Introduction
Prostate cancer (PCa) is the most common
non-cutaneous cancer and the second leading cause of cancer death
in American males (1). Despite the
initial regression/stabilization brought by androgen deprivation
therapy, a large number of PCa eventually relapses and progresses
to castration-resistant prostate cancer (CRPC), with a median
survival of only 7–12 months (2),
for which, better understanding and newer management is urgently
needed. Chemotherapy with new agents such as taxol analogs, and new
combinatorial regimens has recently made some progress (3). However, the overall outcome of
current chemotherapeutic strategies is still unsatisfactory
(4).
Camptothecin (CPT) and its analogs are a promising
class of anticancer drugs, which have advanced to the forefront of
chemotherapy when used alone or in combination with others
(5). CPT inhibits DNA
topoisomerase I (Topo I) by blocking the rejoining step of the
cleavage/religation reaction, resulting in accumulation of the
covalent reaction intermediate, the cleavable complex, which leads
to apoptosis and cell cycle arrest (6). The first two CPT derivatives,
irinotecan (Camptosar) and topotecan (Hycamtin), were approved by
US Food and Drug Administration (FDA) in 1996 for the treatment of
colon/rectum cancer, and lung cancer (7). Other analogs, such as exatecan
(8), have also shown very
promising results in clinical trials. However, the effects of CPT
analogs on PCa are controversial. Most preclinical studies have
shown that CPTs are effective for both androgen-dependent and
androgen-independent PCa (9,10).
Unfortunately, a couple of clinical trials (phase I/II) with
different CPT analogs have only shown very limited benefits on CRPC
patients (11–13). Recently, we have demonstrated that
NSC606985, a highly water-soluble CPT analog that has rarely been
studied for its anticancer activity, produces a significant
dose-dependent induction of cell apoptosis at nanomolar
concentrations in DU145, LNCaP and PC3 PCa cells (14).
Previous studies have shown that CPT, and apoptotic
effects of its analogs may involve protein kinase Cδ (PKCδ)
signaling pathway (14–16). PKCδ is a member of the
serine/threonine-specific PKC family, which has been found to be
involved in diverse signaling pathways. From an oncology
standpoint, PKCδ is generally considered as an anti-proliferative
and pro-apoptotic protein kinase (17). Previous studies have shown that
PKCδ activates both intrinsic and extrinsic apoptotic pathways in
PCa cells under phorbol 12-myristate 13-acetate (PMA) stimulation
(18,19). However, recent studies have
gradually revealed that PKCδ can also function as an anti-apoptotic
protein and it is critical for the survival of several cancer cells
(20,21). It has been proposed that these
apparent opposite actions of PKCδ are tightly regulated by
epigenetic mechanisms including post-translational modifications
and cellular compartmentalization (22).
In the present study, we aimed to investigate the
effects of CPT analogs, mainly NSC, in LAPC4 cells, elucidate its
potential molecular mechanism, and reveal its impact on future CRPC
management.
Materials and methods
Materials
NSC, and topotecan were kindly provided by the Drug
Synthesis and Chemistry Branch, Developmental Therapeutic Program,
National Cancer Institute (Bethesda, MD, USA). Tissue culture
medium, rottlerin, protease inhibitors, propidium iodide (PI), and
anti-β-actin primary antibody were purchased from Sigma-Aldrich
(St. Louis, MO, USA). Fetal bovine serum (FBS), L-glutamine,
penicillin and streptomycin were from Gemini Bio-Products
(Calabasas, CA, USA). Z-VAD-fluoromethylketone (FMK), RNase A,
protease K, and M-MLV reverse transcriptase were from Promega
(Madison WI, USA). Antibodies against cyclin A, and PKCδ were from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
cytochrome c antibody (clone 7H8.2C12) was obtained from BD
Pharmingen (San Diego, CA, USA). The SYBR-Green Real-Time PCR
master mix was from Life Technologies (Grand Island, NY, USA).
TriPure Isolation reagent was from Roche Applied Science (Mannheim,
Germany).
Cell culture
LAPC4 cells (kindly provided by Dr C. Sawyer) were
grown in Iscove's modified Dulbecco's medium supplemented with 15%
FBS, 2 mM L-glutamine, 1 nM R1881, 50 U/ml of penicillin, and 50
µg/ml of streptomycin. R1881 was withdrawn 48 h before cell
passage to conduct experiments. PC3, LNCaP and DU145 cells (ATCC,
Rockville, MD, USA) were grown in RPMI-1640 and Dulbecco's modified
Eagle's medium, respectively, supplemented with 10% FBS, 2 mM
L-glutamine, 50 U/ml of penicillin and 50 µg/ml
streptomycin. Cells were maintained in a 5% CO2 - 95%
air humidified atmosphere at 37°C, and cultured in phenol red-free
medium with 5% stripped FBS for 24 h before each experiment. The
expression of androgen receptor (AR) was verified with real-time
PCR whenever a new vial of LAPC4 was thawed. Treatment with 10 nM
dihydrotestosterone was used as a positive control for stimulation
of LAPC4 cell growth.
Cell proliferation assays
Cells were seeded in 96-well plates at ~30% density,
and treated with various regimens as indicated in each experiment
at 24 h after plating. The viable cell number was determined using
the CellTiter AQueous One Solution Cell Proliferation assay kit
from Promega, following the manufacturer's instructions. DNA
biosynthesis was determined using the BrdU Cell Proliferation assay
kit obtained from Calbiochem (San Diego, CA, USA) following the
manufacturer's instructions.
Flow cytometry
Approximately 1×106 LAPC4 cells were
plated in 60-mm plates and treated with vehicle control or various
regimens as indicated in each experiment. At the end of the
treatment, cells were collected, washed with phosphate-buffered
saline (PBS) and fixed with 70% ethanol. For DNA content, cells
were stained with PI (50 µg/ml) plus RNase A (20
µg/ml) in PBS, and analyzed using the FACScan Flow Cytometer
(Becton-Dickinson, Heidelberg, Germany). The data from
1.0×104 cells/sample were collected and analyzed using
the CellQuest software (Becton-Dickinson). For cell apoptosis
analysis, cells were stained with Annexin V in combination with PI
using the Annexin V-FITC apoptosis detection kit I from BD
Pharmingen according to the manufacturer's instructions.
Determination of DNA fragmentation
NSC treated and untreated cells were harvested and
incubated in a lysis buffer [50 mM Tris-HCl (pH 8.0), 20 mM
ethylenediaminetetraacetic acid (EDTA), 10 mM NaCl, 1% sodium
dodecyl sulfate (SDS)] for 20 min on ice. Samples were then
centrifuged and treated with DNase-free RNase A and proteinase K.
Following phenol and chloroform extraction, DNA was precipitated by
ethanol and dissolved in 1X Tris-EDTA buffer. The DNA samples were
then electrophoresed in a 2% agarose gel, visualized by ethidium
bromide staining under ultraviolet light.
Western blot analysis
LAPC4 cells were harvested and total cellular
proteins were extracted using a lysis buffer (62.5 mM Tris-HCl pH
6.8, 100 mM dithiothreitol, 2% SDS, 10% glycerol). The cytosolic,
nucleus and mitochondrial protein extracts were prepared as
described (14). The protein
concentrations were determined using the Bio-Rad Protein assay
(Bio-Rad Laboratories, Hercules, CA, USA) following the
manufacturer's instructions. Protein (25 µg) was loaded in
each lane, electrophoresed on a 15% SDS-PAGE, and transferred to a
nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway,
NJ, USA). The blots were blocked with TBST buffer [500 mM NaCl, 20
mM Tris-HCl (pH 7.4), and 0.1% Tween-20] containing 5% nonfat dry
milk (Bio-Rad Laboratories) and then incubated with specific
primary antibody in TBST buffer containing 1% nonfat dry milk at
4°C overnight. Following secondary antibody incubation for 1 h at
room temperature, bands were visualized using a Super Signal
Chemiluminescence kit (Millipore, Billerica, MA, USA), and exposed
to Kodak X-Max film. β-actin was used as the internal control after
stripping off the original membrane. The images were scanned and
relative band intensities were calculated using the NIH ImageJ
software.
PKCδ Steath™ RNAi transfections
Based upon the sequence of human PRKCD (GenBank™
accession no. NM_006254), custom steath™ RNAi oligos (Invitrogen,
Carlsbad, CA, USA) at 25-base-pair in length,
5′-CCACUACAUCAAGAACCAUGAGUUU-3′ was designed. A non-specific
Steath™ RNAi (NS-RNAi) control, 5′-CCAUGGCGCCAAUUCCAAACAGUUU-3′ was
also synthesized. All RNAi transfections at doses from 25 to 200 nM
were performed using Lipofectamine 2000 (Life Technologies)
following the manufacturer's instructions. For the analysis of PKCδ
knockdown following RNAi transfection, LAPC4 cells
(5×105 cells/well) were seeded in 6-well plates and
transfected with various concentrations of RNAi at 24 h after
plating. The cells were harvested at 96 h of transfection. Total
cellular proteins were extracted and subjected to western blot
analysis as described above. For cell proliferation and flow
cytometric analysis, LAPC4 cells were seeded in 96-well and 60 mm
plates, respectively as described above. Cells were treated with
various regimens for 72 h after 24-h RNAi transfection (100 nM) as
indicated in each experiment. Cell proliferation was determined by
BrdU assay and cell apoptosis and cell cycle were analyzed by flow
cytometry as described above.
