Introduction
Osteosarcoma is the primary cancer of leaf tissue,
which mostly occurs in the long bone metaphysis of teenagers,
especially in the distal femur and proximal tibia. Because of its
high malignancy degree and rapid development, the morbidity and
mortality are high. Emerging evidence suggests that osteosarcoma
should be regarded as a differentiation disease caused by genetic
and epigenetic changes which interrupt the osteoblast
differentiation from mesenchymal stem cells (1), but the specific pathological
processes and molecular mechanism remains unclear.
The enhancer of zeste homolog 2 (EZH2) is a
catalytic subunit of polycomb repressive complex 2 (PRC2) and is
upregulated in different human cancers, such as osteosarcoma
(2), prostate cancer (3), breast cancer (4) and liver cancer (5). EZH2 has been demonstrated to be
involved in a variety of biological processes, such as cell
proliferation and program cell death (6). It has been suggested that EZH2
contributes to gene silencing by mediating DNA methylation and
histone modifications (7). EZH2
impairs gene expression by catalyzing the tri-methylation of
histone H3 lysine 27 (H3K27me3) that interacts with its target
genes (8,9). H3K27me3 is supposed to be a
repressive histone modification to control gene transcription
epigenetically. Prior studies have shown that c-Myc co-expression
with EZH2 is used in determining the prognosis of prostate cancer
patients after surgery (10) and
confirmed that c-Myc is a direct target of EZH2 in glioblastoma
cancer stem cells (11). EZH2
induces c-Myc expression via the inhibition of Myc targeting
miR-494 (12,13). As feedback, c-Myc contributes to
EZH2 upregulation via the repression of EZH2 targeting miR-26a
(12,13). Thus EZH2 and c-Myc regulate each
other in a loop manner.
c-Myc is one of the most commonly overexpressed
oncogenes in cancer (14) and is a
transcription factor involved in DNA replication, cell
proliferation, protein synthesis, chromatin structure,
differentiation and stem cell fate by regulating target gene
expressions (14–16). Except EZH2, the far upstream
element (FUSE) binding protein 1 (FBP1) is another critical
regulator of c-Myc and was discovered because of its functions on
c-Myc (17). FBP1 is a single
stranded DNA and RNA-binding protein that plays an important role
in cell proliferation, survival, metastasis and RNA virus
replication (18–24). Aberrant expression of FBP1 has been
found in a variety of malignant tissues (25,26),
including in ovarian cancer tissue (our unpublished data).
In the past few years, several potent inhibitors of
EZH2 have been discovered. Roughly, they can be divided into
S-adenosyl-L-homocysteine (SAH) hydrolase inhibitor, such as
3-deazaneplanocin A (DZNep); or S-adenosyl-L-methionine (SAM)
competitive inhibitor, such as GSK343 (27,28).
SAM is a universal methyl donor for catalytic reaction of histone
methyltransferase (28,29).
In order to increase our understanding on GSK343 as
a molecule for clinical osteosarcoma treatment, we investigated the
influence of GSK343 on cell viability and program cell death in
osteosarcoma cells and the potential mechanisms underlying its
activity. We found that GSK343 treatment inhibited cell viability
and promotes programmed cell death of the osteosarcoma cells.
Attenuating EZH2 and FBP1 expression are potential underlying
mechanisms.
Materials and methods
Cell culture
Human osteosarcoma cells, Saos2, were cultured in
complete Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% (v/v) fetal bovine serum (FBS), 2 mmol/l L-glutamine, 100
U/ml penicillin, 100 μg/ml streptomycin. All cells were maintained
at 37°C and 5% CO2 in a humidified incubator.
Antibodies and reagents
LC3, H3K27me3, GAPDH antibodies were purchased from
Cell Signaling Technology (Danvers, MA, USA). EZH2 antibody was
obtained from BD Technology (Research Triangle Park, NA, USA). All
antibodies were used at the concentrations recommended by the
supplier. DMEM medium, FBS and L-glutamine were obtained from Gibco
(Gaithersburg, MD, USA). GSK343, FBP1, c-Myc and 3-methyladenine
(3-MA) were provided by Sigma Chemical (St. Louis, MO, USA). The
CellTiter 96® AQueous One Solution proliferation assay
kit
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt; MTS) was purchased from Promega (Madison, WI, USA).
