Introduction
Tumor microenvironments have become hotspots in
cancer research; of these environments, the inflammatory
microenvironment plays a decisive role in tumorigenesis. The
accumulated compelling evidence over the last decade indicate the
role for inflammation in tumorigenesis, which has attracted
increased research attention. Collectively, 15–20% of all cancer
deaths are linked to underlying infections and inflammation
(1,2). In particular, hepatocellular
carcinoma (HCC), with a high percentage of >90%, develops
because of chronic liver damage and inflammation (3,4).
Sustainable inflammation causes the overproduction of various
cytokines, growth factors, and cytotoxic media. Cytotoxic media,
such as reactive oxygen species (ROS), nitrogen oxygen species
(NOS), and metalloproteases, can induce oxidative DNA damage, DNA
methylation, and hepatocyte injury. Moreover, the compensatory
hepatocyte regeneration triggered by cytokines and growth factors
enhances the accumulation of genomic damage, thereby accelerating
hepatocarcinogenesis (4–6). The observation that the inflammatory
microenvironment promotes tumorigenesis is supported by
considerable evidence. However, the mechanism through which the
inflammatory microenvironment alters cellular gene expression and
phenotype to promote tumorigenesis has not been elucidated.
Diethylnitrosamine (DEN) is widely used as a potent
hepatocarcinogenic initiator in animal models of carcinogenesis;
this compound induces DNA adduct formation, resulting in DNA
mutation (7,8). To explore the relationship between
hepatitis and liver cancer development, we performed
intraperitoneal injection of DEN on C57/BL mouse to induce in
vivo hepatocellular tumorigenesis. Parallel experiments with a
combination of pyrrolidine dithiocarbamate (PDTC) or
lipopolysaccharide (LPS) with DEN were performed to mimic an
anti-inflammation or pro-inflammation situation, respectively. In
our previous study, we explored the dynamic metabolic changes
during hepatitis and liver cirrhosis; the inflammatory environment
induces remarkable changes in carbohydrate and lipid metabolism,
and d-glucose and
d-mannitol can be
used as potential early diagnostic biomarkers of HCC (9). In this study, we analyzed the
influence of the inflammatory environment on hepatocellular gene
expression and functions through LFQ proteomic technology coupled
with bioinformatics analysis. The present study aimed to reveal
possible regulatory mechanisms of hepatocellular pathological
changes during the precancerous inflammation stage.
Materials and methods
Establishment of hepatitis mouse
model
A mouse model of hepatitis was established using
previously described methods (9).
All chemical compounds used for animal treatment were purchased
from Sigma (USA). These compounds included DEN, phenobarbital (PB),
PDTC, and LPS. PB was used as tumor promoter to increase the
occurrence of cancer. C57BL/6 mice (males, six weeks of age) were
obtained from the Xiamen University Laboratory Animal Center.
C57BL/6 mice were randomly assigned to four groups and
intraperitoneally injected once a week with the following: PBS
(control group, n=12); DEN (DEN group, n=24); DEN and PDTC
(DEN+PDTC group, n=24); or DEN and LPS (DEN+LPS, n=24) for up to 24
weeks. The concentrations of the administered chemicals were based
on the body weight of the mice: DEN, 80 mg/kg; PDTC, 50 mg/kg; and
LPS, 1.25 mg/kg. PB (0.05%) was added to the drinking water on the
10th week since the first DEN injection to enhance DEN-induced
tumor formation. After 12 weeks of treatment, four mice from each
treatment group and two mice from the control group were sacrificed
per month to collect liver samples. All animal handling procedures
were approved by the Institutional Review Board of Xiamen
University.
Hematoxylin and eosin staining
Liver tissues from mice were fixed in 4%
paraformaldehyde for 12–24 h and embedded in paraffin.
Subsequently, 5-μm sections were prepared with a microtome, placed
on clean polylysine-coated slides, and then heated at 60°C for 2 h.
After deparaffinization and rehydration, the sections were stained
by hematoxylin and eosin (H&E) according to standard
protocols.
Western blot analysis
Liver tissues were homogenized and lysed in RIPA
buffer (50 mM Tris-HCl, pH 8.0; 150 mM sodium chloride; 1.0% Triton
X-100; 0.5% SDS; 0.5% sodium deoxycholate; 1 mM PMSF; 1X protease
inhibitor cocktail, Roche) at 4°C for 30 min. After sonication in
an ice bath, the lysates were clarified by centrifugation at 4°C
and 12,000 × g for 15 min. Protein concentration was determined by
Bradford assay. The proteins were resolved by SDS-PAGE, transferred
to a PVDF membrane, and immunoblotted with specific primary and
secondary antibodies. The signals were detected by ECL.
Label-free quantitative proteomics
analysis
For sample preparation, 100 mg of liver tissues were
homogenized and lysed in lysis buffer containing 8 M urea, 50 mM
Tris-base (pH 8.0), and 1X protease inhibitor cocktail (Roche) in
an ice bath. After sonication, the lysates were clarified by
centrifugation at 4°C and 12,000 × g for 30 min and then purified
via acetone precipitation. The samples were redissolved in urea
lysis buffer. Protein concentration was determined by Bradford
assay. After reduction in 30 mM DTT and alkylation in 50 mM
iodoacetamide (IAA), the proteins were digested by incubation with
trypsin (1:50, enzyme to protein) at 37°C for 16 h. The samples
were desalted by passing them through a SepPak C18 column (Waters).
The peptides were analyzed by LC-MS/MS (AB Sciex TripleTOF 5600+).
The MS data were analyzed with Maxquant software.
Bioinformatics analysis
Bioinformatics analysis with ingenuity pathway
analysis (IPA; Ingenuity® Systems, www.ingenuity.com) included tox list analysis,
protein-protein interacting network analysis and canonical pathway
analysis. The entire protein list containing the protein accession
number (UniprotKB) and the corresponding fold changes was uploaded
as an Excel spreadsheet. The protein accession numbers were mapped
to their corresponding genes in the Ingenuity Pathways Knowledge
Base. The gene list was used for bioinformatics analysis. The
cut-off for the fold-change was set to 2.0. KEGG pathway enrichment
by DAVID was performed according to the instructions for the
database. Given that the database did not identify fold-change
data, we only used the differentially expressed proteins for the
analysis.