Real-Time PCR
Approximately 5×105 LAPC4 cells were
seeded in 6-well plates and treated with different regiments, and
harvested at different time-points as indicated in each experiment.
Total cellular RNA was extracted with TriPure Isolation reagent
following the manufacture's instructions. One microgram of RNA per
sample was used for reverse transcription using M-MLV reverse
transcriptase with RNase inhibitor. Real-time PCR was done with an
Eco Real-Time PCR system (Illumina, San Diego, CA, USA). CCNA2
expression level was detected using 2X SYBR™-Green Real-Time PCR
master mix. The expression level was normalized with human ACTB
using ΔΔCt method. The primers used were as follows: for CCNA2
forward, 5′-TGGACCTTCACCAGACCTAC-3′ and reverse,
5′-GGTTGAGGAGAGAAACACCA-3′; for ACTB forward,
5′-CTAGAAGCATTTGCGGTGGACGATG-3′ and reverse,
5′-TCATGAAGTGTGACGTGGACATCCG-3′. The final concentration of forward
and reverse primer in each reaction system was 200 nM,
respectively, and the final concentration of total RNA templates
was 40 ng/well. The cycling conditions are listed in Table I.
 | Table IReal-time PCR conditions. |
Table I
Real-time PCR conditions.
| Cycle no. | Step |
Temperature
(°C) | Time
(sec) |
|---|
| 1X | UDG incubation | 50 | 120 |
| 1X | Polymerase
activation | 95 | 600 |
| 40X | Denature | 95 | 15 |
| Anneal/extend | 60 | 60 |
| 1X | Melt curve
analysis | 95 | 15 |
| | 55 | 15 |
| | 95 | 15 |
Statistics
The data are presented as mean ± standard error of
the mean. One-way analysis of variance (ANOVA) following post-hoc
Student-Newman-Keuls test was used to determine the differences
among multiple groups. A p-value <0.05 was considered
statistically significant. The data and statistical analysis comply
with the recommendations on experimental design and analysis in
pharmacology (23). All analyses
were performed using SigmaPlot 12.0 from Systat Software (Chicago,
IL, USA).
Results
CPT and its analogs produce a dose- and
time-dependent dual action on cell growth and death in LAPC4
cells
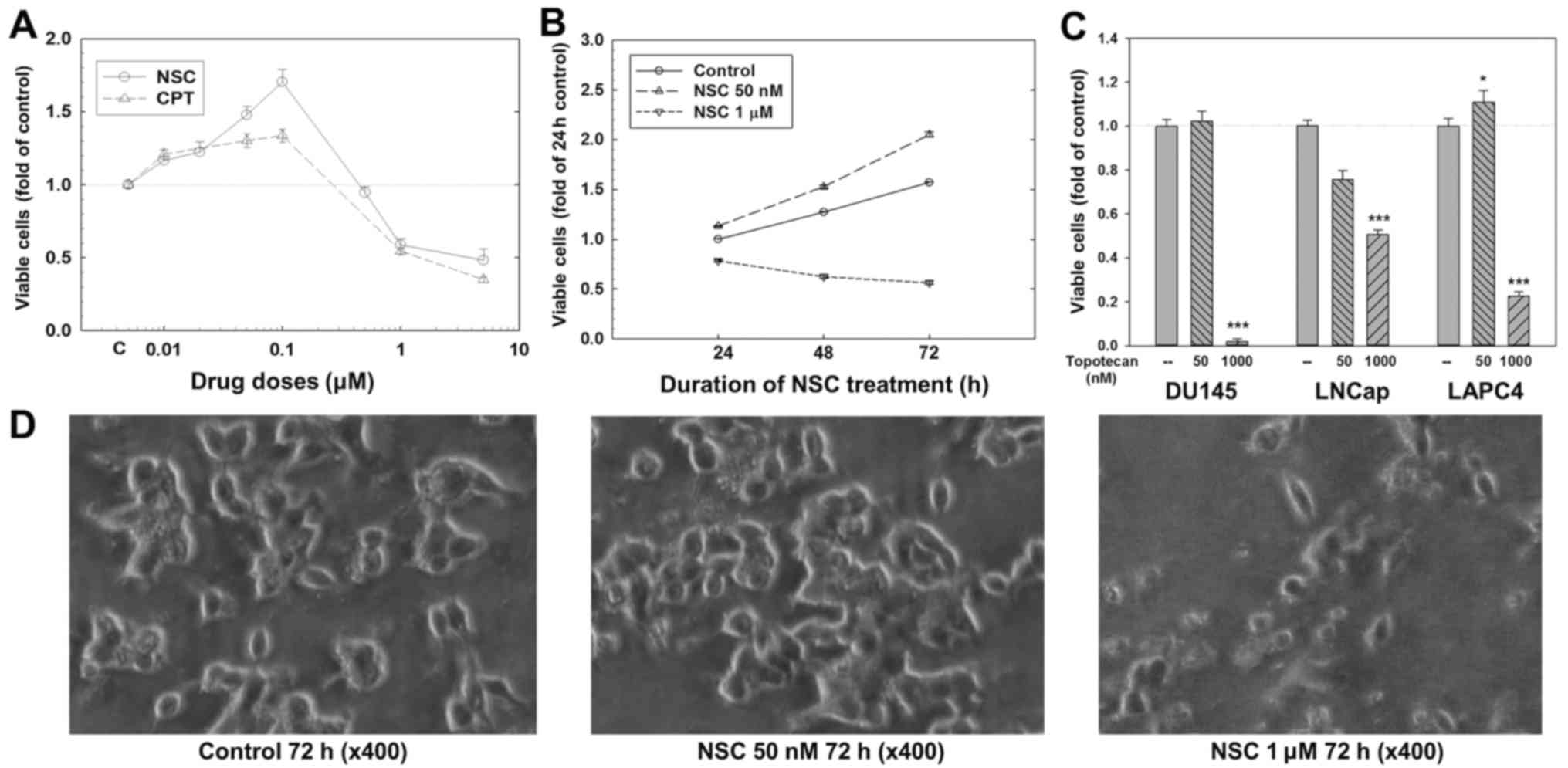
LAPC4 cells were treated with various doses of CPT
and NSC for 24, 48 or 72 h. As shown in Fig. 1A, at 72 h, NSC at doses ranging
from 10 to 100 nM produced a dose-dependent increase in viable cell
number (1.7-folds of control at 100 nM, p<0.01); while at doses
ranging from 500 nM to 5 µM, NSC produced a dose-dependent
decrease in viable cell number (0.48-fold of control at 5
µM, p<0.01). This biphasic effect of NSC was also
time-dependent (Fig. 1B). An
increase in viable cell number was observed at 24 h and more
obvious at 48 and 72 h with 50 nM NSC treatment, while a
time-dependent decrease in viable cell number was observed when
cells were treated with 1 µM NSC. Similar dose-dependent
biphasic effect on viable cell number was observed after 72 h
treatment with CPT (Fig. 1A) and
topotecan (Fig. 1C) in LAPC4
cells, although the stimulation by CPT and topotecan was relatively
weaker compared to NSC. Like NSC and CPT (14), topotecan at a low-dose (50 nM) did
not stimulate cell growth, while significantly decreased viable
cell numbers at a high-dose (1 µM) in DU145 and LNCaP cells
(p<0.001) (Fig. 1C).
Morphological analysis showed that there were more
viable cells with higher cell density in NSC 50 nM group than in
control group after 72 h treatment (Fig. 1D). In contrast, 1 µM NSC
induced significant decrease in cell number with apoptotic
characteristics such as shrunken cells and apoptotic bodies
(Fig. 1D).