Penicillin and streptomycin sulfate were purchased from Hyclone
(Logan, UT, USA).
Cell viability assay
Saos-2 cells were seeded at 5.0×103
cells/well in a 96-well plate and treated with different
concentrations of GSK343 for 48 h. Cell viability was then measured
using the MTS assay following the manufacturer's manual. The
absorbance at 490 nm was measured on a 96-well plate reader. The
experiment was repeated at least three times.
Flow cytometric analysis
Flow cytometry was used for cell cycle distribution
analysis and apoptosis detection. For apoptosis detection, the
Saos-2 cells were cultured in 6-well plates. After 48 h of
treatment with different concentrations of GSK343, cells were
collected and incubated with Annexin V-fluorescein isothiocyanate
(FITC) (Beijing Biosea Biotechnology Co., Ltd., Beijing, China) for
30 min at 4°C in the dark, and then incubated with propidium iodide
(PI) (Beijing Biosea Biotechnology Co., Ltd.) for 5 min. Analysis
was immediately performed by flow cytometry (FACSAria II,
Becton-Dickinson, Franklin Lakes, NJ, USA). For cell cycle
analysis, the Saos-2 cells were trypsinized and fixed in 70% (v/v)
ethanol overnight at 4°C. Then cells were collected and resuspended
in phosphate-buffered saline (PBS) containing 50 μg/ml of PI. Cell
cycle analyses were performed using FACS Aria II
(Becton-Dickinson).
Western blot analysis
Cells were lysed for western blotting in modified
RIPA buffer [150 mmol/l NaCl, 1% NP-40, 50 mmol/l Tris-Cl (pH 8.0),
0.1% SDS] supplemented with protease and phosphatase inhibitor,
PMSF (1 mmol/l). After rapid homogenization, the homogenate was
incubated in ice for 30 min and centrifuged at 12,000 g for 15 min
at 4°C. Protein concentration was determined by bicinchoninic acid
protein assay (Pierce, MO, USA). The protein samples (30–50 μg)
were resolved by SDS-PAGE (12%) and transferred to PVDF membranes
(Millipore, Boston, MI, USA). Following transfer, membranes were
blocked with 5% skim milk in TBS-Tween (0.05 M Tris, 0.15 M NaCl,
pH 7.5, and 0.2% Tween-20) for 1 h, and then incubated at 4°C
overnight with primary antibodies. Membranes were incubated with
anti-rabbit/mouse HRP-labeled secondary antibodies for 1 h at room
temperature after washing, and detected with ECL-Plus (Pierce).
Relative abundance was quantified by densitometry using Quantity
One 4.6.7 software (Bio-Rad, Philadelphia, PA, USA).
Laser confocal immunofluorescence
microscopy
The cells were treated with different concentrations
(0–20 μmol/l) of GSK343 for 48 h, and then fixed in 4%
paraformaldehyde (PFA) for 15 min, washed in PBS, blocked using 10%
normal goat serum, and incubated with a 1:100 dilution of rabbit
polyclonal antibody against LC3 overnight at 4°C. FITC-conjugated
anti-rabbit IgG were applied at a 1:200 dilution for 1 h. The cells
were counterstained with DAPI and observed immediately using a
Zeiss confocal microscope (Oberkochen, Germany).
