Introduction
Ovarian cancer ranks fifth in cancer deaths among
women in the United States, accounting for ~5% of all cancer deaths
diagnosed among women (1). Among
the gynecologic cancers (uterine, cervical and ovarian), ovarian
cancer has the highest rate of deaths. The conventional course of
therapy is maximal surgical resection of the tumor mass, followed
by chemotherapy based on taxane and platinum (2). Despite 70% of patients responding
well to first-line platinum-based therapy, the emergence of side
effects and drug resistance has rendered a variety of the currently
available chemotherapeutic drugs ineffective (3). The 5-year survival rate for patients
with advanced ovarian cancer remains <40% because of acquired
drug resistance and adverse side effects (4,5).
Hence, there is an urgent need to explore novel therapeutic
interventions and overcome drug resistance for this disease.
Natural products have played a beneficial role in
cancer treatment for >50 years (6–8).
They have already afforded some clinically used chemotherapeutic
agents, and are a proven resource to explore new anticancer drugs
for further cancer research. Thus, out of 175 small-molecule
chemotherapy drugs in Western countries over a period of ~70 years,
~49% were either obtained from organisms directly or derived from
natural products (8).
Black tea is one of the most widely consumed
beverages around the world. A prospective cohort study showed that
black tea consumption appeared to be inversely correlated with some
cancer risks induced by smoking and a reduced intake of vegetables
and fruits (9). In particular, US
women mainly ingested dietary flavonols from black tea, and
flavonol intake could lower the risk of ovarian cancer (10). Theaflavins are the major bioactive
components in black tea. They are orange or orange-red in color and
possess a benzotropolone skeleton that is formed from the
co-oxidation of selected pairs of catechins during black tea
production (11). The major
theaflavins in black tea are theaflavin (TF1), theaflavin-3-gallate
(TF2a), theaflavin-3′-gallate (TF2b) and theaflavin-3, 3′-digallate
(TF3). Theaflavins have been demonstrated to inhibit lung
tumorigenesis in A/J mice (12)
and a variety of cancer cells including SV40 transformed WI38 human
cells (13), Caco-2 colon cancer
cells (13), human stomach cancer
Kato III cells (14) and human
breast cancer cells (15). We have
previously reported that TF3 could induce apoptosis and cell cycle
arrest (16) and inhibit
angiogenesis (17) in human
ovarian carcinoma cells. TF2a and TF2b showed similar inhibitory
effect on human ovarian carcinoma cells to TF3 (18), but their effect against ovarian
cancer is not yet clear. Therefore, we aimed to investigate the
inhibitory effect of TF2a and TF2b on the platinum-resistant
ovarian cancer cell line A2780/CP70 and a normal ovarian surface
epithelial IOSE-364 cell line. The possible mechanisms by which
TF2a and TF2b-induced apoptosis and cell cycle arrest and the
detailed molecular signaling pathway in the ovarian cancer cells
were explored.
Materials and methods
Cell culture and reagents
The platinum-resistant human ovarian cancer cell
line A2780/CP70 (p53 wild-type) was presented by Dr Binghua Jiang
at West Virginia University. The normal ovarian surface epithelial
cell line, IOSE-364 was a gift from Dr Nelly Auersperg at
University of British Columbia. The cells were cultured in
rPMI-1640 medium (Sigma, St. Louis, MO, USA) supplemented with 10%
fetal bovine serum (FBS) (Invitrogen, Rockford, IL, USA) at 37°C in
a humidified incubator with 5% CO2. TF2a and TF2b
monomers were isolated and purified using a previous method
(19). Primary antibodies to
caspase-3, cleaved caspase-3 (Asp175), caspase-7, cleaved caspase-7
(Asp198), cyclin D1, cyclin E1 (D7T3U), CDK2 (78B2), CDK4 (D9G3E),
p21Waf1/Cip1 (12D1), p53 (7F5), ATM (D2E2), p-ATM
(Ser1981 (D6H9), histone H2AX (D17A3), p-histone H2AX (Ser139),
Akt, p-Akt (Ser473), p38, p-p38 (Thr180/Tyr182) (28B10), JNK and
JNK (Thr183/Tyr185) were purchased from Cell Signaling Inc.
(Danvers, MA, USA). Primary antibodies PARP-1 (F-2), chk1 (G4),
p-chk1 (Ser345), chk2 (H-300), p-chk2 (Thr68), p-p53 (Ser15), ERK1
(K-23), p-ERK1/2 (Thr202), GAPDH (0411) and the secondary
antibodies were purchased from Santa Cruz Biotechnology Inc.
(Mariposa, CA, USA).
Cell viability assay
The cell viability was assessed using
[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay. A2780/CP70 cells were seeded into 96-well plates at a
density of 2×104 cells per well and incubated overnight.
Then cells were treated with different concentrations of TF2a and
TF2b (0–40 µM) or an equal amount of dimethyl sulfoxide
(DMSO) (as vehicle) (Sigma) for 24 h. Cell viability was measured
using CellTiter 96® Aqueous One Solution Cell
Proliferation assay kit (Promega, St. Louis, MO, USA), according to
the manufacturer's instructions. Cell viability was expressed as a
percentage compared to that of control cells (vehicle
treatment).
LDH cytotoxicity assay
A2780/CP70 cells were seeded in 96-well plates with
the density of 2×104 cells per well and incubated
overnight. Then cells were treated with different concentrations of
TF2a and TF2b (0–40 µM) or an equal amount of DMSO (as
vehicle) for 24 h. After incubation, LDH was determined by LDH
cytotoxicity assay kit (Thermo Fisher Scientific, Waltham, MA, USA)
according to the manufacturer's instructions.
Hoechst 33342 staining
A2780/CP70 cells were seeded in 24-well plates at
2×104 cells/well and incubated overnight. Cells were
treated with TF2a and TF2b (0, 5, 10 and 20 µM) for 24 h.
Then the cells were stained with 10 µg/ml Hoechst 33342
(Sigma) in PBS for 10 min in the dark at 37°C. Cellular morphology
was examined under a fluorescence microscope (ZEISS).
Flow cytometric analysis of apoptotic
cells
Cell apoptosis was determined using an Alexa
Fluor® 488 Annexin V/Dead Cell Apoptosis kit
(Invitrogen). After treatment with TF2a and TF2b (0, 5, 10 and 20
µM) for 24 h, A2780/CP70 cells were collected, centrifuged
for 10 min at 1,500 rpm and then washed twice with PBS. Cells were
stained in binding buffer with Alexa Fluor 488 Annexin V and
propidium iodide (PI) for 15 min. The stained cells were determined
with flow cytometry (FACSCalibur system; BD Biosciences), measuring
the fluorescence emission at 530 and 575 nm using 488 nm
excitation.
Flow cytometry analysis of the cell
cycle
A2780/CP70 cells treated with TF2a and TF2b (0, 5,
10 and 20 µM) for 24 h were collected, suspended in 70%
ethanol and stored at −20°C overnight. Then the cells were washed
twice with PBS and incubated with 180 µg/ml RNase A
(Invitrogen) at 37°C for 15 min. After 15 min staining with 50
µg/ml PI (Sigma) in the dark at 37°C, the cells were
analyzed by flow cytometry (FACSCalibur system; BD
Biosciences).
Caspase-3/7 assay
A2780/CP70 cells were seeded in 96-well plates at
the density of 2×104 cells per well, incubated
overnight. Then the cells were treated with TF2a and TF2b (0, 5, 10
and 20 µM) for 24 h. After 24-h treatment, 100 µl of
caspase-3/7 reagent was added to each well, mixed well and
incubated for 1 h at 37°C. The caspase-3/7 activities in cells were
determined using a Caspase-Glo 3/7 assay kit (Promega) according to
the manufacturer's instructions. A Synergy™ HT Multi-Mode
Microplate Reader (BioTek) was used to measure luminescence. Total
protein levels were used to normalize the caspase-3/7 activities.
The caspase-3/7 activities were expressed as a percentage compared
to that of the untreated control. A BCA assay kit (Pierce, St.
Louis, MO, USA) was used to measure total protein levels in the
cells.
Western blotting
A2780/CP70 cells were seeded in 60-mm dishes at the
density of 1×106 cells/dish, incubated overnight, and
treated with TF2a and TF2b (0, 5, 10 and 20 µM) for 24 h.
Then the cells were collected with M-PER Mammalian Protein
Extraction reagent (Pierce) supplemented with Halt™ Protease and
Phosphatase Inhibitor Single-Use Cocktail (Life Technologies, Grand
Island, NY, USA). The total protein levels were detected with BCA
Protein assay kit (Pierce). Equal amounts of protein were separated
by sodium dodecyl sulfate polyacrylamide gel electrophoresis and
transferred onto nitrocellulose membranes. The membrane was blocked
with 5% skim milk in Tris-buffer saline containing 0.1% Tween-20
for 1 h at room temperature, and then incubated with specific
primary antibodies and appropriate secondary antibodies conjugated
with horseradish peroxidase. The antigen-antibody complex was
visualized with Super Signal West Dura Extended Duration Substrate
(Life Technologies) and ChemiDoc™ MP System (Bio-Rad, Hercules, CA,
USA). Protein bands were analyzed with ImageJ software and
normalized with GAPDH.
Silence with small interfering RNA
(siRNA)
A2780/CP70 cells were seeded in 60-mm dishes at
5×105 cells/dish and incubated overnight. Then the cells
were transfected with p53 siRNA (Santa Cruz Biotechnology, Inc.,
Danvers, MA, USA) in Lipofectamine 2000 transfection reagent
(Invitrogen) for 24 h according to the manufacturer's protocol. The
control cells were transfected with control siRNA (Santa Cruz,
Danvers, MA, USA). Then cells were treated with TF2a and TF2b (0,
5, 10 and 20 µM) for 24 h. Cell viability was determined and
western blot analysis was carried out.
