Introduction
Glioblastoma (GBM) is the most malignant brain tumor
in humans and it is characterized by diffuse and infiltrative
growth into the surrounding brain tissue, enhanced cell growth, the
ability to overcome cell death and antitumor immune responses and
therapy resistance. The main reason for the high malignancy of GBM
is the highly infiltrative phenotype, which is why complete
surgical removal of all tumor cells is impossible and recurrence
inevitable. During disease progression a shift toward a more
mesenchymal phenotype is common. Especially the mesenchymal subtype
of GBM is associated with an aggressive phenotype, and GBM cells
specifically use a mesenchymal mode of cell migration (1). Enhanced expression of epithelial to
mesenchymal transition (EMT) proteins like SLUG or ZEB2 correlates
with an invasive phenotype of GBM, high tumor grade and faster
tumor progression (2,3). Beside the involvement of EMT
proteins, migration and invasion of glioma cells are regulated by
other processes, including changes in cell metabolism, destruction
of the extracellular matrix (ECM) and regulation of cell adhesion
and cell architecture. Non-coding RNAs, transcription factors, cell
surface proteins, secreted cyto- and chemokines and other proteins
with different functions are involved in the enhancement of
motility of GBM cells (reviewed in ref. 4).
CPE was originally described as a
neuropeptide-processing enzyme. Numerous studies have shown that
CPE, which is present in different variants that are products of
differential splicing, proteolytic cleavage or glycosylation, is a
multifunctional protein that plays roles in the endocrine and
nervous systems and also in cancer (5). An alternatively spliced CPE
transcript (ΔN-CPE) has been found in human tumor cells. It encodes
a cytoplasmic protein lacking a part of the N-terminal region due
to alternative splicing of the first exon. ΔN-CPE was described to
increase the metastatic potential of tumor cells by upregulating
metastasis-related genes (6). We
recently identified a secreted version of CPE (sCPE) that, in
contrast to ΔN-CPE, provides an anti-migratory and anti-invasive,
but pro-proliferative function in glioma cells. sCPE is released
into the extracellular space, where it, at least partially by
modulating the cell architecture and increasing cell adhesion,
transmits its anti-migratory function (7).
One key signaling element in the malignant behavior
of glioma is its high invasive phenotype which involves the
stimulation of a variety of motility-associated signaling cascades.
Modulation of these cascades by external and internal stimuli
results into changes in gene expression and in the execution of
glioma cell migration and invasion. While it has been described
that in hippocampal neurons CPE induces the expression of the known
pro-invasive and anti-apopotic B-cell lymphoma 2 (BCL-2)
protein via ERK1/2 signaling (8,9),
information on additional mechanisms, pathways or factors by which
sCPE provides its anti-migratory effect in glioma cells are still
rare. The aim of the present study therefore, was to identify
targets of sCPE as well as signaling cascades that are affected by
sCPE and that are specific for transmitting its anti-migratory
effects in glioma cells.
Our findings show that sCPE modifies the expression
of a panel of mRNAs involved in the modulation of migration,
invasion and cell architecture or that are part of
motility-associated signaling cascades integrating the transforming
growth factor beta (TGF-β), the integrin, the focal adhesion kinase
(FAK), the cell division cycle 42 (Cdc42), the p21-activated kinase
(PAK)-1 as well as the signal transducer and activator of
transcription 3 (STAT3) and the mitogen-activated protein kinase
(MAPK/ERK) pathways. One prominent factor that was downregulated in
sCPE-overexpressing glioma cells was the oncogenic protein SLUG
that has been described as an invasion-related transcription factor
overexpressed in many cancers including glioma (10). In our in vitro cell culture
model reduced SLUG expression mediated by sCPE was, at least
partially, responsible for the observed anti-migratory phenotype of
sCPE-overexpressing glioma cells.
Materials and methods
Cells, viruses and reagents
LNT-229 and LN-308 human malignant glioma cells were
kindly provided by N. de Tribolet (Lausanne, Switzerland), T98G
cells were purchased from the American Type Culture Collection
(ATCC; Manassas, VA, USA). Tu-132 and Tu-140 GBM primary cells were
established from human GBM tissue and used at passages 5–10. The
use of primary glioma cells was approved by the ethics committee of
the University of Tübingen (125/2007BO1). All cells were maintained
in Dulbecco's modified Eagle's medium (DMEM; Gibco-Life
Technologies, Eggenstein, Germany) containing 10% fetal calf serum
(FCS; Gibco), penicillin (100 U/ml), streptomycin (100
µg/ml) and the appropriate selection antibiotic in a
humidified atmosphere containing 5% CO2. The generation
of sCPE-overexpressing (LNT-229-rCPE, from Rattus
norvegicus) or neo control cells (LNT-229-neo) has been
previously described (7). Besides
this, stable glioma cell lines overexpressing human CPE under
control of the CMV promoter were generated by transduction of the
cells with the lentivirus pReceiver LV105-CPE or its empty
counterpart (GeneCopeia, Inc., Rockville, MD, USA) followed by the
selection with puromycin. Rat and human mature CPE protein shows
96% identity and 98% similarity (5) in the amino acid (AA) sequence and a
total conservation of the Zn-carboxypeptidase domain, indicating
that the enzymatic activity is not different between the species.
Besides, a correct procession and maturation of sCPE is conserved
in both species, since the penta-arginine sequence
(RRRRR42) is identical. Changes are present in the AA
sequence in closest distance to the prohormone sorting signal
binding site (the region between R255 and
K260). To avoid 'off target' effects, CPE-knockdown in
Tu-140 cells was induced by transient transfection with
CPE-specific or non-target POOL siRNA (siTOOLs Biotech GmbH,
Martinsried, Germany) and Lipofectamine 2000 (Thermo Fisher
Scientific, Waltham, MA, USA) or by lentiviral transduction using
pGIPZ vectors carrying either control or CPE-specific shRNA. The
SLUG knockdown in LNT-229, LN-308, T98G and Tu-132 cells was
generated by transfection of the cells with SLUG-specific or
control POOL siRNA (MISSON esiRNA huSNAI2; MISSION esiRNA EGFP;
Sigma-Aldrich, Darmstadt, Germany) using the Viromer BLUE
transfection kit (Biozym Scientific GmbH, Hessisch Oldendorf,
Germany). The construction of Ad-EGFP has been described (11). For the generation of Ad-SLUG, human
SLUG cDNA was cloned into pTRACK-CMV using the Ad-Easy system
provided by B. Vogelstein (Baltimore, MD, USA) (12). Ad-SLUG additionally codes for EGFP
in a second expression cassette. Recombinant adenoviral genomes
were transfected into HEK-293 cells (ATCC). Viruses were
CsCl-purified, dialysed and titrated using the Clontech Rapid
Titration system (Takara Bio Europe SAS, Saint-Germain-en-Laye,
France). Transgene expression was verified by immunoblot. Infection
with recombinant adenoviruses was accomplished by exposing cells to
100 MOI of the appropriate adenovirus in serum-free DMEM for 15 min
followed by the addition of serum-containing medium. To inhibit
ERK1/2 activity, glioma cells were treated with the MAPK inhibitor
U0126 (10 µM, 24 h; R&D Systems GmbH, Wiesbaden,
Germany). If not mentioned otherwise, all other reagents were from
Sigma-Aldrich.
Measurement of cell migration
The Transwell migration assay have been previously
described (7). Briefly, cells were
seeded in the upper layer of 8 µm pore-sized Boyden Trans
well chambers and were allowed to actively migrate for 18 h towards
FCS containing DMEM as attractant medium placed in the bottom
chamber. Migrated cells on the lower layer of the membrane were
fixed, stained with hematoxylin/eosin and counted. Number of
migrated cells was normalized to cell density at the end of the
migration period, assessed in parallel by crystal violet staining
as previously described (13).
Immunofluorescence
For the assessment of cytoskeleton changes, the
cells were seeded on poly-L-lysin coated coverslips, allowed to
grow and fixed in 4.5% formaldehyde. Staining was accomplished
using the Actin Cytoskeleton and Focal Adhesion Staining kit
(Millipore, Schwalbach, Germany) which consists of TRITC-conjugated
phalloidin allowing the detection of filamentous actin in the
cytoskeleton, anti-vinculin as a universal focal adhesion marker
and DAPI to stain the nucleus. Quantification of cell adhesion was
done by measuring the vinculin positive area using the ImageJ
software (National Institute of Health, Bethesda, MD, USA) and
counting the number of large focal adhesion complexes (LFA) per
cell.
Immunoblot analysis and detection of
receptor phosphorylation
The general immunoblot procedure has been described
elsewhere (11). For the detection
of phospho-EGFR, LNT-229 cells were serum starved for 24 h followed
by the addition of conditioned medium produced by either LNT-229
control or CPE-overexpressing cells. For generation of
supernatants/conditioned medium, the cells were treated as
indicated and serum-free medium was added. Supernatants were
harvested 24 or 48 h later, clarified from cell debris by
centrifugation and concentrated using Amicon concentrators
(Millipore). Protein contents were analyzed according to Bradford.
Lysates collected for the detection of receptor phosphorylation
were lysed in RIPA buffer and protein determination was performed
using the BCA assay (Pierce BCA protein assay kit; Thermo Fisher
Scientific, Darmstadt, Germany) according to the manufacturer's
protocol. Following antibodies were used: CPE (#610758, 1:2,000; BD
Biosciences, Heidelberg, Germany), MT1-MMP/MMP14 (#2010-1, 1:1,000;
Epitomics Burlingame CA, USA) and TIMP-2 (Mab971, 1:1,000; R&D
Systems). SLUG (#9585, 1:1,000), MMP-2 (#4022, 1:1,000), the
phospho-EGFR antibody sampler kit (#9922, 1:1,000), the PathScan
Multiplex Western Cocktail I (for the detection of phospho-ERK1/2,
#5301, 1:1,000), STAT3 (#9139, 1:2,000) and
phospho-STAT3S727 (#9134, 1:1,000) were purchased from
Cell Signaling Technology (Frankfurt am Main, Germany). ERK1/2
(sc-135900, 1:2,000), Bcl-2 (sc-509, 1:1,000) and α-tubulin
(sc-12462-R, 1:2,000) were from Santa Cruz Biotechnology
(Heidelberg, Germany). Actin was provided by Abcam (ab8227,
1:2,000; Cambridge, UK) and GAPDH from Chemicon (Billerica, MA,
USA; AB2302, 1:2,000). Protein expression was quantified using the
ChemiDoc MP system and ImageLab 5.1 software (Bio-Rad Laboratories,
Munich, Germany). For normalization of protein expression in
lysates, either GAPDH or α-tubulin was used as indicated. For
normalization of protein expression in supernatants, the membranes
were stained with Ponceau S and total stained protein was used. For
the detection of phosphorylated receptors, the receptor tyrosine
kinase phosphorylation array (Human Phospho-Receptor Tyrosine
Kinase Array kit; R&D Systems) was executed according to the
manufacturer's protocol.
