Introduction
Circular RNAs (circRNAs) are a recently rediscovered
class of non-coding RNA (1,2).
They are formed by backsplicing events that involve a downstream 3'
splice donor site joining an upstream 5' splice acceptor site in
the primary transcript. Since the discovery of the first two
circular RNAs (DCC in humans and SRY in mice) in the 1990s
(3,4), numerous circRNAs have been identified
in silico and validated in experiments (1,2).
CircRNAs are remarkably stable, conserved, highly abundant and
predominantly cytoplasmic (2).
They are generated through several distinct mechanisms that rely on
complementary sequences within flanking introns (2,5,6),
exon skipping (6,7), and exon-containing lariat precursors
(8). CircRNA expression is
regulated by an RNA editing enzyme or RNA binding proteins, such as
ADAR (6) and Quaking (9). Similar to linear RNA, circRNAs are
generated from exons or introns at canonical splice sites and
require typical spliceosomal machinery (10–12).
Computational biologists have developed several
alignment algorithms to identify circRNAs using RNA-seq data. Two
main approaches are used to detect circRNAs. One approach uses the
annotated genome to build a reference scrambled exon-exon junction
database. The scrambled exome includes all possible pairs of
intragenic exons in a non-canonical order and the circularization
of a single exon. The backsplice junction reads are aligned
contiguously along their full length to databases, including KNIFE
and other pipelines (13,14). The second strategy improves the
alignment algorithms and the pipeline, identifying the backsplice
aligned reads to the genome or transcriptome, and examples include
mapsplice, find_circ, segemehl, circExplorer, circRNA_finder, CIRI,
DCC and acfs (5,15–21).
These algorithms differ in accuracy and sensitivity, and there is
little overlap in their predictions (22).
Several studies have revealed that circRNAs are
substantially enriched in the brain tissues of humans and mice. Of
note, the expression levels of circRNAs are dynamic during brain
development and are independent of the linear transcript that
originates from the same gene locus (23–25).
In epithelial ovarian carcinoma, circRNAs display altered
expression patterns between primary ovarian tumors and metastatic
tumors (14). In heart-specific
circRNA candidates, there is a lack of differential expression of
circRNAs between normal and diseased human heart (26). CircRNAs may also have an impact on
aging and multiple disorders. CDR1as (ciRS-7) can serve as a miRNA
'sponge', arresting miR-7 function. In addition, miR-7 is a vital
regulatory miRNA in Parkinson's disease (16). Genome-wide association studies
linked a newly identified circRNA species, called cANRIL, with
atherosclerosis risk (27).
To systematically investigate the intra- and
inter-individual variation in circRNA expression profiles and the
role of circRNAs in humans, we collected a large set of total
RNA-seq data from NCBI Sequence Read Archive and ENCODE (28,29).
A pipeline named RAISE (circRNA ReAlign Internal Structure and
Expression) was developed to analyze circRNA candidates in these
samples. Using RAISE, we identified 59,128 circRNA candidates in
HCC and adjacent non-tumor tissues. Only a small portion of
circRNAs is universally expressed in the recruited HCC samples. The
expression of circRNAs in HCC shows inter-individual variations. In
advance, we estimated whether circRNA expression varies in other
tissues, especially in circRNA high-abundance tissues. Similar to
liver cases, only 0.5% of the 50,631 brain circRNA candidates are
shared among the 30 recruited brain samples. Moreover, we found
inter- and intra-individual diversity in circRNA expression in
granulocyte RNA-seq data from seven individuals sampled 3 times at
one-month intervals. Our results suggest that the majority of
circRNAs exhibit inter-and intra-individual variations. When
proposing variable circRNAs that are naturally highly expressed as
prognostic markers, it is necessary to collect a sufficient number
of individuals to confirm that these circRNAs are robustly
expressed in humans.
Materials and methods
RNA-seq datasets
We downloaded publicly available human and mouse
RNA-seq data set samples from NCBI SRA (28) and ENCODE (29). Human samples included
hepatocellular carcinoma (accession no. GSE65485) (30), granulocyte samples (accession no.
GSE70390) (31), brain (accession
nos. GSE53697 and GSE71315) (32,33).
Mouse samples included liver and brain (accession no. PRJEB5489)
(Table I).
 | Table ISummary of RNA-seq data set. |
Table I
Summary of RNA-seq data set.
| Accession no. | Liver | Brain | Granulocytes | Other | Total | Description |
|---|
| GSE65485 | 55 | 0 | 0 | 0 | 55 | 50 HCC and 5
adjacent non-tumor |
| GSE77661 | 4 | 2 | 0 | 20 | 26 | In 4 liver samples,
2 are normal, 1 is HCC, 1 is HCC adjacent non-tumor tissue, 2
heart |
| GSE73570 | 0 | 0 | 0 | 6 | 6 | Blood 1 repeat |
| encode | 2 | 3 | 0 | 38 | 43 | 2 liver sample, 3
brain samples |
| GSE53697 | 0 | 17 | 0 | 0 | 17 | 8 control, 9
Alzheimer's diseased |
| GSE71315 | | 16 | 0 | 0 | 16 | 8 ribozero total
RNA, 8 polyA+ RNA |
| PRJEB5489 | 12 | 12 | 0 | 0 | 24 | Mouse normal |
| GSE70390 | 0 | 0 | 21 | 0 | 21 | Human granulocytes
7x3 |
RAISE: the workflow of circRNA
identification
RAISE is a pipeline designed as a shell script to
run after circRNA backsplice sites have been identified. This
pipeline can identify circRNA internal structures and alternative
exon usage. In this study, we combined four circRNA prediction
algorithms, including mapsplice (2,15,34),
find_circ (16), acfs (20,24),
and circRNA_finder (18). The main
steps of RAISE are briefly described in Fig. 1. The first step is to obtain the
unmapped reads. RNA-seq reads were aligned to the reference genome
and transcriptome by hisat2, filtering contiguous and canonical
splice reads. Meanwhile, circRNA_finder was employed to identify
circRNAs. The second step is applying mapsplice, acfs and find_circ
to detect circRNAs from unmapped reads by combining the three tools
and the circRNA_finder results. The third step is to extract the
genomic region sequence of the circRNA backsplice site, tandemly
duplicating this sequence to create a pseudo circRNA reference. The
fourth step is to obtain backsplice junction site coverage and
depth. The step 1 unmapped reads are realigned to the pseudo
circRNA reference by hisat2. The fifth step is conducting
paired-end read analysis with one read aligned to the backsplice
site. If the mate reads are mapped to the circRNA region, they
could be circRNA paired-end reads. If the mate reads are mapped out
to a range of circRNA regions, they could be decoys. Then, the
proper mapped paired-end reads and decoy reads were counted. The
sixth step is to predict the circRNA transcript based on the proper
mapped paired-end reads. With one read aligned to the backsplice
site, the mate alignment read contains the circRNA inner splice
site, which is used to detect circRNA internal structure and exon
usage. The alternative step is to detect the support of circRNAs
paired-end reads. There are several linear discordant paired-end
alignment reads within the genomic region of the circRNA backsplice
site that could be the potentially supported reads of circRNA
candidates (Fig. 1). This pipeline
is available as git repository: https://github.com/liaoscience/RAISE.