Statistical analysis
In the present study, all samples were analyzed in
triplicate. The data are presented as mean ± standard deviations
(SD). Multiple comparisons were analyzed by least significant
difference (LSD) test. Statistical differences between two groups
were analyzed with Student's t-test. All statistical analysis was
carried out using Statistical Analysis system. p<0.05 and
p<0.01 were considered statistically significant and highly
significant, respectively.
Results
Effect of TF2a and TF2b on A2780/CP70
cell viability
Ovarian cancer A2780/CP70 cell line and normal
ovarian epithelial IOSE-364 cell line were used to investigate the
effect of TF2a and TF2b on ovarian cells. The anti-proliferation
effect of TF2a and TF2b on A2780/CP70 and IOSE-364 was evaluated
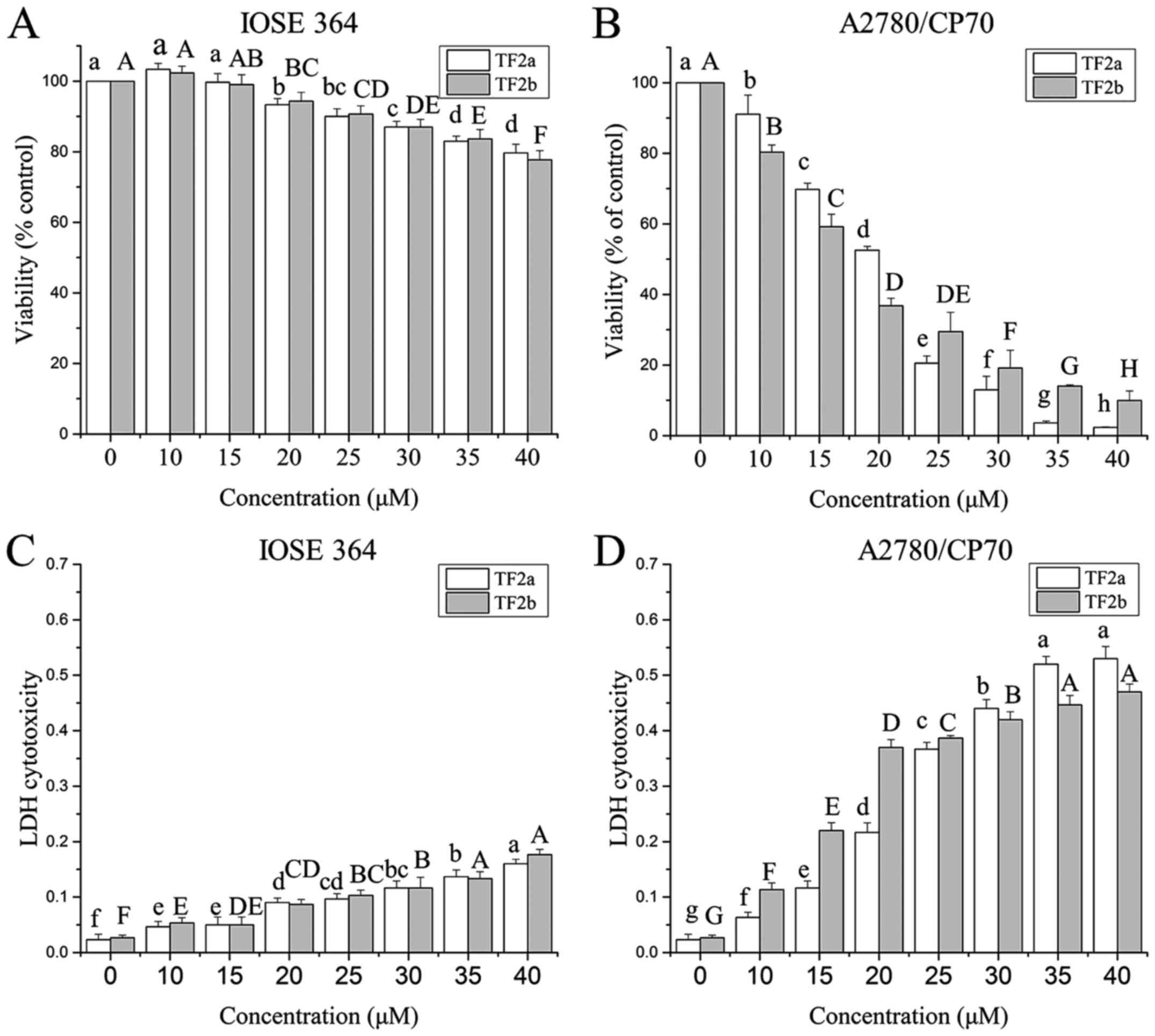
using the MTS assay. As shown in Fig.
1A and B, TF2a and TF2b inhibited the proliferation of the
cells in a dose-dependent manner. The cell viability ranged from
100 to 2.32% for TF2a treatment, and from 100 to 9.95% for TF2b
treatment. The IC50 values of TF2a and TF2b against
A2780/CP70 cells were 18.1 and 17.2 µM, respectively. In
contrast, TF2a and TF2b had relatively moderate cytotoxic effect on
IOSE-364 cells. The least viability of IOSE 364 cells was 79.67%
for TF2a and 77.67% for TF2b after 40 µM TF2a and TF2b
treatment for 24 h.
To further investigate the cytotoxicity effect of
TF2a and TF2b against A2780/CP70 and IOSE-364 cells, LDH
cytotoxicity was evaluated. As shown in Fig. 1C and D, TF2a and TF2b induced
cytotoxicity of the cells after treatment (0–40 µM) for 24 h
in a dose-dependent manner. TF2a and TF2b induced dramatic LDH
release in A2780/CP70 cells as the treatment concentration
increased. TF2a and TF2b induced 23.0 and 17.4 times more LDH
release than that of control in A2780/CP70 cells at the
concentration of 40 µM. However, TF2a and TF2b induced
slight LDH release in IOSE-364. TF2a and TF2b induced 6.7 and 6.5
times more LDH release than that of control in IOSE364 cells at the
concentration of 40 µM. As the viability and cytotoxicity
results showed, TF2a and TF2b had higher cytotoxic effect against
ovarian cancer A2780/CP70 cells than those against normal ovarian
IOSE-364 cells.
TF2a and TF2b induce apoptosis in
A2780/CP70 cells
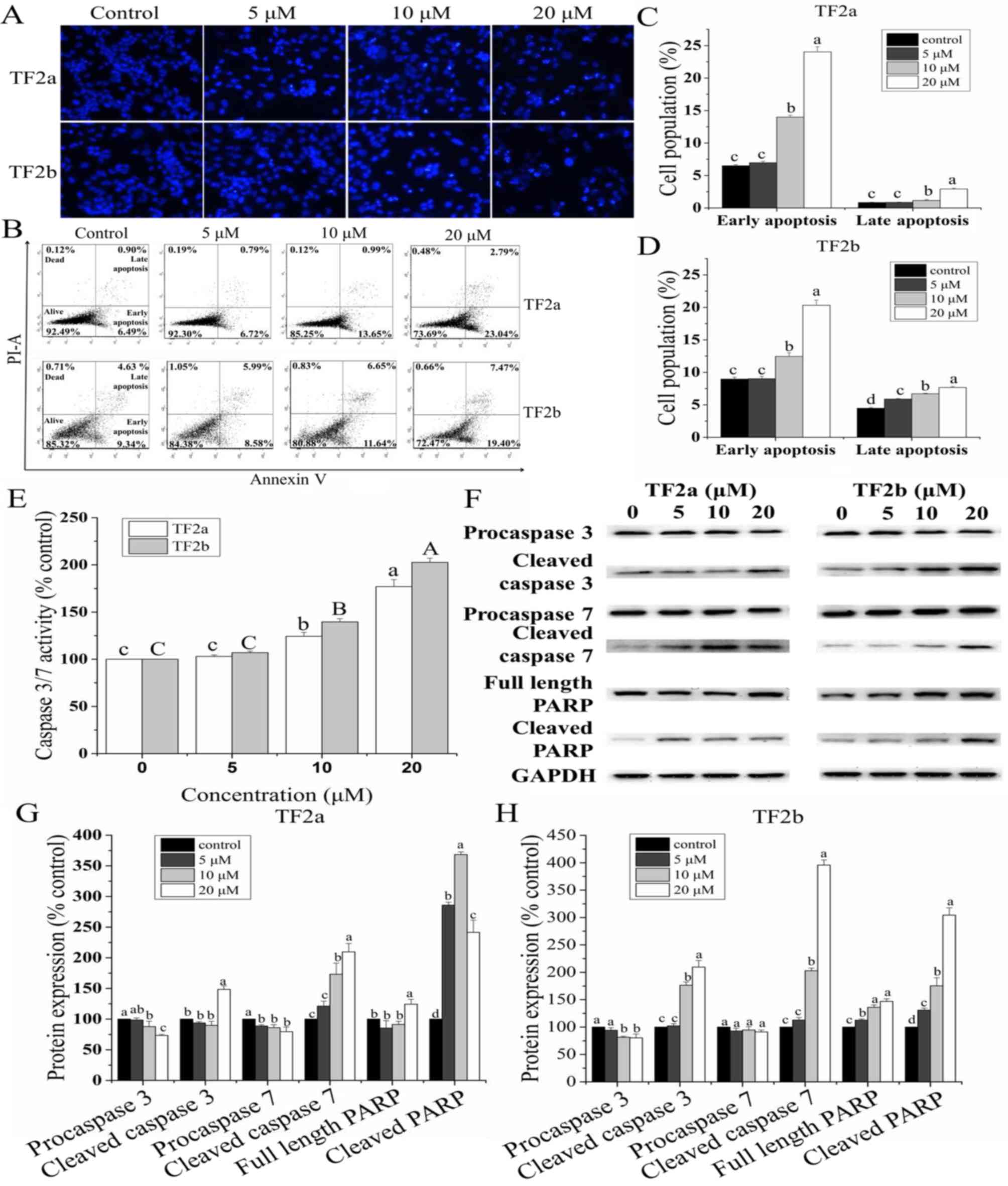
To study whether TF2a and TF2b induced apoptosis
resulting in cell viability inhibition and cell cytotoxicity, the
changes of nuclear morphology in A2780/CP70 cells treated with TF2a
and TF2b (0, 5, 10 and 20 µM) were evaluated using Hoechst
33342 DNA staining. As showed in Fig.
2A, the nuclei in control cells were intact, and appeared less
bright. With treatment of TF2a and TF2b, the apoptotic cells were
stained much brighter with condensed or fragmented nuclei.