Transcriptome profiling experiments
Total RNAs were extracted using the standard TRIzol
(Invitrogen) protocol provided by the manufacturer for preparation
of mRNA. RNA purity and integrity were monitored using
NanoDrop® ND-1000 spectrophotometer and Agilent 2100
Bioanalyzer with RNA 6000 Nano assay kit. Only RNAs with no sign of
contamination or marked degradation (RIN >9) were considered
good quality and used for further analysis. Transcriptome profiles
were determined in the same triplicate of RNAs using the Affymetrix
Human Transcriptome Array 2.0. For whole-transcript expression
analysis, 100 ng of total RNAs were processed and labeled using the
GeneChip WT PLUS Reagent kit (Affymetrix, Santa Clara, CA, USA).
Upon hybridization of labelled products, arrays were washed and
stained using the Affymetrix GeneChip WT Terminal Labeling and
Hybridization kit, before being scanned using a GeneChip Scanner
3000.
Microarray data analysis
CEL files generated upon array scanning were
imported into Partek® Genomics Suite™ (GS) 6.6 for
preprocessing. Partek was set up to run standard RMA at the
probeset level. Resulting log2 probeset intensities were then
imported into R statistical environment (http://www.R-project.org/) for further analysis.
First, log2 intensity values were summarized to estimate the
expression level of each transcript cluster (TC) by averaging the
intensity signals from the corresponding probeset regions. Matching
between probesets, TCs and targeted genes was verified through
Affymetrix annotation files (HTA-2_0 probeset and transcript hg19
na33.1 csv file). The quality of the data was then evaluated by
assessing repeatability Pearson's correlation coefficients, and
through visual inspection of density plots and relative log
expression plots. Principal component analysis was also used to
reduce dimensionality of the data, visualize the concordance
between biological replicates, and assess if the variability in
data actually reflected what was expected from the experimental
design. Finally, the Limma package (R/Bioconductor) was used to
estimate the statistical significance of TC expression level
differences between LNT-229-rCPE and LNT-229-neo samples as the
reference. Resulting P-values were adjusted for multiple testing
error using the Benjamini and Hochberg's false discovery rate (FDR)
(14). Elements with a FDR
<0.05 were considered as differentially expressed (DE),
irrespective of the fold-change. Microarray expression data are
available at ArrayExpress (http://www.ebi.ac.uk/arrayexpress) under the accession
number E-MTAB-5297. The Qiagen's Ingenuity® Pathway
Analysis software (IPA; Qiagen Redwood City, Inc., Redwood City,
CA, USA; www.qiagen.com/ingenuity) was used for
transcript cluster mapping and for data mining, including
functional analyses, upstream analysis and gene network
reconstruction. Heatmaps were generated corresponding to
significant genes with an FDR of <0.01. Right-tailed Fisher's
exact test was used to calculate a P-value for functional
enrichment analysis (threshold: -log(P-value) >1.301,
corresponding to a P-value <0.05).
Quantitative RT-PCR
RNA was reverse-transcribed using SuperScript II
(Invitrogen GmbH, Karlsruhe, Germany). Target gene expression was
determined using the SYBR-Green Master Mix (Thermo Fisher
Scientific) on an ABI 7200 system. Relative mRNA expression was
quantified ([EDCT (gene of interest)/EDCT
(housekeeping gene)]). The following primers were used: huCPE-f,
CCACCATGTCGCAAGAATGA and huCPE-r, AAGCTCCACGGTGATCTCAAA; rCPE-f,
ATGGGAATGAGGCTGTTGGAC and rCPE-r, GGCATGATGTGAATGCGGGTA; RPLP0-f,
GAGTCCTGGCCTTGTCTGTGG and RPLP0-r, TCCGACTCTTCCTTGGCTTCA; SPP1-f,
GCCGAGGTGATAGTGTGGTT and SPP1-r, ACGGCTGTCCCAATCAGAAG; SLUG-f,
CATACCACAACCAGAGATCC and SLUG-r, GAGGAGTATCCGGAAAGAGG; STC1-f,
AAGATGGCGACCACCAAAGT and STC1-r, GCAGTGACGCTCATAAGGGA; MGST1-f,
GGTTTTGTTTATGGTACTTCAGAGT and MGST1-r, TGTGAATTGTTCATTTAGATGTGCC;
CD9-f, AAACGCTGAAAGCCATCCAC and CD9-r, GATGGCATCAGGACAGGACTT;
MGAT4A-f, TGGTGTTGCAGAAGGAATGGT and MGAT4A-r,
TCAGATGATCAGTTGGTGGCT; ADAMTS4-f, GACAAGTGCATGGTGTGCG and
ADAMTS4-r, GCCGGACAAGAATGTGGGT; PCDH17-f, AGTTTGTTCAAAGTAGCTCCACG
and PCDH17-r, TCACAGCAGGAGCCTTTGTT. RPLP0 primers were used for
normalization.
Mouse experiments
Athymic FoxN1-deficient NMRI nude mice were
purchased from Janvier Labs (Saint Berthevin, France). Deficiency
in the FoxN1 gene provides thymic aplasia which results in
immunodeficiency regarding lack of T cells, whereas B and NK cells
remain. The mutation also leads to a defect of the hair follicule
leading to transient downy hair. When this duvet is gone, mice
appear nude. Female mice of 5 weeks of age were used in all
experiments. Mice were held in groups of 4 to 5 mice in filter top
cages under sterile conditions in the animal facility of the Hertie
Institute at a temperature of 24°C and 50% humidity according to
the regulations of the Society for Laboratory Animal Science. For
tumor growth, mice were anaesthetized using fentanyl, midazolam and
medetomidin and 100,000 cells were implanted in the right striatum
of the mice. After surgery, anesthesia was neutralized using
naloxon, flumazenil and atipazemol. Analgetics (carpofen) were
added intraoperatively. The mice were sacrified when neurological
symptoms like tremor, tilt of the head, hemiparesis, hemiplegia or
uncoordinated motion appeared of if the mice lost weight (fixed
end-point experiment). Tumor burden was determined optically.
Survival analyses were done by generating Kaplan-Meier survival
curves. The animal experiments were licensed by the regional board
Tübingen and were performed according to the German law, Guide for
Care and Use of Laboratory Animals.
Statistical analysis
The figures show data obtained in at least three
independent experiments as indicated. Statistical analyses were
performed using GraphPad Prism version 6.0, (GraphPad Software,
Inc., La Jolla, CA, USA). Quantitative data were assessed for
significance by t-test (P<0.05; P<0.01; P<0.001). Survival
of mice was analyzed by Kaplan-Meier live table and for comparison
of survival, the Wilcoxon and log-rank tests were used
(significance level a=0.05; JMP 11.0 software; SAS Institute, Inc.,
Cary, NC, USA).
Results
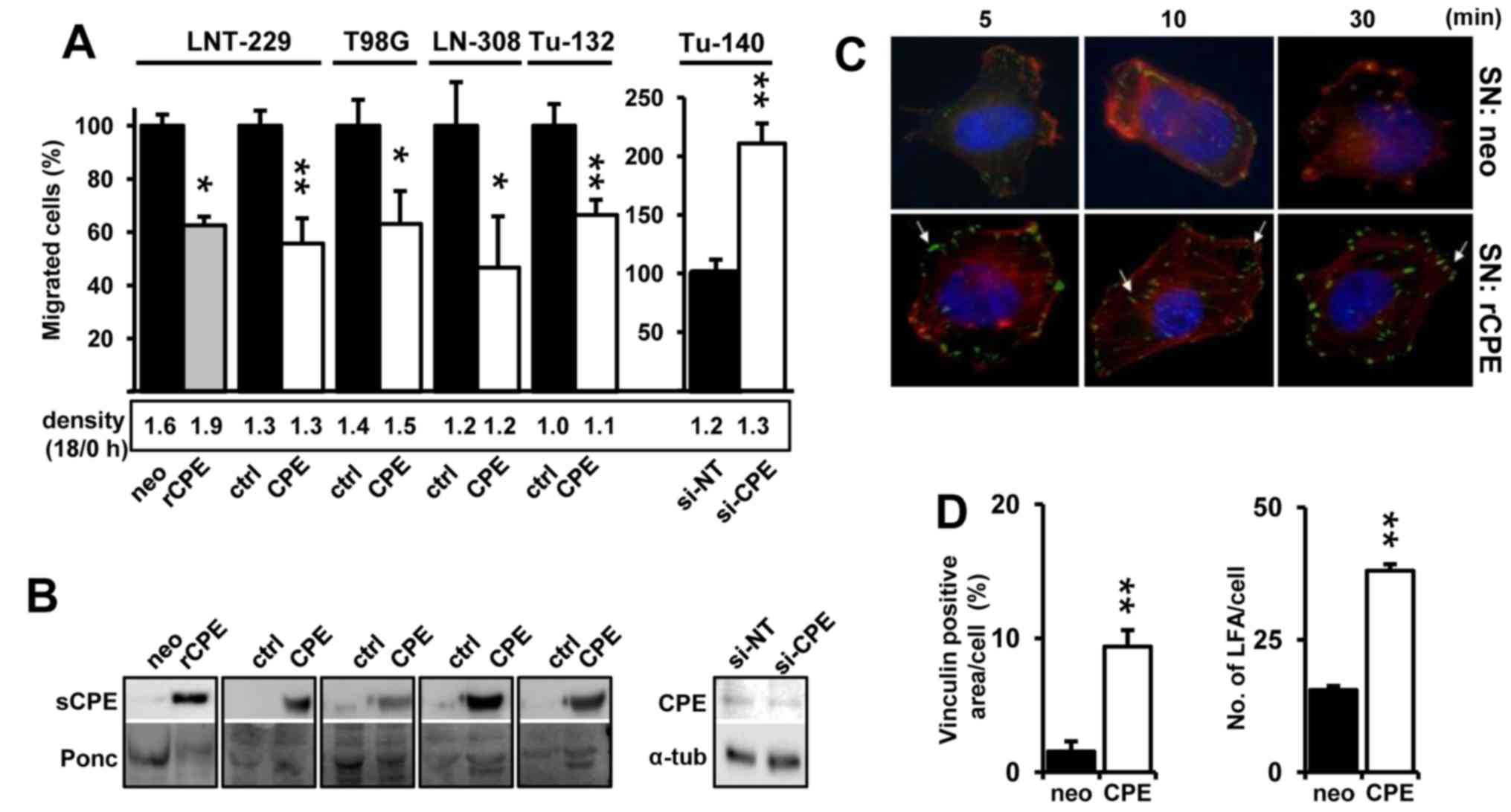
sCPE mitigates cell motility and enhances
cell adhesion in established and primary glioma cells
We have previously shown that sCPE reduced cell
motility in LNT-229 and LN-308 glioma cell lines (7). To demonstrate a general
anti-migratory function of sCPE in glioma, we generated additional
sCPE-overexpressing established and a primary low passage glioma
cell lines by lentiviral transduction using pReceiver LV105-CPE.
