2013-09) from the First People's
Hospital of Chenzhou City in China.
Abbreviations:
|
4-PBA
|
4-phenylbutyric acid
|
|
ALLN
|
N-acetyl-L-leucyl-L-leucylnorleucinal
|
|
ATF6
|
activating transcription factor 6
|
|
CHOP/GAD153
|
growth arrest- and DNA
damage-inducible gene 153
|
|
CRPC
|
castration-resistant prostate
cancer
|
|
EPO
|
epoxomicin
|
|
ER
|
endoplasmic reticulum
|
|
FBS
|
fetal bovine serum
|
|
GRP78
|
glucose regulated protein 78
|
|
HIF-1
|
hypoxia-inducible factor 1
|
|
IRE1
|
inositol-requiring enzyme 1
|
|
NSC
|
NSC606985
|
|
PERK
|
pancreatic ER kinase (PKR)-like ER
kinase
|
|
Top1
|
topoisomerase 1
|
|
TUDCA
|
tauroursodeoxycholic acid
|
|
UPR
|
unfold protein response
|
|
VEGF
|
vascular endothelial growth factor
|
|
XBP1s
|
splicing X-box-binding protein-1
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hellerstedt BA and Pienta KJ: The current
state of hormonal therapy for prostate cancer. CA Cancer J Clin.
52:154–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bubendorf L, Schöpfer A, Wagner U, Sauter
G, Moch H, Willi N, Gasser TC and Mihatsch MJ: Metastatic patterns
of prostate cancer: An autopsy study of 1,589 patients. Hum Pathol.
31:578–583. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heidenreich A, Aus G, Bolla M, Joniau S,
Matveev VB, Schmid HP and Zattoni F; European Association of
Urology. EAU guidelines on prostate cancer. Eur Urol. 53:68–80.
2008. View Article : Google Scholar
|
|
5
|
Antonarakis ES and Eisenberger MA:
Expanding treatment options for metastatic prostate cancer. N Engl
J Med. 364:2055–2058. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shah RB, Mehra R, Chinnaiyan AM, Shen R,
Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA,
et al: Androgen-independent prostate cancer is a heterogeneous
group of diseases: Lessons from a rapid autopsy program. Cancer
Res. 64:9209–9216. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Petrylak DP, Tangen CM, Hussain MH, Lara
PN Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M,
et al: Docetaxel and estramustine compared with mitoxantrone and
prednisone for advanced refractory prostate cancer. N Engl J Med.
351:1513–1520. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tannock IF, de Wit R, Berry WR, Horti J,
Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, et
al; TAX 327 Investigators. Docetaxel plus prednisone or
mitoxantrone plus prednisone for advanced prostate cancer. N Engl J
Med. 351:1502–1512. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wall ME, Ward EC, Cook CE, Palmer KH,
McPhail HT and Sim GA: Plant antitumor agents. I. The isolation and
structure of camptothecin, a novel alkaloidal leukemia and tumor
inhibitor from Camptotheca acuminata. J Am Chem Soc. 88:3888–3890.
1966. View Article : Google Scholar
|
|
10
|
Wall ME: Camptothecin and taxol: Discovery
to clinic. Med Res Rev. 18:299–314. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pizzolato JF and Saltz LB: The
camptothecins. Lancet. 361:2235–2242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pommier Y: Topoisomerase I inhibitors:
Camptothecins and beyond. Nat Rev Cancer. 6:789–802. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rapisarda A, Uranchimeg B, Scudiero DA,
Selby M, Sausville EA, Shoemaker RH and Melillo G: Identification
of small molecule inhibitors of hypoxia-inducible factor 1
transcriptional activation pathway. Cancer Res. 62:4316–4324.
2002.PubMed/NCBI
|
|
14
|
Song MG, Gao SM, Du KM, Xu M, Yu Y, Zhou
YH, Wang Q, Chen Z, Zhu YS and Chen GQ: Nanomolar concentration of
NSC606985, a camptothecin analog, induces leukemic-cell apoptosis
through protein kinase Cdelta-dependent mechanisms. Blood.
105:3714–3721. 2005. View Article : Google Scholar
|
|
15
|
Tan C, Cai LQ, Wu W, Qiao Y,
Imperato-McGinley J, Chen GQ and Zhu YS: NSC606985, a novel
camptothecin analog, induces apoptosis and growth arrest in
prostate tumor cells. Cancer Chemother Pharmacol. 63:303–312. 2009.
View Article : Google Scholar
|
|
16
|
Harding HP, Calfon M, Urano F, Novoa I and
Ron D: Transcriptional and translational control in the Mammalian
unfolded protein response. Annu Rev Cell Dev Biol. 18:575–599.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rutkowski DT and Kaufman RJ: A trip to the
ER: Coping with stress. Trends Cell Biol. 14:20–28. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Binet F and Sapieha P: ER stress and
angiogenesis. Cell Metab. 22:560–575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schröder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Marciniak SJ and Ron D: Endoplasmic
reticulum stress signaling in disease. Physiol Rev. 86:1133–1149.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu YS, Cai LQ, You X, Cordero JJ, Huang Y
and Imperato-McGinley J: Androgen-induced prostate-specific antigen
gene expression is mediated via dihydrotestosterone in LNCaP cells.