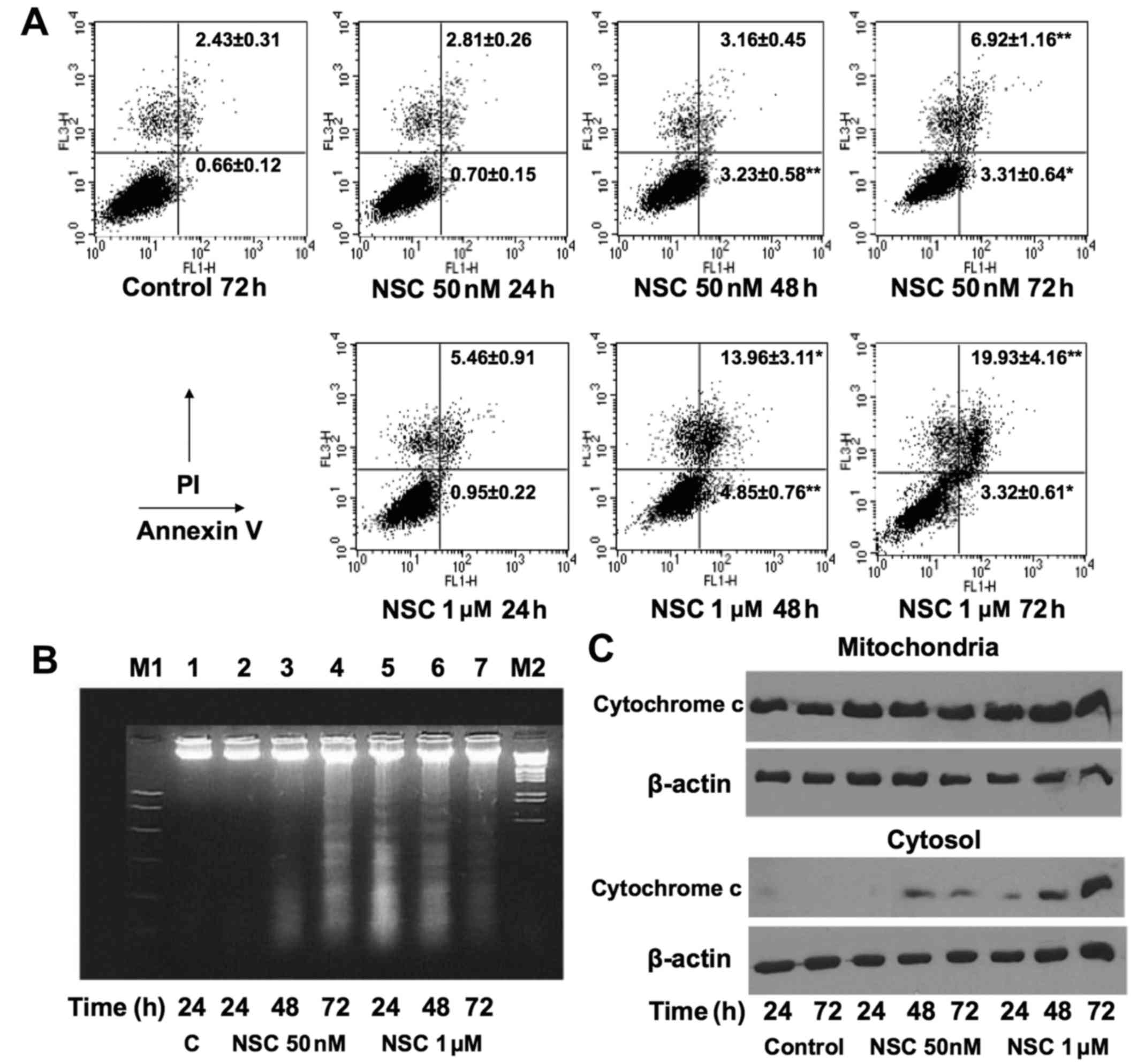
NSC induces apoptosis in LAPC4 cells
To investigate whether the NSC-induced decrease in
viable cell number is associated with cell apoptosis, Annexin V/PI
staining was performed to determine the apoptotic and/or necrotic
cells after NSC treatment in LAPC4. Treatment with NSC produced a
time- and dose-dependent increase in early (Annexin
V+/PI−), late apoptotic (Annexin
V+/PI+) and necrotic cells (Annexin
V−/PI+) as shown in Fig. 2A. The fractions of Annexin
V+ cells were significantly increased at 48 and 72 h of
NSC treatment, and the changes were much greater at 1 µM
than 50 nM dose. The pro-apoptosis effect was further confirmed by
demonstrating that NSC produced a time- and dose-dependent
induction of DNA fragmentation (Fig.
2B), and cytosolic cytochrome c release (Fig. 2C).
The dual action of NSC in LAPC4 cells
involves PKCδ
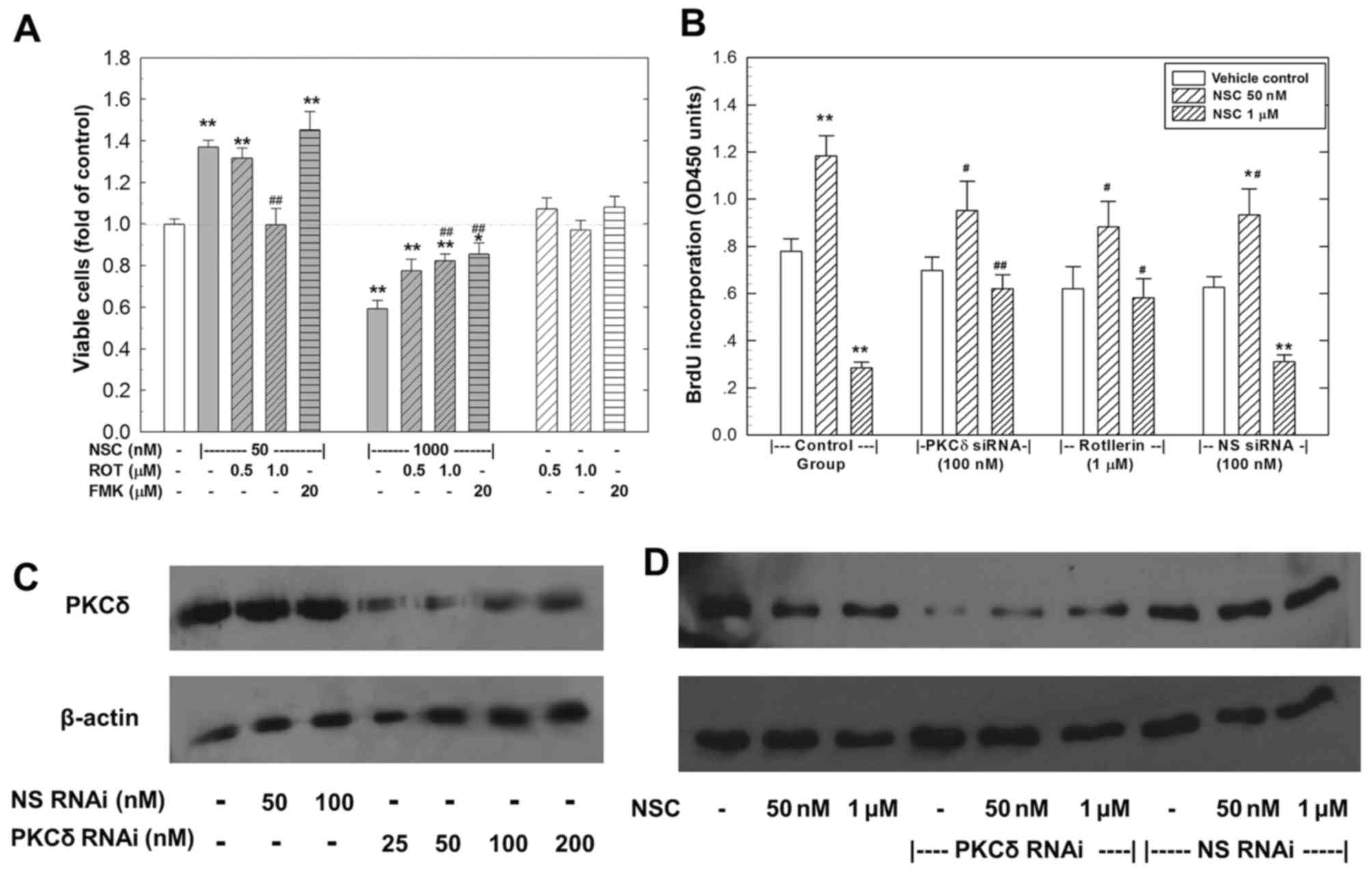
To investigate whether the NSC-caused dual action
involves PKCδ activation, rottlerin was used to inhibit PKCδ
activity. As shown in Fig. 3A, at
1 µM, rottlerin completely blocked the low-dose (50 nM)
NSC-induced cell growth and significantly rescued the high-dose (1
µM) NSC-caused decrease in viable cell number. On the other
hand, FMK, a pan-caspase inhibitor, significantly rescued the
high-dose NSC-induced decrease in viable cell number without any
effect on the low-dose NSC-induced cell growth. Moreover, PKCδ
knockdown by specific RNAi (100 nM) blocked NSC dual action in
LAPC4 cells as shown in Fig. 3B.