Overexpression of both human or rat sCPE mitigates migration in
glioma cell lines (LNT-229, T98G and LN-308) and primary low
passage Tu-132 glioma cells. In comparison and as demonstrated
before for LNT-229 cells (7), the
knockdown of sCPE induces cell motility in Tu-140 that possess
higher endogenous CPE levels compared to Tu-132 cells (Fig. 1A and B). According to the 'Grow or
Go' hypothesis, proliferation and migration are inversely regulated
(15). For this reason and to
avoid that migration rates were influenced by proliferation, we
also measured cell density at the start and end of the migration
period, and also for longer time periods. Only rat CPE
overexpressing LNT-229 cells showed slightly elevated proliferation
[(7) and data not shown]. Even if
the differences in cell density were negligible at the end of the
migration period (Fig. 1A, lower
panel) we used these values to normalize cell migration. We have
previously shown that overexpression of CPE induced the generation
of large focal adhesion complexes (LFA) in LNT-229 cells suggesting
a more adhesive phenotype (7).
This effect is most probably mediated by sCPE since addition of
sCPE-containing supernatants induced the formation of
vinculin-positive LFA and actin stress fibers in LNT-229 cells
(Fig. 1C and D).
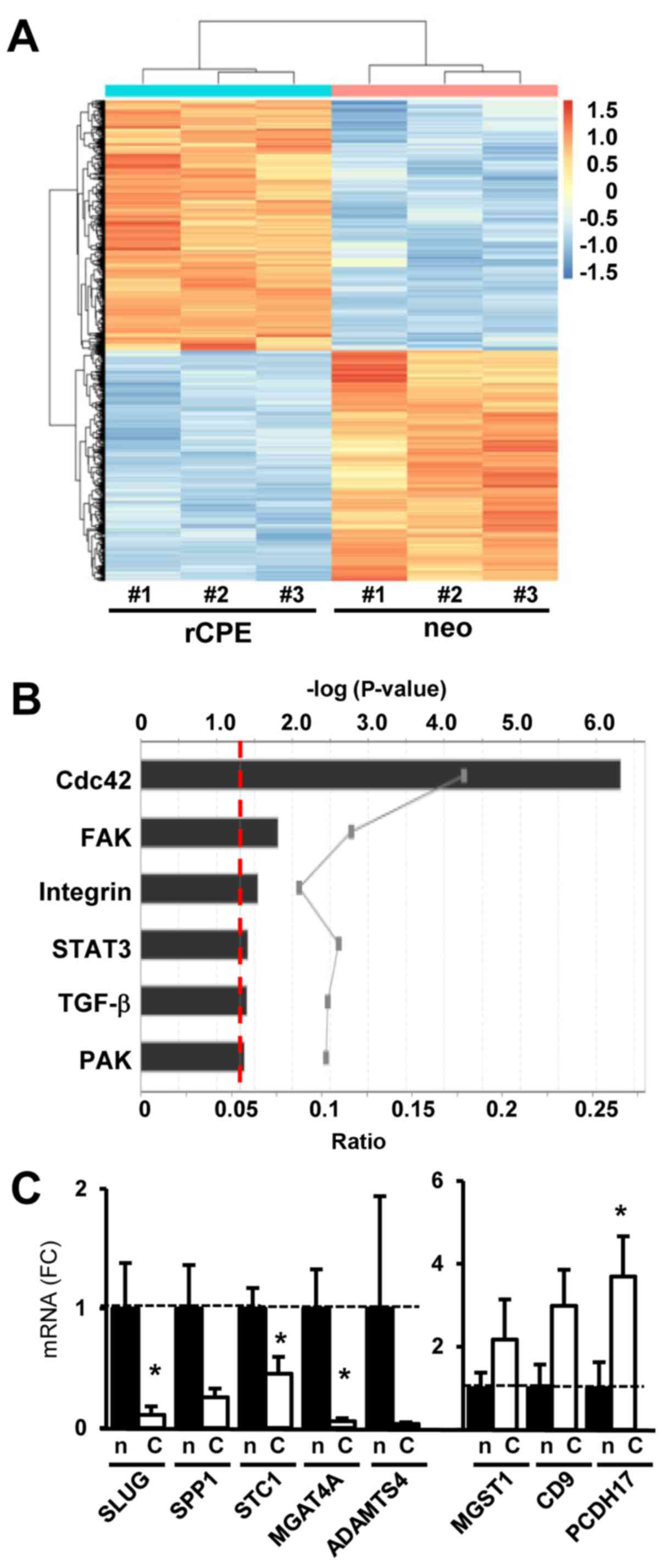
sCPE affects the expression of genes
mostly related to cell architecture and motility
Considering that extracellular sCPE could serve as a
ligand or signaling factor, or might compete with the binding of
extracellular ligands to their receptors, finally leading to
changes in gene expression, we were interested whether
overexpression of sCPE may alter the expression of
motility-associated genes in glioma cells. We therefore analyzed
differential gene expression in less migratory sCPE-overexpressing
LNT-229 (LNT-229-rCPE) and its sibling cell line (LNT-229-neo) that
migrates faster and secrets only a very low amount of sCPE
(Fig. 1A). Using mRNA expression
data, we found 1065 mRNAs with a false discovery rate (FDR)
<0.01 to be differentially expressed in sCPE-overexpressing
compared to neo control cells. In this group a heatmap plot
represents the intensity of significant genes (FDR <0.01)
deciphering that the two cell lines properly cluster in a balanced
number of upregulated and downregulated genes (Fig. 2A). IPA, further detailed analyses
and a broad literature search showed that in the panel of these
differentially expressed genes at least 100 genes were connected to
the regulation of cell motility. Besides, IPA demonstrated an
enrichment of differentially expressed mRNAs in sCPE-overexpressing
LNT-229 cells that are associated to the Cdc42-, FAK-, STAT3-,
TGF-β-, PAK- and integrin-signaling pathways (Fig. 2B and Table I), with a tendency to a reduced
activation of the Cdc42-, TGF-β-, PAK- and integrin-signaling
pathways in these cells (data not shown). After activation, these
pathways are known to induce cell motility.
 | Table IMotility-associated genes
differentially expressed in rat sCPE overexpressing LNT-229
cells. |
Table I
Motility-associated genes
differentially expressed in rat sCPE overexpressing LNT-229
cells.