Results
Pathological changes in mouse liver
during the precancerous inflammation stage
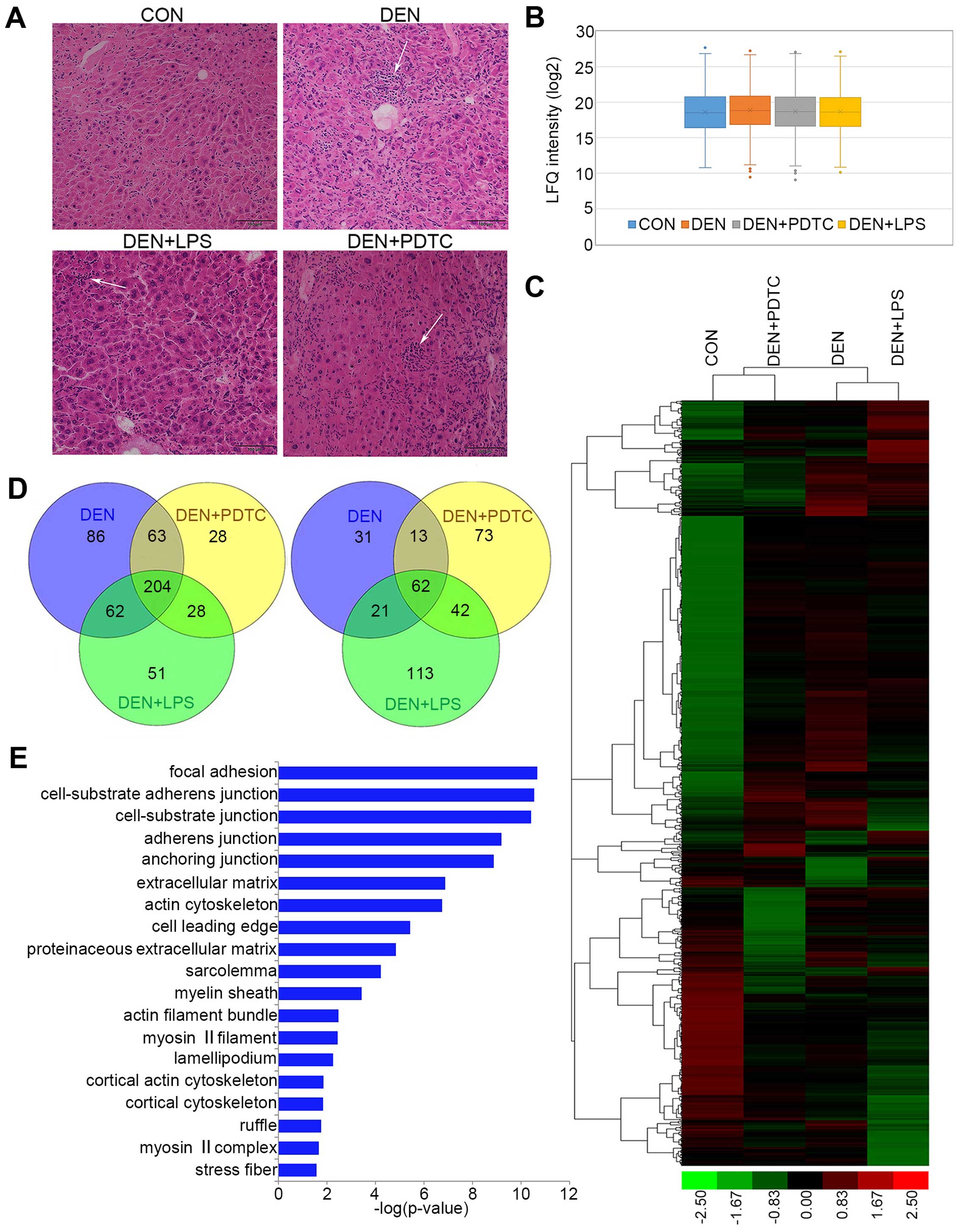
To identify pathological changes in mouse liver, we
subjected the paraffin sections of mouse liver tissues to H&E
staining. As shown in Fig. 1A
(9), the hepatic lobule in normal
livers (control group) contained an intact structure, which was
surrounded by cords of liver cells that radiated in all directions.
Among the liver tissues derived from the treatment groups (DEN,
DEN+PDTC, and DEN+LPS groups), a large number of infiltrated
inflammatory cells appeared and partially formed focal
inflammation.
Identification of differentially expressed proteins
during the precancerous inflammation stage was by LFQ proteomic
analysis. To compare differences in protein expression between
normal and inflammatory liver tissues, we obtained the protein
expression profiles of different groups by LFQ proteomic analysis.
A total of 2,666 proteins were identified and quantified. To
estimate the quantitative capacity of our data set, we first
applied box-plot analysis in comparing the average LFQ intensity of
the four groups. As shown in Fig.
1B, the average LFQ intensity was similar across all groups;
this finding indicated the unbiased result of our data set. To
discern the differences in global protein expression across the
four groups, the LFQ intensity of differentially expressed proteins
were subjected to hierarchical clustering analysis (HCA) by Cluster
3.0. The result was visualized as a heat map (Fig. 1C) with Treeview. The expression
profiles of the three treatment groups significantly differed from
the control group.
A Venn diagram (Fig.
1D) was applied to show the distribution of differentially
expressed proteins in the DEN, DEN+PDTC, and DEN+LPS groups. A
total of 204 upregulated proteins and 62 downregulated proteins
were shared by the three treatment groups. Cellular component
enrichment toward 204 upregulated proteins was performed with the
Gene Ontology database (http://geneontology.org/; powered by PANTHER). These
proteins were mainly associated with cell adhesion, cell junctions,
and cytoskeleton (Fig. 1E).