In contract, transfection of an NS-RNAi failed to alter NSC effect
on DNA biosynthesis (Fig. 3B). The
knockdown of PKCδ by RNAi transfection was confirmed using western
blot analysis (Fig. 3C and D).
In flow cytometry analysis, as shown in Table II, co-administration of rottlerin
at 1 µM for 72 h, which did not have any significant impact
on cell apoptosis per se, markedly inhibited the NSC-induced
apoptosis. PKCδ-knockdown significantly attenuated the 1 µM
NSC-induced late (Annexin V+/PI+) and early
(Annexin V+/PI−) apoptotic cells from 19.93
and 3.32% to 7.81 and 1.68%, respectively (Table II). In contrast, transfection of
the NS-RNAi did not significantly affect NSC-induced apoptosis.
| Table IIBlockade of NSC-induced cell
apoptosis by rottlerin and specific PKCδ RNAi using flow cytometry
analysis (72 h). |
Table II
Blockade of NSC-induced cell
apoptosis by rottlerin and specific PKCδ RNAi using flow cytometry
analysis (72 h).
Cell types
| Early apoptotic
cells Late apoptotic cells (Annexin V+/PI−, %
of total cells) | (Annexin
V+/PI+, % of total cells) |
|---|
| Treatment |
|---|
| Control | 0.66±0.12 | 2.4±0.31 |
| NSC 50 nM | 3.31±0.64a | 6.92±1.16b |
| NSC 1
µM | 3.32±0.61a | 19.93±4.16b |
| Rottlerin 1
µM | 0.66±0.18 | 3.91±0.75 |
| Rottlerin+NSC 50
nM | 0.13±0.06 | 3.94±0.87c |
| Rottlerin+NSC 1
µM | 5.11±1.28b | 6.81±1.72b,d |
| PKCδ RNAi | 2.59±0.81 | 4.60±0.81 |
| PKCδ RNAi+NSC 50
nM | 2.97±0.88 | 5.91±0.11 |
| PKCδ RNAi+NSC 1
µM | 1.68±0.43d | 7.81±1.42a,c |
| NS RNAi | 4.16±0.77 | 5.25±1.22 |
| NS RNAi+NSC 50
nM | 5.58±1.03 | 9.4±1.92a |
| NS RNAi+NSC 1
µM | 14.85±3.89b | 19.71±4.50b |
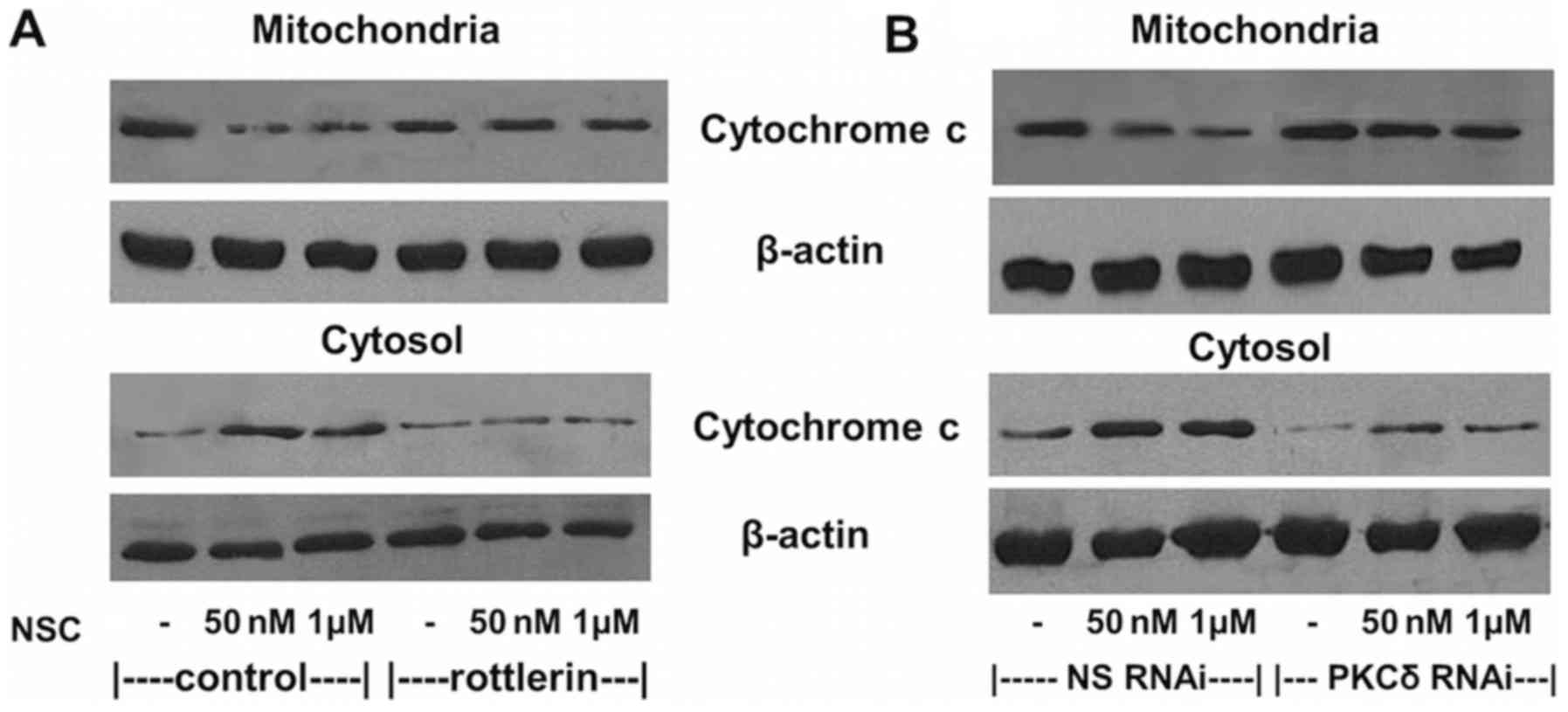
Consistent with the inhibition of NSC-induced cell
apoptosis, NSC-induced cytochrome c release from
mitochondria to cytosol was also greatly attenuated by the
co-administration of 1 µM rottlerin (Fig. 4A) and the transfection of 100 nM
PKCδ RNAi (Fig. 4B). However, the
transfection of an NS-RNAi did not change the NSC-induced
cytochrome c release as shown in Fig. 4B.
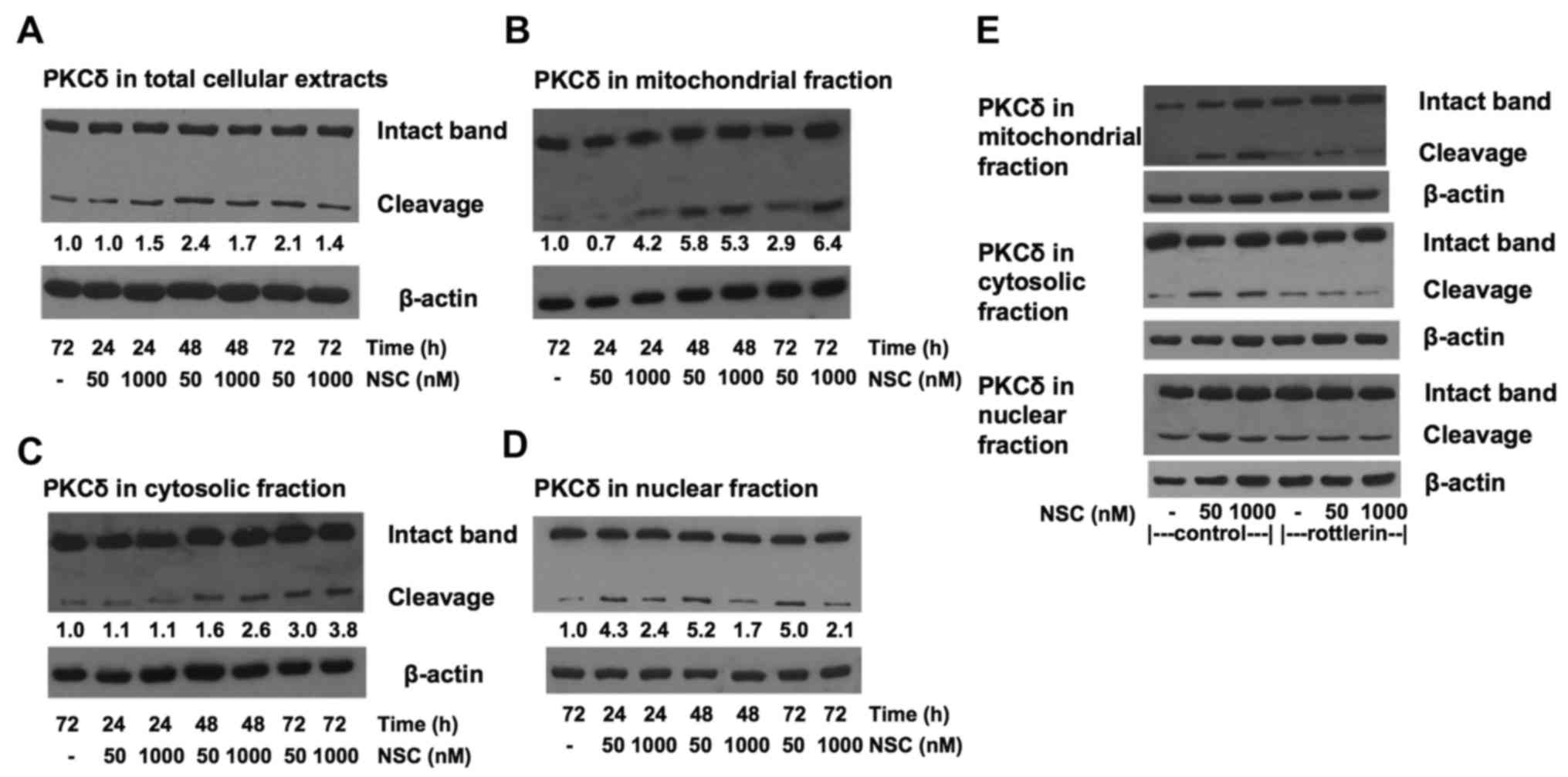
NSC produces a dose-dependent
differential PKCδ cleavage in subcellular compartments
To explore the potential mechanism of NSC dual
action on cell growth and apoptosis, the proteolytic cleavage of
PKCδ in various subcellular compartments were analyzed by western
blot analysis. The total PKCδ expression level was not altered with
NSC treatment, but a slight increase of PKCδ cleavage was observed
in total cellular protein after NSC treatment as shown in Fig. 5A. Most interestingly, NSC treatment
resulted in a dose- and time-dependent differentiated change of
PKCδ proteolytic cleavage in different subcellular compartments as
shown in Fig. 5B–D. Treatment with
a high-dose (1 µM) NSC resulted in a more rapid and robust
PKCδ cleavage in the membrane/mitochondrial fraction than those
treated with a low-dose (50 nM) NSC (Fig. 5B). The level of mitochondrial PKCδ
cleavage was elevated >4-fold at 24 h of 1 µM NSC
treatment and sustained for at least 72 h. Similar time-dependent
PKCδ cleavage was observed at either a low or a high-dose NSC
treatment in the cytosol (Fig.