| Gene | Protein |
Pro/anti-migratory | Fold-change in
microarray (CPE/neo) | Function | Association to
signaling pathway |
|---|
| ADAMTS1 | A disintegrin and
metalloproteinase with thrombospondin motifs 1 | pro | 2.9x down
(P<10−4) | Metalloproteinase,
contributes to IGF-II-mediated IGF1R phosphorylation and cellular
migration in glioma cells; semaphorin 3C cleavage induced by
ADAMTS1 promotes cell migration; marker for poor prognosis in
glioma | FAK, Integrin,
TGF-β |
| ADAMTS4 | A disintegrin and
metalloproteinase with thrombospondin motifs 4 | pro | 3.3x down
(P<10−11) | Metalloproteinase;
degradation of aggrecan; matrix degrading enzyme | FAK, Integrin,
TGF-β |
| ARRDC3 | Arrestin
domain-containing 3 | anti | 1.5x up
(P<10−4) | Overexpression
represses cancer cell proliferation, migration, invasion, growth in
soft agar and in vivo tumorigenicity; downregulation has the
opposite effects; controls the cell surface adhesion molecule β4
integrin; often epigenetically silenced | Integrin |
|
CD9/MRP-1 | Tetraspanin | anti | 3.5x up
(P<10−11) | Inhibits CD26
mediated enhancement of invasive potential of mesenthelioma;
migration regulating effects of CD9 have been linked to
adhesion-dependent signaling pathways including FAK, PI3K, p38 MAPK
and Jun N-terminal kinase (JNK) | Cdc42, FAK,
Integrin, STAT3, TGF-β |
| CTSD | Cathepsin D | pro | 1.7x down
(P<10−4) | Involved in cancer
cell invasion; cancer cell invasion is also induced by cathepsin B
which is activated by cathepsin D | Cdc42, STAT3 |
| CTSH | Cathepsin H | pro | 2.3x down
(P<10−4) | Induces glioma cell
invasion; correlates with glioma malignancy; promotes hepatoma cell
migration and invasion | Integrin |
| ENPP2 | Autotaxin | pro | 4x down
(P<10−10) | Multifunctional
phosphodiesterase; potent cell motility-stimulating factor in GBM;
promotes MMP-3 production | STAT3, Cdc42, FAK,
Integrin, TGF-β |
| IGFBP7 | Insulin-like growth
factor binding protein 7 | pro | 3.3x down
(P<10−5) | Induces migration
in glioma cells; knockdown restores TGF-β induced EMT | TGF-β |
| L1-CAM2/CHL1 | Cell adhesion
molecule L1-like | pro | 5.1x down
(P<10−8) | Overexpressed in
glioma stem cells; L1-CAM stimulates glioma cell motility; SLUG
binds to both L1-CAM promoters and is essential for its induction
by TGF-β | FAK Integrin,
TGF-β |
| MAP7D3/MDP3 |
Microtubule-associated protein 7 domain
containing protein 3 | pro | 4.3x down
(P<10−8) | Involved in
microtubule polymerization; transendothelial B cell migration; loss
of MAP7D3 reduces the motility of malignant cancer cells by
altering the turnover of actin | STAT3 |
| MGAT4A |
N-acetyl-glucosamyl-transferase IV A | pro | 3.2x down
(P<10−9) | Transfers GlcNAc in
specific linkage to N-glycans; upregulared in breast cancer tissue;
high expression promotes invasion in choriocarcinoma | Integrin,
TGF-β |
| MGST1 | Microsomal
glutathione-S-transferase 1 | anti | 5.4x up
(P<10−12) | Involved in
laminin-dependent migration in PC-12 cells; lower expression in
PC12 cell show weaker adhesion; upregulated in glioma-derived glial
progenitor cells | TGF-β |
| MST4 | Member of the
sterile 20 serine/threonine kinase family | pro | 13.2x down
(P<10−13) | Involved in cell
migration; promotes EMT in hepatocellular carcinoma (HCC) | Cdc42 |
| PAK3 | p21 protein
activated kinase 3 | pro | 1.98x down
(P<10−5) | Stimulates cell
migration and anchorage-independent growth | Cdc42, FAK,
Integrin, |
| PCDH17 | Procadherin 17 | anti | 4.7x up
(P<10−11) | Inhibits cell
migration and invasion of esophageal squamous cell carcinoma;
silenced in many cancers; regulates actin dynamics; loss promotes
metastasis and invasion in HCC cells | Integrin |
| PPARG | Peroxisome
proliferator-activated receptor γ | anti | 1.9x up
(P<10−6) | PPARγ agonists
block glioma motility and invasiveness | Cdc42, FAK,
STAT3, |
| PTGS2/COX-2 | Cyclooxygenase
2 | pro | 4.2x down
(P<10−6) | Enzyme involved in
prostaglandin (including PGE2) biosynthesis; promotes glioma cell