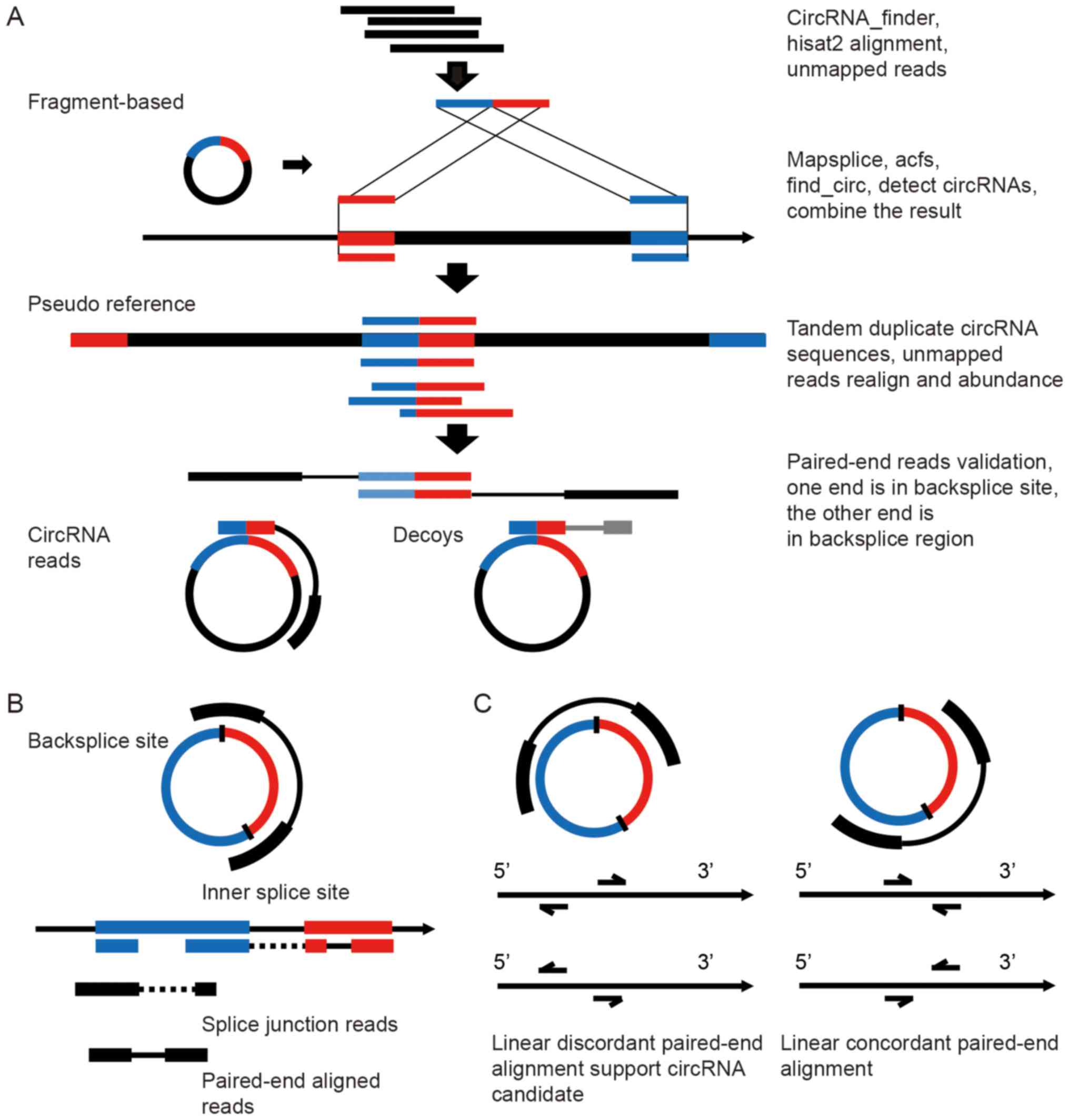 | Figure 1The RAISE workflow for the
identification and quantification of circRNA candidates. (A) The
pipeline of circRNA detection: 1, Hisat2 alignment and filtering of
mapped reads, STAR alignment and circRNA_finder identification. 2,
Detection of circRNAs from unmapped reads using mapsplice, acfs and
find_circ tools, and merger of their outputs with the
circRNA_finder results. 3, Extraction of the genomic region
corresponding to the circRNA backsplice site from the previous four
tools' output, tandem duplication of the sequences to create a
pseudo circRNA reference, realignment of the unmapped reads to the
pseudo reference, and estimation of the abundance of circRNA
candidates. 4, From the paired-end data, if one read is aligned to
a circRNA backsplice site and if the mate read is aligned to the
circRNA region, these paired-end reads are classified among circRNA
reads, whereas if the mate read is aligned outside the circRNA
backsplice site, the paired-reads are classified as decoys. (B)
Prediction of circRNA internal exon usage by paired-end and splice
junction reads. (C) Discordant linear alignment reads that could be
potential circRNA candidate paired-end reads. |
CircRNA annotation and
quantification
CircRNA annotation is based on the Gencode (35) human genome (v38) and mouse genome
(v10). We intersected the circRNA donator/acceptor site to annotate
gene regions, including coding RNA, non-coding RNA, intron,
antisense and intergenic regions, with the BEDTools suite (v2.16.2)
(36). The number of reads aligned
to the circRNA-specific head-to-tail junctions was used as a
measurement of circRNAs expression. Normalization with circRNA
Spliced backsplice Reads Per Billion Mapped Reads (SRPBM) in each
library was performed to enable the comparison of relative
expression among samples (37).
Circular to linear ratios were calculated using the backsplice
reads of circRNAs by dividing the mean value of reads that span the
linear splice junction reads, including the left and right sides of
the circRNA splice sites (38).
Mapping and quantification of linear mRNA
and lncRNA expression
Sequencing quality was assessed by FASTQC (39). After removing adaptor and
low-quality reads using cutadapt (40) (-q 10 -e 0.1 -O 10 -m 50), the clean
reads were aligned to the human (hg38) genome reference sequences
using hisat2 (41) with the
default parameters. The bam files were generated, sorted, and
deduplicated using SAMTOOLS (v1.3) (42). Read counts were tabulated with
HT-Seq (43) in 'union' mode with
the Gencode human v24 GTF file as a reference. Stringtie (v1.3)
(44) was also used to estimate
the total transcriptional output based on the Gencode human gene
annotation (HG38 version 24) (35).
Gene Ontology enrichment analysis
Gene ontology (GO) term enrichment analysis was
performed using DAVID (45), by
inputting the list of circRNA derived host locus genes.
Statistical analysis
The raw counts were first normalized using trimmed
mean of M-values (TMM). Differential circRNA or gene expression was
estimated using the edgeR package, and a negative binomial model
was used to estimate differential expression between tumor and
adjacent non-tumor tissues (FDR <0.02, 2-fold change) (46). Statistical analyses were performed
using R 3.3.1 (http://www.r-project.org/).
Results
RAISE: a cocktail of circRNA analysis
pipeline
Numerous computational pipelines use backsplice
reads to identify circRNAs; however, there is little overlap in
these circRNA detection methods. Each algorithm has bias and 'blind
spots', so we combined several different read aligners to identify
more circRNAs and increase the robustness of circRNA identification
(22). Previous algorithms detect
circRNA backsplice sites, but they do not include internal
structure information. We combined four available circRNA detection
algorithms (mapsplice, acfs, circRNA_finder, and find_circ) to
develop an integrated pipeline called RAISE to improve the
prediction accuracy and detect the internal exon usage of circRNAs
(Fig. 1). RAISE is an easy-to-use
shell script pipeline. The four selected tools chosen are based on
previous reviews and research (22,34).
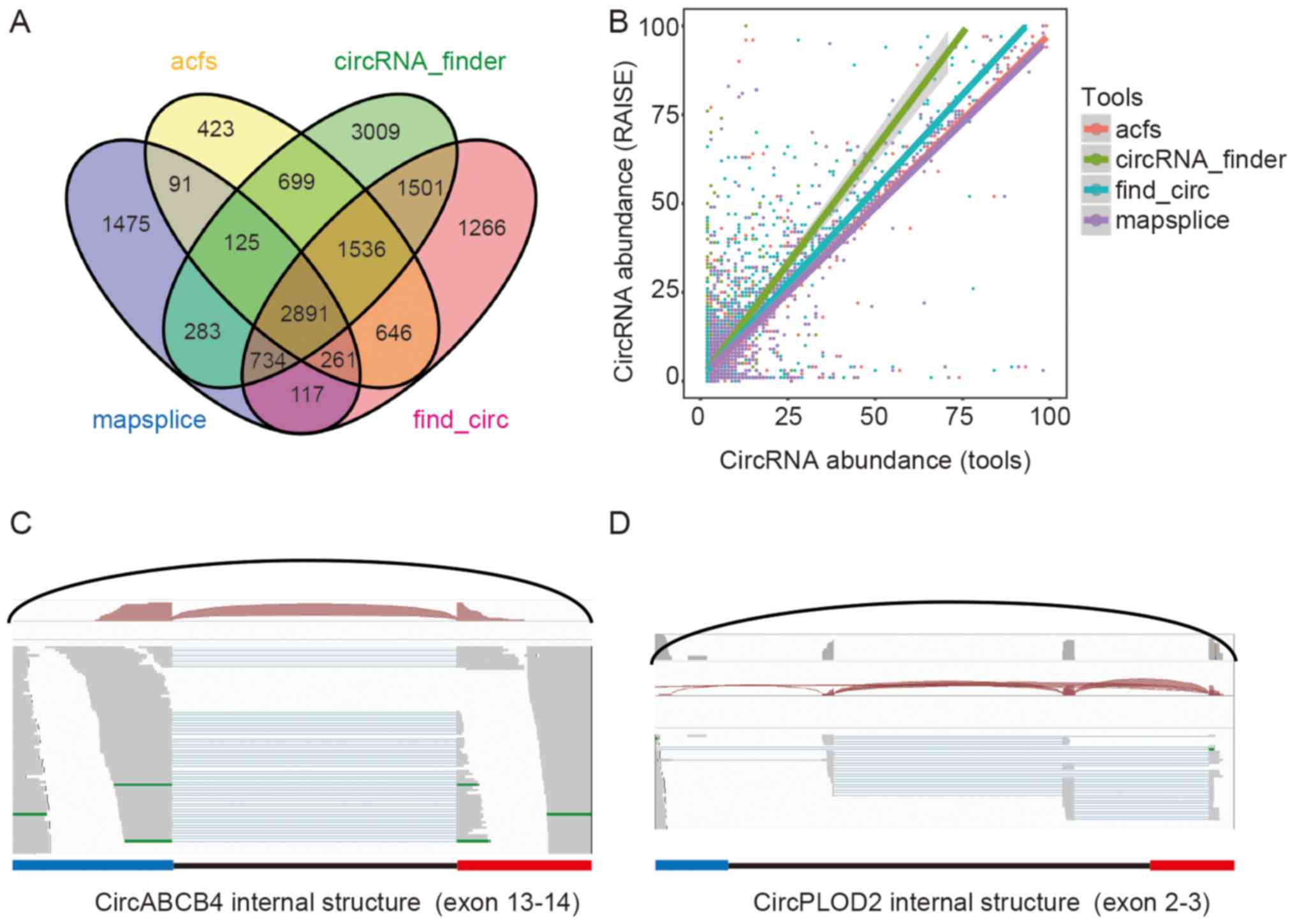
We tested RAISE on human liver rRNA depleted samples
from ENCODE. In this RNA-seq library, 5,977 circRNA candidates were
detected by mapsplice, 6,672 circRNA candidates by acfs, 10,778
circRNA candidates by circRNA_finder, and 8,952 circRNA candidates
by find_circ. There were 2,891 circRNA candidates that were
detected by all four tools. After application of the RAISE
pipeline, 14,145 circRNA candidates were detected, and when we
filtered out candidates with less than two backsplice reads, there
were 8,270 circRNA candidates for advanced analysis. We compared
the abundance of circRNA candidates between these tools and RAISE.