5C). Whereas in the nuclear compartment, NSC-induced increase
in PKCδ cleavage was more rapid, intense and sustainable at
low-dose (>4-fold) compared to high-dose treatment (~2-fold)
(Fig. 5D). Moreover, the addition
of 1 µM rottlerin greatly reduced both the low-dose and
high-dose NSC-induced proteolytic cleavage of PKCδ in LAPC4 cells
(Fig. 5E). Taken together, these
data indicate that NSC produced a dose-dependent differential PKCδ
cleavage in the subcellular compartments of LAPC4 cells.
NSC produces a dose-dependent alteration
in cell cycle in LAPC4 cells
Analysis of nuclear DNA distribution showed that NSC
produced a time- and dose-dependent increase in hypoploid cells
(sub-G1 cells) as shown in Table
III, in agreement with the Annexin V/PI staining analysis as
described above (Fig. 2A and
Table II). The fraction of sub-G1
cells, an important indicator of cell apoptosis, was markedly
increased in cells treated with 1 µM NSC for 72 h, while it
only had slight increase in cells treated with 50 nM NSC as shown
in Table III. NSC at 50 nM
produced a significant elevation in the fraction of G2/M cells.
Consistent with the concept that NSC-induced cell apoptosis
involves PKCδ activation, the addition of rottlerin and PKCδ RNAi
transfection both markedly inhibited the high-dose (1 µM)
NSC-induced sub-G1 cells (p<0.01) without significant
alterations in NSC-induced other cell cycle changes. Whereas, a
non-specific RNAi had no effect on NSC-induced cell cycle changes
(Table III).
| Table IIINSC-induced cell cycle changes and
the blockade by rottlerin and specific PKCδ RNAi using flow
cytometry analysis. |
Table III
NSC-induced cell cycle changes and
the blockade by rottlerin and specific PKCδ RNAi using flow
cytometry analysis.
Cell cycle
| Sub-G1 (% of total
cells) | G1 (% of total
cells) | S (% of total
cells) | G2/M (% of total
cells) |
|---|
| Treatment |
|---|
| Control | 2.54±0.35 | 58.74±2.27 | 11.07±0.60 | 28.63±1.61 |
| NSC 50 nM | 4.18±0.43 | 45.13±2.11b | 10.24±1.16 | 38.96±1.88a |
| NSC 1
µM | 16.44±1.63b | 42.28±2.10b | 17.25±1.67 | 23.18±2.02 |
| Rottlerin 1
µM | 4.60±0.29 | 54.78±2.03 | 10.31±1.34 | 30.62±1.65 |
| Rottlerin+NSC 50
nM | 4.87±0.45 | 47.28±1.98b | 10.02±1.14 | 37.64±1.67a |
| Rottlerin+NSC 1
µM | 7.14±0.72a,d | 41.16±2.34b | 18.31±1.35 | 33.77±1.79 |
| PKCδ RNAi | 3.76±0.53 | 53.82±2.02 | 11.94±1.31 | 32.68±1.87 |
| PKCδ RNAi+NSC 50
nM | 4.58±0.45 | 42.17±2.01b | 12.73±1.20 | 39.54±1.74a |
| PKCδ RNAi+NSC 1
µM | 6.42±0.74a,d | 41.5±2.02b | 18.39±1.55 | 32.85±2.07 |
| NS RNAi | 4.09±0.48 | 54.16±2.06 | 10.59±1.32 | 31.75±1.61 |
| NS RNAi+NSC 50
nM | 4.60±0.57 | 42.36±2.12b | 13.19±1.22 | 38.82±2.02a |
| NS RNAi+NSC 1
µM | 17.35±1.47b | 42.57±2.36b | 17.12±1.46 | 22.64±1.52 |
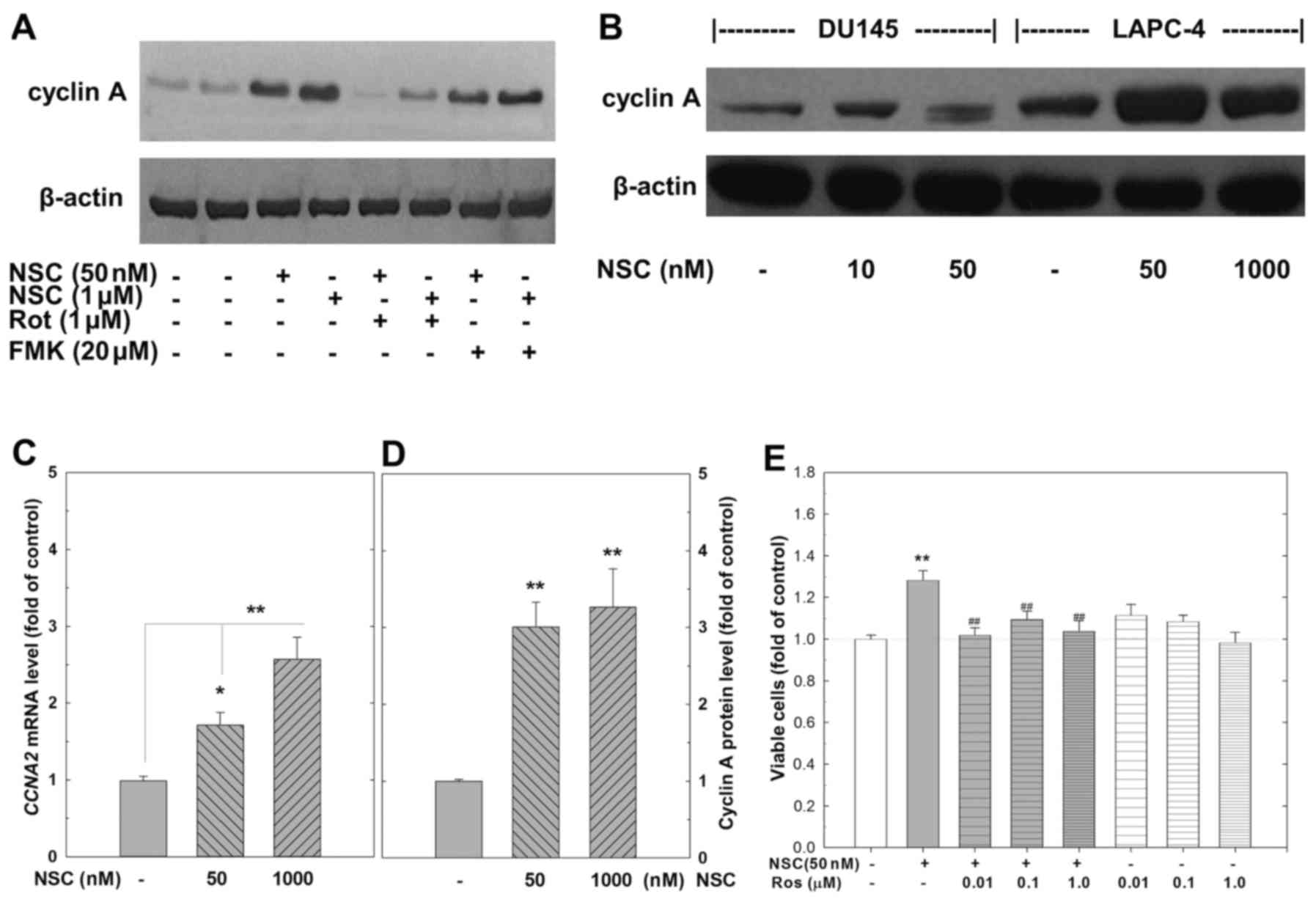
NSC-induced cell proliferation is
associated with an induction of cyclin A expression and
cyclin-dependent kinase activity
The activity of cyclin/cyclin-dependent kinase (CDK)
complexes that regulate the progression of cell cycle is controlled
by the synthesis of appropriate cyclins during a specific phase of
the cell cycle. Western blot analysis demonstrated that treatment
with NSC at either a low (50 nM) or a high-dose (1 µM)
dramatically increased cyclin A expression in LAPC4, but not in
DU145 cells (Fig. 6A, B and D).