migration | STAT3, Cdc42, FAK,
Integrin, TGF-β, PAK |
| PTPRD | Protein tyrosine
phosphatase, receptor type, D | anti | 3.2x up
(P<10−7) | Loss in high grade
GBM; reintroduction enhances cell adhesion of GBM cells; suppresses
cancer cell migration | STAT3, FAK,
TGF-β |
| PXDN | Peroxidasin | pro | 8.7x down
(P<10−13) | Regulator of cell
plasticity and extracellular matrix remodeling; glioma endothelial
marker gene; upregulated in the tumor vasculature ECM | Integrin,
TGF-β |
| SDC2 | Syndecan-2 | pro | 2.7x down
(P<10−9) | Promotes membrane
protrusion and migration; involved in cell adhesion; induces cell
migration and invasion in human colon and pancreatic cancer
cells | Cdc42, FAK,
Integrin, STAT3, TGF-β |
|
SLUG/SNAI2 | Slug | pro | 8.0x down
(P<10−8) | Transcription
factor involved in EMT processes | STAT3, Cdc42, FAK,
Integrin, TGF-β |
| SPP1 | Osteopontin
(OPN) | pro | 10.1x down
(P<10−7) | Matricellular
protein; promotes glioma cell migration and invasion | STAT3, Cdc42/Rho,
FAK, Integrin, TGF-β |
| STC1 |
Stanniocalcin-1 | pro | 1.6x down
(P=0.004) | Secreted
glycoprotein; biomarker of glioma progression; hypoxia-dependent
migration factor in glioma | STAT3, Cdc42, FAK,
TGF-β |
| TGFBR2 | TGF-β receptor Type
II | pro | 1.9x down
(P<10−5) | Receptor for TGF-β;
promotes migration in glioma cells | STAT3, Integrin,
TGF-β |
| ZFPM2/FOG-2 | Zinc finger
protein, FOG family member 2 | pro | 2.3x down
(P<10−9) | Involved in
post-mitotic neuronal migration; found to interact with STAT3 in
liver | STAT3 |
The expression of assorted genes identified by
microarray analysis that are known to be involved in different
steps of the regulation of cell motility was validated by qRT-PCR.
Motility-and cell adhesion-associated genes such as SNAI2/SLUG (8x
down), osteopontin (SPP1/OPN, 10.1x down), stanniocalcin-1 (STC,
1.6x down), a disintegrin and metalloprotease with thrombospondin
motifs (ADAMTS4, 3.3x down), procaherin-17 (PCDH17, 4.7x up),
tetraspanin/motility related protein-1 (CD9/MRP-1, 3.5x up),
N-acetyl-glucosamyl-transferase IV A (MGAT4A, 5.4x down),
microsomal glutathione-S-transferase 1 (MGST1, 4.2x down) and
others were differently expressed in sCPE-overexpressing compared
to neo control LNT-229 cells (Table
I and Fig. 2C).
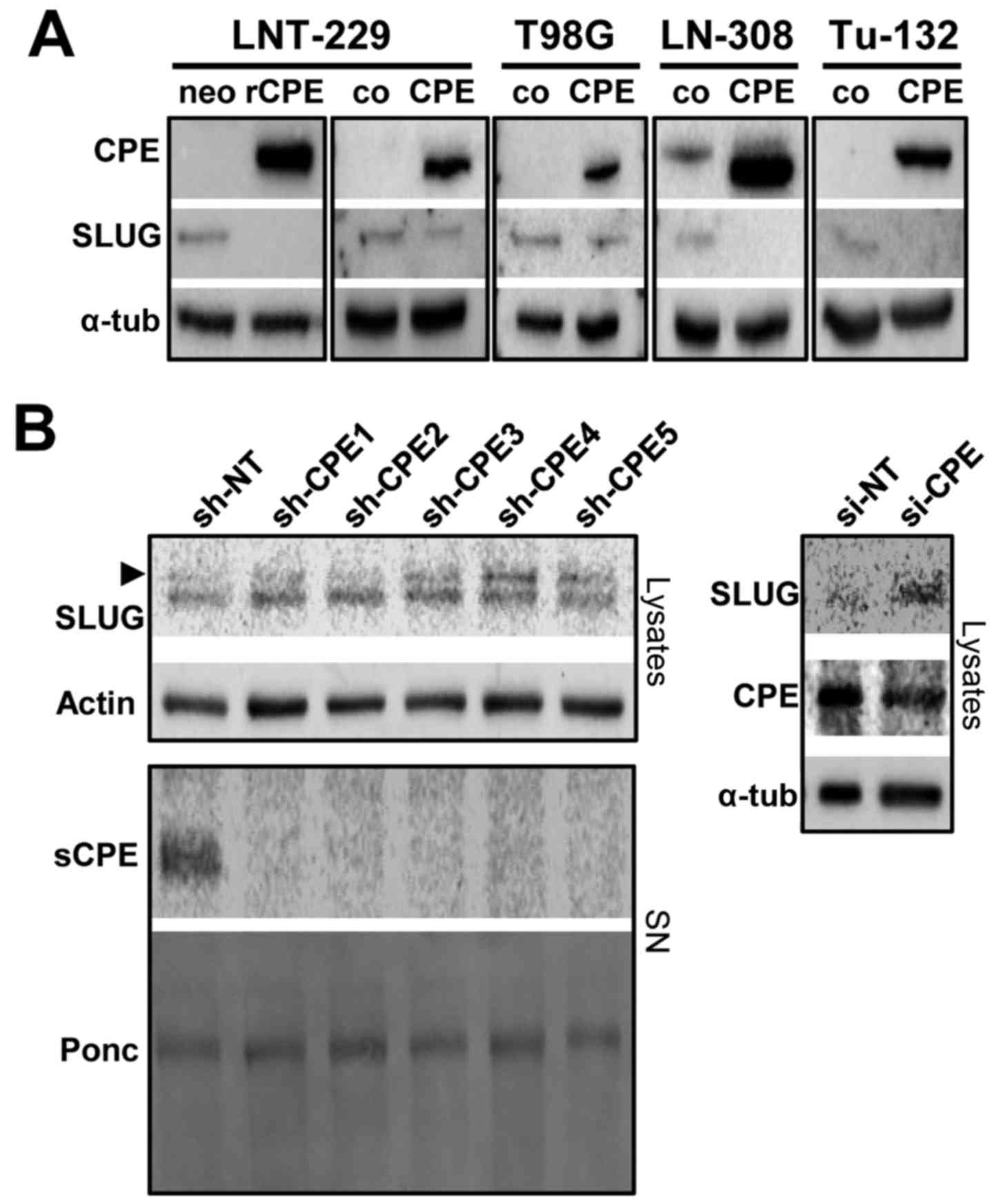
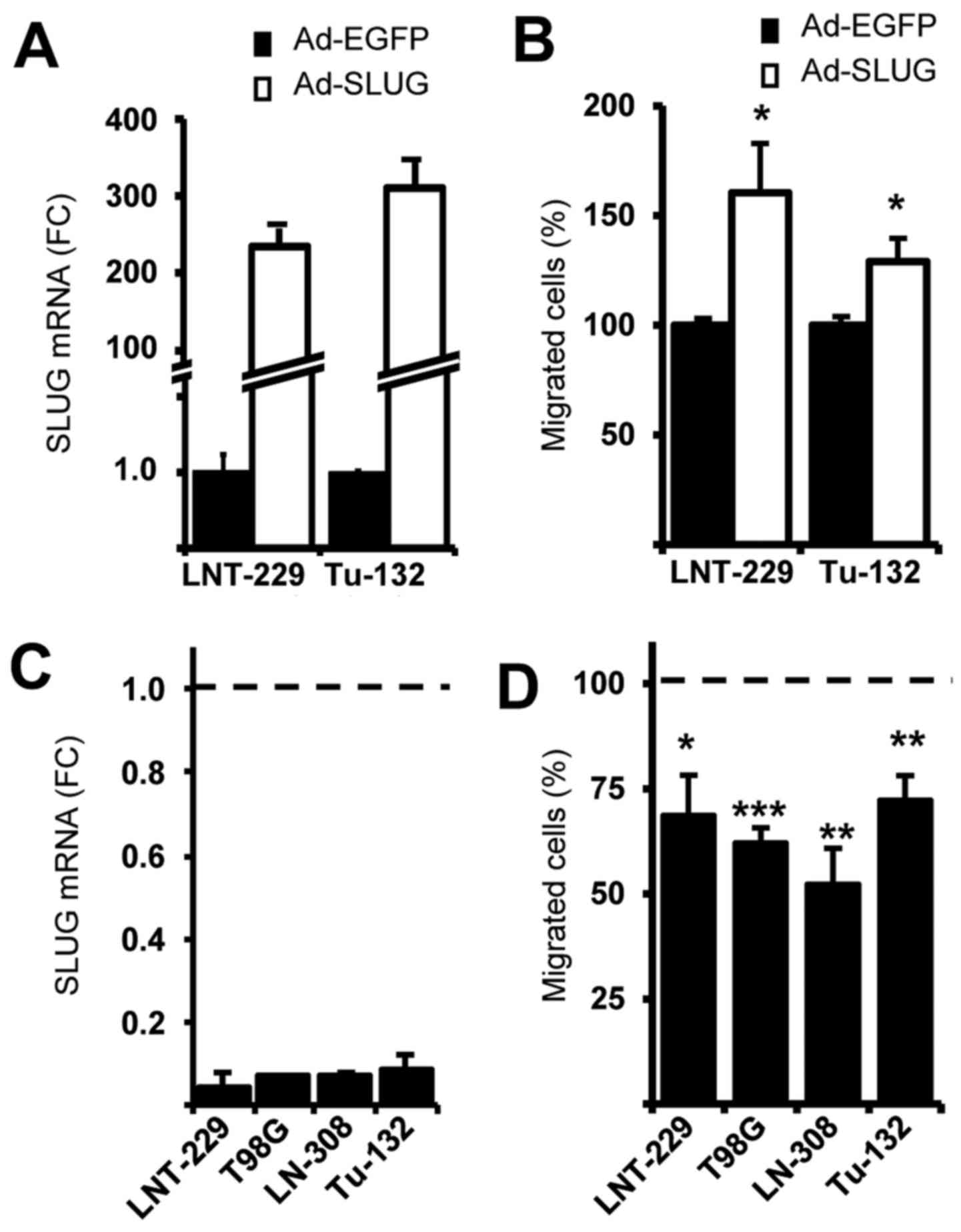
SLUG expression negatively correlates
with sCPE and glioma cell migration
Since we found SLUG as one prominent factor being
downregulated in rat sCPE-overexpressing LNT-229 cells, we tested
whether overexpression of human sCPE provides the same effect.
Indeed, reduced expression of SLUG was detected in all human
sCPE-overexpressing low passage primary and established glioma cell
lines (Fig. 3A). Vice versa,
knockdown of CPE in Tu-140 cells that provide elevated CPE
secretion but no basal SLUG, induced the expression of SLUG
(Fig. 3B). We therefore tested
whether SLUG modulates glioma cell migration. Adenovirus-based
overexpression of SLUG induces migration whereas knockdown of SLUG
mitigates migration (Fig. 4).
In our microarray data we identified several
differentially expressed genes that are involved in the remodeling
of the ECM. Therefore, we tested whether matrix metalloproteinases
(MMPs), the main players in invasive EMT-like processes and
destructors of the ECM, were differentially regulated. Even if
there was no regulation of MMPs on mRNA level (data not shown),
MMP-2 was downregulated in those sCPE-overexpressing cell lines
that harbor MMP-2 (LNT-229 and T98G). With the exception of LN-308
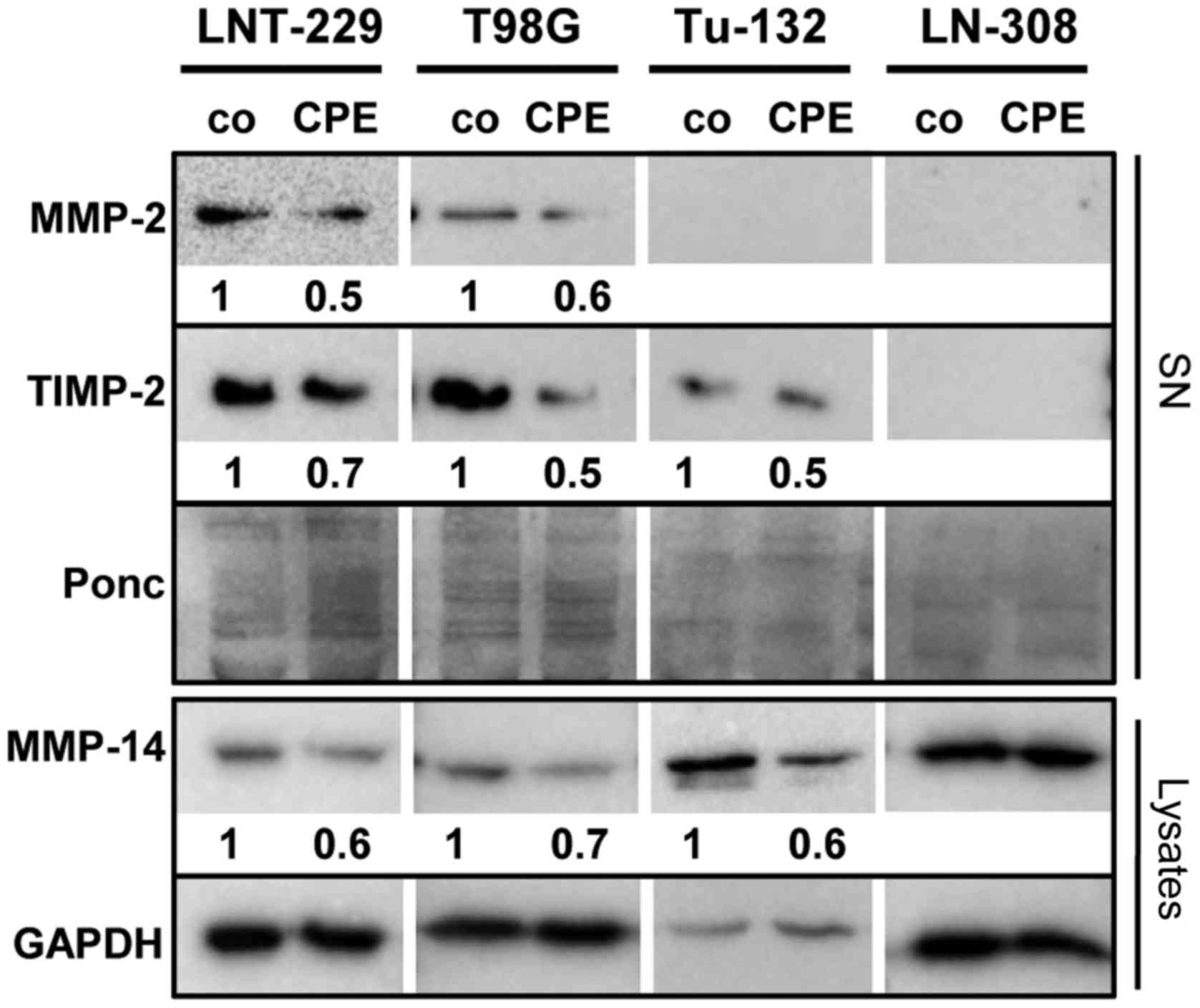
cells, also MT1-MMP/MMP-14 was reduced. It has been described that
the net MMP-2 activity correlates with the level of TIMP-2
expression (16). Knowing that
under certain conditions TIMP-2 activates MMP-2, we analyzed TIMP-2
expression, demonstrating that TIMP-2 was also downregulated in
sCPE-overexpressing LNT-229-, T98G- and Tu-132 cells (Fig. 5).