The abundance in acfs and mapsplice was close to that of RAISE,
whereas the abundance in find_circ and circRNA_finder was less than
that in RAISE (Fig. 2A and B).
Furthermore, the internal exon usage of 3,052 high-abundance
circRNA candidates was predicted. For example, the exon composition
of circABCB4 contains exon 13 and exon 14 of the ABCB transcript,
with one read aligned to the backsplice sites and the mate
alignment read containing cis-junction splice information, which is
consistent with the linear RNA junction site (Fig. 2C). A total of 1,255 alternative
splice sites were also detected in the library; for example,
circPLOD2 was derived from exons 2 and 3 of the PLOD2 gene, and
there were two cis-splice junctions in intron 2 (Fig. 2D). Exon usage was different with
the host locus linear RNAs.
CircRNAs display only minor alterations
in expression in HCC and adjacent non-tumor tissue
In order to investigate the circRNA expression
profile in HCC, we collected 61 human liver samples. All of the
RNA-seq data are non-poly(A)-selected, and are downloaded from NCBI
and ENCODE, GSE65485 included 50 HCC samples and 5 adjacent
non-tumor tissues; GSE77661 included 4 liver samples; ENCODE
included 2 liver samples (Table
I). These data included 51 HCC samples, 6 adjacent non-tumor
samples and 4 normal liver samples. The sequencing depths of these
samples ranged from 29.3 to 122.3 million reads. Approximately 95%
of these reads were aligned to the human reference genome (hg38).
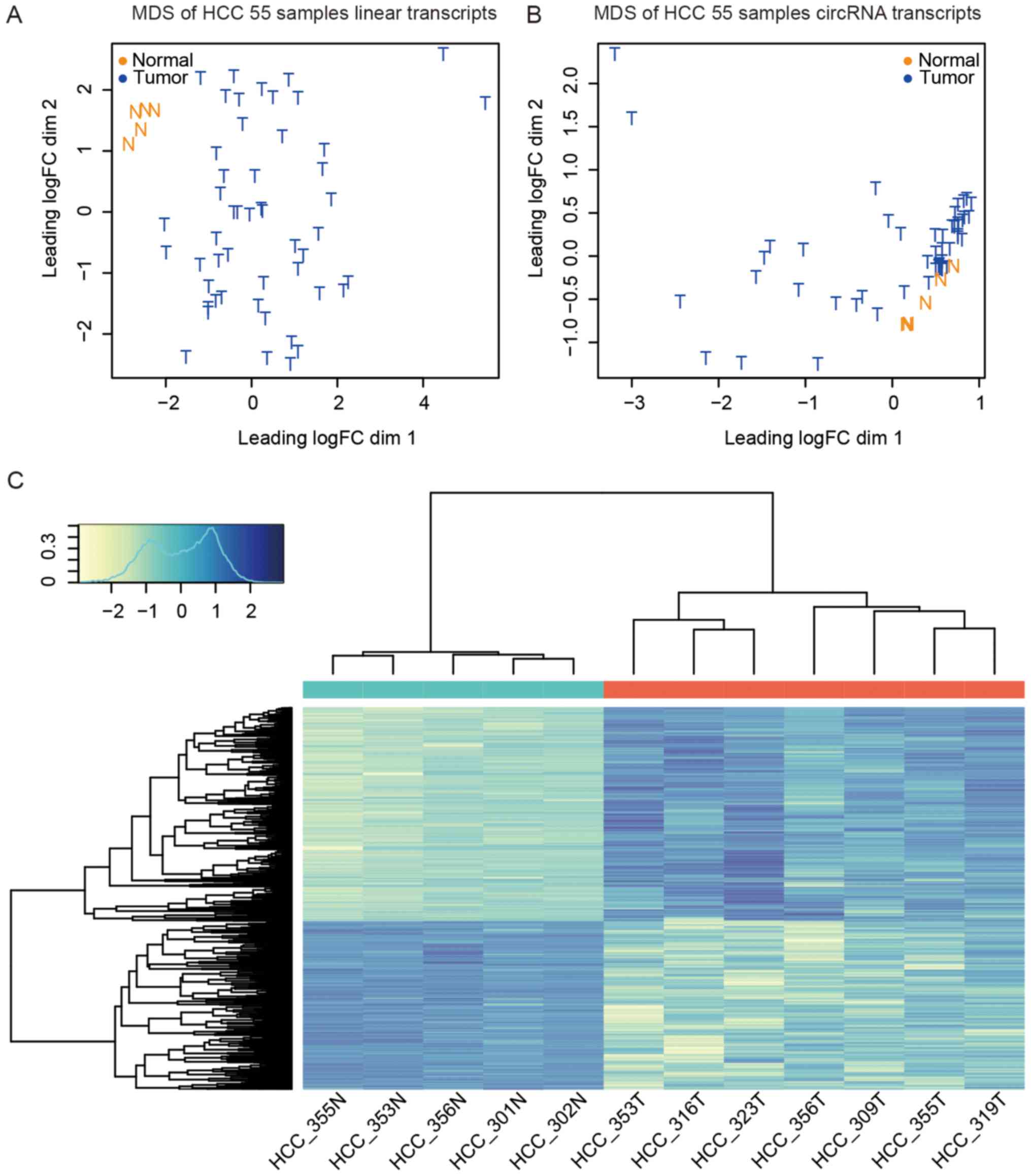
Multidimensional scaling (MDS) (47) analysis showed that the HCC samples
were distinct from normal and adjacent non-cancerous tissue samples
(Fig. 3A).
The RAISE pipeline was applied on the unmapped reads
(5%), and 59,128 distinct circRNA candidates were identified in 61
liver samples. We chose seven HCC samples and five adjacent
non-tumor samples (unpaired samples) to compare the expression
patterns of circRNAs between tumor and adjacent non-tumor tissues.
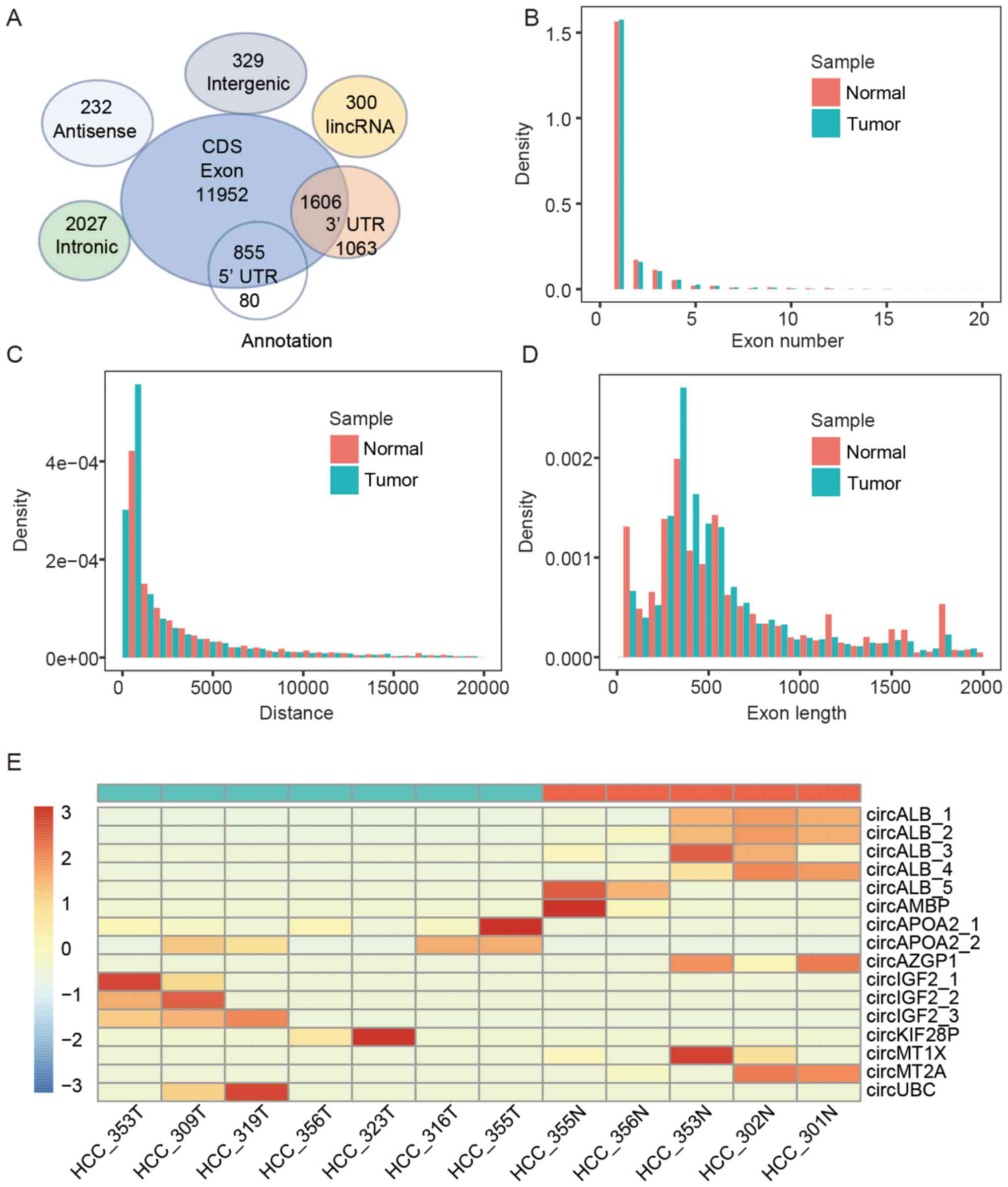
First, we found that 80% of the circRNA candidates in these 12 HCC
samples were derived from the protein-coding exonic regions while
other smaller fractions were antisense, long non-coding RNAs,
intergenic regions and intronic regions (Fig. 4A). The genomic features of these
circRNAs, e.g., genomic origins, exon numbers, exon length and
genomic distance, were compared between HCC and adjacent non-tumor
tissue. The exon numbers of most circRNAs were less than five. The
length of most exonic circRNAs was ~300–500 nt with a genomic
distance of ~1,000–3,000 bp. There was no significant difference in
the genomic features between HCC and adjacent non-tumor tissue
(Fig. 4B–D).