J Androl. 24:681–687. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai J, Hong Y, Weng C, Tan C,
Imperato-McGinley J and Zhu YS: Androgen stimulates endothelial
cell proliferation via an androgen receptor/VEGF/cyclin A-mediated
mechanism. Am J Physiol Heart Circ Physiol. 300:H1210–H1221. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wen J, Zhao Y, Li J, Weng C, Cai J, Yang
K, Yuan H, Imperato-McGinley J and Zhu YS: Suppression of
DHT-induced paracrine stimulation of endothelial cell growth by
estrogens via prostate cancer cells. Prostate. 73:1069–1081. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baydas G, Reiter RJ, Akbulut M, Tuzcu M
and Tamer S: Melatonin inhibits neural apoptosis induced by
homocysteine in hippocampus of rats via inhibition of cytochrome c
translocation and caspase-3 activation and by regulating pro- and
anti-apoptotic protein levels. Neuroscience. 135:879–886. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kisselev AF and Goldberg AL: Proteasome
inhibitors: From research tools to drug candidates. Chem Biol.
8:739–758. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Villalpando Rodriguez GE and Torriglia A:
Calpain 1 induce lysosomal permeabilization by cleavage of
lysosomal associated membrane protein 2. Biochim Biophys Acta.
1833:2244–2253. 2013. View Article : Google Scholar
|
|
29
|
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M,
Vaillancourt E, Smith RO, Görgün CZ and Hotamisligil GS: Chemical
chaperones reduce ER stress and restore glucose homeostasis in a
mouse model of type 2 diabetes. Science. 313:1137–1140. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Benz C, Angermüller S, Töx U,
Klöters-Plachky P, Riedel HD, Sauer P, Stremmel W and Stiehl A:
Effect of tauroursodeoxycholic acid on bile-acid-induced apoptosis
and cytolysis in rat hepatocytes. J Hepatol. 28:99–106. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kubota K, Niinuma Y, Kaneko M, Okuma Y,
Sugai M, Omura T, Uesugi M, Uehara T, Hosoi T and Nomura Y:
Suppressive effects of 4-phenylbutyrate on the aggregation of Pael
receptors and endoplasmic reticulum stress. J Neurochem.
97:1259–1268. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luo ZF, Feng B, Mu J, Qi W, Zeng W, Guo
YH, Pang Q, Ye ZL, Liu L and Yuan FH: Effects of 4-phenylbutyric
acid on the process and development of diabetic nephropathy induced
in rats by streptozotocin: Regulation of endoplasmic reticulum
stress-oxidative activation. Toxicol Appl Pharmacol. 246:49–57.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wiley JC, Meabon JS, Frankowski H, Smith
EA, Schecterson LC, Bothwell M and Ladiges WC: Phenylbutyric acid
rescues endoplasmic reticulum stress-induced suppression of APP
proteolysis and prevents apoptosis in neuronal cells. PLoS One.
5:e91352010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zinszner H, Kuroda M, Wang X, Batchvarova
N, Lightfoot RT, Remotti H, Stevens JL and Ron D: CHOP is
implicated in programmed cell death in response to impaired
function of the endoplasmic reticulum. Genes Dev. 12:982–995. 1998.
View Article : Google Scholar
|
|
35
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Watanabe Y, Tsuchiya H, Sakabe T, Matsuoka
S, Akechi Y, Fujimoto Y, Yamane K, Ikeda R, Nishio R, Terabayashi
K, et al: CD437 induces apoptosis in ovarian adenocarcinoma cells
via ER stress signaling. Biochem Biophys Res Commun. 366:840–847.
2008. View Article : Google Scholar
|
|
37
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar
|
|
38
|
Logue SE, Cleary P, Saveljeva S and Samali
A: New directions in ER stress-induced cell death. Apoptosis.