sCPE mediated reduction of SLUG and
mitigation of glioma cell migration are transmitted via ERK1/2
It has been described that in some cancer cell lines
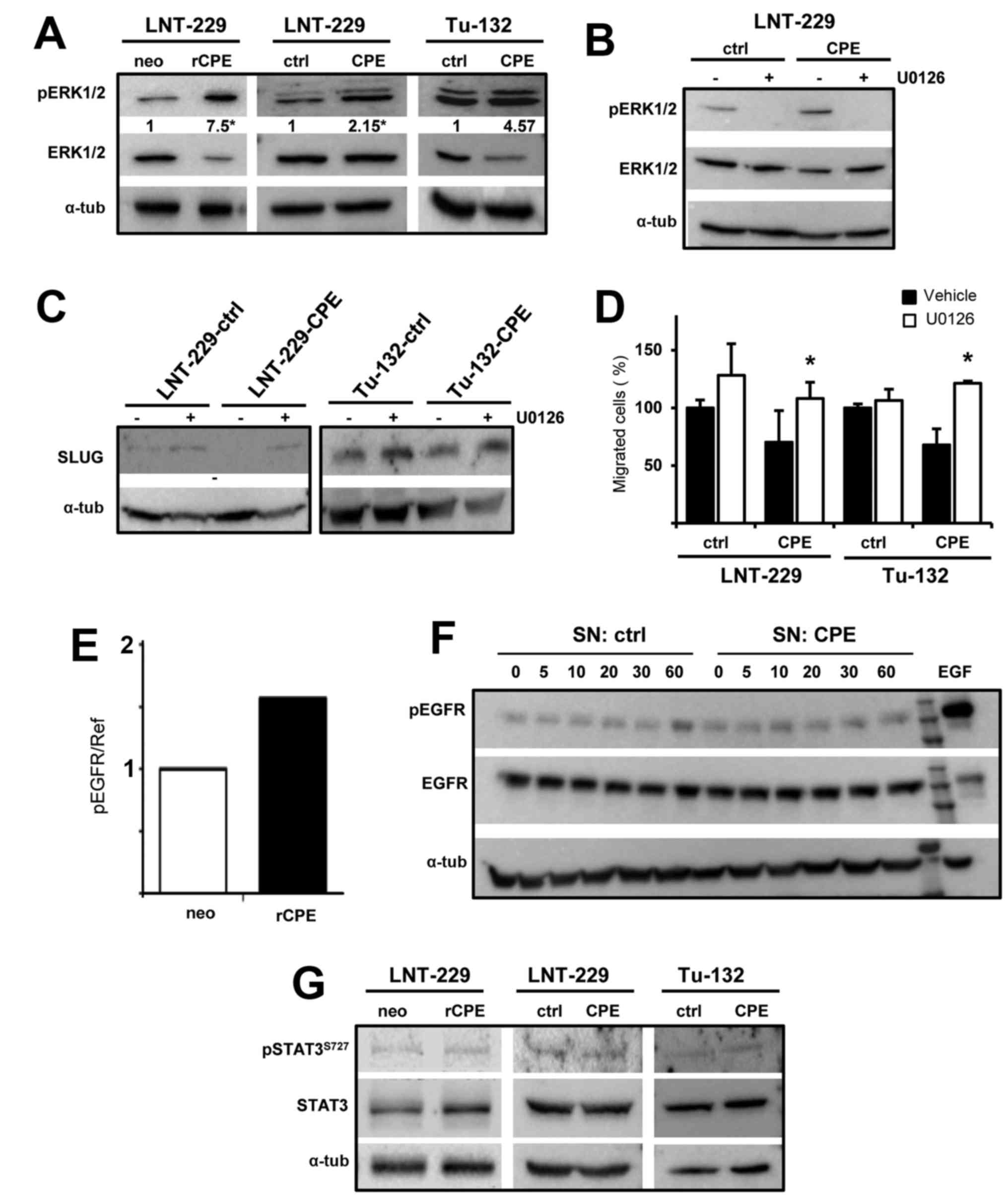
CPE can also transmit its activity via ERK1/2 (17). We analyzed whether ERK
phosphorylation was altered in both rat and human
sCPE-overexpressing human glioma cells and found elevated ERK
phosphorylation in LNT-229 and Tu-132 sCPE-overexpressing cells
(Fig. 6A). After inhibition of
ERK1/2 activation using the MEK inhibitor U0126 no reduction of
SLUG was detectable anymore in sCPE-overexpressing LNT-229 and
Tu-132 cells (Fig. 6C). Whereas no
significant effect of U0126 on cell migration was detectable in
LNT-229 and Tu-132 control cells, addition of U0126 abolished the
anti-migratory activity of sCPE in the sCPE-overexpressing cells
(Fig. 6D). To determine the
upstream cell surface receptor by which extracellular sCPE mediates
ERK activation we performed a membrane-based assay that allows the
detection of 49 phosphorylated receptor tyrosine kinases. We found
a 1.5-fold upregulation of epidermal growth factor receptor (EGFR)
phosphorylation (Fig. 6E),
however, this could not be confirmed by immunoblot analysis of
phospho-EGFR in LNT-229 glioma cells cultivated in sCPE-containing
conditioned medium (Fig. 6F and
data not shown) indicating that phosphorylation of the EGFR may not
be responsible for sCPE-mediated ERK phosphorylation.
SLUG expression can also be regulated by STAT3.
Since many of the mRNAs we found to be differentially expressed by
microarray expression analysis are associated to STAT3, we also
analyzed STAT3 phosphorylation, but did not detect any changes in
phospho-STAT3 in sCPE-overexpressing cells (Fig. 6G).
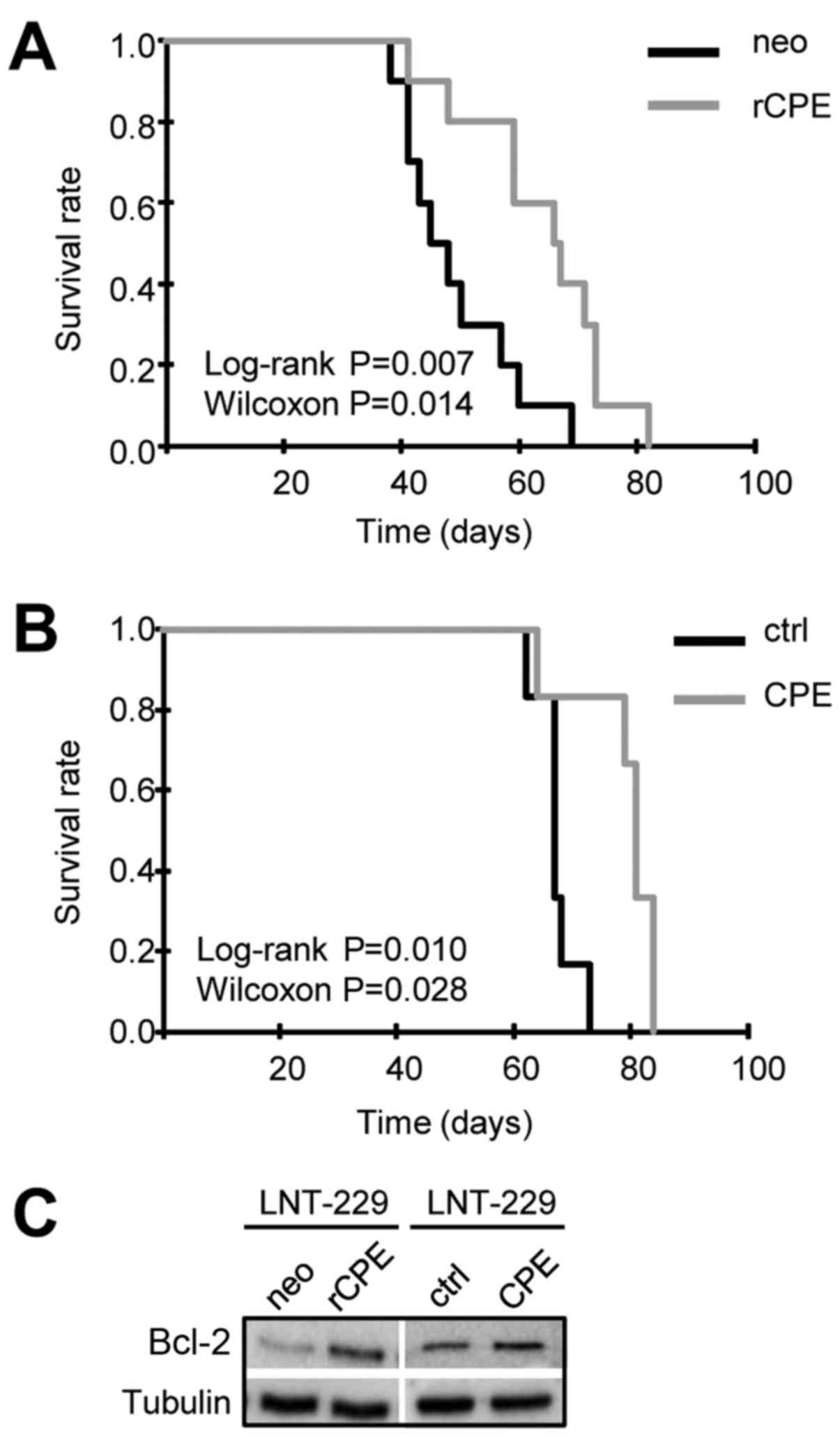
sCPE overexpression prolongs the survival
of glioma-bearing mice
In order to consider a possible translational
application of sCPE and knowing that CPE, among 311 proteases, is
the only peptidase that is downregulated in GBM (18), we were interested whether the
anti-migratory effects induced by sCPE we observed in vitro
is associated to the survival of glioma-bearing mice. For this we
implanted either rat or human sCPE-overexpressing LNT-229 or their
sibling control cells into the right striatum of nude mice. The
time-point the mice developed neurological symptoms they were
sacrificed. At this time-point all mice developed large tumors.
Survival analyses demonstrate that mice harboring tumors derived
from sCPE-overexpressing cells survived significantly longer than
mice harboring control tumors (Fig. 7A
and B). Prolonged survival of CPE-tumor bearing mice was not an
effect of reduced proliferation in sCPE expressing cells (Fig. 1A, lower panel and data not shown)
and even not an effect of elevated apoptosis in these cells since
no differences in caspase activity were detectable (7). Besides and due to the data shown by
Murthy et al (8,17), who demonstrated that CPE
upregulates BCL-2 we analyzed BCL-2 expression in control-and
sCPE-overexpressing glioma cells lines and found even enhanced
BCL-2 protein expression in all sCPE expressing cell lines
(Fig. 7C and data not shown).
Discussion
The effect of CPE on tumor cell migration has been
described for several tumor entities and also for glioma (7,17).
We have demonstrated that a secreted form CPE is responsible for
its anti-migratory effect in at least three established and two
primary glioma cell lines. Primary GBM cells normally grow as
neurospheres and at least a subpopulation of these cells provide
glioma stem cell characteristics. In our experiments primary GBM
cells were grown in the presence of FCS leading to a more
differentiated and adherently growing phenotype. However, this
allows the comparison of the anti-migratory function of sCPE in
adherently growing established and primary cells.