Co-immunoprecipitation (Co-IP)
Cultured Saos-2 cells were collected in lysis buffer
(150 mmol/l NaCl, 50 mmol/l Tris-HCl, 1 mmol/l EDTA, pH 7.5, 1%
NP-40) containing protease inhibitor (Roche, CA, USA) and kept in
ice for 30 min after washed twice with PBS. After sonication and
centrifugation, the whole-cell lysate was made. Whole-cell lysates
were incubated with either anti-EZH2 antibody or IgG (Abcam,
Cambridge, UK) at 4°C with rotation for 2 h, then were incubated
with prepared Protein A+G agarose beads (Santa Cruz, CA, USA) at
4°C overnight. The precipitations were washed four times with lysis
buffer, and were eluted in SDS-PAGE loading buffer by boiling for 5
min. The supernatants were then resolved by SDS-PAGE and
transferred to PVDF membranes. Immunoblotting using appropriate
antibodies was conducted using standard protocol.
Statistical analysis and data
presentation
All data shown represented one of at least three
independent experiments and were expressed as mean ± SD. Statistics
was performed using Students't test. P<0.05 was considered
statistically significant difference.
Results
GSK343 inhibits the viability of
osteosarcoma Saos-2 cells
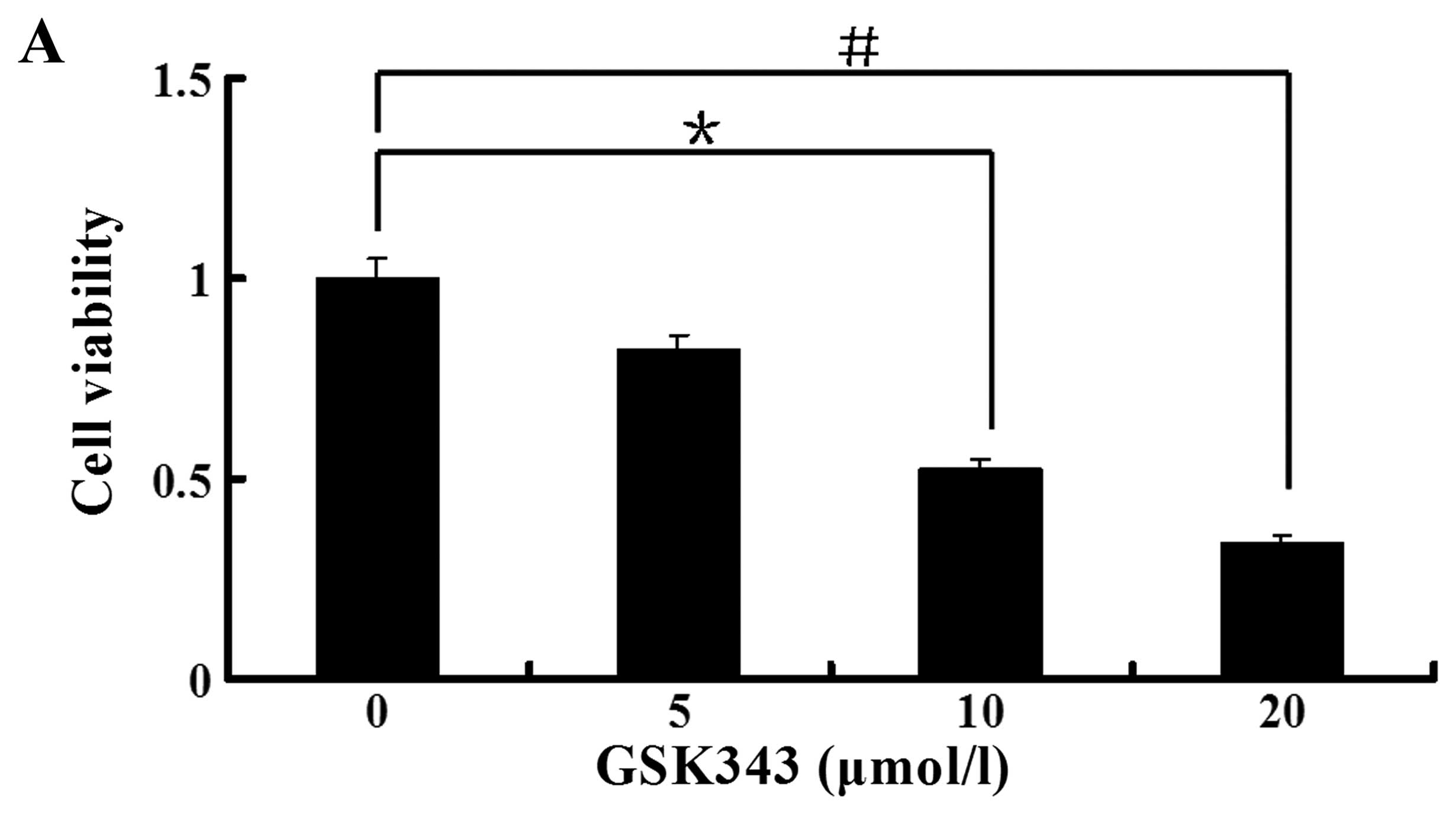
We assessed the effect of GSK343 on the viability of
osteosarcoma Saos-2 cells. Cell viability was examined with MTS
assay. Ten and 20 μmol/l of GSK343 inhibited significantly Saos-2
cell viability, and the cell viability decreased to 52 and 34% for
10 and 20 μmol/l of GSK343, respectively (Fig. 1A). GSK343 inhibited cell viability
in a dose-dependent manner by the concentration of 20 μmol/l.
GSK343 induces cell cycle arrest and
apoptosis in osteosarcoma Saos-2 cells
In order to elucidate the factors which decreased
cell viability, we investigated cell cycle transition and
apoptosis, namely type I programmed cell death, in Saos-2 cells
treated with GSK343 by flow cytometry. A significant elevation in
G1-phase and an evident decline in S-phase cells were observed
(Fig. 1B). GSK343 (20 μmol/l)
increased the percentage of G1-phase Saos-2 cells from 45.66 to
53.85% (p<0.05), and reduced the percentage of S-phase and