18:537–546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar
S, Nie D, Myers MG Jr and Ozcan U: Endoplasmic reticulum stress
plays a central role in development of leptin resistance. Cell
Metab. 9:35–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Deldicque L, Cani PD, Philp A, Raymackers
JM, Meakin PJ, Ashford ML, Delzenne NM, Francaux M and Baar K: The
unfolded protein response is activated in skeletal muscle by
high-fat feeding: Potential role in the downregulation of protein
synthesis. Am J Physiol Endocrinol Metab. 299:E695–E705. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nivala AM, Reese L, Frye M, Gentile CL and
Pagliassotti MJ: Fatty acid-mediated endoplasmic reticulum stress
in vivo: Differential response to the infusion of Soybean and Lard
Oil in rats. Metabolism. 62:753–760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nakamura S, Takizawa H, Shimazawa M,
Hashimoto Y, Sugitani S, Tsuruma K and Hara H: Mild endoplasmic
reticulum stress promotes retinal neovascularization via induction
of BiP/ GRP78. PLoS One. 8:e605172013. View Article : Google Scholar
|
|
45
|
Pierre N, Deldicque L, Barbé C, Naslain D,
Cani PD and Francaux M: Toll-like receptor 4 knockout mice are
protected against endoplasmic reticulum stress induced by a
high-fat diet. PLoS One. 8:e650612013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee AS: GRP78 induction in cancer:
Therapeutic and prognostic implications. Cancer Res. 67:3496–3499.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
D'Hertog W, Maris M, Ferreira GB,
Verdrengh E, Lage K, Hansen DA, Cardozo AK, Workman CT, Moreau Y,
Eizirik DL, et al: Novel insights into the global proteome
responses of insulin-producing INS-1E cells to different degrees of
endoplasmic reticulum stress. J Proteome Res. 9:5142–5152. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rosengren V, Johansson H, Lehtiö J,
Fransson L, Sjöholm A and Ortsäter H: Thapsigargin down-regulates
protein levels of GRP78/BiP in INS-1E cells. J Cell Biochem.
113:1635–1644. 2012.
|
|
49
|
Scheuner D and Kaufman RJ: The unfolded
protein response: A pathway that links insulin demand with
beta-cell failure and diabetes. Endocr Rev. 29:317–333. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Meng L, Mohan R, Kwok BH, Elofsson M, Sin
N and Crews CM: Epoxomicin, a potent and selective proteasome
inhibitor, exhibits in vivo antiinflammatory activity. Proc Natl
Acad Sci USA. 96:10403–10408. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bird PI, Trapani JA and Villadangos JA:
Endolysosomal proteases and their inhibitors in immunity. Nat Rev
Immunol. 9:871–882. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Turk V, Stoka V, Vasiljeva O, Renko M, Sun
T, Turk B and Turk D: Cysteine cathepsins: From structure, function
and regulation to new frontiers. Biochim Biophys Acta. 1824:68–88.
2012. View Article : Google Scholar
|
|
53
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lu M, Lawrence DA, Marsters S,
Acosta-Alvear D, Kimmig P, Mendez AS, Paton AW, Paton JC, Walter P
and Ashkenazi A: Opposing unfolded-protein-response signals
converge on death receptor 5 to control apoptosis. Science.
345:98–101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shiraishi T, Yoshida T, Nakata S, Horinaka
M, Wakada M, Mizutani Y, Miki T and Sakai T: Tunicamycin enhances
tumor necrosis factor-related apoptosis-inducing ligand-induced
apoptosis in human prostate cancer cells. Cancer Res. 65:6364–6370.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Engin F and Hotamisligil GS: Restoring
endoplasmic reticulum function by chemical chaperones: An emerging
therapeutic approach for metabolic diseases. Diabetes Obes Metab.
12(Suppl 2): 108–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ben Mosbah I, Alfany-Fernández I, Martel
C, Zaouali MA, Bintanel-Morcillo M, Rimola A, Rodés J, Brenner C,
Roselló-Catafau J and Peralta C: Endoplasmic reticulum stress
inhibition protects steatotic and non-steatotic livers in partial
hepatectomy under ischemia-reperfusion. Cell Death Dis. 1:e522010.
View Article : Google Scholar
|
|
59
|
Arrojo E, Drigo R, Fonseca TL, Castillo M,
Salathe M, Simovic G, Mohácsik P, Gereben B and Bianco AC:
Endoplasmic reticulum stress decreases intracellular thyroid
hormone activation via an eIF2a-mediated decrease in type 2
deiodinase synthesis. Mol Endocrinol. 25:2065–2075. 2011.
View Article : Google Scholar
|
|
60
|
Duricka DL, Brown RL and Varnum MD:
Defective trafficking of cone photoreceptor CNG channels induces
the unfolded protein response and ER-stress-associated cell death.
Biochem J. 441:685–696. 2012. View Article : Google Scholar :
|
|
61
|
Koyama M, Furuhashi M, Ishimura S, Mita T,
Fuseya T, Okazaki Y, Yoshida H, Tsuchihashi K and Miura T:
Reduction of endoplasmic reticulum stress by 4-phenylbutyric acid
prevents the development of hypoxia-induced pulmonary arterial
hypertension. Am J Physiol Heart Circ Physiol. 306:H1314–H1323.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Reddy RK, Mao C, Baumeister P, Austin RC,
Kaufman RJ and Lee AS: Endoplasmic reticulum chaperone protein
GRP78 protects cells from apoptosis induced by topoisomerase
inhibitors: Role of ATP binding site in suppression of caspase-7
activation. J Biol Chem. 278:20915–20924. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fu HY, Minamino T, Tsukamoto O, Sawada T,
Asai M, Kato H, Asano Y, Fujita M, Takashima S, Hori M, et al:
Overexpression of endoplasmic reticulum-resident chaperone
attenuates cardiomyocyte death induced by proteasome inhibition.
Cardiovasc Res. 79:600–610. 2008. View Article : Google Scholar : PubMed/NCBI
|