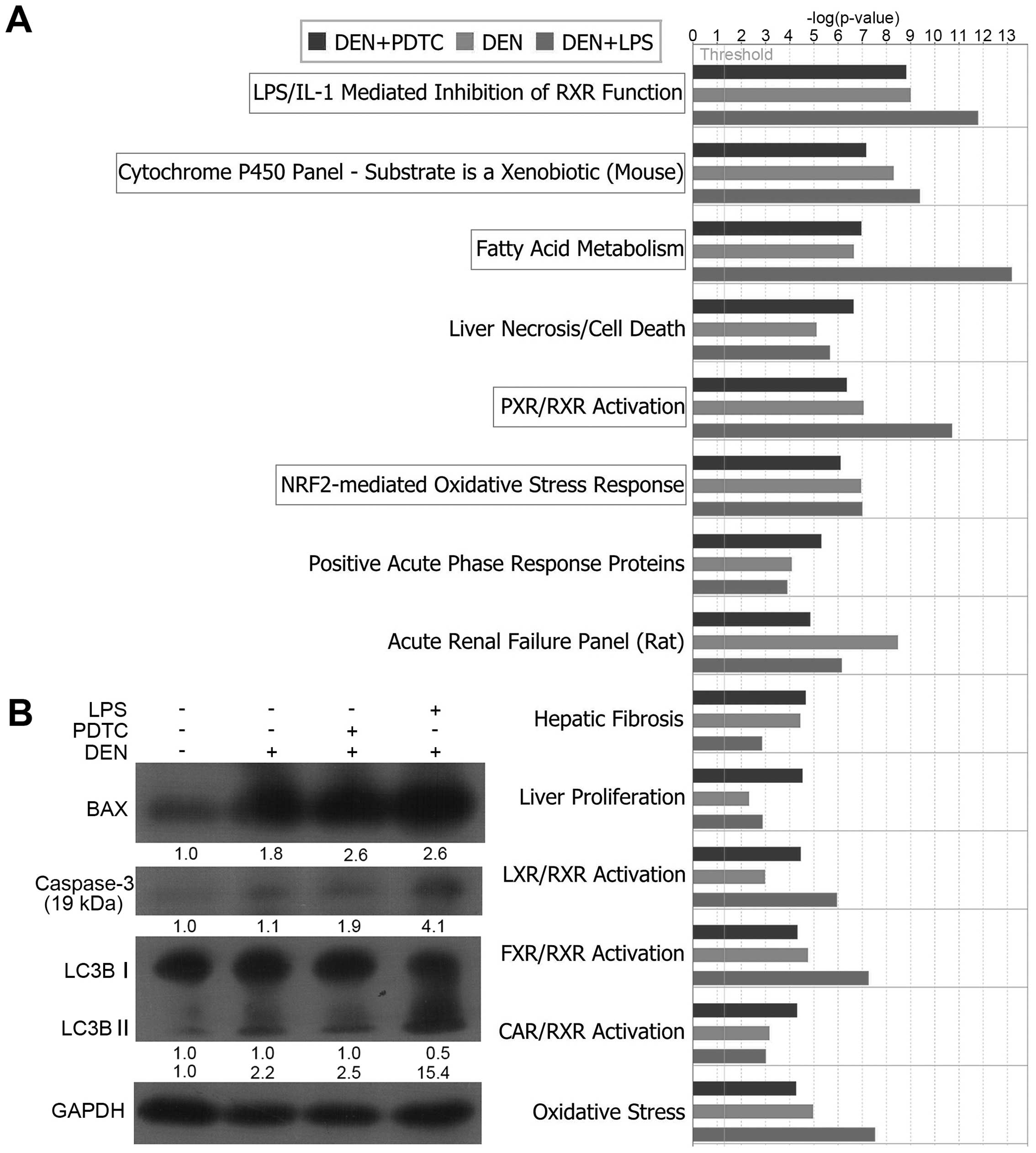
Tox list analysis by IPA revealed
oxidative stress-induced hepatocellular injury and death during the
precancerous inflammation stage
IPA furnishes the analysis of Tox lists to correlate
experimental data with clinical pathology endpoints. In IPA, a
p-value is calculated using right-tailed Fischer’s exact test; this
value is always used to measure the likelihood that the association
between a set of focus genes in our data and a given term is due to
random chance. Low p-values indicate that the association is less
likely random but is significant. We selected the threshold of
p<0.05 (−log p>1.3, as indicated by a line in the figures) as
the statistically significant level (the same was used in the
following canonical pathway analysis). Our data set showed a
significant association with the Tox lists of LPS/IL-1-mediated
inhibition of RXR function, cytochrome P450 panel-substrate nature
as xenobiotic (mouse), fatty acid metabolism, PXR/RXR activation,
and NRF2-mediated oxidative stress response (Fig. 2A). All these pathological endpoints
were related to oxidative stress.
The pathological endpoint of liver necrosis/cell
death (−log p>5) indicated that hepatic cells might experience a
certain degree of damage and death. We detected the expression
levels of some proteins associated with cell apoptosis and
autophagy. As shown in Fig. 2B,
the levels of the activated fragment of Caspase-3, BAX, and the
small fragment of LC3B significantly increased in the DEN,
DEN+PDTC, and DEN+LPS groups.