Double staining with Annexin V FITC and
PI followed by flow cytometry analysis was also used to investigate
the apoptosis induced by TF2a and TF2b
As shown in Fig.
2B–D, the treatment of TF2a and TF2b significantly increased
the percentage of early apoptotic cells and late apoptotic cells in
a dose-dependent manner (p<0.05). The portion of early apoptotic
cells increased from 6.51 to 24.01% for TF2a and from 8.96 to
20.30% for TF2b. The portion of late apoptotic cells increased from
0.85 to 2.93% for TF2a and from 4.48 to 7.66% for TF2b. The total
percent of apoptotic cells increased from 7.36 to 26.94% for TF2a
and 13.44 to 27.96% for TF2b, respectively.
Caspase-3/7 plays a central role in the
execution-phase of cell apoptosis
To confirm that TF2a and TF2b induced apoptosis, we
evaluated the caspase-3/7 activities in A2780/CP70 cells. As showed
in Fig. 2E, treatment with TF2a
and TF2b enhanceed the caspase-3/7 activity by 1.77- and 2.03-fold
compared to that in controls, respectively.
Furthermore, the expression of proteins related to
apoptosis in A2780/CP70 cells was analyzed by western blot
(Fig. 2F–H). As the treatment
concentrations of TF2a and TF2b increased, the expression of
procaspase-3/7 proteins for TF2a and procaspase-3 proteins for TF2b
decreased significantly and the expression of cleaved caspase-3/7,
full length PARP and cleaved PARP proteins increased significantly.
There was no significant difference in the expression of
procaspase-7 proteins for TF2b. Taken together, these results
indicated that TF2a and TF2b can induce apoptosis in A2780/CP70
cells.
TF2a and TF2b induce G1 cell cycle arrest
in A2780/CP70 cells
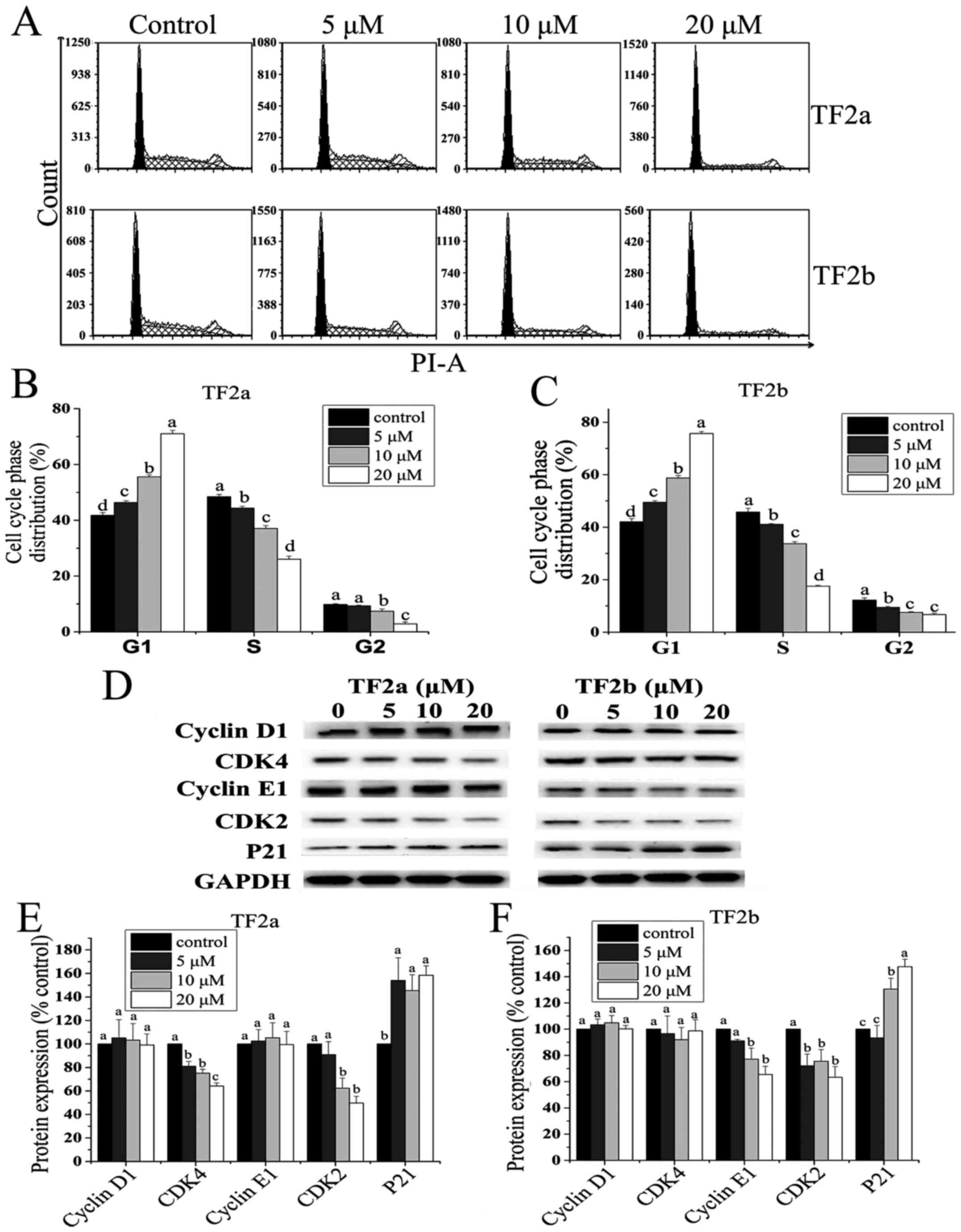
To investigate whether the anti-proliferative effect
of TF2a and TF2b against A2780/CP70 cells was triggered by cell
cycle arrest, we analyzed the distribution of cell cycle phase in
cells treated with TF2a and TF2b (0, 5, 10 and 20 µM) and
stained with PI using flow cytometry. As shown in Fig. 3A–C, the cell population at the G1
phase was increased in a dose-dependent manner (p<0.05). The
percentage of the cells at G1 phase increased from 41.77 to 71.07%
for TF2a and from 42.06 to 75.68% for TF2b. In addition, a
significant decrease in the proportion of cells at both S and G2
phase was observed. These results suggested that TF2a and TF2b
induced G1 cell cycle arrest in A2780/CP70 cells.
To investigate the underlying mechanism of TF2a and
TF2b-induced G1 phase arrest in A2780/CP70 cells, the effect of
TF2a and TF2b on G1 phase regulatory proteins was evaluated by
western blot analysis. As shown in Fig. 3D–F, TF2a could effectively
downregulate CDK2 and CDK4 protein expression (p<0.05),
upregulate p21 protein expression (p<0.05), but had no effect on
the expression of cyclin D1 and cyclin E1 proteins (p>0.05).
TF2b could effectively downregulate CDK2 and cyclin E1 protein
expression (p<0.05), upregulate p21 protein expression
(p<0.05), but had no effect on the expression of CDK4 and cyclin
D1 proteins (p>0.05). These western blot results implied that
TF2a and TF2b induced G1 phase arrest in A2780/CP70 cells by
downregulating CDK2 and CDK4 protein expression and CDK2 and cyclin
E1 protein expression, respectively.
Role of p53 in TF2a and TF2b-induced
apoptosis and cell cycle arrest
The p53 signaling pathway plays an important role in
cellular response to DNA damage and other genomic aberrations.