G2-phase cells from 37.02 to 31.04%, and 17.32 to 15.11%,
respectively. These data indicated that GSK343 blocked Saos-2 cell
cycle progression at the G1-phase and inhibited DNA synthesis.
The Annexin V-FITC/PI assay revealed that GSK343
treatment significantly enhanced Saos-2 apoptosis comparing with
the untreated Saos-2 cells. Treatment with 20 μmol/l of GSK343
increased apoptosis 4.6-fold (Fig. 1C
and D, p<0.01). These data indicated that GSK343 induced
apoptosis.
GSK343 induces autophagy in osteosarcoma
Saos-2 cells
Autophagic cell death is another important
programmed cell death and is known as type II program cell death.
It plays an important role in cell fate. LC3, a hallmark of
mammalian autophagy, is essential for the formation of
autophagosomes and for the completion of macroautophagy (30). LC3-II is an
LC3-phosphatidylethanolamine conjugate and a promising
autophagosomal marker (31). Since
the conversion rate of LC3-I to LC3-II is a hallmark of mammalian
autophagy, we examined the expression of LC3-I and LC3-II and the
conversion rate of LC3-I to LC3-II. As shown in Fig. 2A and B, western blot analysis
showed that LC3-II was significantly upregulated by GSK343
treatment in a dose-dependent manner from 5 to 20 μmol/l.
Furthermore, immunofluorescence assay confirmed LC3-II puncta
formation (red staining, Fig. 2C).
Same as the results of western blot analysis, GSK343 also promoted
LC3-II puncta formation in a dose-dependent manner.
3-MA has been identified as a classical autophagy
inhibitor (32) and widely used in
pharmacokinetic studies of autophagy. To investigate the effects of
3-MA on GSK343-induced autophagy, we treated Saos-2 cells with 5
mmol/l of 3-MA for 2 h before the treatment of 20 μmol/l of GSK343.
Western blot analysis showed that 3-MA attenuated GSK343-induced
LC3-II accumulation while 3-MA alone had no any effect on LC3-II
accumulation (Fig. 2D and E). This
result implied that 3-MA treatment significantly attenuated
GSK343-induced autophagy. Combined together, these findings
indicate that GSK343 treatment induces autophagic cell death in
Saos-2 cells.