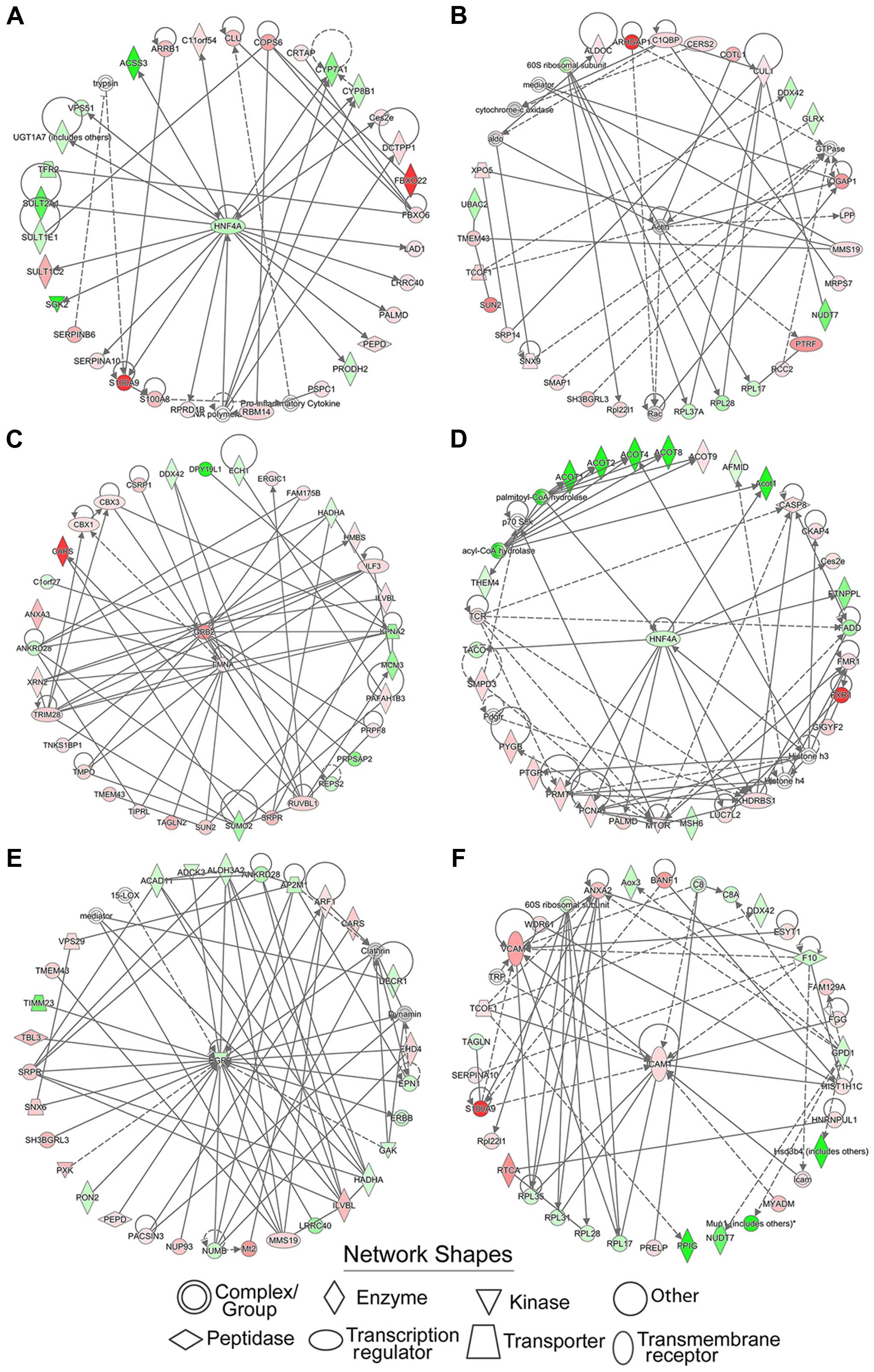
Identification of significant protein
functional groups by establishing protein-interacting networks
The functions of proteins highly rely on their
interactions. By establishing protein-interacting networks,
thousands of proteins can be assigned to different functional
groups, which facilitate the analysis of regulatory events in the
proteomics data. We used the IPA software to map the differentially
expressed proteins into corresponding protein-protein interaction
networks. The networks of corresponding molecules were
algorithmically generated based on their connectivity and assigned
a score. This score took into account the number of focus molecules
derived from our input dataset and the size of the network to
approximate how relevant this network was to our original dataset.
However, the score might not be an indication of the quality or
significance of the network. The intensity of the color in the
network represented the degree of upregulation (red) or
downregulation (green) of molecules in our dataset. The uncolored
genes did not belong to our dataset, but were integrated into the
network because of their relevance as indicated by IPA.
A total of 25 protein-interacting networks were
acquired from each treatment group. The top two networks of each
group with the highest scores are shown in Fig. 3, and the involved functions are
listed in Table I. Centered on
HNF4A, actin, GRB2, LMNA, EGFT, and ICAM1, these networks were
involved in cellular assembly and organization, DNA replication,
recombination, and repair, developmental disorders, hereditary
disorders, the cell cycle, lipid metabolism, nucleic acid
metabolism, and organismal injury and abnormalities.
 | Table ICharacterization of protein
interacting networks. |
Table I
Characterization of protein
interacting networks.