Activation of p53 leads to either cell cycle arrest and DNA repair
or apoptosis (20). To clarify the
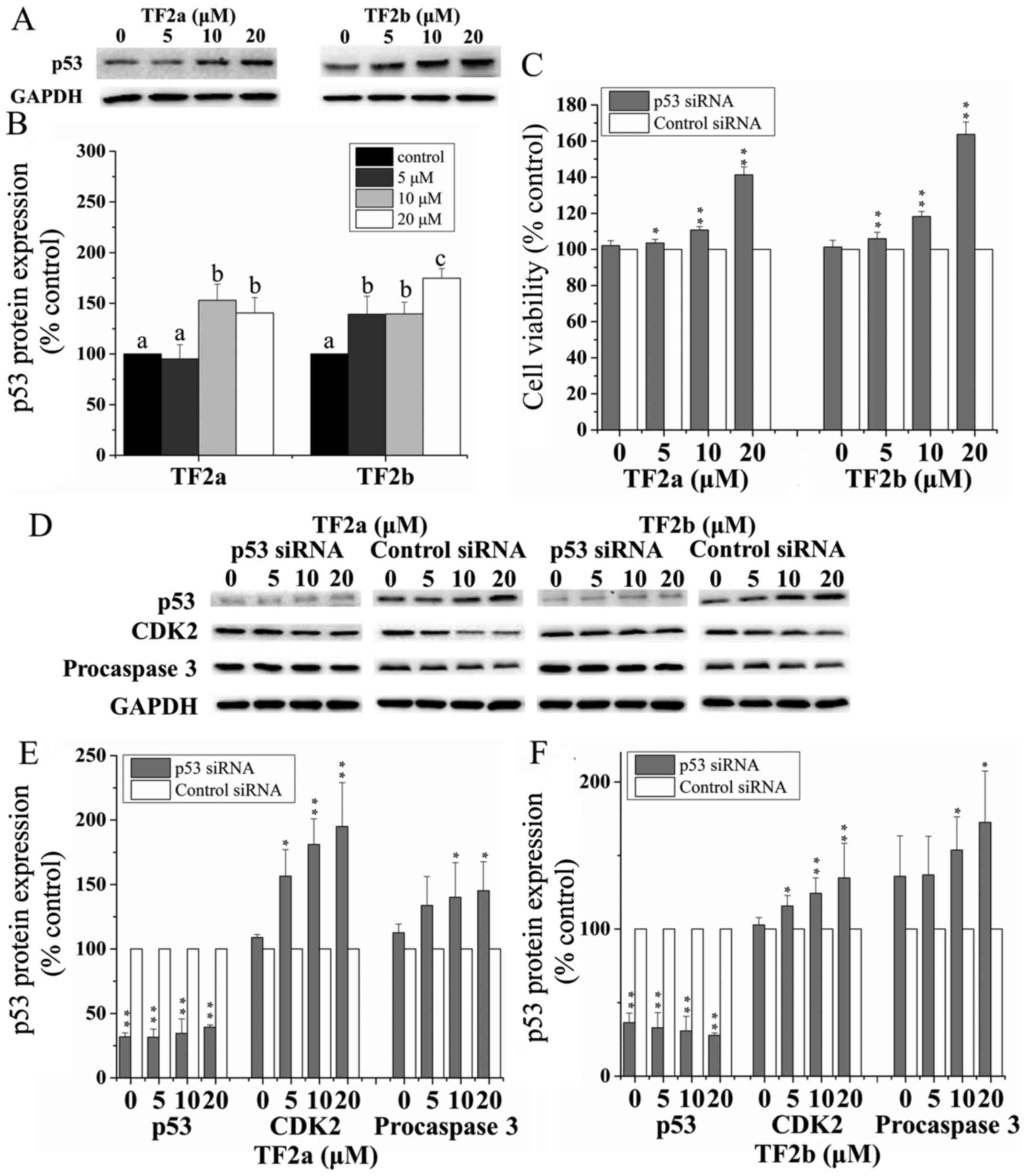
role of p53 in apoptosis and cell cycle arrest induced by TF2a and
TF2b in A2780/CP70 cells after treatment for 24 h, p53 protein
expression was determined by western blotting. As shown in Fig. 4A and B, TF2a and TF2b significantly
upregulated the protein expression of p53 (p<0.05).
To further analyze the effect of p53 on apoptosis
and cell cycle G1 phase arrest induced by TF2a and TF2b, p53 was
silenced by siRNA. Pre-incubation of 50 nM p53 siRNA significantly
attenuated the inhibitory effect of TF2a and TF2b on A2780/CP70
cells (p<0.05 or 0.01) (Fig.
4C). The expression of p53 protein was markedly inhibited after
treatment with 50 nM p53 siRNA (p<0.01) (Fig. 4D–F). The p53 protein depletion
weakened the effect of TF2a and TF2b-induced decrease in CDK2 and
procaspase-3 protein expression (p<0.05). These western blot
results suggested that p53 played an important role in apoptosis
and G1 cell cycle arrest induced by TF2a and TF2b in A2780/CP70
cells.
TF2a and TF2b induce DNA damage in
A2780/CP70 cells
The p53 protein plays a major role in cellular
response to DNA damage. The cell cycle can be held at the G1
regulation point on DNA damage recognition (21). To determine whether TF2a and TF2b
induced DNA damage in A2780/CP70 cells after treatment for 24 h,
the levels of DNA damage related proteins including histone H2A.X,
p-histone H2A.X (Ser139), ATM, p-ATM (Ser1981), Chk1, Chk2, p-Chk1
(Ser345), p-Chk2 (Thr68) and p-p53 (Ser15) were determined.
The phosphorylation of histone H2A.X at
Ser139 indicates DNA double-strand breaks
ATM is autophosphorylated on Ser1981 in response to
DNA damage (22). As shown in
Fig. 5, both TF2a and TF2b
significantly increased the protein levels of p-histone H2A.X
(Ser139), and p-ATM (Ser1981) (p<0.05), but had no effect on the
protein level of total histone H2A.X (p>0.05). TF2a
significantly increased the protein level of ATM (p<0.05), but
TF2b had no significant effect on the protein expression of ATM
(p>0.05). Chk1 and Chk2 can be phosphorylated by p-ATM. Both
TF2a and TF2b significantly upregulated the protein levels of
p-Chk1 (Ser345) and p-Chk2 (Thr68) (p<0.05), but had no
significant effect on the protein levels of total Chk1/2
(p>0.05). The p53 protein can be phosphorylated by ATM, ATR, and
DNA-PK at Ser15, promoting both the accumulation and activation of
p53 in response to DNA damage (23). Both TF2a and TF2b significantly
increased the protein levels of p-p53 (Ser15) (p<0.05). It was
concluded that TF2a and TF2b induced DNA damage by activating
ATM-Chk1 and ATM-Chk2 pathways.
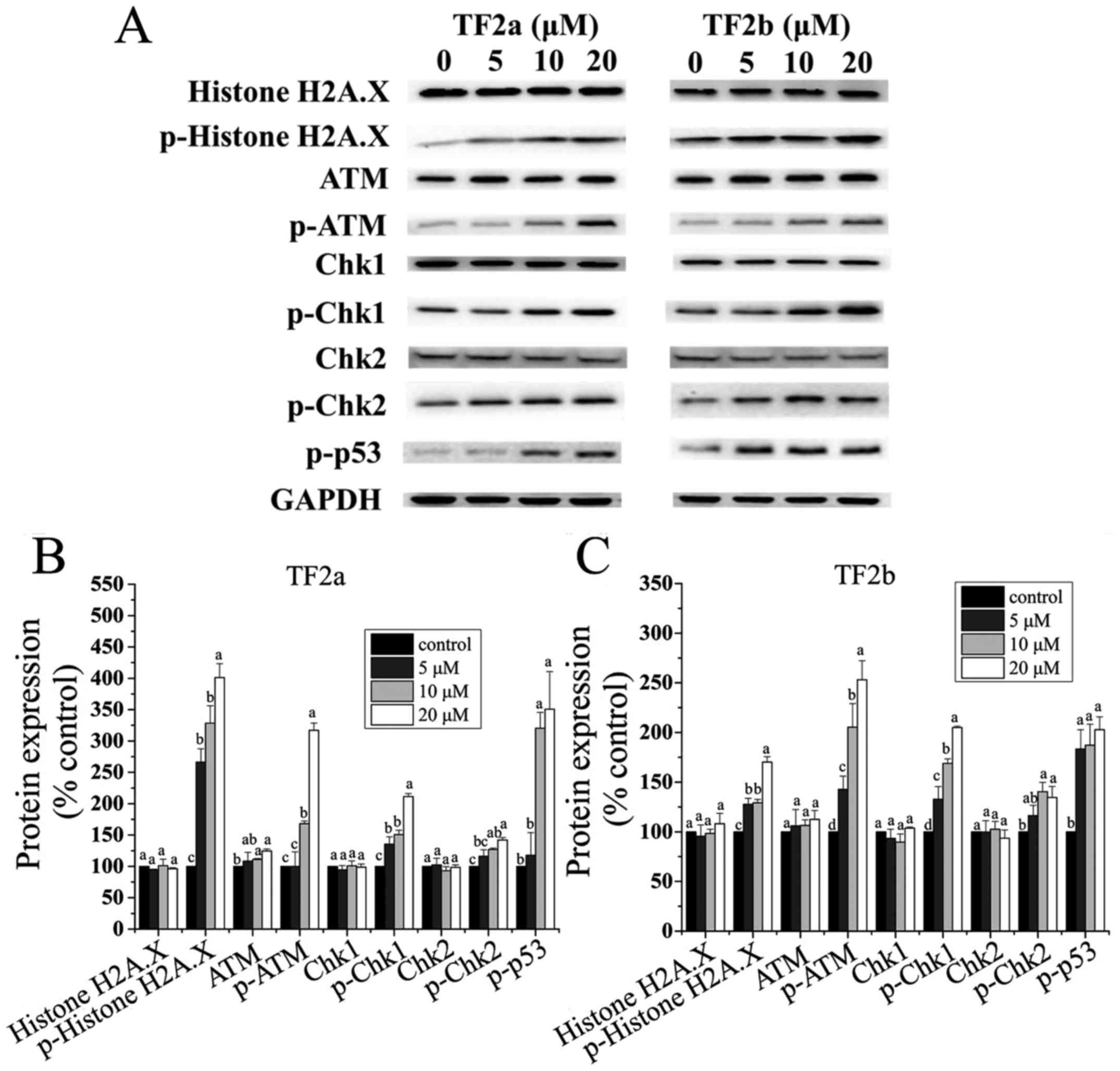 | Figure 5TF2a and TF2b induce DNA damage in
A2780/CP70 cells. (A) Protein expression levels of DNA
damage-related proteins, including histone H2A.X, p-histone H2A.X
(Ser139), ATM, p-ATM (Ser1981), Chk1, Chk2, p-Chk1 (Ser345), p-Chk2
(Thr68) and p-p53 (Ser15) were analyzed by western blotting.