The NSC-induced cyclin A expression was greatly attenuated by
rottlerin, but not by FMK (Fig.
6A). RT-PCR analysis confirmed that the levels of CCNA2 mRNA,
the somatic type of cyclin A, were significantly elevated at 72 h
of NSC treatment as shown in Fig.
6C. Co-administration of roscovitine (ROS), a selective CDKs
inhibitor, at doses ranging from 0.01 to 1 µM completely
blocked the low-dose NSC (50 nM) inducing cell growth in LAPC4
cells (Fig. 6E).
Discussion
CPT, a unique pentacyclic quinoline alkaloid
isolated from Camptotheca acuminate (24), a native tree from Tibet, China, is
one of the prominent leading compounds in anticancer drug
development (6). Unfortunately,
CPT analogs have only shown limited benefits in the treatment of
CRPC (11–13). Previous studies indicate that NSC,
a synthetic CPT analogue, induces cell apoptosis and growth
inhibition in PCa cells (14),
leukemia cells (16), and ovarian
cancer cells (25) through an
activation of the intrinsic apoptotic pathway and induction of cell
cycle arrest. In the present study, we demonstrated, for the first
time, that both CPT and its analogs such as NSC and topotecan
produced a dose-dependent dual action on cell growth and cell
apoptosis in LAPC4 PCa cells (Fig.
1). At low-doses (10–100 nM), both CPT and its analogs produced
an overall favorable effect on cell proliferation, whereas, at
high-doses (500 nM-5 µM), CPT and its analogs predominately
caused cell apoptosis. Using NSC as an example, we further
demonstrated that this dual action was mediated, at least in part,
via PKCδ activation on both ends. This conclusion is supported by
the results that co-administration of rottlerin, or transfection of
PKCδ RNAi, both significantly inhibited the proliferative effect
induced by low-dose NSC (Fig.
3A–B), and blocked the apoptotic effect induced by high-dose
NSC (Figs. 3A and B and 4 and Table
II).
PKCδ is abundantly expressed in mammalian tissues,
and has been proved to be involved in the regulation of both
intrinsic and extrinsic apoptosis for a wide range of stimuli
through various cell-type specific pathways (18,26).
It can be activated mainly by four mechanisms, one of which is
proteolytic cleavage (22,27). Generally, when the
intrinsic/mitochondrial apoptosis pathway is induced by cell stress
such as DNA damage by UV, cisplatin and CPT analogs, PKCδ can be
proteolytically cleaved by caspase-3 into a 38 kDa regulatory
fragment, and a 41 kDa constantly activated catalytic fragment
which translocates to mitochondria, leading to an amplification of
the apoptosis cascade in various cell types (27–30).
Furthermore, PKCδ also acts as a 'gatekeeper' to inhibit cell cycle
progression through G1/S and G2/M checkpoints by mediating p21CIP1,
a G1 CDK inhibitor, and decreasing the expression of cyclins
(31). Our previous studies in
leukemic cells have demonstrated that NSC rapidly induces the
proteolytic activation of PKCδ with a loss of mitochondrial
transmembrane potential (Δψm) and caspase-3 activation, which is
completely blocked by co-administration of rottlerin (16). In the present study, we have
demonstrated that NSC produced a time- and dose-dependent induction
of cell apoptosis (Fig. 2) and
PKCδ proteolytic activation (Fig.
5) in LAPC4 cells. These NSC-induced changes were blocked by
co-administration of rottlerin or knockdown of PKCδ with a specific
RNAi (Figs. 3, 4 and 5
and Tables II and III). Our results suggest that NSC
induced DNA damage and PKCδ proteolytic activation, leading to an
activation of the intrinsic apoptosis pathway in LAPC4 cells.
Most surprisingly, for the very first time, we found
that both CPT and CPT analogs (NSC and topotecan) induced cell
growth in LAPC4 cells at nanomolar doses. Unlike the NSC-induced
apoptotic effect, this proliferative effect of NSC was involved in
PKCδ, but not caspase activation (Figs. 3 and 6). Although PKCδ has long been proposed
as a pro-apoptotic gene and a tumor suppressor, growing evidence
suggests that it is also involved in cell proliferation and
survival pathways (21,32,33).
PKCδ activation has been reported to induce the insulin-like growth
I factor (IGF-I) proliferative signaling in renal carcinoma cells
(34), increase the
anchorage-independent growth of metastatic breast cancer cells
(35), and be correlated with
reduced overall breast cancer patient survival (36). In contrast, downregulation of PKCδ
in breast cancer cells results in impairment in cell survival and a
potentiation of chemotherapeutic agent-induced apoptosis (37). Although not fully understood, PKCδ
has been reported to regulate multiple molecular signal pathways
including NF-κB, Akt-PI3K, mTOR, and Ras/Raf/MEK/MAPK mitogenic
signal transduction pathways (20,21,38),
which may lead to cell survival and proliferation. In the present
study, we demonstrated that NSC greatly induced cyclin A expression
in LAPC4 cells (Fig. 6), a
well-known factor in accelerating cell proliferation via
interacting with CDK2 (39). The
NSC-induced cyclin A expression was associated with NSC-induced
cell growth since roscovitine, a specific CDK inhibitor, completely
blocked NSC-induced cell growth (Fig.