| Group | Score | Focus molecule
number | Top diseases and
functions |
|---|
| DEN | 54 | 32 | Small molecule
biochemistry, developments disorder, hereditary disorder |
| DEN | 44 | 28 | Lipid metabolism,
nucleic acid metabolism, small molecule biochemistry |
| DEN+PDTC | 64 | 35 | Cellular assembly and
organization, DNA replication, recombination, and repair, cardiac
arrythmia |
| DEN+PDTC | 44 | 28 | Lipid metabolism,
nucleic acid metabolism, small molecule biochemistry |
| DEN+LPS | 48 | 30 | Cell cycle, cellular
assembly and organization, infectious diseases |
| DEN+LPS | 42 | 31 | Cardiac stenosis,
cardiovascular disease, organismal injury and abnormalities |
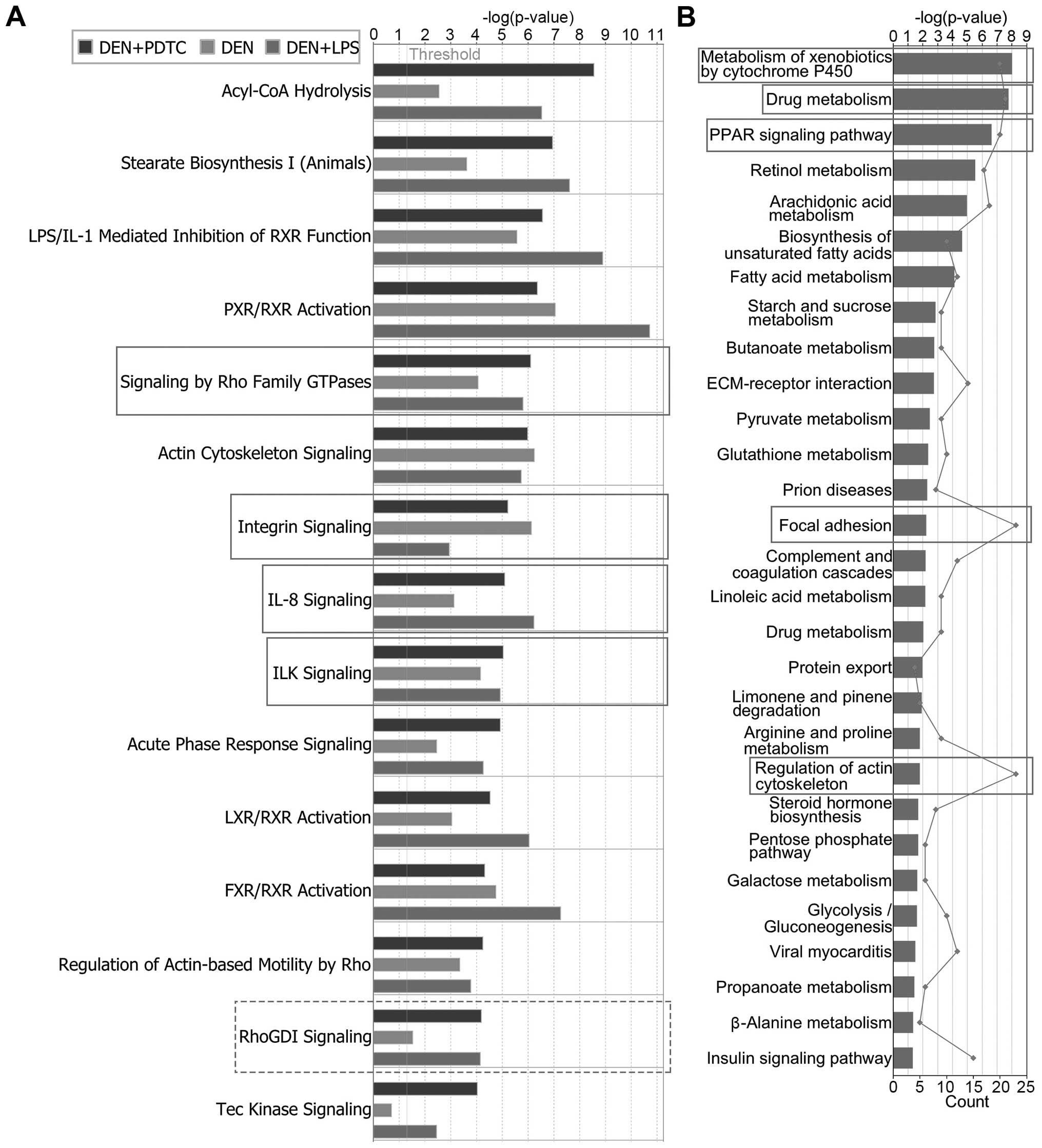
Pathway analysis by IPA and DAVID
revealed that sustainable inflammation enhanced the EMT ability in
hepatic cells
The signaling pathways of the differentially
expressed proteins can explain how the inflammatory
microenvironment transforms hepatic cells. Consequently, we
performed canonical pathway analysis with the IPA software.
Additional to the p-values calculated by the Fischer’s exact test,
IPA gave a Z-score to indicate if the pathway was in an activated
state (Z >0) or inhibited state (Z <0). The Z-score was
calculated according to the expression level (up or down) of a
corresponding molecule in the data set and the expected expression
value (up or down) in the pathway. We selected Z ≥2 and Z ≤−2 as
indicators of the activated and inhibited states, respectively. As
shown in Table II and Fig. 4A, integrin signaling, signaling by
Rho family GTPases, IL-8 signaling, and ILK signaling were the
common activated pathways among the DEN, DEN+PDTC, and DEN+LPS
groups. By contrast, RhoGDI signaling was the common inhibited
pathway. All these pathways had a close relationship with
EMT-related cell adhesion and migration.
| Table IIThe common activated or inhibited
signaling pathways in three treatment groups indicated by IPA. |
Table II
The common activated or inhibited
signaling pathways in three treatment groups indicated by IPA.
| DEN | DEN+PDTC | DEN+LPS |
|---|
|
|
|
|
|---|
| Ingenuity canonical
pathways | State | Z-score | State | Z-score | State | Z-score |
|---|
| Integrin
signaling | Activated | 2.828 | Activated | 3 | Activated | 2.496 |
| Signaling by Rho
family GTPases | Activated | 2.5 | Activated | 2.5 | Activated | 3.3 |
| IL-8 signaling | Activated | 2.496 | Activated | 2 | Activated | 2.982 |
| ILK signaling | Activated | 2.496 | Activated | 2.138 | Activated | 2.84 |
| RhoGDI
signaling | Inhibited | −2.121 | Inhibited | −2.53 | Inhibited | −2.309 |
To obtain precise results, we further uploaded the
differentially expressed proteins into the DAVID database
(https://david.ncifcrf.gov/) for KEGG
signaling pathway enrichment analysis. A total of 29 pathways with
p<0.05 were enriched (Fig. 4B).