GSK343 reduces the expression of EZH2,
H3K27me3 and c-Myc in Saos-2 cells
The expression of EZH2 protein was tested in GSK343
untreated and treated Saos-2 cells. As shown in Fig. 3A, GSK343 reduced the expression of
EZH2 protein in a dose-dependent manner. In addition, we
investigated the expression of EZH2 target protein, H3K27me3, which
was also reduced by GSK343 in a dose-dependent manner.
A recent study demonstrated that EZH2 promoted c-Myc
expression by attenuating miR-494 which was shown to inhibit c-Myc
expression in aggressive B-cell lymphomas (13). Here, we found that GSK343 not only
inhibited EZH2 expression, but also inhibited c-Myc expression
(Fig. 3B). In addition, we
confirmed the physical interaction between EZH2 and c-Myc by Co-IP
assay. As shown in Fig. 3C,
immunoblot assay detected c-Myc in EZH2 antibody pull-down
solution.
GSK343 attenuates FBP1 expression
As we know, FBP1 was discovered because its function
on c-Myc expression. FBP1 promotes c-Myc expression by binding with
the c-Myc promoter. We found that GSK343 treatment decreased FBP1
expression in Saos-2 cells in a dose-dependent manner (Fig. 3B).
Since c-Myc is the target of EZH2 and FBP1, we
wished to clarify the relationship between EZH2 and FBP1. With the
Co-IP assay, we also confirmed the physical interaction between
EZH2 and FBP1 (Fig. 3D).
Discussion
EZH2, the catalytic subunit of polycomb repressive
complex 2 (PRC2), catalyzes trimethylation of lysine 27 on histone
H3 (H3K27me3) and mediates transcriptional silencing (33,34).
H3K27 trimethylation suppresses transcription of specific genes
that are proximal to the site of histone modification (35). EZH2 is directly implicated in
various cancers as well as osteosarcoma. In this study, we found
that GSK343, one EZH2 methyltransferase inhibitor, inhibited
osteosarcoma cell viability. This result is consistent with the
finding in human hepatocellular carcinoma cells (36).
Low cell viability may be induced by cell cycle
restrain and cell death activation. By flow cytometry, we found
GSK343 treatment significantly suppressed cell cycle transition
from G1 phase to S/G2 phase and activated apoptosis in osteosarcoma
Saos-2 cells (Figs. 1 and 2). Except for activating apoptosis,
GSK343 treatment also activated autophagic cell death. As it is
known, LC3 is a hallmark of autophagy that contains LC3-I and
LC3-II forms. LC3-II is an LC3-phosphatidylethanolamine conjugate
and a promising autophagosomal marker (31). In this study, LC3-II was
significantly upregulated by GSK343 in a dose-dependent manner
(Fig. 2). It was consistent with
our expectation that the pretreatment with 3-MA, an autophagy
inhibitor, decreased GSK343-induced autophagic cell death (Fig. 2). GSK343 treatment induced
programmed cell death in osterosarcoma, including apoptosis and
autophagic cell death.
As it is known, EZH2 can positively regulate c-Myc
expression (37). c-Myc is a
pleiotropic transcription factor that both activates and represses
a broad range of target genes and is indispensable for cell growth
(35). In addition, c-Myc is able
to bind to the promoter of EZH2 and provoke EZH2 overexpression
through suppression of miR-26a (38) and has been identified as a key
regulator of EZH2 expression (12,39).
In this study, we found that GSK343 treatment inhibited the
expression of EZH2 and c-Myc in osteosarcoma cells. Further, we
found the physical interaction between EZH2 and c-Myc.
FBP1 is a critical regulator of the proto-oncogene
c-Myc through binding with the c-Myc promoter and plays important
roles in cell proliferation, survival and metastasis (18). We found that GSK343 treatment not
only inhibited EZH2 expression, but also inhibited FBP1 expression
in osteosarcoma cells (Fig. 3).