6E). Moreover, rottlerin, but not FMK, completely blocked
NSC-induced cyclin A expression and cell growth, indicating that
PKCδ activation is upstream of cyclin A expression (Fig. 6A). The NSC-induced cyclin A
upregulation was not observed in DU145 cells, consistent with our
precious study (14), in which we
revealed a potent apoptotic but not proliferative effect of NSC in
DU145 cells, indicating a cell-dependent differential effect of NSC
in PCa cells. Taken together, our data strongly indicate that
administration of NSC in LAPC4 cells produced a proteolytic
activation of PKCδ leading to an upregulation of cyclin A,
resulting in a promotion of cell survival and proliferation
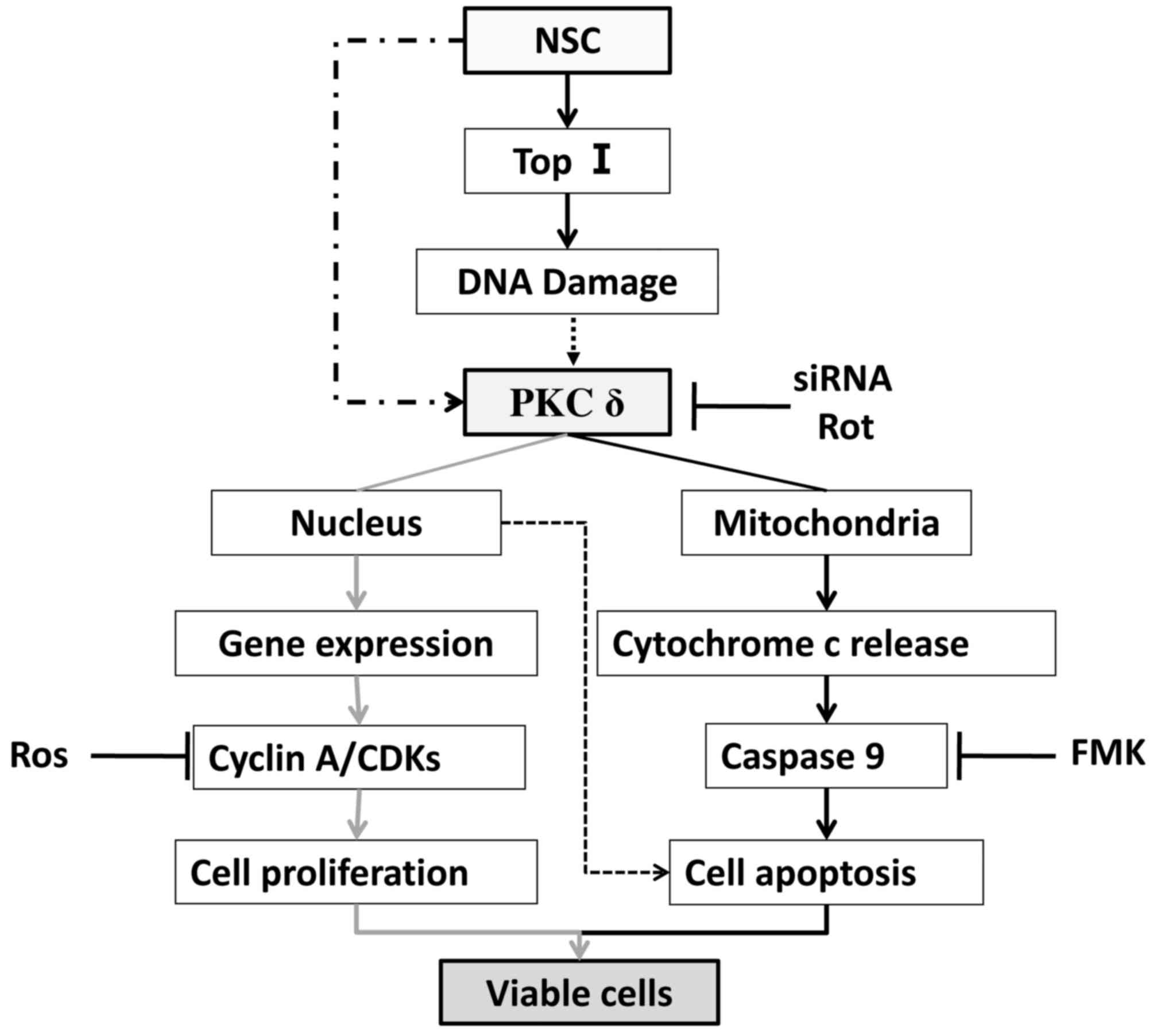
(Fig. 7). We therefore, for the
first time, demonstrated that PKCδ activation is associated with
both cell proliferation and apoptosis in the same cell line.
It is intriguing why PKCδ activation by NSC results
in this dose-dependent biphasic effect. This unconventional
dose-response effect has traditionally been attributed to a
functional antagonism model, which proposed that the drug interacts
with two independent receptor-effector systems, causing effects
that counteract each other in the same system (40–42).
The overall outcome would therefore be governed by the sum of the
two independent dose-response effects. However, unlike the two
independent systems predicted in the model, we observed that the
stimulation of both cell proliferation and apoptosis involve PKCδ
activation. We have therefore hypothesized that this NSC biphasic
effect may be mediated through a differential activation of PKCδ in
subcellular compartments. This hypothesis is supported by our
demonstration that a low-dose (50 nM) of NSC predominately
increased the proteolytic PKCδ cleavage in nuclei, while a
high-dose (1 µM) of NSC mainly induced PKCδ cleavage in
mitochondria (Fig. 5). Although
the functional significance of this subcellular differential
activation of PKCδ remains to be elucidated, this is in agreement
with previous demonstrations that PKCδ activation in mitochondria
causes cell apoptosis (28,30),
while its activation in nuclei may induce cell proliferation
(43). Studies have shown that
PKCδ nucleic trans-location is a central step mediating
IGF-I-induced mitogenic and proliferative signaling in primary
human skeletal cells (43), and in
hypoxia-induced cell proliferation and differentiation in human
lung epithelial cells (44). Based
on previous studies and our present data, we propose that the
dose-dependent biphasic effect of NSC on LAPC4 cells is an
integration of cell proliferation and cell apoptosis induced by the
differential subcellular activation of PKCδ as illustrated in
Fig. 7. At low-doses of NSC, PKCδ
activity is predominately elevated in the nucleus, presumably due
to a relatively low-level DNA damage, leading to a more prominent
upregulation of pro-proliferation genes such as cyclin A and
consequently cell growth. On the other hand, PKCδ activation is
dramatically increased in the mitochondrion at high NSC doses,
presumably due to a higher level DNA damage, which leads to a more
prominent activation of the intrinsic apoptotic pathway, outweighs
its proliferative effect, and eventually causes cell death.
Finally, the biological and clinical significance of
differential response to CPT analogs among PCa cells should not be
underestimated. Among the cell lines tested, we have observed that
the order of cell sensitivity to NSC-induced cell death is DU145
> PC3 > LNCaP > LAPC4 (14). DU145 cells are the most sensitive
PCa cells to NSC with a dramatic apoptosis and cell death at
nanomolar concentrations. However, at the same concentration, NSC
and topotecan significantly induced cell viability in LAPC4 cells
(Fig. 1). Moreover, unlike the
molecular alterations observed in LAPC4 cells, NSC neither
activates PKCδ (14), nor alters
cyclin A expression (Fig. 6B) in
DU145 cells. This cell-differential effect suggests that the
response of individual patients to CPT chemotherapy is variable,
which may account, at least in part, for the failure of CPT-related
clinical trials in PCa patients (11–13).
In conclusion, the present study demonstrates that
CPT and its analogs, such as NSC and topotecan, produce a
dose-dependent biphasic effect on cell growth and apoptosis in
LAPC4 PCa cells, which may be mediated through a differential
subcellular activation of PKCδ. This atypical biphasic effect of
CPT analogs has clear cell specificity. The differential response
of different prostate cancer cells to CPT analogs underscores the
importance of identifying specific biomarker(s) associated with
drug sensitivity, which will guide clinical trials and patient
management, leading to an individualized chemotherapy for PCa
patients.
Acknowledgments
We are very grateful to Dr Charles Sawyer (Memorial
Sloan-Kettering Cancer Center) for the LAPC4 cells. This study is
supported in part by grants from the National Institute of Health
(no. UL1-TR000457), the Argenbright Research Fund and the Cohen
Research Fund.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Petrylak DP: The treatment of
hormone-refractory prostate cancer: Docetaxel and beyond. Rev Urol.
8(Suppl 2): S48–S55. 2006.PubMed/NCBI
|
|
3
|
Silvestris N, Leone B, Numico G, Lorusso V
and De Lena M: Present status and perspectives in the treatment of
hormone-refractory prostate cancer. Oncology. 69:273–282. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aziz MH, Dreckschmidt NE and Verma AK:
Plumbagin, a medicinal plant-derived naphthoquinone, is a novel
inhibitor of the growth and invasion of hormone-refractory prostate
cancer. Cancer Res. 68:9024–9032. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Frese S, Schüller A, Frese-Schaper M,
Gugger M and Schmid RA: Cytotoxic effects of camptothecin and
cisplatin combined with tumor necrosis factor-related
apoptosis-inducing ligand (Apo2L/TRAIL) in a model of primary
culture of non-small cell lung cancer. Anticancer Res.
29:2905–2911. 2009.PubMed/NCBI
|
|
6
|
Legarza K and Yang LX: Novel camptothecin
derivatives. In Vivo. 19:283–292. 2005.PubMed/NCBI
|
|
7
|
Srivastava V, Negi AS, Kumar JK, Gupta MM
and Khanuja SP: Plant-based anticancer molecules: A chemical and
biological profile of some important leads. Bioorg Med Chem.
13:5892–5908. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
National Cancer (NC) Institute: NCI Drug
Dictionary. https://www.cancer.gov/publications/dictionaries/cancer-drug.
|
|
9
|
Hörmann V, Kumi-Diaka J, Durity M and
Rathinavelu A: Anticancer activities of genistein-topotecan
combination in prostate cancer cells. J Cell Mol Med. 16:2631–2636.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Minelli R, Cavalli R, Ellis L, Pettazzoni
P, Trotta F, Ciamporcero E, Barrera G, Fantozzi R, Dianzani C and
Pili R: Nanosponge-encapsulated camptothecin exerts anti-tumor
activity in human prostate cancer cells. Eur J Pharm Sci.
47:686–694. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klein CE, Tangen CM, Braun TJ, Hussain MH,
Peereboom DM, Nichols CR, Rivkin SE, Dakhil SR and Crawford ED:
SWOG-9510: evaluation of topotecan in hormone refractory prostate
cancer: a Southwest Oncology Group study. Prostate. 52:264–268.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reese DM, Tchekmedyian S, Chapman Y,
Prager D and Rosen PJ: A phase II trial of irinotecan in
hormone-refractory prostate cancer. Invest New Drugs. 16:353–359.