Next, we inquired whether liver circRNAs are
differentially expressed between tumor and adjacent non-tumor
tissues. Unlike mRNAs and lncRNAs, hundreds of linear RNA genes are
significantly differentially expressed in these two groups. We did
not detect any differentially expressed circRNA candidates with
statistical significance (Fig.
4E). The circRNA circALB (chr4:73405119-73408712) from exon 2–4
of the ALB gene was in high abundance in adjacent non-tumor tissue,
but it was not detected in 84% of a total of 50 HCC samples. Of
note, ALB mRNA was highly abundant in the liver samples.
CircRNAs show inter- and intra-individual
expression diversity
In addition to assessing circRNA expression patterns
in HCC, we turned our attention to the liver-specific expression of
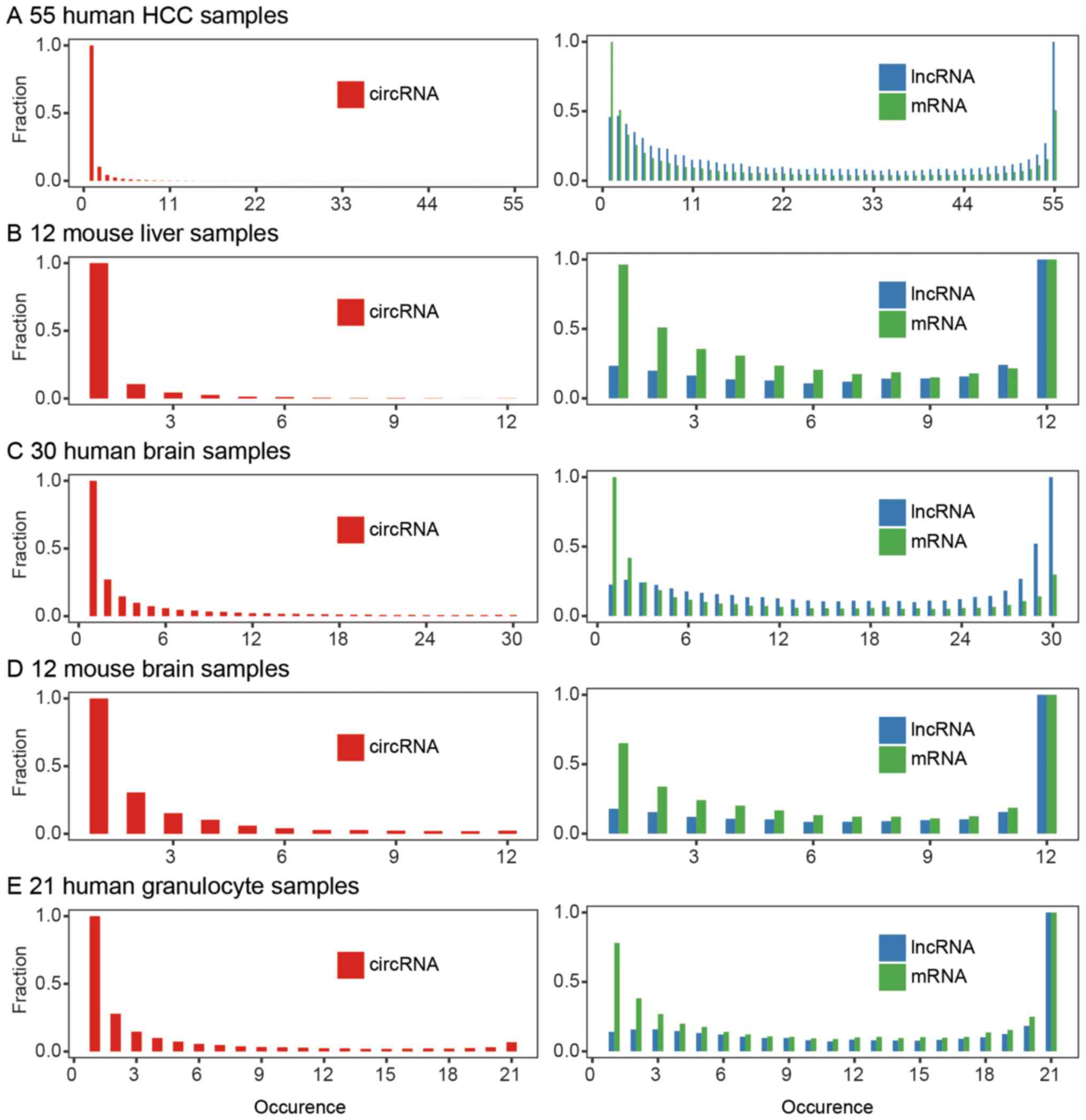
circRNAs. We calculated the occurrence of each circRNA in these
samples to discover the shared circRNAs and unique circRNAs. For
example, circAPOA2 was detected in 28 HCC samples; hence, its
occurrence was 28. In 55 HCC samples, the occurrence of circRNAs
ranged from 1 to 55. We analyzed the total detected circRNA
candidates and linear gene occurrences. Unlike linear RNAs, ~10% of
the protein-coding RNAs and non-coding RNAs were expressed in all
55 HCC samples, whereas almost no detected circRNAs were shared by
the 55 samples (Fig. 5A).
Consequently, a single gene locus can transcribe multiple circular
isoforms (48,49). We asked whether the diversity among
the samples is due to variations in circular RNA isoform selection
within the gene locus. We set the circRNAs derived from the same
transcript as 'transcript circRNA' and those from the same gene
locus as 'gene circRNA'. For example, there are 329 circRNA
isoforms in the ALB gene locus; we set these circRNAs as circALB.
Furthermore, we compared the diversity in gene-and transcript-level
circular RNA expression. Seven of the 16,133 transcript circRNAs
and 9 of the 9,696 gene circRNAs were shared among the 55 HCC
samples (data not shown). These nine genes expressed circRNAs in
all the samples independently of the circRNA backsplice sites. The
ratio of transcript to gene circRNAs remained low in contrast to
the ratio of transcript to linear RNAs. To test whether a similar
expression pattern exists in mice, we downloaded 12 mouse liver
RNA-seq datasets from NCBI SRA, analyzed them with the same
pipeline (Table I), and identified
3,801 circRNA candidates in these samples. Only 0.1% of circRNAs
were shared in the mouse liver samples, i.e., 0.1% of the
transcripts as well as 0.1% of the gene circRNAs were shared in
each sample. This result is consistent with the human liver circRNA
expression profile (Fig. 5B).
We then asked whether inter-individual circRNA
expression diversity is also common in other human tissues. Since
circRNAs are highly abundant in the brain (data not shown)
(24), we downloaded several
batches of brain ribosomal RNA depleted RNA-seq data from NCBI SRA
and analyzed the brain circRNA expression profile with our pipeline
(Table I). We identified 50,631
circRNA candidates in 30 human brain samples and found ~0.5% shared
in each brain sample (Fig. 5C).
The same pipeline was used on the mouse brain samples, and its
results showed similarities to those of the human brain (Fig. 5D). Circular RNAs display greater
variation than linear genes in both brain and liver.