A2780/CP70 cells were treated with designated concentrations (0, 5,
10 and 20 µM) of TF2a and TF2b for 24 h. The changes of the
protein levels induced by TF2a (B) and TF2b (C) were expressed as
quantification histograms with error bars. Results are expressed as
mean ± SD from three independent experiments. Significant
differences among different treatments are marked with different
letters (p<0.05). |
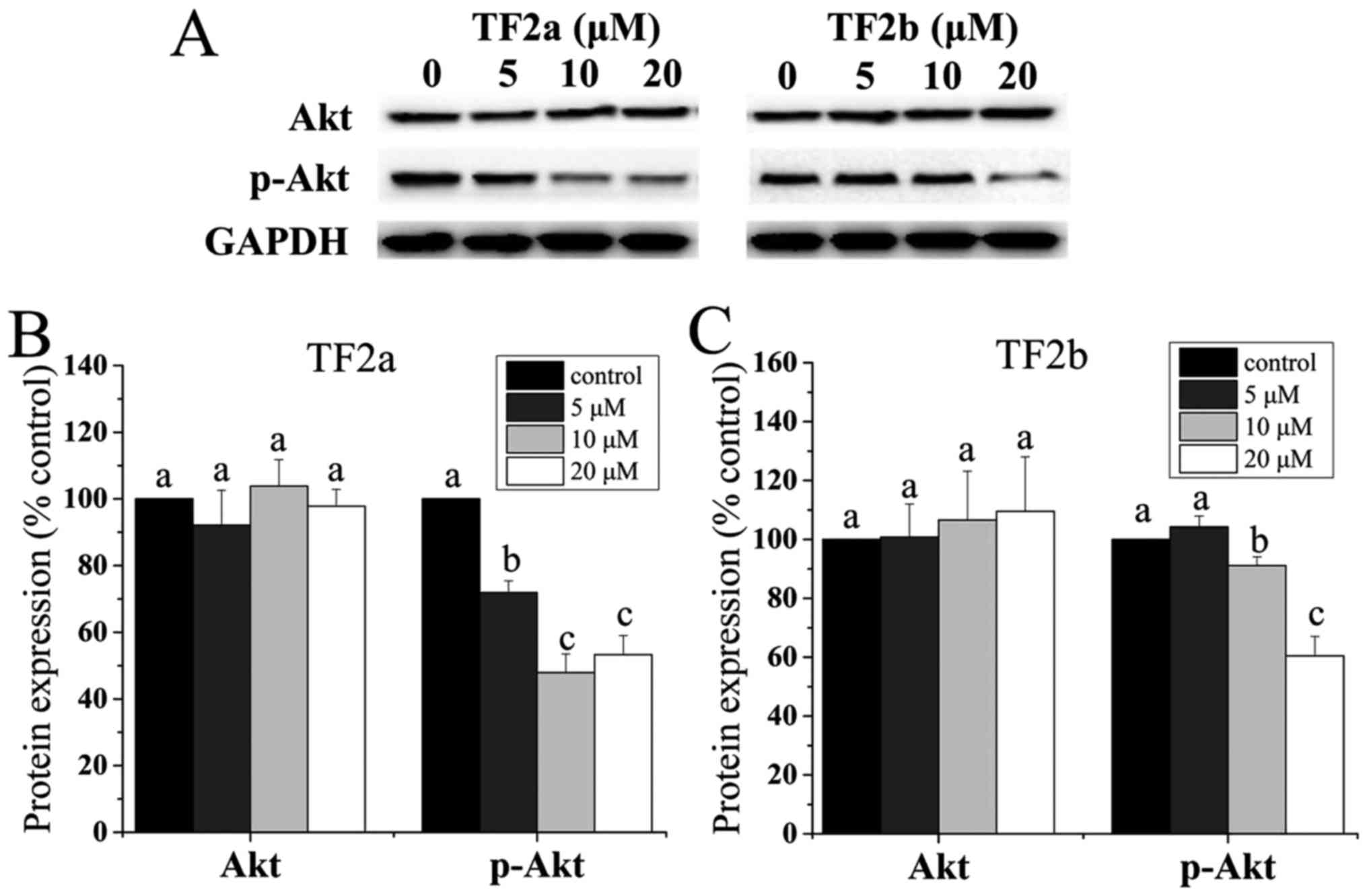
Role of Akt in TF2a and TF2b-induced
apoptosis and cell cycle arrest
It was demonstrated that the p53 level is negatively
controlled by Akt (24). Akt plays
a critical role in controlling survival and apoptosis which is
activated through phosphorylation by insulin and various growth and
survival factors (25,26). In addition to its role in survival,
Akt is involved in cell cycle regulation by preventing
GSK-3β-mediated degradation of cyclin D1 (27). Therefore, the protein levels of Akt
and p-Akt in A2780/CP70 cells were determined. As shown in Fig. 6, TF2a and TF2b significantly
reduced the phosphorylation of Akt and had no influence on the
level of total Akt protein (p<0.05).
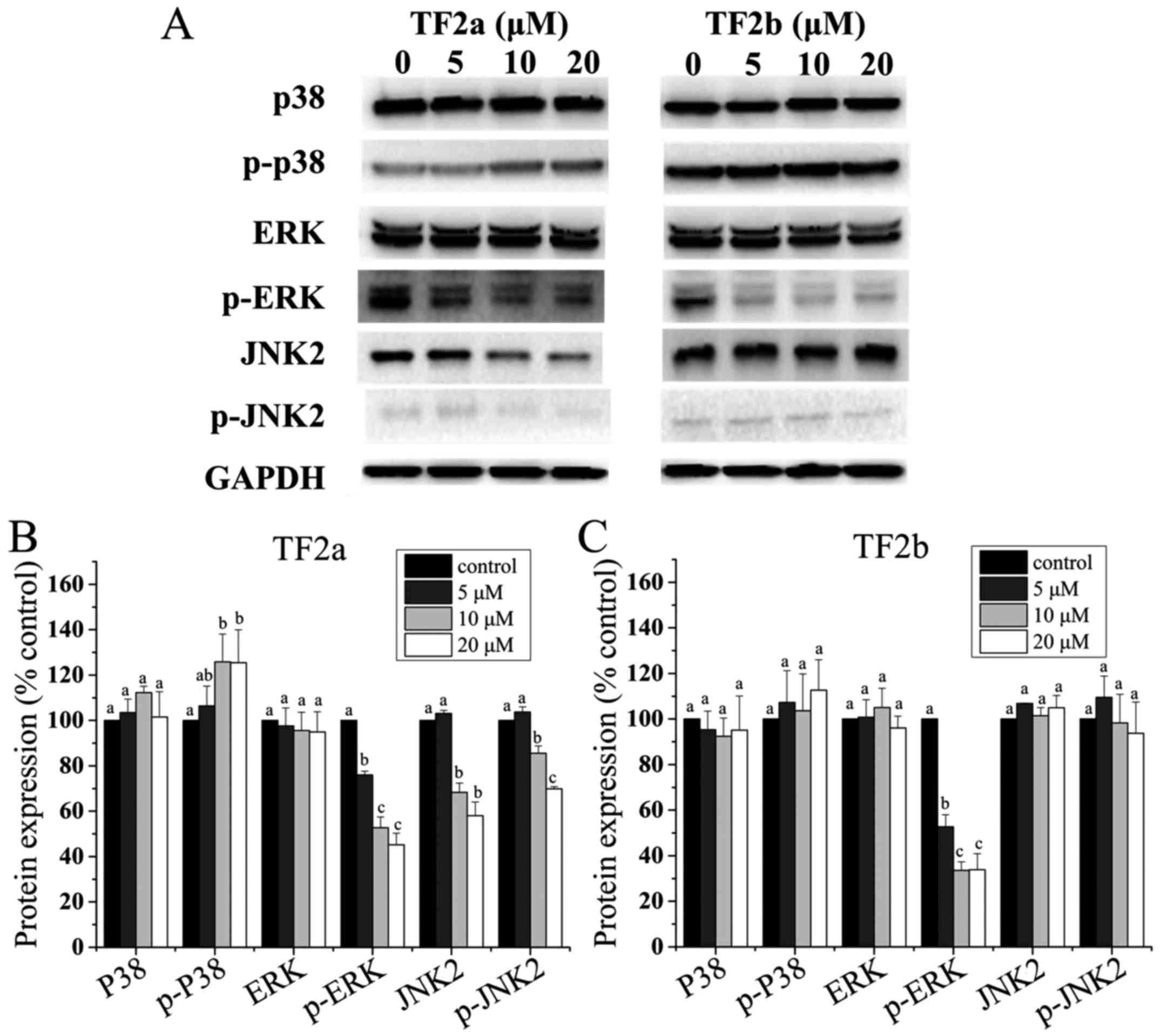
Role of MAPK in TF2a and TF2b-induced
apoptosis and cell cycle arrest
It is clear that the p53 protein can functionally
interact with the mitogen-activated protein kinase (MAPK) (28). Therefore, to clarify whether MAPKs
mediated apoptosis and cell cycle arrest induced by TF2a and TF2b
in A2780/CP70 cells, the levels of p38, p-p38, ERK1/2, p-ERK1/2,
JNK1/2 and p-JNK1/2 were determined. As shown in Fig. 7, TF2a increased the phosphorylation
of p38 (p<0.05), but had no effect on the protein expression of
p38 (p>0.05). TF2b had no effect on the protein expression of
p38 and the phosphorylation of p38 (p>0.05). Both TF2a and TF2b
suppressed the phosphorylation of ERK1/2 (p<0.05), but had no
effect on the protein expression of ERK1/2 (p>0.05). TF2a
suppressed the protein expression of JNK2 and the phosphorylation
of JNK2 (p<0.05), but TF2b had no effect on the protein
expression of JNK2 and the phosphorylation of JNK2 (p>0.05). The
protein expression of JNK1 and p-JNK1 was so low in A2780/CP70
cells that they were not detected by western blotting (Fig. 7).