1998–1999. View Article : Google Scholar
|
|
13
|
Hudes GR, Kosierowski R, Greenberg R,
Ramsey HE, Fox SC, Ozols RF, McAleer CA and Giantonio BJ: Phase II
study of topotecan in metastatic hormone-refractory prostate
cancer. Invest New Drugs. 13:235–240. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan C, Cai LQ, Wu W, Qiao Y,
Imperato-McGinley J, Chen GQ and Zhu YS: NSC606985, a novel
camptothecin analog, induces apoptosis and growth arrest in
prostate tumor cells. Cancer Chemother Pharmacol. 63:303–312. 2009.
View Article : Google Scholar
|
|
15
|
Albihn A, Mo H, Yang Y and Henriksson M:
Camptothecin- induced apoptosis is enhanced by Myc and involves
PKCdelta signaling. Int J Cancer. 121:1821–1829. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song MG, Gao SM, Du KM, Xu M, Yu Y, Zhou
YH, Wang Q, Chen Z, Zhu YS and Chen GQ: Nanomolar concentration of
NSC606985, a camptothecin analog, induces leukemic-cell apoptosis
through protein kinase Cdelta-dependent mechanisms. Blood.
105:3714–3721. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parker PJ and Murray-Rust J: PKC at a
glance. J Cell Sci. 117:131–132. 2004. View Article : Google Scholar
|
|
18
|
Gonzalez-Guerrico AM and Kazanietz MG:
Phorbol ester-induced apoptosis in prostate cancer cells via
autocrine activation of the extrinsic apoptotic cascade: A key role
for protein kinase C delta. J Biol Chem. 280:38982–38991. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yin L, Bennani-Baiti N and Powell CT:
Phorbol ester-induced apoptosis of C4-2 cells requires both a
unique and a redundant protein kinase C signaling pathway. J Biol
Chem. 280:5533–5541. 2005. View Article : Google Scholar
|
|
20
|
Jackson DN and Foster DA: The enigmatic
protein kinase Cdelta: Complex roles in cell proliferation and
survival. FASEB J. 18:627–636. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basu A and Pal D: Two faces of protein
kinase Cδ: The contrasting roles of PKCδ in cell survival and cell
death. Sci World J. 10:2272–2284. 2010. View Article : Google Scholar
|
|
22
|
Steinberg SF: Distinctive activation
mechanisms and functions for protein kinase Cdelta. Biochem J.
384:449–459. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Curtis MJ, Bond RA, Spina D, Ahluwalia A,
Alexander SP, Giembycz MA, Gilchrist A, Hoyer D, Insel PA, Izzo AA,
et al: Experimental design and analysis and their reporting: New
guidance for publication in BJP. Br J Pharmacol. 172:3461–3471.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Afzal O, Kumar S, Haider MR, Ali MR, Kumar
R, Jaggi M and Bawa S: A review on anticancer potential of
bioactive heterocycle quinoline. Eur J Med Chem. 97:871–910. 2015.
View Article : Google Scholar
|
|
25
|
Zhang N, Zhang H, Xia L, Zheng Y, Yu Y,
Zhu Y, Chen G and Di W: NSC606985 induces apoptosis, exerts
synergistic effects with cisplatin, and inhibits hypoxia-stabilized
HIF-1alpha protein in human ovarian cancer cells. Cancer Lett.
278:139–144. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gavrielides MV, Gonzalez-Guerrico AM,
Riobo NA and Kazanietz MG: Androgens regulate protein kinase Cdelta
transcription and modulate its apoptotic function in prostate
cancer cells. Cancer Res. 66:11792–11801. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao M, Xia L and Chen GQ: Protein kinase
cδ in apoptosis: A brief overview. Arch Immunol Ther Exp (Warsz).
60:361–372. 2012. View Article : Google Scholar
|
|
28
|
Majumder PK, Pandey P, Sun X, Cheng K,
Datta R, Saxena S, Kharbanda S and Kufe D: Mitochondrial
translocation of protein kinase C delta in phorbol ester-induced
cytochrome c release and apoptosis. J Biol Chem. 275:21793–21796.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li L, Lorenzo PS, Bogi K, Blumberg PM and
Yuspa SH: Protein kinase Cdelta targets mitochondria, alters
mitochondrial membrane potential, and induces apoptosis in normal
and neoplastic keratinocytes when overexpressed by an adenoviral
vector. Mol Cell Biol. 19:8547–8558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sumitomo M, Ohba M, Asakuma J, Asano T,
Kuroki T, Asano T and Hayakawa M: Protein kinase Cdelta amplifies
ceramide formation via mitochondrial signaling in prostate cancer
cells. J Clin Invest. 109:827–836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toyoda M, Gotoh N, Handa H and Shibuya M:
Involvement of MAP kinase-independent protein kinase C signaling
pathway in the EGF-induced p21(WAF1/Cip1) expression and growth
inhibition of A431 cells. Biochem Biophys Res Commun. 250:430–435.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mauro LV, Grossoni VC, Urtreger AJ, Yang
C, Colombo LL, Morandi A, Pallotta MG, Kazanietz MG, Bal de Kier
Joffé ED and Puricelli LL: PKC Delta (PKCdelta) promotes tumoral
progression of human ductal pancreatic cancer. Pancreas.
39:e31–e41. 2010. View Article : Google Scholar
|
|
33
|
Clark AS, West KA, Blumberg PM and Dennis
PA: Altered protein kinase C (PKC) isoforms in non-small cell lung
cancer cells: PKCdelta promotes cellular survival and
chemotherapeutic resistance. Cancer Res. 63:780–786.
2003.PubMed/NCBI
|
|
34
|
Datta K, Nambudripad R, Pal S, Zhou M,
Cohen HT and Mukhopadhyay D: Inhibition of insulin-like growth
factor-I-mediated cell signaling by the von Hippel-Lindau gene
product in renal cancer. J Biol Chem. 275:20700–20706. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kiley SC, Clark KJ, Duddy SK, Welch DR and
Jaken S: Increased protein kinase C delta in mammary tumor cells:
Relationship to transformtion and metastatic progression. Oncogene.
18:6748–6757. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McKiernan E, O'Brien K, Grebenchtchikov N,
Geurts-Moespot A, Sieuwerts AM, Martens JW, Magdolen V, Evoy D,
McDermott E, Crown J, et al: Protein kinase Cdelta expression in
breast cancer as measured by real-time PCR, western blotting and
ELISA. Br J Cancer. 99:1644–1650. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCracken MA, Miraglia LJ, McKay RA and
Strobl JS: Protein kinase C delta is a prosurvival factor in human
breast tumor cell lines. Mol Cancer Ther. 2:273–281.
2003.PubMed/NCBI
|
|
38
|
Kumar V, Pandey P, Sabatini D, Kumar M,
Majumder PK, Bharti A, Carmichael G, Kufe D and Kharbanda S:
Functional interaction between RAFT1/FRAP/mTOR and protein kinase
cdelta in the regulation of cap-dependent initiation of
translation. EMBO J. 19:1087–1097. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
De Boer L, Oakes V, Beamish H, Giles N,
Stevens F, Somodevilla-Torres M, Desouza C and Gabrielli B: Cyclin
A/cdk2 coordinates centrosomal and nuclear mitotic events.
Oncogene. 27:4261–4268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ariens EJ, Simonis AM and Van Rossum JM:
Theory and experiments concerning the effects of drugs. Pharm
Weekbl. 91:617–635. 1956.In Dutch. PubMed/NCBI
|
|
41
|
Szeto HH, Zhu YS, Umans JG, Dwyer G, Clare
S and Amione J: Dual action of morphine on fetal breathing
movements. J Pharmacol Exp Ther. 245:537–542. 1988.PubMed/NCBI
|
|
42
|
Zhu YS and Szeto HH: Morphine-induced
tachycardia in fetal lambs: A bell-shaped dose-response curve. J
Pharmacol Exp Ther. 249:78–82. 1989.PubMed/NCBI
|
|
43
|
Czifra G, Tóth IB, Marincsák R, Juhász I,
Kovács I, Acs P, Kovács L, Blumberg PM and Bíró T: Insulin-like
growth factor-I-coupled mitogenic signaling in primary cultured
human skeletal muscle cells and in C2C12 myoblasts. A central role
of protein kinase Cdelta. Cell Signal. 18:1461–1472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang H, Okamoto M, Panzhinskiy E, Zawada
WM and Das M: PKCδ/midkine pathway drives hypoxia-induced
proliferation and differentiation of human lung epithelial cells.
Am J Physiol Cell Physiol. 306:C648–C658. 2014. View Article : Google Scholar : PubMed/NCBI
|