There are hints that CPE modulates the ERK1/2, AKT
and WNT signaling pathways (8,19,20),
by this also regulating gene expression, however, it still remains
unclear whether changes in the activation of these pathways are
responsible for reduced glioma cell motility. In
sCPE-overexpressing cells we found altered expression of at least
100 genes involved in the regulation of cell motility (data not
shown). Among those regulated genes the main part is involved in
the regulation of the TGF-β, Ccd42, PAK, FAK, STAT3 and integrin
signaling pathways which we have found to be negatively regulated
by sCPE using IPA (Table I and
Fig. 2). qRT-PCR validation of
assorted genes that are known to regulate cell motility at
different stages led to the identification of at least three
anti-migratory genes significantly upregulated by sCPE (Fig. 2C). CD9/MRP-1 is closely linked to
plateled derived growth factor mediated glioma cell migration
(21), downregulation of MGST1
weakens cell adhesion in PC12 cells (22) whereas PCDH17 regulates actin
dynamics and inhibits migration and invasion (23,24).
In contrast, many pro-migratory genes like ADAMTS4, MGAT4A, SPP1,
STC1 and SNAI2/SLUG are downregulated by sCPE (Fig. 2C). ADAMTS4 is a matrix degrading
protease important for glioma migration (25), STC1, a biomarker of glioma
progression, has been described as a hypoxia-dependent migration
factor (26), SPP1 is a
matricellular protein that promotes glioma cell migration and
invasion (27) and MGAT4A is
upregulated in several tumors and has been suggested to promote
invasion and metastasis (28–31).
One prominent factor we found to be highly
downregulated in sCPE-overexpressing glioma cells was the SNAIL
family transcriptional regulator SLUG. SLUG is upregulated during
the epithelial to mesenchymal transition (EMT) in epithelial
cancers and is a prominent metastatic factor (32). GBM is not an epithelial tumor, but
EMT-like processes have been described for GBM, and enhanced SLUG
expression is linked to the more invasive and migratory mesenchymal
phenotype of gliomas (10,33–35).
SLUG seemed to be directly regulated by sCPE, since overexpression
of sCPE led to reduced SLUG expression and migration, whilst
knockdown of CPE to enhanced SLUG expression and migration
(Figs. 2 and 3). SLUG is a highly unstable protein with
a half-life of <1 h (36). This
might explain why the adenoviral mediated upregulation of SLUG did
not enhance migration as prominently as the SLUG knockdown reduced
migration (Fig. 4). SLUG provides
its pro-migratory function by different pathways. Besides, it binds
to the promoters of the L1-CAM gene family that augment glioma cell
motility and invasion and correlate with FAK activity (37,38).
We identified L1-CAM2/CHL1 to be 5.1x downregulated in
sCPE-overexpressing cells. MMPs are also involved in EMT-like
processes and their expression is regulated by SNAIL family members
(39). In glioma, MMPs are mainly
regulated by TGF-β, a pathway we identified to be negatively
regulated in sCPE-overexpressing glioma cells (Fig. 2). We found reduced levels of MMP-2,
MT1-MMP/MMP-14 and TIMP-2 in those CPE-overexpressing cell that
express these enzymes (Fig. 5),
suggesting a direct correlation of sCPE, TGF-β, SLUG and MMPs. In
this regard it is known that in mesenchymal, but not in glial areas
of brain tumors, SLUG, TGF-β, MMP-2 and MMP-9 are highly expressed
or activated (35,40). Consistently, we found high CPE
expression in the more adhesive and lesser migratory epithelial
like-cell fraction of a gliosarcoma, whereas the sarcoid fraction
of this tumor was negative for CPE (unpublished data). Notably, the
level of MMP reduction did not strictly correlate with the level of
sCPE-expression, sCPE-mediated reduction of SLUG and mitigation of
migration. This indicates that processes such as sCPE-mediated
differential expression of other migration modulating genes,
influencing sCPE on migratory pathways or even an interaction
between these pathways and genes might play pivotal roles in the
anti-migratory effect of sCPE.
It has been recently described that ERK1/2 is
phosphorylated and activated in CPE overexpressing cells (17,19).
Elevated ERK phosphorylation in sCPE-overexpressing glioma cells is
paralleled by reduced SLUG expression and migration. Consistently,
inhibition of ERK1/2 activation completely abolishes the
sCPE-mediated downregulation of SLUG and led to elevated cell
migration, indicating that sCPE-dependent SLUG expression and cell
motility is regulated via ERK1/2 (Fig.
6). The typical activation of ERK1/2 by EGFR leads to enhanced
cell migration in many tumor cells. However, no phosphorylation of
EGFR was detectable in sCPE-overexpressing cells or by addition of
sCPE-containing supernatants to parental glioma cells (Fig. 6). Besides, inhibition of ERK1/2
activation by U0126 does not alter the migratory potential of
several glioma cell lines (41,42)
as we have also seen in LNT-229 and Tu-132 control cells.
Therefore, we suggest that in glioma cells and in the context of
sCPE overexpression, the activation of ERK1/2 by a yet unknown
receptor provides anti-migratory effects whereas under other
circumstances when no or only very low amounts of sCPE are present,
phosphorylation of ERK1/2 can induce migration. Identification of
the signals or factors that determine ERK1/2 to work in a pro- or
anti-migratory fashion needs further intensive investigation. It
will also be a challenge for the future to identify the upstream
factors that led to the observed activation of ERK1/2 by sCPE.
One possible mechanism how CPE might also mitigate
glioma cell migration beside downregulating SLUG is the effect of
CPE on the WNT-pathway as described by Skalka et al
(43). WNT regulates the
architecture of the cytoskeleton and in this regard is responsible
for cell shape, cell adhesion and cell motility. In HEK cells, CPE
aggregates with and refrains WNT3a in vesicle-like structures,
leading to reduced WNT secretion and by this mechanism putatively
inhibits WNT3a effects. This assumption and our observation that
sCPE negatively regulates Rac1 (unpublished data), might explain
why sCPE-overexpression (7), or
addition of sCPE-containing supernatants to parental glioma cells
(Fig. 1), induce the formation of
LFA complexes. To what extent a WNT3a-CPE complex is involved in
the reduction of migration in glioma cells needs further
investigation. Another study has shown that CPE deposited on the
plasma membrane is rapidly internalized and recycled back to the
transgolgi network by ARF6 (44).
ARF6 is necessary for tumor cell invasion and inhibition of ARF6
reduces cell migration. Therefore, one can hypothesize that most
ARF6 will be bound to overexpressed CPE and this way is not able
any more to transmit its pro-migratory function (45). However, in the sCPE-overexpressing
glioma cells CPE is presented mainly in a secreted extracellular
and not in a membrane bound form. Therefore, it is still
speculative whether an interaction of CPE and ARF6 in glioma cells
occurs and by this interaction CPE reduces ARF6-mediated cell
motility.
Using an in vivo orthotopic mouse glioma
model we demonstrated that sCPE-overexpression in the growing tumor
resulted in a prolonged survival of tumor-bearing mice (Fig. 7), indicating that
sCPE-overexpressing glioma cells presenting a lower migratory
capacity might not be as malignant as their counterparts
(presenting only low or even no CPE expression) that exhibit a
highly migratory and invasive potential. In this model we could not
completely reason that the prolonged survival is solely a result of
the reduced migration we observed in vitro since many
pathways are modulated by sCPE-overexpression that also might
influence other processes involved in tumor malignancy such as
clonogenic survival, TGF-β-mediated immunosuppression, changes in
tumor cell metabolism and others. However, prolonged survival of
CPE glioma-bearing mice was not caused by sCPE-mediated reduction
of proliferation (Fig. 1A) or
induction of apoptosis since CPE-overexpressing cells provided no
enhanced caspase activity (7), but
showed elevated Bcl-2 levels (Fig.
7C).
To conclude, in glioma cells sCPE is a regulator of
the expression of motility-associated genes and pathways.
Especially SLUG was downregulated by sCPE. By down-regulation of
SLUG, sCPE at least partially provides its anti-migratory function.
There are multiple other signaling cascades and targets by which
sCPE might also transmit its anti-migratory capacity. How far and
to what extent single pathways, other differentially expressed
genes beside SLUG, or the interconnection of sCPE-regulated
pathways are involved in the anti-migratory effect of CPE needs
further investigation. Last but not least, the expression of CPE in
glioma cells resulted into a survival benefit in a mouse glioma
model.
Acknowledgments
The present study was supported by the German Cancer
Foundation (Deutsche Krebshilfe) to U.N. (no. 110450) and M.M. (no.
110451).
References
|
1
|
Zhong J, Paul A, Kellie SJ and O'Neill GM:
Mesenchymal migration as a therapeutic target in glioblastoma. J
Oncol. 2010:4301422010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Phillips HS, Kharbanda S, Chen R, Forrest
WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, et
al: Molecular subclasses of high-grade glioma predict prognosis,
delineate a pattern of disease progression, and resemble stages in
neurogenesis. Cancer Cell. 9:157–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iser IC, Pereira MB, Lenz G and Wink MR:
The epithelial-to-mesenchymal transition-like process in
glioblastoma: An updated systematic review and in silico
investigation. Med Res Rev. 37:271–313. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Naumann U, Harter PN, Rubel J, Ilina E,
Blank AE, Esteban H and Mittelbronn M: Glioma cell migration and
invasion as potential target for novel treatment strategies. Transl
Neurosci. 4:314–329. 2013. View Article : Google Scholar
|
|
5
|
Cawley NX, Wetsel WC, Murthy SR, Park JJ,
Pacak K and Loh YP: New roles of carboxypeptidase E in endocrine
and neural function and cancer. Endocr Rev. 33:216–253. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee TK, Murthy SR, Cawley NX, Dhanvantari
S, Hewitt SM, Lou H, Lau T, Ma S, Huynh T, Wesley RA, et al: An
N-terminal truncated carboxypeptidase E splice isoform induces
tumor growth and is a biomarker for predicting future metastasis in
human cancers. J Clin Invest. 121:880–892. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Höring E, Harter PN, Seznec J,
Schittenhelm J, Bühring HJ, Bhattacharyya S, von Hattingen E,
Zachskorn C, Mittelbronn M and Naumann U: The 'go or grow'
potential of gliomas is linked to the neuropeptide processing
enzyme carboxypeptidase E and mediated by metabolic stress. Acta
Neuropathol. 124:83–97. 2012. View Article : Google Scholar
|
|
8
|
Murthy SR, Thouennon E, Li WS, Cheng Y,
Bhupatkar J, Cawley NX, Lane M, Merchenthaler I and Loh YP:
Carboxypeptidase E protects hippocampal neurons during stress in
male mice by up-regulating prosurvival BCL2 protein expression.