CircRNA expression profiles are varied and diverse
between individuals, whether or not circRNAs are reproducibly
expressed within one individual. We downloaded 21 human granulocyte
ribo-zero RNA-seq datasets from NCBI SRA (Table I). The data came from seven healthy
individuals, with three samples at least one month apart (31). We used the previous pipeline to
identify circRNAs and found that 3% were shared among these 21
samples. Within the individuals, circRNAs showed less reproducible
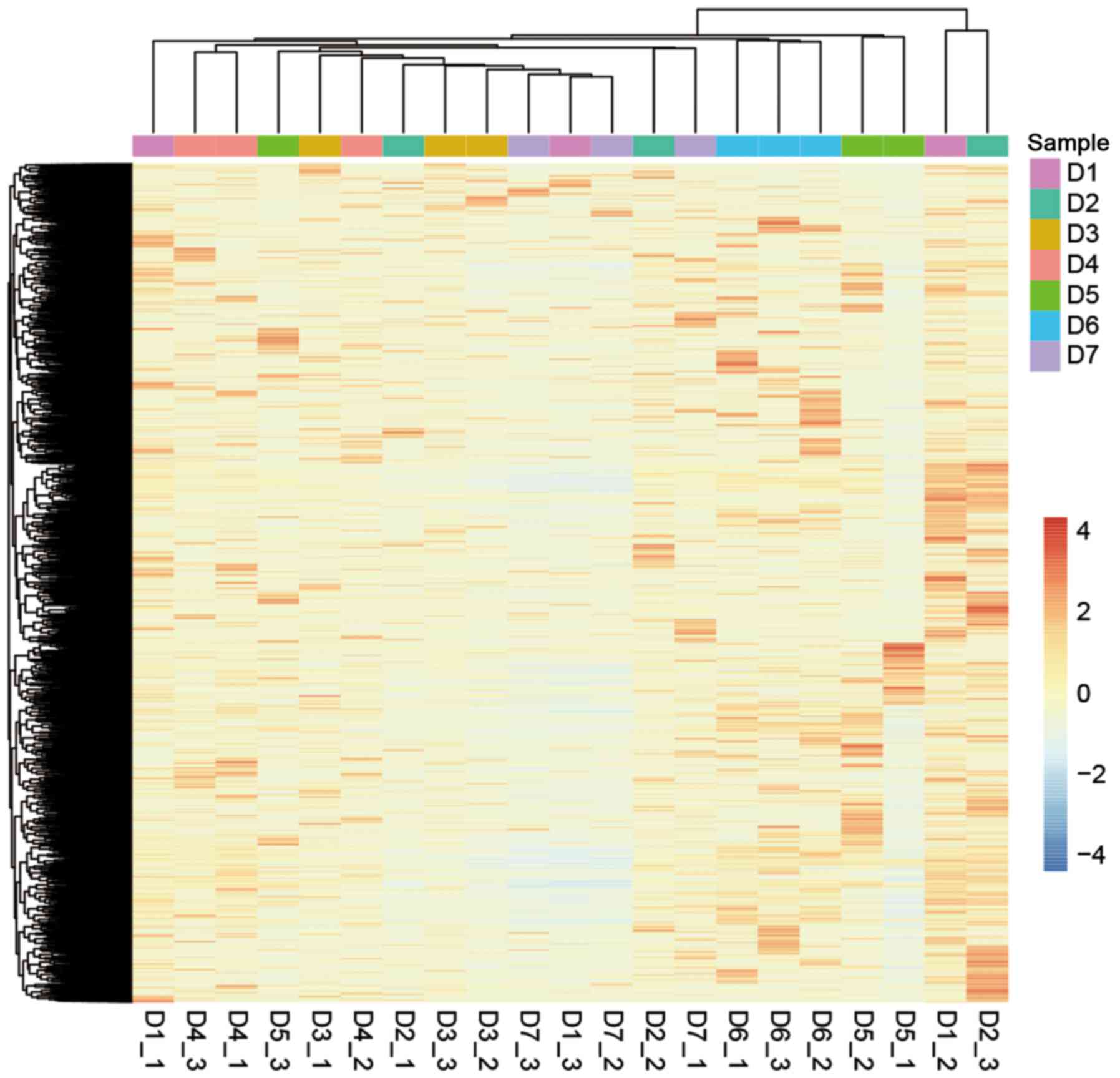
expression than linear RNAs between different individuals (Fig. 5E and Table II). The unsupervised hierarchical
clustering of the circRNA expression profiles of the 21 samples
displayed inconsistent results within each donor (Fig. 6).
| Table IICircRNA distribution in different
tissues. |
Table II
CircRNA distribution in different
tissues.
| Specie tissue | Human brain | Mouse brain | Human liver | Mouse liver | Human
granulocytes |
|---|
| Samples number | 30 | 12 | 61 | 12 | 21 |
| CircRNA
candidates | 50,631 | 7,124 | 59,128 | 3,801 | 31,063 |
| Prediomant | 5,017 | 516 | 2,743 | 244 | 3,893 |
| Alter splice | 3,042 | 182 | 5,228 | 55 | 1,528 |
| Shared
(>20%) | 8,910 | 1,983 | 469 | 353 | 8,509 |
| Shared
prediomant | 405 | 157 | 8 | 19 | 984 |
| Shared alter
splice | 232 | 33 | 3 | 4 | 268 |
| Unique
(<20%) | 41,721 | 5,141 | 58,659 | 3,448 | 22,178 |
| Unique
prediomant | 4,612 | 359 | 2,735 | 225 | 2,909 |
| Unique alter
splice | 2,869 | 156 | 5,224 | 55 | 1,318 |
| Transcript
circRNA | 19,544 | 4,177 | 20,530 | 2,788 | 12,809 |
| Co-exist | 8,086 | 1,695 | 8,516 | 1,291 | 4,584 |
| Shared
co-exist | 2,082 | 634 | 333 | 192 | 1,849 |
In short, for the tissues with low circRNA
abundance, e.g., liver, the ratio of shared circRNAs was lower. For
the tissues with high circRNA abundance, e.g., brain and
granulocytes, the ratio increased.
Shared circRNAs are highly abundant and
are derived from circRNA hotspot gene loci
We further investigated the characteristics of
shared circRNAs and unique circRNAs. We observed that with an
increase in the occurrence of circRNAs, their abundance also
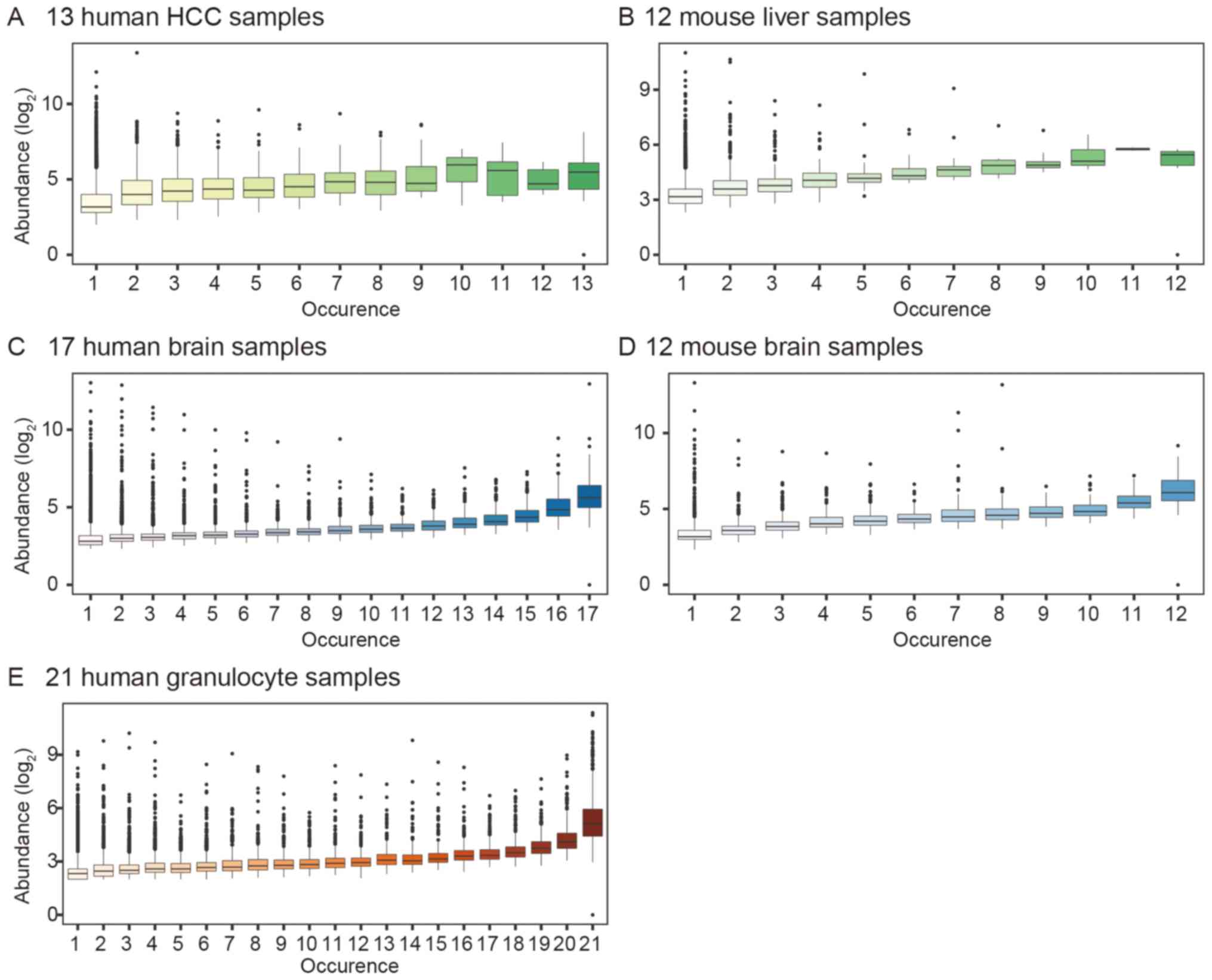
increased in all the studied samples. In 13 HCC and 17 brain
samples, the abundance of the highest occurring circRNA was 5 times
more than that of the lowest occurring circRNA. We also analyzed
the human granulocyte dataset and found that its circRNA expression
profile was similar to that of the human brain samples (Fig. 7).