Discussion
Ovarian cancer is the most lethal gynecological
malignancy. Cisplatin and its derivatives are first-line
chemotherapeutic agents in the treatment of ovarian cancer, and
their resistance and adverse side effects are major barriers in
successful ovarian cancer treatment (2). Theaflavins showed effective
inhibition of ovarian cancer and were less cytotoxic to normal
ovarian IOSE-364 cells (18).
Hence, the possible mechanisms underlying these modulations of TF2a
and TF2b against ovarian cancer cells stimulated our interest. In
the present study, we found that the IC50 values of TF2a
and TF2b against A2780/CP70 cells were 18.1 and 17.2 µM,
which were much lower than those against IOSE-364 cells (Fig. 1). In LDH assay, significant LDH
leakage was observed in ovarian cancer cell lines while slight LDH
release was induced in IOSE-364 cells (Fig. 1). The MTS and LDH assays suggested
the effect of TF2a and TF2b against ovarian cancer cells and normal
ovarian cells was different. Conventional chemotherapy although
cytotoxic, does not discriminate cancer and normal cells. An ideal
anticancer drug is expected to be selective and cytotoxic to cancer
cells but less cytotoxic to normal cells (29). TF2a and TF2b were demonstrated to
be selective and cytotoxic to ovarian carcinoma A2780/CP70 cell
line and less cytotoxic to normal ovarian surface epithelial
IOSE-364 cell line. The selective cell growth inhibitory effect
maybe partly attributed to the apoptosis and G1 cell cycle arrest
induced by TF2a and TF2b.
One mechanism by which cancer cells develop
resistance to chemotherapeutic agents and radiation is correlated
with their resistance to apoptosis. Thus, apoptosis induced by TF2a
and TF2b in ovarian cancer cells was evaluated using several
methods. Morphological hallmarks of apoptosis in the nucleus are
chromatin condensation and nuclear fragmentation (30). The changes were clearly observed in
ovarian cancer cells after treatment with TF2a and TF2b by Hoechst
33342 staining. TF2a and TF2b rendered condensed and fragmented
nuclei brighter in a dose-dependent manner (Fig. 2A). Loss of plasma membrane
asymmetry is another morphological hallmark of apoptosis (31) which was observed in ovarian cancer
cells after treatment with TF2a and TF2b by Annexin V/PI staining.
TF2a and TF2b increased the percent of both early and late
apoptotic cells in a dose-dependent manner (Fig. 2B–D). Caspases are central to the
mechanism of apoptosis as they are both the initiators and
executioners. Caspases involved in apoptosis have been classified
by their mechanism of action and are either initiator caspases
(caspase-8 and -9) or executioner caspases (caspase-3 and -7)
(32). Activation of caspase-3 and
-7 requires proteolytic processing of the inactive zymogen into
activated fragments. Caspase-3 and -7 activities and protein levels
of caspase-3 and -7 in ovarian cancer cells after treatment with
TF2a and TF2b were determined. TF2a and TF2b increased the
caspase-3 and -7 activities (Fig.
2E) and protein levels of cleaved caspase-3 and -7 (Fig. 2F–H). As cleavage of procaspase-3
and -7 increased, the protein levels of procaspase-3 and -7
decreased. PARP is involved in a number of cellular processes such
as DNA repair, genomic stability, and programmed cell death.
Cleaved PARP serves as a marker of cells undergoing apoptosis
(33–35). The protein level of cleaved PARP
increased after treatment with TF2a and TF2b in a dose-dependent
manner. Taken together, all these results indicated that TF2a and
TF2b inhibited ovarian cancer cells by inducing apoptosis.
Cell cycle regulation plays an important role in
tumorigenesis and tumor progression (36). Cell cycle arrest is important for
determining toxicities and responses to current cancer therapies
(37). Cell cycle arrest may be
attributed partly to the inhibitory effect of TF2a and TF2b on
ovarian cancer cells. Flow cytometric analysis demonstrated that
TF2a and TF2b induced cell cycle G1 phase arrest in A2780/CP70
cells after TF2a and TF2b treatment (Fig. 3A–C). Previous studies have reported
that theaflavins induced cell cycle G1 phase arrest in a variety of
cancer cells such as rat hepatoma AH109A cells (38) and murine B16 melanoma cells
(38) and cell cycle G2 phase
arrest in a variety of cancer cells including human prostate
carcinoma PC-3 cells (39) and
human ovarian cancer A2780/CP70 cells (16). To further investigate the
underlying mechanism of TF2a and TF2b-induced cell cycle G1 arrest,
the protein levels of G1-related regulatory proteins were
determined. The G1 checkpoint arrests the cell cycle by inhibiting
this G1-S transition machinery. It usually arrests the cell cycle
G1 phase by inhibiting cyclin E-CDK2 and cyclin D-CDK4 complex
(40). The p21 protein is a potent
cyclin-dependent kinase inhibitor, which is necessary for the
p53-mediated G1 arrest (41) and
functions as a regulator of cell cycle progression at G2 phase
(42). In the present study, TF2a
and TF2b induced G1 arrest in A2780/CP70 cells by downregulating
CDK2 and CDK4 protein expression and CDK2 and cyclin E1 protein
expression, respectively (Fig.
3D–F). The p21 protein level was upregulated by TF2a and TF2b.
TF2a could not influence the protein expression of cyclin D1 and
cyclin E1 and TF2b could not influence the protein expression of
CDK4 and cyclin D1. The results indicated CDK2 and CDK4 for TF2a
and CDK2 and cyclin E for played an important role in cell cycle G1
arrest of A2780/CP70 cells.
The p53 tumor suppressor protein plays a major role
in cellular response to DNA damage and other genomic aberrations.
Since over 50% of human cancers carry loss of function mutations in
p53 gene, p53 is considered to be one of the classical type tumor
suppressors. Activation of wild-type p53 can lead to either cell
cycle arrest and DNA repair or apoptosis that help to prevent tumor
development (43,44). It is known that the p53 gene
sequence in A2780/CP70 cell line is wild-type (45). Our data showed TF2a and TF2b
significantly increased the protein expression of p53 (Fig. 4A and B). Furthermore, silencing by
the p53 siRNA significantly attenuated the inhibitory effect of
TF2a and TF2b on A2780/CP70 cells and abrogated TF2a and
TF2b-induced decrease in CDK2 and procaspase-3 protein expression.
These results suggested that p53 played an important role in
apoptosis and G1 cell cycle arrest induced by TF2a and TF2b in
A2780/CP70 cells. It has been reported that p53 could mediate
apoptosis induced by theaflavins in human breast and prostate
cancer cells which was consistent with results in the present study
(46,47).
Activation of p53 can occur in response to DNA
damage. Several forms of DNA damage have been shown to activate
p53, including those generated by ionising radiation, radio-mimetic
drugs, ultraviolet light and chemicals (48). ATM is a sensor of DNA damage caused
by ionizing radiation, UV-light, or radio-mimetic agents, of which
activation by autophosphorylation at Ser1981 occurs in response to
exposed DNA double-stranded breaks (49). Chk1/2 acts downstream of ATM and
plays an important role in DNA damage checkpoint control. Chk1 at
Ser345 (50) and Chk2 at Thr68
(51,52) are phosphorylated by ATM in response
to DNA damage, histone H2A.X is required for checkpoint-mediated
cell cycle arrest and DNA repair following DNA double-stranded
breaks (53). DNA damage results
in rapid phosphorylation of histone H2A.X at Ser139 by ATM
(22,54). ATM can phosphorylate p53 at Ser15,
which impaired the ability of MDM2 to bind p53 and promoted both
the accumulation and activation of p53 in response to DNA damage
(23,55). In the present study, TF2a and TF2b
upregulated the phosphorylation of ATM at Ser1981, histone H2A.X at
Ser139, Chk1 at Ser345 and Chk2 at Thr68 (Fig. 5) which indicated DNA
double-stranded break occurred in A2780/CP70 cells. Total p53
protein and phosphorylation of p53 at Ser15 were markedly increased
in a dose-dependent manner, which also suggested DNA damage
occurred in A2780/CP70 cells. Previous studies reported that
activation of ATM/Chk/p53 pathway could induce apoptosis in human
pancreatic cancer BxPC-3 cells (56) and cell cycle G1 arrest in A549 lung
cancer cells (57). Our results
suggested that this pathway was involved in TF2a and TF2b-induced
apoptosis and cell cycle G1 phase arrest in A2780/CP70 cells.