Endocrinology. 154:3284–3293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weiler M, Bähr O, Hohlweg U, Naumann U,
Rieger J, Huang H, Tabatabai G, Krell HW, Ohgaki H, Weller M, et
al: BCL-xL: Time-dependent dissociation between modulation of
apoptosis and invasiveness in human malignant glioma cells. Cell
Death Differ. 13:1156–1169. 2006. View Article : Google Scholar
|
|
10
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Naumann U, Kügler S, Wolburg H, Wick W,
Rascher G, Schulz JB, Conseiller E, Bähr M and Weller M: Chimeric
tumor suppressor 1, a p53-derived chimeric tumor suppressor gene,
kills p53 mutant and p53 wild-type glioma cells in synergy with
irradiation and CD95 ligand. Cancer Res. 61:5833–5842.
2001.PubMed/NCBI
|
|
12
|
He TC, Zhou S, da Costa LT, Yu J, Kinzler
KW and Vogelstein B: A simplified system for generating recombinant
adenoviruses. Proc Natl Acad Sci USA. 95:2509–2514. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Naumann U, Huang H, Wolburg H, Wischhusen
J, Weit S, Ohgaki H and Weller M: PCTAIRE3: A putative mediator of
growth arrest and death induced by CTS-1, a dominant-positive
p53-derived synthetic tumor suppressor, in human malignant glioma
cells. Cancer Gene Ther. 13:469–478. 2006. View Article : Google Scholar
|
|
14
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerfull approach to
multiple testing. J R Stat Soc B. 57:289–300. 1995.
|
|
15
|
Giese A, Loo MA, Tran N, Haskett D, Coons
SW and Berens ME: Dichotomy of astrocytoma migration and
proliferation. Int J Cancer. 67:275–282. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bernardo MM and Fridman R: TIMP-2 (tissue
inhibitor of metal-loproteinase-2) regulates MMP-2 (matrix
metalloproteinase-2) activity in the extracellular environment
after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem J.
374:739–745. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murthy SR, Dupart E, Al-Sweel N, Chen A,
Cawley NX and Loh YP: Carboxypeptidase E promotes cancer cell
survival, but inhibits migration and invasion. Cancer Lett.
341:204–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verbovšek U, Motaln H, Rotter A, Atai NA,
Gruden K, Van Noorden CJ and Lah TT: Expression analysis of all
protease genes reveals cathepsin K to be overexpressed in
glioblastoma. PLoS One. 9:e1118192014. View Article : Google Scholar
|
|
19
|
Cheng Y, Cawley NX and Loh YP:
Carboxypeptidase E/NFα1: A new neurotrophic factor against
oxidative stress-induced apoptotic cell death mediated by ERK and
PI3-K/AKT pathways. PLoS One. 8:e715782013. View Article : Google Scholar
|
|
20
|
Skalka N, Caspi M, Caspi E, Loh YP and
Rosin-Arbesfeld R: Carboxypeptidase E: A negative regulator of the
canonical Wnt signaling pathway. Oncogene. 32:2836–2847. 2013.
View Article : Google Scholar :
|
|
21
|
Jeibmann A, Halama K, Witte HT, Kim SN,
Eikmeier K, Koos B, Klämbt C and Paulus W: Involvement of CD9 and
PDGFR in migration is evolutionarily conserved from Drosophila glia
to human glioma. J Neurooncol. 124:373–383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sobczak M, Boczek T, Ferenc B, Taha J,
Kozaczuk A, Wiktorska M, Sacewicz-Hofman I, Niewiarowska J and
Zylinska L: Functional characteristic of PC12 cells with reduced
microsomal glutathione transferase 1. Acta Biochim Pol. 57:589–596.
2010.PubMed/NCBI
|
|
23
|
Haruki S, Imoto I, Kozaki K, Matsui T,
Kawachi H, Komatsu S, Muramatsu T, Shimada Y, Kawano T and Inazawa
J: Frequent silencing of protocadherin 17, a candidate tumour
suppressor for esophageal squamous cell carcinoma. Carcinogenesis.
31:1027–1036. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dang Z, Shangguan J, Zhang C, Hu P, Ren Y,
Lv Z, Xiang H and Wang X: Loss of protocadherin-17 (PCDH-17)
promotes metastasis and invasion through hyperactivation of
EGFR/MEK/ERK signaling pathway in hepatocellular carcinoma. Tumour
Biol. 37:2527–2535. 2016. View Article : Google Scholar
|
|
25
|
Held-Feindt J, Paredes EB, Blömer U,
Seidenbecher C, Stark AM, Mehdorn HM and Mentlein R:
Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and
metalloproteinases with thrombospondin motifs 4 and 5) are
expressed in human glioblastomas. Int J Cancer. 118:55–61. 2006.
View Article : Google Scholar
|
|
26
|
Lee JK, Joo KM, Lee J, Yoon Y and Nam DH:
Targeting the epithelial to mesenchymal transition in glioblastoma:
The emerging role of MET signaling. Onco Targets Ther. 7:1933–1944.
2014.PubMed/NCBI
|
|
27
|
Lu DY, Yeh WL, Huang SM, Tang CH, Lin HY
and Chou SJ: Osteopontin increases heme oxygenase-1 expression and
subsequently induces cell migration and invasion in glioma cells.
Neuro-oncol. 14:1367–1378. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Potapenko IO, Haakensen VD, Lüders T,
Helland A, Bukholm I, Sørlie T, Kristensen VN, Lingjaerde OC and
Børresen-Dale AL: Glycan gene expression signatures in normal and
malignant breast tissue; possible role in diagnosis and
progression. Mol Oncol. 4:98–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Niimi K, Yamamoto E, Fujiwara S, Shinjo K,
Kotani T, Umezu T, Kajiyama H, Shibata K, Ino K and Kikkawa F: High
expression of N-acetylglucosaminyltransferase IVa promotes invasion
of choriocarcinoma. Br J Cancer. 107:1969–1977. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan J, Wang S, Yu S, He J, Zheng W and
Zhang J: N-acetyl-glucosaminyltransferase IVa regulates metastatic
potential of mouse hepatocarcinoma cells through glycosylation of
CD147. Glycoconj J. 29:323–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ide Y, Miyoshi E, Nakagawa T, Gu J,
Tanemura M, Nishida T, Ito T, Yamamoto H, Kozutsumi Y and Taniguchi
N: Aberrant expression of N-acetylglucosaminyltransferase-IVa and
IVb (GnT-IVa and b) in pancreatic cancer. Biochem Biophys Res
Commun. 341:478–482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tania M, Khan MA and Fu J: Epithelial to
mesenchymal transition inducing transcription factors and
metastatic cancer. Tumour Biol. 35:7335–7342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kahlert UD, Nikkhah G and Maciaczyk J:
Epithelial-to-mesenchymal(-like) transition as a relevant molecular
event in malignant gliomas. Cancer Lett. 331:131–138. 2013.
View Article : Google Scholar
|
|
34
|
Chesnelong C and Luchman AH: STAT3 is a
key regulator of an 'EMT-like' process mediated by Slug in GBM.
Cancer Res. 76:25242016. View Article : Google Scholar
|
|
35
|
Iwadate Y: Epithelial-mesenchymal
transition in glioblastoma progression. Oncol Lett. 11:1615–1620.
2016.PubMed/NCBI
|
|
36
|
Shih JY and Yang PC: The EMT regulator
slug and lung carcinogenesis. Carcinogenesis. 32:1299–1304. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Geismann C, Arlt A, Bauer I, Pfeifer M,
Schirmer U, Altevogt P, Müerköster SS and Schäfer H: Binding of the
transcription factor Slug to the L1CAM promoter is essential for
transforming growth factor-β1 (TGF-β)-induced L1CAM expression in
human pancreatic ductal adenocarcinoma cells. Int J Oncol.
38:257–266. 2011.
|
|
38
|
Yang M, Li Y, Chilukuri K, Brady OA,
Boulos MI, Kappes JC and Galileo DS: L1 stimulation of human glioma
cell motility correlates with FAK activation. J Neurooncol.
105:27–44. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nagaishi M, Paulus W, Brokinkel B, Vital
A, Tanaka Y, Nakazato Y, Giangaspero F and Ohgaki H:
Transcriptional factors for epithelial-mesenchymal transition are
associated with mesenchymal differentiation in gliosarcoma. Brain
Pathol. 22:670–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Goldberg L and Kloog Y: A Ras inhibitor
tilts the balance between Rac and Rho and blocks
phosphatidylinositol 3-kinase-dependent glioblastoma cell
migration. Cancer Res. 66:11709–11717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stepanenko AA, Andreieva SV, Korets KV,
Mykytenko DO, Baklaushev VP, Chekhonin VP and Dmitrenko VV: mTOR
inhibitor temsirolimus and MEK1/2 inhibitor U0126 promote
chromosomal instability and cell type-dependent phenotype changes
of glioblastoma cells. Gene. 579:58–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Skalka N, Caspi M, Lahav-Ariel L, Loh YP,
Hirschberg K, Rosin-Arbesfeld R and Carboxypeptidase E:
Carboxypeptidase E (CPE) inhibits the secretion and activity of
Wnt3a. Oncogene. 35:6416–6428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arnaoutova I, Jackson CL, Al-Awar OS,
Donaldson JG and Loh YP: Recycling of Raft-associated prohormone
sorting receptor carboxypeptidase E requires interaction with ARF6.
Mol Biol Cell. 14:4448–4457. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sabe H: Requirement for Arf6 in cell
adhesion, migration, and cancer cell invasion. J Biochem.
134:485–489. 2003. View Article : Google Scholar : PubMed/NCBI
|