This is the first study reporting that GSK343, one of EZH2
inhibitors, inhibits FBP1 expression. Interestingly, we detected a
physical interaction between EZH2 and FBP1 in vivo by Co-IP
assay.
FBP1, EZH2 and c-Myc are all important cell fate
regulators. It is demonstrated that FBP1-c-Myc and EZH2-c-Myc
signal pathways play important roles in cell fate regulation.
Combining our results, we believe that suppression of cell cycle
transition and the activation of programmed cell death of GSK343
are, at least in part, through the inhibition on FBP1-c-Myc and
EZH2-c-Myc signal pathways. It is reported that FBP1 promote
tumor-relevant functions by at least partly employing stathmin, a
microtubule-destabilizing factor (40,41).
On the contrary, EZH2 is reported to inhibit cofilin
phosphorylation, another microtubule stabilization regulator
(42). Except our finding on the
physical interaction between FBP1 and EZH2, there is no other study
on them. Therefore, considering the physical interaction of FBP1
and EZH2 and their functions on microtubule stabilization, it is
reasonable to assume there is a link between FBP1-c-Myc and
EZH2-c-Myc signal pathways. Further research regarding this
connection will be meaningful.
In conclusion, our observations indicate that
GSK343, the EZH2 inhibitor, is a potential molecule for
osteosarcoma clinical treatment. GSK343 inhibits cancer cell
viability by attenuating cell cycle promotion and promoting
programmed cell death. We postulate that the inhibition of GSK343
on c-Myc, EZH2 and FBP1 is one of the potential underlying
mechanisms.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (nos. 81272222, to Z.L.) and Medical
and Health Science and Technology Project of Guangzhou (no.
20161A010019, to X.X.). The authors would like to thank Siyu Liu
from Rutgers, the State University of New Jersey, USA for her help
in manuscript writing. Flow cytometry work was supported by
Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics
and Gene Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen
University.
References
|
1
|
Tang N, Song WX, Luo J, Haydon RC and He
TC: Osteosarcoma development and stem cell differentiation. Clin
Orthop Relat Res. 466:2114–2130. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lv YF, Yan GN, Meng G, Zhang X and Guo QN:
Enhancer of zeste homolog 2 silencing inhibits tumor growth and
lung metastasis in osteosarcoma. Sci Rep. 5:129992015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Varambally S, Dhanasekaran SM, Zhou M,
Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt
RG, Otte AP, et al: The polycomb group protein EZH2 is involved in
progression of prostate cancer. Nature. 419:624–629. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kleer CG, Cao Q, Varambally S, Shen R, Ota
I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al: EZH2
is a marker of aggressive breast cancer and promotes neoplastic
transformation of breast epithelial cells. Proc Natl Acad Sci USA.
100:11606–11611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Au SL, Wong CC, Lee JM, Wong CM and Ng IO:
EZH2-mediated H3K27me3 is involved in epigenetic repression of
deleted in liver cancer 1 in human cancers. PLoS One. 8:e682262013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simon JA and Lange CA: Roles of the EZH2
histone methyl-transferase in cancer epigenetics. Mutat Res.
647:21–29. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schlesinger Y, Straussman R, Keshet I,
Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E,
Reubinoff BE, et al: Polycomb-mediated methylation on Lys27 of
histone H3 pre-marks genes for de novo methylation in cancer. Nat
Genet. 39:232–236. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cao R, Wang L, Wang H, Xia L,
Erdjument-Bromage H, Tempst P, Jones RS and Zhang Y: Role of
histone H3 lysine 27 methylation in Polycomb-group silencing.