By ranking the number (line chart in Fig. 4B) of differentially expressed
proteins in each pathway, the top five signaling pathways were
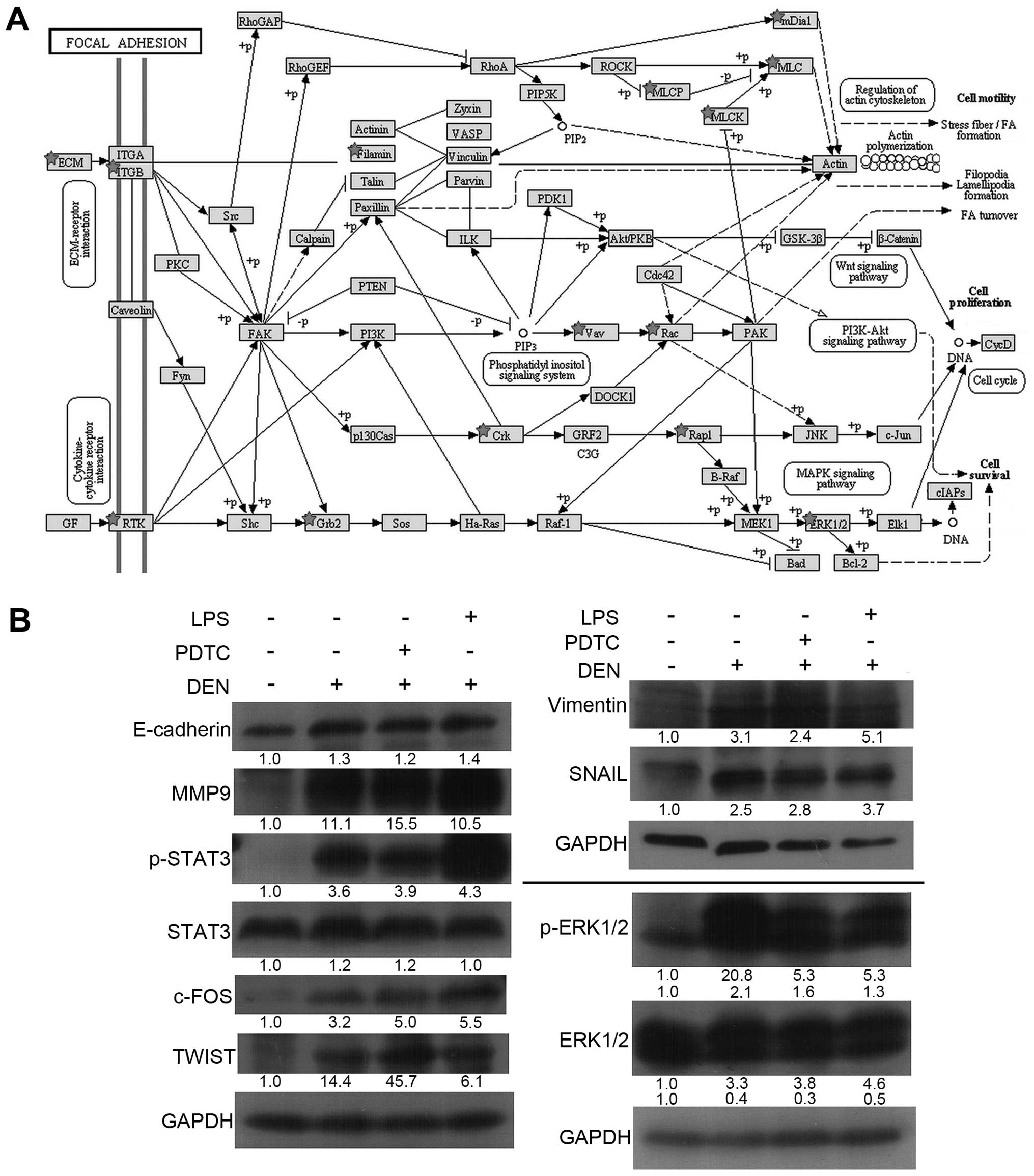
focal adhesion (Fig. 5A),
regulation of actin cytoskeleton, drug metabolism by cytochrome
P450, metabolism of xenobiotics by cytochrome P450, and PPAR
signaling pathway (data not shown). Drug metabolism, metabolism of
xenobiotics by cytochrome P450, and PPAR signaling pathway are
closely related to inflammatory responses and oxidative stress,
which is consistent with the results of the IPA tox list analysis.
Moreover, focal adhesion and the regulation of actin cytoskeleton
are closely associated with cell adhesion and migration, which
supported the results of the IPA canonical pathway analysis.
To further verify the enhanced EMT ability in
hepatic cells from proteomics data, we detected the expression
levels of some key factors in EMT by western blot analysis. As
shown in Fig. 5B, EMT-associated
key proteins, such as p-STAT3, TWIST, SNAIL, Vimentin, and MMP-9,
were all upregulated in the three treatment groups, which
demonstrated that hepatic cells had access to EMT. E-cadherin is a
key protein that was expected to be downregulated in EMT.
Interestingly, E-cadherin also increased in hepatic cells during
the inflammation stage. According to the focal adhesion pathway
(Fig. 5A), the ERK signaling
pathway was activated as a downstream target of integrin signaling.
This trend was confirmed by the increasing levels of phosphorylated
ERK1/2 and its downstream transcription factor, c-FOS (Fig. 5B).
Discussion
HCC is the third leading cause of cancer-related
death worldwide (10). The
condition often develops in the context of chronic hepatitis, which
is mainly caused by viruses or toxic compounds (11,12).
In-depth research has provided a clear understanding of the
relationship between inflammation and liver cancer. However, the
precise underlying mechanism has not been completely elucidated.
The study of cellular pathological changes and their regulatory
mechanisms during the early inflammatory period will contribute
toward accelerating HCC diagnosis and prevention. Having
established a mouse inflammation-cancer model successfully, we
performed a systemic exploration on the molecular pathogenic
mechanisms during the precancerous inflammatory period via LFQ
proteomics technology combined with bioinformatics analysis.
Our results demonstrated a common trend of
inflammation in the three treatment groups. DEN has been widely
recognized to cause liver cell injury and induce hepatocellular
carcinoma. The role of LPS in promoting inflammation has also been
confirmed. However, PDTC impairs the inflammatory response by
inhibiting the synthesis of NOS and the activation of NF-κB.