Akt is a serine/threonine kinase that plays an
important role in the development and progression of cancers
(58). This protein kinase is
activated by insulin and various growth and survival factors
(59). The AKT pathway is one of
the most frequently hyperactivated signaling pathways in human
cancers. Inhibition of Akt pathway may be a helpful and potential
cancer therapy (60). Akt could
mediate negative control of p53 levels through enhancing
MDM2-mediated targeting of p53 for degradation (61). It was reported that inhibition of
p-Akt sensitized cancer cells to cisplatin in a p53-dependent
manner (62). Our data showed that
TF2a and TF2b significantly inhibited the phosphorylation of Akt
(Fig. 6), indicating that TF2a and
TF2b might increase the protein expression of p53 by inactivating
Akt in A2780/CP70 cells. It has been reported that TF3 targeted Akt
pathways to induce apoptosis in human ovarian cancer A2780/CP70
cells (16). Our results suggest
Akt pathway was involved in TF2a and TF2b-induced apoptosis and G1
phase arrest in A2780/CP70 cells.
The p53 protein can functionally interact with the
MAPK pathways (28). Upon exposure
to stressful stimuli, MAPKs phosphorylate and activate p53, leading
to p53-mediated cellular responses. MAPKs function in protein
kinase cascades that play a critical role in the regulation of cell
growth and differentiation, and control of cellular responses to
cytokines and stress. In this study, TF2a increased the
phosphorylation of p38, but TF2b had no effect on the
phosphorylation of p38 (Fig. 7).
Both TF2a and TF2b suppressed the phosphorylation of Erk1/2. TF2a
suppressed the phosphorylation of JNK2 (p<0.05), but TF2b had no
effect on the phosphorylation of JNK2. It was reported that
dysregulation of p38 MAPK levels in patients were associated with
advanced stages and short survival in cancer patients (63). Inhibition of p38 MAPK was
correlated with the resistance to anoikis, which allowed
circulating cancer cells to survive (64). The p38 activation in breast cancer
cells inhibited tumor metastasis (65). The Erk pathway is activated in
>30% of human cancers. Inhibition of Erk pathway is an
attractive strategy for cancer therapy. It was reported that
small-molecule ERK pathway inhibitors such as BAY 43-9006,
PD184352, PD0325901 and ARRY-142886 have reached the clinical trial
stage (66). A previous study
reported theaflavins inhibited tumor promoter-induced activator
protein 1 activation and cell transformation through the inhibition
of a JNK-dependent pathway (67).
It was also found that JNK inhibition led to antitumor activity in
ovarian cancers (68) and was
associated with longer progression-free survival of patients with
ovarian cancer (69). Our results
suggest TF2a could induce apoptosis and G1 phase arrest through
p38, Erk and JNK pathways and TF2b could induce apoptosis and G1
phase arrest through ERK pathway.
Adverse side effects and acquired resistance to
conventional chemotherapy based on platinum have become major
barriers in successful ovarian cancer treatment, and drive the
development of other selective anticancer drugs. Our study
demonstrated that TF2a and TF2b exhibited a potent growth
inhibitory effect in cisplatin-resistant ovarian cancer A2780/CP70
cells and were less cytotoxic to the normal ovarian cell line
IOSE-364. TF2a and TF2b induced apoptosis and cell cycle G1 phase
arrest in ovarian cancer A2780/CP70 cells. Downregulation of CDK2
and CDK4 for TF2a and CDK2 and cyclin E1 for TF2b led to the
accumulation of cells in G1 phase. TF2a and TF2b induced apoptosis
and G1 through p53-dependent pathways. TF2a and TF2b induced DNA
damage through ATM/Chk/p53 pathway. TF2a and TF2b induced
inhibition of Akt pathway. As for MAPK pathways, TF2a activated p38
pathway and suppressed Erk and JNK pathways and TF2b only
suppressed Erk pathway without activating the p38 pathway. Our
findings help elucidate the mechanisms by which TF2a and TF2b may
contribute to the prevention and treatment of platinum-resistant
ovarian cancer. TF2a and TF2b would be potential compounds for
treating platinum-resistant ovarian cancer.
Acknowledgments
We thank Dr Kathy Brundage from the Flow Cytometry
Core at the West Virginia University for providing technical help
on apoptosis analysis. This study was supported by NIH grants
P20RR016477 from the National Center for research resources and
P20GM103434 from the National Institute for General Medical
Sciences (NIGMS) awarded to the West Virginia IDeA Network of
Biomedical research Excellence. This study was also supported by
grant no. P20GM104932 from NIGMS, a component of the National
Institutes of Health (NIH) and its contents are solely the
responsibility of the authors and do not necessarily represent the
official view of NIGMS or NIH; COBRE grant GM102488/RR032138, ARIA
S10 grant RR020866, FORTESSA S10 grant OD016165 and INBRE grant
GM103434; the Natural Science Foundation of Zhejiang Province
(grant no. LY15C200007), the National Natural Science Foundation of
China (grant no. 31501474), and Oolong Tea Industry Collaborative
Innovation Center.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ali AY, Farrand L, Kim JY, Byun S, Suh JY,
Lee HJ and Tsang BK: Molecular determinants of ovarian cancer
chemoresistance: New insights into an old conundrum. Ann NY Acad
Sci. 1271:58–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Limtrakul P, Pitchakarn P and Suzuki S:
Kuguacin J, a Triterpenoid from Momordica charantia Linn: A
Comprehensive Review of Anticarcinogenic Properties. INTECH Open
Access Publisher; View
Article : Google Scholar : 2013
|
|
4
|
Al Rawahi T, Lopes AD, Bristow RE, Bryant
A, Elattar A, Chattopadhyay S and Galaal K: Surgical cytoreduction
for recurrent epithelial ovarian cancer. Cochrane Database Syst
Rev. Feb;28(2): CD008765 View Article : Google Scholar : 2013.
|
|
5
|
Wang V, Li C, Lin M, Welch W, Bell D, Wong
YF, Berkowitz R, Mok SC and Bandera CA: Ovarian cancer is a
heterogeneous disease. Cancer Genet Cytogenet. 161:170–173. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cragg DJ, Kingston GM and Newman DG:
Anticancer Agents from Natural Products. CRC Press; 2011,
View Article : Google Scholar
|
|
7
|
Butler MS, Robertson AA and Cooper MA:
Natural product and natural product derived drugs in clinical
trials. Nat Prod Rep. 31:1612–1661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Newman DJ and Cragg GM: Natural products
as sources of new drugs from 1981 to 2014. J Nat Prod. 79:629–661.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goldbohm RA, Hertog MG, Brants HA, van
Poppel G and van den Brandt PA: Consumption of black tea and cancer
risk: A prospective cohort study. J Natl Cancer Inst. 88:93–100.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cassidy A, Huang T, Rice MS, Rimm EB and
Tworoger SS: Intake of dietary flavonoids and risk of epithelial
ovarian cancer. Am J Clin Nutr. 100:1344–1351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Finger A: In vitro studies on the effect
of polyphenol oxidase and peroxidase on the formation of
polyphenolic black tea constituents. J Sci Food Agric. 66:293–305.
1994. View Article : Google Scholar
|
|
12
|
Yang GY, Liu Z, Seril DN, Liao J, Ding W,
Kim S, Bondoc F and Yang CS: Black tea constituents, theaflavins,
inhibit 4- (methylnitrosamino)-1- (3-pyridyl)-1-butanone
(NNK)-induced lung tumorigenesis in A/J mice. Carcinogenesis.
18:2361–2365. 1997. View Article : Google Scholar
|
|
13
|
Lu J, Ho C-T, Ghai G and Chen KY:
Differential effects of theaflavin monogallates on cell growth,
apoptosis, and Cox-2 gene expression in cancerous versus normal
cells. Cancer Res. 60:6465–6471. 2000.PubMed/NCBI
|
|
14
|
Hibasami H, Komiya T, Achiwa Y, Ohnishi K,
Kojima T, Nakanishi K, Sugimoto Y, Hasegawa M, Akatsuka R and Hara
Y: Black tea theaflavins induce programmed cell death in cultured
human stomach cancer cells. Int J Mol Med. 1:725–727.
1998.PubMed/NCBI
|
|
15
|
Lahiry L, Saha B, Chakraborty J, Adhikary
A, Mohanty S, Hossain DM, Banerjee S, Das K, Sa G and Das T:
Theaflavins target Fas/caspase-8 and Akt/pBad pathways to induce
apoptosis in p53-mutated human breast cancer cells. Carcinogenesis.
31:259–268. 2010. View Article : Google Scholar
|
|
16
|
Tu Y, Kim E, Gao Y, Rankin GO, Li B and
Chen YC: Theaflavin-3, 3′-digallate induces apoptosis and G2 cell
cycle arrest through the Akt/MDM2/p53 pathway in
cisplatin-resistant ovarian cancer A2780/CP70 cells. Int J Oncol.
48:2657–2665. 2016.PubMed/NCBI
|
|
17
|
Gao Y, Rankin GO, Tu Y and Chen YC:
Theaflavin-3, 3′-digallate decreases human ovarian carcinoma
OVCAR-3 cell-induced angiogenesis via Akt and Notch-1 pathways, not
via MAPK pathways. Int J Oncol. 48:281–292. 2016. View Article : Google Scholar
|
|
18
|
Gao Y, Rankin GO, Tu Y and Chen YC:
Inhibitory Effects of the Four Main Theaflavin Derivatives Found in
Black Tea on Ovarian Cancer Cells. Anticancer Res. 36:643–651.