Science. 298:1039–1043. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Czermin B, Melfi R, McCabe D, Seitz V,
Imhof A and Pirrotta V: Drosophila enhancer of Zeste/ESC complexes
have a histone H3 methyltransferase activity that marks chromosomal
Polycomb sites. Cell. 111:185–196. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li K, Chen MK, Situ J, Huang WT, Su ZL, He
D and Gao X: Role of co-expression of c-Myc, EZH2 and p27 in
prognosis of prostate cancer patients after surgery. Chin Med J
(Engl). 126:82–87. 2013.
|
|
11
|
Suvà ML, Riggi N, Janiszewska M,
Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino
D, Cironi L, et al: EZH2 is essential for glioblastoma cancer stem
cell maintenance. Cancer Res. 69:9211–9218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Koh CM, Iwata T, Zheng Q, Bethel C,
Yegnasubramanian S and De Marzo AM: Myc enforces overexpression of
EZH2 in early prostatic neoplasia via transcriptional and
post-transcriptional mechanisms. Oncotarget. 2:669–683. 2011.
View Article : Google Scholar
|
|
13
|
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T,
Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al: Coordinated
silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic
target of histone modification in aggressive B-Cell lymphomas.
Cancer Cell. 22:506–523. 2012. View Article : Google Scholar
|
|
14
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kuser-Abali G, Alptekin A and Cinar B:
Overexpression of MYC and EZH2 cooperates to epigenetically silence
MST1 expression. Epigenetics. 9:634–643. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dang CV, O'Donnell KA, Zeller KI, Nguyen
T, Osthus RC and Li F: The c-Myc target gene network. Semin Cancer
Biol. 16:253–264. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Duncan R, Bazar L, Michelotti G, Tomonaga
T, Krutzsch H, Avigan M and Levens D: A sequence-specific,
single-strand binding protein activates the far upstream element of
c-myc and defines a new DNA-binding motif. Genes Dev. 8:465–480.
1994. View Article : Google Scholar
|
|
18
|
Zhang J and Chen QM: Far upstream element
binding protein 1: A commander of transcription, translation and
beyond. Oncogene. 32:2907–2916. 2013. View Article : Google Scholar
|
|
19
|
Matsushita K, Tomonaga T, Shimada H,
Shioya A, Higashi M, Matsubara H, Harigaya K, Nomura F, Libutti D,
Levens D, et al: An essential role of alternative splicing of c-myc
suppressor FUSE-binding protein-interacting repressor in
carcinogenesis. Cancer Res. 66:1409–1417. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zubaidah RM, Tan GS, Tan SB, Lim SG, Lin Q
and Chung MC: 2-D DIGE profiling of hepatocellular carcinoma
tissues identified isoforms of far upstream binding protein (FUBP)
as novel candidates in liver carcinogenesis. Proteomics.
8:5086–5096. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dixit U, Liu Z, Pandey AK, Kothari R and
Pandey VN: Fuse binding protein antagonizes the transcription
activity of tumor suppressor protein p53. BMC Cancer. 14:9252014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dixit U, Pandey AK, Liu Z, Kumar S,
Neiditch MB, Klein KM and Pandey VN: FUSE binding protein 1
facilitates persistent hepatitis C virus replication in hepatoma
cells by regulating tumor suppressor p53. J Virol. 89:7905–7921.
2015. View Article : Google Scholar :
|
|
23
|
Zhang Z, Harris D and Pandey VN: The FUSE
binding protein is a cellular factor required for efficient
replication of hepatitis C virus. J Virol. 82:5761–5773. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chien HL, Liao CL and Lin YL: FUSE binding
protein 1 interacts with untranslated regions of Japanese
encephalitis virus RNA and negatively regulates viral replication.