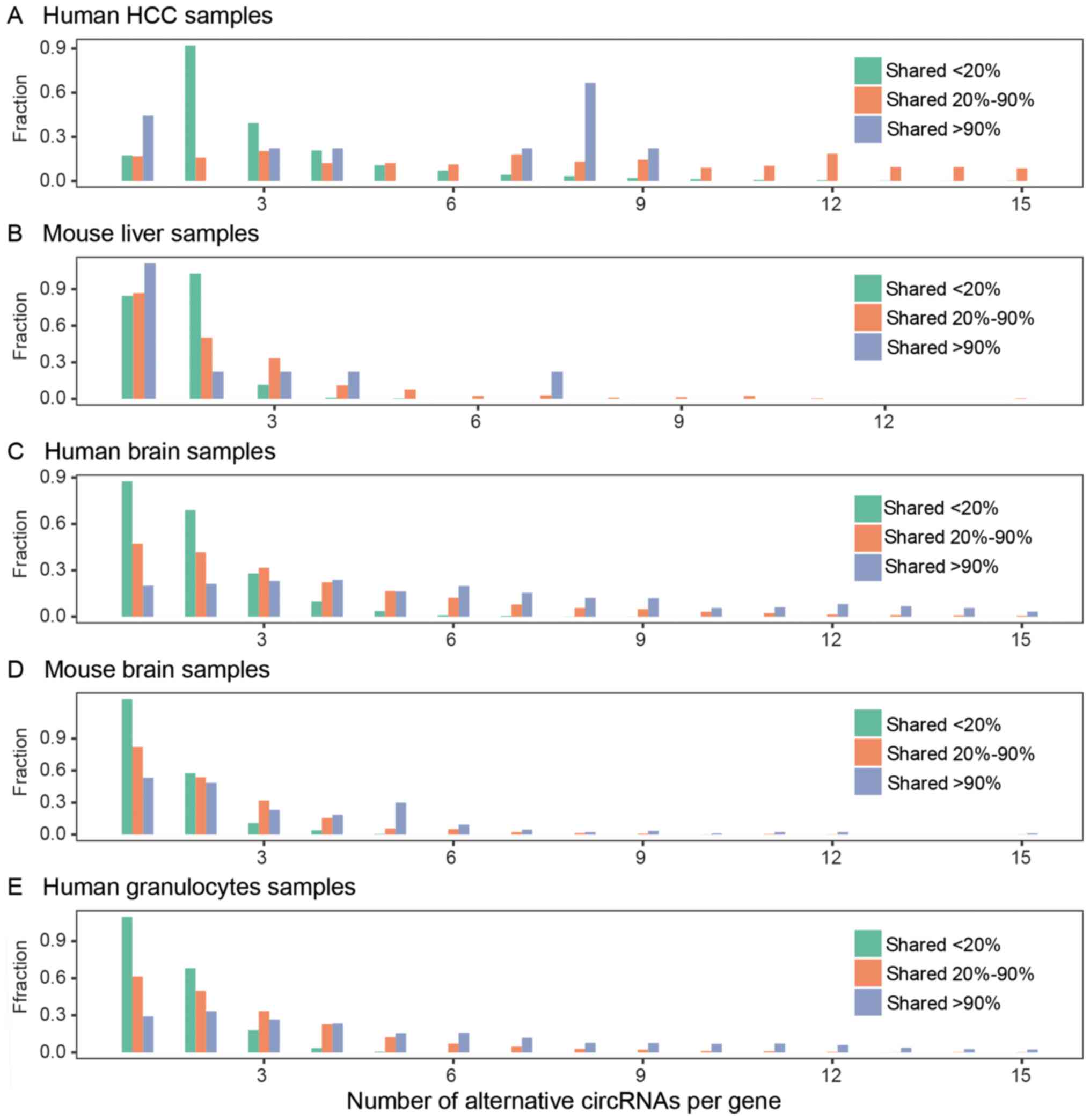
Then, we discovered that a single gene locus can
produce multiple circRNAs. We investigated whether the different
occurrences of gene circRNAs corresponded to different numbers of
circular isoforms. Based on the occurrence of the circRNA gene loci
in the recruited samples, these circRNA gene loci were assigned to
one of three categories: shared 20%, shared 20–90%, and shared 90%.
Shared 20% are the gene loci of expressed circRNAs in <20% of
the recruited samples. Shared 20–90% are the gene locus-expressed
circRNAs in >20% and <90% of the recruited samples. Shared
90% are the gene locus-expressed circRNAs in >90% of recruited
samples. Most of the shared 20% circRNA gene loci have one or two
circRNA isoforms. Shared 90% circRNA gene loci have multiple
circRNA isoforms. A total of 230 of these gene loci gave rise to
>10 circRNAs in human granulocytes (Fig. 8). These gene loci are circRNA
hotspot gene loci (38). The
shared 90% circRNA loci have not only more distinct circRNA
isoforms but also highly abundant circRNA isoforms compared to
those in the other categories. We then analyzed the alternative
splicing and alternative backsplicing of highly shared circRNA gene
loci. Typically, the shared circRNA genes express more than two
circRNAs in their gene locus; only one or two are highly abundant,
whereas the others are lowly abundant and diverse. For example, two
of the four circular isoforms of the circRNA UBXN7 were highly
abundant. CircRNA UBXN7-1, derived from exon 3–5, was universally
expressed in all the human granulocyte samples. CircRNA UBXN7-2
derived from exon 2–5 was detected in 14 of 21 granulocyte samples.
Its other two circular isoforms were low abundance and were
detected in less than five samples. The alternative backsplicing
and alternative cis-splicing of circRNAs were diverse.
Furthermore, we investigated the relative circular
to linear transcript abundance. We suggested that the predominant
circRNAs have a circular to linear ratio >1. We found 2,045
circRNA isoforms that are predominant transcripts among the 55 HCC
samples, but only 8 predominant circRNAs, were shared in the HCC
samples. Moreover, in the 30 brain samples, there were 5,017
predominant circRNAs and 405 shared circRNAs (Table II). Predominant circRNAs were not
significantly enriched in shared circRNAs.
In summary, shared circRNAs are highly abundant. The
shared circRNA gene loci have multiple distinct circRNAs. In these
shared circRNA gene loci, circRNA expression demonstrates diverse
alternative cis-splicing and alternative backsplicing.
Comparison of the tissue-specific shared
circRNAs in humans
Even though a large number of circRNAs are
inter-individually diverse and vary among the samples, a small
proportion of circular RNAs is shared. Previous studies indicated
that the expression of circular RNA is related to the genomic
origin of the linear transcripts (5,23,24)
and that circular RNAs regulate the transcription of host mRNAs
(9,50,51).
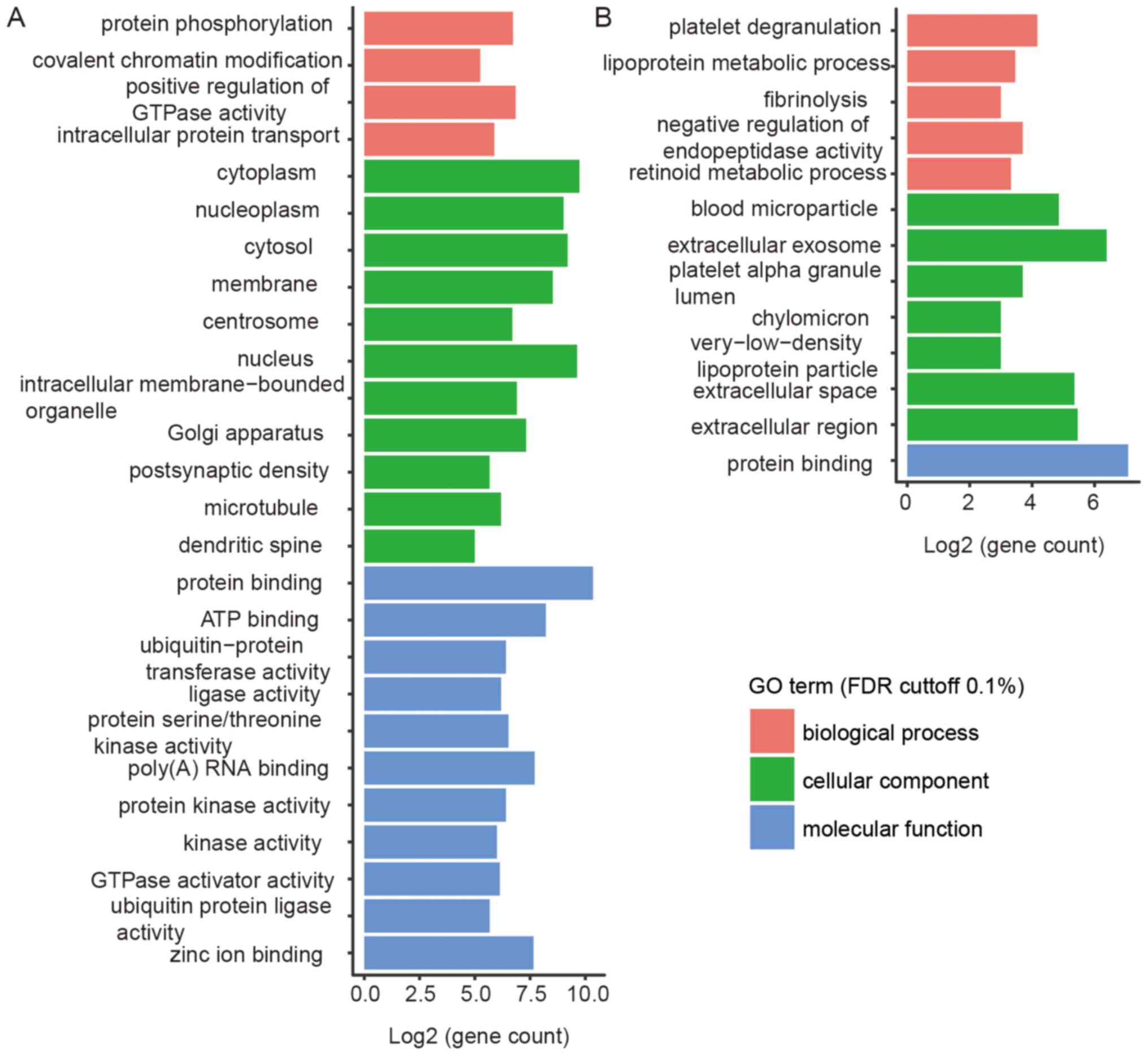
We conducted a Gene Ontology analysis on the linear transcripts
derived from the shared liver and brain circRNAs, which revealed
significant differences between them. Since there are almost no
shared circRNAs in the liver, we considered the circRNAs detected
in >20% of the samples as shared circRNAs in the liver. We found
that liver samples were enriched with lipoprotein metabolic process
and extracellular exosome while brain samples were enriched with
protein phosphorylation, postsynaptic density, and protein kinase
activity (Fig. 9). Both liver and
brain samples contained numerous protein binding genes, and most of
the GO terms were related to tissue-specific functions. Highly
represented gene categories included ApoE and ALB genes in the
liver and RIMS1, HTT and KLHL24 in the brain (Fig. 9 and Table III).