2016.PubMed/NCBI
|
|
19
|
Xu Y, Jin Y, Wu Y and Tu Y: Isolation and
purification of four individual theaflavins using semi-preparative
high performance liquid chromatography. J Liq Chromatogr Relat
Technol. 33:1791–1801. 2010. View Article : Google Scholar
|
|
20
|
Levine AJ: p53 the cellular gatekeeper for
growth and division. Cell. 88:323–331. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lakin ND and Jackson SP: Regulation of p53
in response to DNA damage. Oncogene. 18:7644–7655. 1999. View Article : Google Scholar
|
|
22
|
Burma S, Chen BP, Murphy M, Kurimasa A and
Chen DJ: ATM phosphorylates histone H2AX in response to DNA
double-strand breaks. J Biol Chem. 276:42462–42467. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tibbetts RS, Brumbaugh KM, Williams JM,
Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C and Abraham RT: A
role for ATR in the DNA damage-induced phosphorylation of p53.
Genes Dev. 13:152–157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gottlieb TM, Leal JFM, Seger R, Taya Y and
Oren M: Cross-talk between Akt, p53 and Mdm2: Possible implications
for the regulation of apoptosis. Oncogene. 21:1299–1303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Franke TF, Kaplan DR and Cantley LC: PI3K:
Downstream AKTion blocks apoptosis. Cell. 88:435–437. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Franke TF, Yang S-I, Chan TO, Datta K,
Kazlauskas A, Morrison DK, Kaplan DR and Tsichlis PN: The protein
kinase encoded by the Akt proto-oncogene is a target of the PDGF-
activated phosphatidylinositol 3-kinase. Cell. 81:727–736. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Diehl JA, Cheng M, Roussel MF and Sherr
CJ: Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and
subcellular localization. Genes Dev. 12:3499–3511. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu GS: The functional interactions between
the p53 and MAPK signaling pathways. Cancer Biol Ther. 3:156–161.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Blagosklonny MV: Overcoming limitations of
natural anticancer drugs by combining with artificial agents.
Trends Pharmacol Sci. 26:77–81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. BioMed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 7:72015. View Article : Google Scholar
|
|
33
|
Knaapen M, De Bie M, Muhring J and Kockx
M: Cleaved PARP as a marker for apoptosis in tissue sections.
Promega Notes. 72:71999.
|
|
34
|
Los M, Mozoluk M, Ferrari D, Stepczynska
A, Stroh C, Renz A, Herceg Z, Wang ZQ and Schulze-Osthoff K:
Activation and caspase-mediated inhibition of PARP: A molecular
switch between fibroblast necrosis and apoptosis in death receptor
signaling. Mol Biol Cell. 13:978–988. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Morales J, Li L, Fattah FJ, Dong Y, Bey
EA, Patel M, Gao J and Boothman DA: Review of poly (ADP-Ribose)
polymerase (PARP) mechanisms of action and rationale for targeting
in cancer and other diseases. Crit Rev Eukaryot Gene Expr.
24:15–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chow A: Cell cycle control by oncogenes
and tumor suppressors: Driving the transformation of normal cells
into cancerous cells. Nature Education. 3:72010.
|
|
37
|
Sherr CJ and Bartek J: Cell cycle-targeted
cancer therapies. Annu Rev Cancer Biol. 1:41–57. 2017. View Article : Google Scholar
|
|
38
|
Zhang G, Miura Y and Yagasaki K: Induction
of apoptosis and cell cycle arrest in cancer cells by in vivo
metabolites of teas. Nutr Cancer. 38:265–273. 2000. View Article : Google Scholar
|
|
39
|
Prasad S, Kaur J, Roy P, Kalra N and
Shukla Y: Theaflavins induce G2/M arrest by modulating expression
of p21waf1/cip1, cdc25C and cyclin B in human prostate
carcinoma PC-3 cells. Life Sci. 81:1323–1331. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kawabe T: G2 checkpoint abrogators as
anticancer drugs. Mol Cancer Ther. 3:513–519. 2004.PubMed/NCBI
|
|
41
|
Waldman T, Kinzler KW and Vogelstein B:
p21 is necessary for the p53-mediated G1 arrest in human cancer
cells. Cancer Res. 55:5187–5190. 1995.PubMed/NCBI
|
|
42
|
Yu W, Park S-K, Jia L, Tiwary R, Scott WW,
Li J, Wang P, Simmons-Menchaca M, Sanders BG and Kline K:
RRR-γ-tocopherol induces human breast cancer cells to undergo
apoptosis via death receptor 5 (DR5)-mediated apoptotic signaling.
Cancer Lett. 259:165–176. 2008. View Article : Google Scholar
|
|
43
|
Ozaki T and Nakagawara A: Role of p53 in
cell death and human cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar
|
|
44
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brown R, Clugston C, Burns P, Edlin A,
Vasey P, Vojtĕsek B and Kaye SB: Increased accumulation of p53
protein in cisplatin-resistant ovarian cell lines. Int J Cancer.
55:678–684. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lahiry L, Saha B, Chakraborty J,
Bhattacharyya S, Chattopadhyay S, Banerjee S, Choudhuri T, Mandal
D, Bhattacharyya A, Sa G, et al: Contribution of p53-mediated Bax
transactivation in theaflavin- induced mammary epithelial carcinoma
cell apoptosis. Apoptosis. 13:771–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kalra N, Seth K, Prasad S, Singh M, Pant
AB and Shukla Y: Theaflavins induced apoptosis of LNCaP cells is
mediated through induction of p53 down-regulation of NF-kappa B and
mitogen-activated protein kinases pathways. Life Sci. 80:2137–2146.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meek DW: The p53 response to DNA damage.
DNA Repair (Amst). 3:1049–1056. 2004. View Article : Google Scholar
|
|
49
|
Lee JH and Paull TT: Activation and
regulation of ATM kinase activity in response to DNA double-strand
breaks. Oncogene. 26:7741–7748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao H and Piwnica-Worms H: ATR-mediated
checkpoint pathways regulate phosphorylation and activation of
human Chk1. Mol Cell Biol. 21:4129–4139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Matsuoka S, Rotman G, Ogawa A, Shiloh Y,
Tamai K and Elledge SJ: Ataxia telangiectasia-mutated
phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA.
97:10389–10394. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Melchionna R, Chen X-B, Blasina A and
McGowan CH: Threonine 68 is required for radiation-induced
phosphorylation and activation of Cds1. Nat Cell Biol. 2:762–765.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yuan J, Adamski R and Chen J: Focus on
histone variant H2AX: To be or not to be. FEBS Lett. 584:3717–3724.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rogakou EP, Pilch DR, Orr AH, Ivanova VS
and Bonner WM: DNA double-stranded breaks induce histone H2AX
phosphorylation on serine 139. J Biol Chem. 273:5858–5868. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shieh S-Y, Ikeda M, Taya Y and Prives C:
DNA damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sahu RP, Batra S and Srivastava SK:
Activation of ATM/Chk1 by curcumin causes cell cycle arrest and
apoptosis in human pancreatic cancer cells. Br J Cancer.
100:1425–1433. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lee H, Kim Y, Jeong JH, Ryu J-H and Kim
W-Y: ATM/CHK/p53 pathway dependent chemopreventive and therapeutic
activity on lung cancer by pterostilbene. PloS One.
11:e01623352016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Altomare DA and Testa JR: Perturbations of
the AKT signaling pathway in human cancer. Oncogene. 24:7455–7464.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Crowell JA, Steele VE and Fay JR:
Targeting the AKT protein kinase for cancer chemoprevention. Mol
Cancer Ther. 6:2139–2148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mabuchi S, Kuroda H, Takahashi R and
Sasano T: The PI3K/AKT/mTOR pathway as a therapeutic target in
ovarian cancer. Gynecol Oncol. 137:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Abraham AG and O'Neill E:
PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans.
42:798–803. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kim CW, Lu JN, Go S-I, Jung JH, Yi SM,
Jeong JH, Hah YS, Han MS, Park JW, Lee WS, et al: p53 restoration
can overcome cisplatin resistance through inhibition of Akt as well
as induction of Bax. Int J Oncol. 43:1495–1502. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Koul HK, Pal M and Koul S: Role of p38 MAP
kinase signal transduction in solid tumors. Genes Cancer.
4:342–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cheng T-L, Symons M and Jou T-S:
Regulation of anoikis by Cdc42 and Rac1. Exp Cell Res. 295:497–511.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hong B, Li H, Zhang M, Xu J, Lu Y, Zheng
Y, Qian J, Chang JT, Yang J and Yi Q: p38 MAPK inhibits breast
cancer metastasis through regulation of stromal expansion. Int J
Cancer. 136:34–43. 2015. View Article : Google Scholar
|
|
66
|
Kohno M and Pouyssegur J: Targeting the
ERK signaling pathway in cancer therapy. Ann Med. 38:200–211. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Dong Z, Ma W, Huang C and Yang CS:
Inhibition of tumor promoter-induced activator protein 1 activation
and cell transformation by tea polyphenols, (−)-epigallocatechin
gallate, and theaflavins. Cancer Res. 57:4414–4419. 1997.PubMed/NCBI
|
|
68
|
Vivas-Mejia P, Benito JM, Fernandez A, Han
H-D, Mangala L, Rodriguez-Aguayo C, Chavez-Reyes A, Lin YG, Carey
MS, Nick AM, et al: JNK-1 inhibition leads to antitumor activity in
ovarian cancer. Clin Cancer Res. 16:184–194. 2010. View Article : Google Scholar
|
|
69
|
Kitanaka C, Sato A and Okada M: JNK
signaling in the control of the tumor-initiating capacity
associated with cancer stem cells. Genes Cancer. 4:388–396. 2013.
View Article : Google Scholar : PubMed/NCBI
|