J Virol. 85:4698–4706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bettegowda C, Agrawal N, Jiao Y, Sausen M,
Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G,
et al: Mutations in CIC and FUBP1 contribute to human
oligodendroglioma. Science. 333:1453–1455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rabenhorst U, Beinoraviciute-Kellner R,
Brezniceanu ML, Joos S, Devens F, Lichter P, Rieker RJ, Trojan J,
Chung HJ, Levens DL, et al: Overexpression of the far upstream
element binding protein 1 in hepatocellular carcinoma is required
for tumor growth. Hepatology. 50:1121–1129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan JZ, Yan Y, Wang XX, Jiang Y and Xu HE:
EZH2: Biology, disease, and structure-based drug discovery. Acta
Pharmacol Sin. 35:161–174. 2014. View Article : Google Scholar :
|
|
28
|
Verma SK, Tian X, LaFrance LV, Duquenne C,
Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley
JA, et al: Identification of potent, selective, cell-active
inhibitors of the histone lysine methyltransferase EZH2. ACS Med
Chem Lett. 3:1091–1096. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Amatangelo MD, Garipov A, Li H,
Conejo-Garcia JR, Speicher DW and Zhang R: Three-dimensional
culture sensitizes epithelial ovarian cancer cells to EZH2
methyltransferase inhibition. Cell Cycle. 12:2113–2119. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N, Yamamoto A, Matsui M,
Yoshimori T and Ohsumi Y: In vivo analysis of autophagy in response
to nutrient starvation using transgenic mice expressing a
fluorescent autophagosome marker. Mol Biol Cell. 15:1101–1111.
2004. View Article : Google Scholar :
|
|
31
|
Asanuma K, Tanida I, Shirato I, Ueno T,
Takahara H, Nishitani T, Kominami E and Tomino Y: MAP-LC3, a
promising autophagosomal marker, is processed during the
differentiation and recovery of podocytes from PAN nephrosis. FASEB
J. 17:1165–1167. 2003.PubMed/NCBI
|
|
32
|
Seglen PO and Gordon PB: 3-Methyladenine:
Specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Radulovi V, de Haan G and Klauke K:
Polycomb-group proteins in hematopoietic stem cell regulation and
hematopoietic neoplasms. Leukemia. 27:523–533. 2013. View Article : Google Scholar
|
|
34
|
Popovic R and Licht JD: Emerging
epigenetic targets and therapies in cancer medicine. Cancer Discov.
2:405–413. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao X, Lwin T, Zhang X, Huang A, Wang J,
Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E and Tao J:
Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell
lymphoma survival and clonogenicity. Leukemia. 27:2341–2350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu TP, Lo HL, Wei LS, Hsiao HH and Yang
PM: S-Adenosyl-L-methionine-competitive inhibitors of the histone
methyltransferase EZH2 induce autophagy and enhance drug
sensitivity in cancer cells. Anticancer Drugs. 26:139–147. 2015.
View Article : Google Scholar
|
|
37
|
Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu
H, Sun L, Zhang Y, Chen Y, Li R, et al: Integration of estrogen and
Wnt signaling circuits by the polycomb group protein EZH2 in breast
cancer cells. Mol Cell Biol. 27:5105–5119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sander S, Bullinger L, Klapproth K,
Fiedler K, Kestler HA, Barth TF, Möller P, Stilgenbauer S, Pollack
JR and Wirth T: MYC stimulates EZH2 expression by repression of its
negative regulator miR-26a. Blood. 112:4202–4212. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kaur M and Cole MD: MYC acts via the PTEN
tumor suppressor to elicit autoregulation and genome-wide gene
repression by activation of the Ezh2 methyltransferase. Cancer Res.
73:695–705. 2013. View Article : Google Scholar :
|
|
40
|
Malz M, Weber A, Singer S, Riehmer V,
Bissinger M, Riener MO, Longerich T, Soll C, Vogel A, Angel P, et
al: Overexpression of far upstream element binding proteins: A
mechanism regulating proliferation and migration in liver cancer
cells. Hepatology. 50:1130–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singer S, Malz M, Herpel E, Warth A,
Bissinger M, Keith M, Muley T, Meister M, Hoffmann H, Penzel R, et
al: Coordinated expression of stathmin family members by far
upstream sequence element-binding protein-1 increases motility in
non-small cell lung cancer. Cancer Res. 69:2234–2243. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ferraro A, Boni T and Pintzas A: EZH2
regulates cofilin activity and colon cancer cell migration by
targeting ITGA2 gene. PLoS One. 9:e1152762014. View Article : Google Scholar : PubMed/NCBI
|