H&E staining of mouse liver tissues showed that the
infiltration of inflammatory cells had occurred in DEN, DEN+PDTC,
and DEN+LPS groups (Fig. 1A). Our
final results proved that mice in all three groups had formed
hepatic cirrhosis (9). In heat map
of HCA, similarities were found among the three treatment groups
against the control group. The Venn diagram showed that 204
upregulated proteins and 62 downregulated proteins were shared by
the three treatment groups. All the results indicated that in the
complex regulatory process, some common regulatory mechanisms
allowed the sustainable inflammatory microenvironment to induce the
conversion of hepatic cells to the malignant phenotype. We
performed cellular component enrichment analysis on the 204 common
upregulated proteins and found that these proteins were mainly
components of the extracellular matrix and cytoskeleton, which were
involved in cell adhesion and cell junctions. This trend was
consistent with our subsequent pathway analysis.
Tox list analysis using IPA revealed the association
of our data set with oxidative stress-related pathological
processes. RXR, also called the retinoid X receptor, is a member of
nuclear receptor superfamily. RXR can repress apoptosis by
impairing oxidative stress in cells (13–16).
Cytochrome P450, LPS, and NRF2 are also closely related to
oxidative stress. In addition, some representative proteins related
to liver injury had significantly changed after experiencing
sustainable inflammation according to our proteomic data. For
instance, major urinary protein (MUP) is a protein family mainly
expressed in the liver; the expression levels of proteins in this
family significantly decreased after a liver injury (17). In our data, the expression levels
of MUP2, MUP3, MUP6, and MUP20 were notably decreased. Other liver
injury-related proteins, such as lactotransferrin (TRFL) and
haptoglobin (HPT) (18), were also
significantly changed (data not shown). Therefore, hepatic cells
suffered serious oxidative stress-induced injury. Subsequent
western blot experiments to detect Caspase-3, BAX, and LC3B
demonstrated the death of hepatic cells.
Protein-interacting networks were constructed by IPA
and depicted some protein functional groups associated with
cellular assembly and organization, DNA replication, recombination,
and repair, developmental disorders, hereditary disorders, the cell
cycle, lipid metabolism, nucleic acid metabolism, and organismal
injury and abnormalities. To further dissect the specific
regulatory events, the central node proteins were subject to
functional retrieval based on UNIPROT (http://www.uniprot.org/) and previously published
study. As the central node of two networks, hepatocyte nuclear
factor 4-α (HNF4A) attracted our attention. Growing evidence
suggested that HNF4A plays a role in the inflammatory-cancer loop
(19), but the exact molecular
mechanisms have yet to be defined. HNF4 is a transcriptional
regulation factor that belongs to a family of hormone receptors.
The protein is involved in regulating lipid and glucose metabolism,
cell junctions, and the differentiation and proliferation of liver
and intestinal epithelial cells (20). Zeisberg et al (21) found that HNF4A was downregulated in
mouse liver cells during EMT induced by TGFβ. Rygiel et al
(22) also proved that the
heterogeneous expression of HNF4A could prevent EMT in hepatic
cells. Our data demonstrated the downregulation of HNF4A during the
inflammation stage. This trend directed our attention to EMT, which
was consistent with the pathways analysis.
Canonical pathway analysis by IPA demonstrated some
common pathways shared by the DEN, DEN+PDTC, and DEN+LPS groups.
Integrin signaling, signaling by Rho family GTPase, IL-8 signaling,
and ILK signaling were activated, whereas RhoGDI signaling was
inhibited. Integrin was reported to regulate EMT via focal adhesion
kinase (FAK) (23) or integrin
linked kinase (ILK) (24). The
signaling of PI3K/AKT (25), WNT
(26), and RAS/MAPK (27) are all downstream targets of
integrin signaling. Members of the Rho GTPase family are mainly
involved in the regulation of cytoskeletal and cell adhesion
(28); RhoGDI is an important
negative regulator of Rho GTPase. These results indicated the
changes of hepatic cells in terms of adhesion, migration, or EMT.
Further KEGG pathway enrichment analysis revealed that the
signaling pathways of focal adhesion and the regulation of the
actin cytoskeleton were significantly changed. As shown in Fig. 5A, the integrin-mediated signaling
played a central role in regulating the cytoskeleton, cell
adhesion, and cell migration, which supported the IPA results. The
upregulation of p-STAT3, TWIST, SNAIL, Vimentin, and MMP-9 strongly
demonstrated the tendency of EMT in hepatic cells. Regardless of
the unexpected increase of E-cadherin, the migration of hepatic
cells during the inflammation stage was enhanced, and the potential
EMT tendency should not be ignored.
In concusion, this study provides a global view on
the transformation of hepatic cells exposed to inflammation.
Despite the complexity of regulatory mechanism by which the
inflammatory microenvironment transforms hepatic cells and the
limitations of bioinformatics analysis, which needs verification by
further experiments, our study still partially reveals the
underlying mechanism of transformation. Under a sustainable
inflammatory microenvironment, hepatic cells suffer serious
oxidative stress-induced injury and death. Downregulated HNF4A may
be a warning marker for liver injury and further hepatic cell
deterioration. The regulatory network centered on integrin
signaling increases migration and the EMT ability of hepatic cells
by regulating the cytoskeleton and focal adhesion. The involvement
of HNF4A in this regulatory network still needs to be defined.
Acknowledgements
This study was supported by National Natural Science
Foundation of China (Grant Nos. 81272921, 81272245, 81272445 and
81471970); Joint Programme by Healthy Care System and Educational
Department in Fujian Province (Grant No. WKJ-FJ-16); The Natural
Science Foundation of Fujian Province of China (Grant No.
2013D004); the Fundamental Research Funds for the Central
Universities (Grant No. 20720140545).
References
|
1
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuper H, Adami HO and Trichopoulos D:
Infections as a major preventable cause of human cancer. J Intern
Med. 248:171–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Allavena P, Garlanda C, Borrello MG, Sica
A and Mantovani A: Pathways connecting inflammation and cancer.