| Table IIICircRNAs shared in different
tissues. |
Table III
CircRNAs shared in different
tissues.
| chr | start | end | gene | transcript | strand | circRNA |
|---|
| chr1 | 26729380 | 26774901 | ARID1A | ARID1A-201 | + |
chr1:26729650-26732792(+) |
| chr1 | 1.17E+08 | 1.18E+08 | MAN1A2 | MAN1A2-001 | + |
chr1:117402185-117442325(+) |
| chr1 | 1.17E+08 | 1.18E+08 | MAN1A2 | MAN1A2-001 | + |
chr1:117402185-117420649(+) |
| chr1 | 1.17E+08 | 1.18E+08 | MAN1A2 | MAN1A2-001 | + |
chr1:117402185-117414831(+) |
| chr1 | 1.17E+08 | 1.18E+08 | MAN1A2 | MAN1A2-001 | + |
chr1:117402185-117405645(+) |
| chr1 | 66958911 | 66960078 | MIER1 | MIER1-005 | + |
chr1:66958058-66963160(+) |
| chr1 | 26921726 | 26946862 | NUDC | NUDC-001 | + |
chr1:26942659-26943065(+) |
| chr1 | 1.81E+08 | 1.81E+08 | STX6 | STX6-002 | − |
chr1:180984676-180993425(−) |
| chr1 | 21715124 | 21721146 | USP48 | USP48-008 | − |
chr1:21715388-21721764(−) |
| chr10 | 5709626 | 5714574 | FAM208B | FAM208B-014 | + |
chr10:5699524-5714207(+) |
| chr10 | 1.02E+08 | 1.02E+08 | FBXW4 | FBXW4-002 | − |
chr10:101667885-101676436(−) |
| chr12 | 1.23E+08 | 1.24E+08 | RILPL1 | RILPL1-001 | − |
chr12:123498543-123499536(−) |
| chr13 | 45962176 | 46052759 | ZC3H13 | ZC3H13-002 | − |
chr13:46003138-46020557(−) |
| chr14 | 45121588 | 45131261 | FKBP3 | FKBP3-003 | − |
chr14:45118027-45130790(−) |
| chr14 | 49820096 | 49852780 | NEMF | NEMF-005 | − |
chr14:49825866-49831361(−) |
| chr14 | 39179090 | 39182750 | PNN | PNN-005 | + |
chr14:39179090-39179462(+) |
| chr14 | 22909490 | 22911792 | RBM23 | RBM23-012 | − |
chr14:22909482-22911403(−) |
| chr14 | 22905585 | 22919149 | RBM23 | RBM23-004 | − |
chr14:22906194-22911403(−) |
| chr18 | 9136805 | 9235820 | ANKRD12 | ANKRD12-009 | + |
chr18:9182381-9221999(+) |
| chr18 | 21704957 | 21864974 | MIB1 | MIB1-004 | + |
chr18:21765771-21779685(+) |
| chr19 | 8461969 | 8465372 H | NRNPM H | NRNPM-014 | + |
chr19:8455404-8463686(+) |
| chr2 | 1.12E+08 | 1.12E+08 | ZC3H6 | ZC3H6-001 | + |
chr2:112299848-112300029(+) |
| chr2 | 1.12E+08 | 1.12E+08 | ZC3H6 | ZC3H6-001 | + |
chr2:112299848-112325197(+) |
| chr22 | 50372072 | 50444391 | PPP6R2 | PPP6R2-004 | + |
chr22:50372019-50394135(+) |
| chr3 | 1.96E+08 | 1.97E+08 | RNF168 | RNF168-001 | − |
chr3:196487398-196488683(−) |
| chr3 | 1.58E+08 | 1.59E+08 | RSRC1 | RSRC1-201 | + |
chr3:158122102-158123991(+) |
| chr3 | 1.7E+08 | 1.7E+08 | SEC62 | SEC62-009 | + |
chr3:169976945-169988359(+) |
| chr4 | 1.52E+08 | 1.53E+08 | FBXW7 | FBXW7-004 | − |
chr4:152411302-152412529(−) |
| chr4 | 1.28E+08 | 1.28E+08 | LARP1B | LARP1B-005 | + |
chr4:128074459-128077962(+) |
| chr6 | 4836098 | 4954373 | CDYL | CDYL-007 | + |
chr6:4891712-4892379(+) |
| chr6 | 18223868 | 18264823 | DEK | DEK-001 | − |
chr6:18236451-18258405(−) |
| chr7 | 1E+08 | 1E+08 | ZKSCAN1 | ZKSCAN1-001 | + |
chr7:100023418-100024307(+) |
| chr8 | 61623710 | 61714596 | ASPH | ASPH-001 | − |
chr8:61618977-61653660(−) |
| chr8 | 67083848 | 67195611 | CSPP1 | CSPP1-003 | + |
chr8:67131950-67137603(+) |
| chr8 | 1.08E+08 | 1.08E+08 | EMC2 | EMC2-004 | + |
chr8:108449822-108455930(+) |
| chr9 | 5919008 | 6007787 | KIAA2026 | KIAA2026-002 | − |
chr9:5968018-5988545(−) |
| chr9 | 33948374 | 33989043 | UBAP2 | UBAP2-004 | − |
chr9:33971650-33973237(−) |
| chr9 | 33994481 | 34048872 | UBAP2 | UBAP2-010 | − |
chr9:33986759-34017189(−) |
| chr9 | 33921693 | 34048901 | UBAP2 | UBAP2-001 | − |
chr9:33960825-33989126(−) |
Furthermore, we also analyzed the liver, brain,
blood, granulocyte and heart shared circRNAs, i.e., 39 shared
circRNAs in these tissues (Table
III). Some of these shared circRNAs have been previously
validated by experiments, e.g., circZKSCAN1 (52) and circMAN1A2 (50,53).
CDR1as was detected in the three other tissues except the blood and
granulocyte samples.
Conservation of the identified circRNA
candidates between human and mouse
Previous research indicated that circular RNAs are
evolutionarily conserved in function (2,23,49,54).
First, most circRNAs originated from CDS regions, which are
evolutionally preserved in the genome. Second, the backsplice sites
of circRNAs are conserved. We compared human and mouse conservation
of circRNAs with a previously described computational method
(55). In total, we identified
169,044 circRNA candidates in 178 human samples and 9,886 circRNA
candidates in 24 mouse samples. In human and mouse, there were
3,579 shared conserved gene locus-detected circRNA candidates.
There were 83,389 gene locus-expressed circRNA candidates in humans
and 8,615 circRNA candidates in mice.
Discussion
Eukaryotic circRNAs are a type of less abundant, but
biochemically stable, transcripts that are expressed in diverse
genomic locations. The abundance of circRNAs is ~1–3% of the level
of poly(A)+ RNAs (49),
and most circRNAs exist in low abundance. Therefore, identifying
all of the expressed circRNAs is difficult. Each circRNA prediction
algorithm brings its own bias and 'blind spots' (22). We solved this problem by combining
the distinct algorithms to yield a more trustworthy and sensitive
output (34). It was reported that
circRNA isoforms from the same host transcript share the same
backsplice sites with different internal exons (24). As a result, the internal exon
composition of circRNAs cannot simply be predicted using junction
exons and linear RNA exon composition (24,48,56).
RAISE is designed to detect circRNA internal exon composition and
predict circRNA transcript sequences (Fig. 2C). The availability of coverage and
splice information helps in the identification of a circRNA and its
exon composition. The internal exon usage of circRNAs was used to
predict circRNA transcript sequences and investigate circRNA
functions.
We used RAISE to compare the circRNA expression
profiles in HCC with adjacent non-tumor tissue samples and did not
find any significant differences. Even though there is no
significant differential expression between tumor and adjacent
non-tumor tissue, we observed that circRNAs expression profiles are
diverse between individuals and are independent of the linear gene
expression. In the case of circRNA expression, this variation means
detected or not, whereas for linear gene expression, it represents
whether the abundance is high or low. Since the circRNAs are not
highly enriched in HCC-affected tissues, we tested whether various
other tissues possessed the same expression patterns. In the brain
rRNA depleted RNA-seq data, the ratio of shared circRNAs was 0.5%,
which is higher than the ratio in the liver but lower than the
ratio of the linear gene. The brain and liver samples displayed
high inter-individual variation in the expression of circRNAs. We
collected 21 granulocytes samples from seven individuals at three
time-points to determine whether circRNAs were reproducibly
expressed within one individual. The results showed that circRNAs
are not reproducible within one individual. Briefly, the circRNAs
fall into two categories: the randomly or variably expressed
circRNAs, and the robustly expressed circRNAs. The biological roles
of either of these types is not yet clear and thus requires
functional studies. Several previous studies have highlighted a few
circRNAs which are highly abundant and ubiquitously expressed
(1,55).