Curr Opin Genet Dev. 18:3–10. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nakagawa H and Maeda S: Inflammation- and
stress-related signaling pathways in hepatocarcinogenesis. World J
Gastroenterol. 18:4071–4081. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Balkwill F and Mantovani A: Inflammation
and cancer: Back to Virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markiewski MM, DeAngelis RA and Lambris
JD: Liver inflammation and regeneration: Two distinct biological
phenomena or parallel pathophysiologic processes? Mol Immunol.
43:45–56. 2006. View Article : Google Scholar
|
|
7
|
Connor JH, Weiser DC, Li S, Hallenbeck JM
and Shenolikar S: Growth arrest and DNA damage-inducible protein
GADD34 assembles a novel signaling complex containing protein
phosphatase 1 and inhibitor 1. Mol Cell Biol. 21:6841–6850. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Poirier MC: Chemical-induced DNA damage
and human cancer risk. Discov Med. 14:283–288. 2012.PubMed/NCBI
|
|
9
|
Peng B, Liu F, Han R, Luo G, Cathopoulis
T, Lu K, Li X, Yang L, Liu GY, Cai JC, et al: Dynamic metabolic
change is indicative of inflammation-induced transformation of
hepatic cells. Int J Biochem Cell Biol. 66:45–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berasain C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
New molecular links. Ann NY Acad Sci. 1155:206–221. 2009.
View Article : Google Scholar
|
|
12
|
Nikolaou K, Sarris M and Talianidis I:
Molecular pathways: The complex roles of inflammation pathways in
the development and treatment of liver cancer. Clin Cancer Res.
19:2810–2816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao XR, Gonzales N and Aronowski J:
Pleiotropic role of PPARγ in intracerebral hemorrhage: An intricate
system involving Nrf2, RXR, and NF-κB. CNS Neurosci Ther.
21:357–366. 2015. View Article : Google Scholar
|
|
14
|
Shan P, Pu J, Yuan A, Shen L, Shen L, Chai
D and He B: RXR agonists inhibit oxidative stress-induced apoptosis
in H9c2 rat ventricular cells. Biochem Biophys Res Commun.
375:628–633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chai D, Wang B, Shen L, Pu J, Zhang XK and
He B: RXR agonists inhibit high-glucose-induced oxidative stress by
repressing PKC activity in human endothelial cells. Free Radic Biol
Med. 44:1334–1347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Elbrecht A, Chen Y, Cullinan CA, Hayes N,
Leibowitz M, Moller DE and Berger J: Molecular cloning, expression
and characterization of human peroxisome proliferator activated
receptors gamma 1 and gamma 2. Biochem Biophys Res Commun.
224:431–437. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elchuri S, Naeemuddin M, Sharpe O,
Robinson WH and Huang TT: Identification of biomarkers associated
with the development of hepatocellular carcinoma in CuZn superoxide
dismutase deficient mice. Proteomics. 7:2121–2129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bell LN, Vuppalanchi R, Watkins PB,
Bonkovsky HL, Serrano J, Fontana RJ, Wang M, Rochon J and Chalasani
N; US Drug-Induced Liver Injury Network (DILIN) Research Group.
Serum proteomic profiling in patients with drug-induced liver
injury. Aliment Pharmacol Ther. 35:600–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hatziapostolou M, Polytarchou C, Aggelidou
E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K,
Hadzopoulou-Cladaras M, et al: An HNF4α-miRNA inflammatory feedback
circuit regulates hepatocellular oncogenesis. Cell. 147:1233–1247.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Babeu JP and Boudreau F: Hepatocyte
nuclear factor 4-alpha involvement in liver and intestinal
inflammatory networks. World J Gastroenterol. 20:22–30. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zeisberg M, Yang C, Martino M, Duncan MB,
Rieder F, Tanjore H and Kalluri R: Fibroblasts derive from
hepatocytes in liver fibrosis via epithelial to mesenchymal
transition. J Biol Chem. 282:23337–23347. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rygiel KA, Robertson H, Marshall HL,
Pekalski M, Zhao L, Booth TA, Jones DEJ, Burt AD and Kirby JA:
Epithelial-mesenchymal transition contributes to portal tract
fibrogenesis during human chronic liver disease. Lab Invest.
88:112–123. 2008. View Article : Google Scholar
|
|
23
|
Figel S and Gelman IH: Focal adhesion
kinase controls prostate cancer progression via intrinsic kinase
and scaffolding functions. Anticancer Agents Med Chem. 11:607–616.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cortez V, Nair BC, Chakravarty D and
Vadlamudi RK: Integrin-linked kinase 1: Role in hormonal cancer
progression. Front Biosci (Schol Ed). 3:788–796. 2011.
|
|
25
|
Ke AW, Shi GM, Zhou J, Huang XY, Shi YH,
Ding ZB, Wang XY, Devbhandari RP and Fan J: CD151 amplifies
signaling by integrin α6β1 to PI3K and induces the
epithelial-mesenchymal transition in HCC cells. Gastroenterology.
140:1629–41.e15. 2011. View Article : Google Scholar
|
|
26
|
Gil D, Ciołczyk-Wierzbicka D,
Dulińska-Litewka J, Zwawa K, McCubrey JA and Laidler P: The
mechanism of contribution of integrin linked kinase (ILK) to
epithelial-mesenchymal transition (EMT). Adv Enzyme Regul.
51:195–207. 2011. View Article : Google Scholar
|
|
27
|
Hwangbo C, Kim J, Lee JJ and Lee JH:
Activation of the integrin effector kinase focal adhesion kinase in
cancer cells is regulated by crosstalk between protein kinase
Calpha and the PDZ adapter protein mda-9/Syntenin. Cancer Res.
70:1645–1655. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fuse T, Kanai Y, Kanai-Azuma M, Suzuki M,
Nakamura K, Mori H, Hayashi Y and Mishina M: Conditional activation
of RhoA suppresses the epithelial to mesenchymal transition at the
primitive streak during mouse gastrulation. Biochem Biophys Res
Commun. 318:665–672. 2004. View Article : Google Scholar : PubMed/NCBI
|