In agreement with previous research, individual
circRNA expression seems to be highly stochastic. However, the
variable expression of circRNAs in different samples may also trap
miRNAs (57). Meanwhile, some
online databases, including Arraystar's circRNA target prediction
software, Circ2Traits, CircInteractome and CircNet (58–61),
have been developed to predict circRNA-miRNA interaction networks.
Circ2Traits is a comprehensive database for circular RNAs with
potential association with disease and traits (58). CircNet and CircInteractome, both
predict the miRNAs target of circRNAs and create the circRNA-miRNA
interaction network (59,60).
We also observed that most of the circRNAs are
detected in only a few cell types and that they are not as
cell-type-specific as mRNAs (55).
The variation and diversity of circRNAs expression profiles may be
due to the large number of circRNA transcripts expressed at a low
level. Interestingly, most of the circRNAs co-exist with linear RNA
transcripts; however, only a small portion of these circRNAs are
predominant transcripts (Table
II). There are several shared circRNAs among the recruited
samples in the same tissue. The shared circRNAs usually have
multiple circular isoforms in the gene locus, and these gene loci
are circRNA hotspots. We conducted a Gene Ontology analysis on the
host genes that gave rise to shared circRNAs and showed that these
genes are related to tissue-specific functions.
We used our RAISE pipeline to show that circRNAs
have both intra-and inter-individual variations in their expression
patterns. Our findings can be helpful in identifying novel circRNAs
and designing better therapeutic approaches. Furthermore, according
to our suggestions only the robustly expressed circRNAs are a
candidate for usage as a biomarker.
Acknowledgments
The authors acknowledge the authors behind the
circRNA detection algorithms for making their source codes publicly
available, NCBI SRA and ENCODE online public dataset. This study
was supported by Beijing Postdoctoral Research Foundation (no.
2016ZZ-33), and Beijing Municipal Science and Technology Commission
Research Fund (no. Z171100000417004).
References
|
1
|
Salzman J, Gawad C, Wang PL, Lacayo N and
Brown PO: Circular RNAs are the predominant transcript isoform from
hundreds of human genes in diverse cell types. PLoS One. 7(e30733):
e307332012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar :
|
|
3
|
Nigro JM, Cho KR, Fearon ER, Kern SE,
Ruppert JM, Oliner JD, Kinzler KW and Vogelstein B: Scrambled
exons. Cell. 64:607–613. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Capel B, Swain A, Nicolis S, Hacker A,
Walter M, Koopman P, Goodfellow P and Lovell-Badge R: Circular
transcripts of the testis-determining gene Sry in adult mouse
testis. Cell. 73:1019–1030. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL
and Yang L: Complementary sequence-mediated exon circularization.
Cell. 159:134–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ivanov A, Memczak S, Wyler E, Torti F,
Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M,
Dieterich C, et al: Analysis of intron sequences reveals hallmarks
of circular RNA biogenesis in animals. Cell Rep. 10:170–177. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kelly S, Greenman C, Cook PR and
Papantonis A: Exon skipping is correlated with exon
circularization. J Mol Biol. 427:2414–2417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barrett SP, Wang PL and Salzman J:
Circular RNA biogenesis can proceed through an exon-containing
lariat precursor. eLife. 4:e075402015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Conn SJ, Pillman KA, Toubia J, Conn VM,
Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA and
Goodall GJ: The RNA binding protein quaking regulates formation of
circRNAs. Cell. 160:1125–1134. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen LL and Yang L: Regulation of circRNA
biogenesis. RNA Biol. 12:381–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Salzman J: Circular RNA expression: Its
potential regulation and function. Trends Genet. 32:309–316. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Starke S, Jost I, Rossbach O, Schneider T,
Schreiner S, Hung LH and Bindereif A: Exon circularization requires
canonical splice signals. Cell Rep. 10:103–111. 2015. View Article : Google Scholar
|
|
13
|
Szabo L, Morey R, Palpant NJ, Wang PL,
Afari N, Jiang C, Parast MM, Murry CE, Laurent LC and Salzman J:
Statistically based splicing detection reveals neural enrichment
and tissue-specific induction of circular RNA during human fetal
development. Genome Biol. 16:1262015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahmed I, Karedath T, Andrews SS, Al-Azwani
IK, Mohamoud YA, Querleu D, Rafii A and Malek JA: Altered
expression pattern of circular RNAs in primary and metastatic sites
of epithelial ovarian carcinoma. Oncotarget. 7:36366–36381. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang K, Singh D, Zeng Z, Coleman SJ, Huang
Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al:
MapSplice: Accurate mapping of RNA-seq reads for splice junction
discovery. Nucleic Acids Res. 38:e1782010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hoffmann S, Otto C, Kurtz S, Sharma CM,
Khaitovich P, Vogel J, Stadler PF and Hackermüller J: Fast mapping
of short sequences with mismatches, insertions and deletions using
index structures. PLOS Comput Biol. 5:e10005022009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Westholm JO, Miura P, Olson S, Shenker S,
Joseph B, Sanfilippo P, Celniker SE, Graveley BR and Lai EC:
Genome-wide analysis of drosophila circular RNAs reveals their
structural and sequence properties and age-dependent neural
accumulation. Cell Rep. 9:1966–1980. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao Y, Wang J and Zhao F: CIRI: An
efficient and unbiased algorithm for de novo circular RNA
identification. Genome Biol. 16:42015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
You X and Conrad TO: Acfs: Accurate
circRNA identification and quantification from RNA-Seq data. Sci
Rep. 6:388202016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng J, Metge F and Dieterich C: Specific
identification and quantification of circular RNAs from sequencing
data. Bioinformatics. 32:1094–1096. 2016. View Article : Google Scholar
|
|
22
|
Szabo L and Salzman J: Detecting circular
RNAs: Bioinformatic and experimental challenges. Nat Rev Genet.
17:679–692. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rybak-Wolf A, Stottmeister C, Glažar P,
Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss
R, et al: Circular RNAs in the mammalian brain are highly abundant,
conserved, and dynamically expressed. Mol Cell. 58:870–885. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
You X, Vlatkovic I, Babic A, Will T,
Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al:
Neural circular RNAs are derived from synaptic genes and regulated
by development and plasticity. Nat Neurosci. 18:603–610. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Venø MT, Hansen TB, Venø ST, Clausen BH,
Grebing M, Finsen B, Holm IE and Kjems J: Spatiotemporal regulation
of circular RNA expression during porcine embryonic brain
development. Genome Biol. 16:2452015. View Article : Google Scholar
|
|
26
|
Tan WL, Lim BT, Anene-Nzelu CG,
Ackers-Johnson M, Dashi A, See K, Tiang Z, Lee DP, Chua WW, Luu TD,
et al: A landscape of circular RNA expression in the human heart.
Cardiovasc Res. 113:298–309. 2017.PubMed/NCBI
|
|
27
|
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z
and Sharpless NE: Expression of linear and novel circular forms of
an INK4/ARF-associated non-coding RNA correlates with
atherosclerosis risk. PLoS Genet. 6:e10012332010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alnasir J and Shanahan HP: Investigation
into the annotation of protocol sequencing steps in the sequence
read archive. Gigascience. 4:232015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Birney E, Stamatoyannopoulos JA, Dutta A,
Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis
ET, Thurman RE, et al: Children's Hospital Oakland Research
Institute: Identification and analysis of functional elements in 1%
of the human genome by the ENCODE pilot project. Nature.
447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dong H, Zhang L, Qian Z, Zhu X, Zhu G,
Chen Y, Xie X, Ye Q, Zang J, Ren Z, et al: Identification of
HBV-MLL4 integration and its molecular basis in Chinese
hepatocellular carcinoma. PLoS One. 10:e01231752015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kornienko AE, Dotter CP, Guenzl PM,
Gisslinger H, Gisslinger B, Cleary C, Kralovics R, Pauler FM and
Barlow DP: Long non-coding RNAs display higher natural expression
variation than protein-coding genes in healthy humans. Genome Biol.
17:142016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scheckel C, Drapeau E, Frias MA, Park CY,
Fak J, Zucker-Scharff I, Kou Y, Haroutunian V, Ma'ayan A, Buxbaum
JD, et al: Regulatory consequences of neuronal ELAV-like protein
binding to coding and non-coding RNAs in human brain. eLife.
5:52016. View Article : Google Scholar
|
|
33
|
Liu SJ, Nowakowski TJ, Pollen AA, Lui JH,
Horlbeck MA, Attenello FJ, He D, Weissman JS, Kriegstein AR, Diaz
AA, et al: Single-cell analysis of long non-coding RNAs in the
developing human neocortex. Genome Biol. 17:672016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hansen TB, Venø MT, Damgaard CK and Kjems
J: Comparison of circular RNA prediction tools. Nucleic Acids Res.
44:e582016. View Article : Google Scholar :
|
|
35
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for The ENCODE Project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Quinlan AR and Hall IM: BEDTools: A
flexible suite of utilities for comparing genomic features.
Bioinformatics. 26:841–842. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen
B, Luo Y, Lyu D, Li Y, Shi G, et al: Circular RNA profiling reveals
an abundant circHIPK3 that regulates cell growth by sponging
multiple miRNAs. Nat Commun. 7:112152016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Memczak S, Papavasileiou P, Peters O and
Rajewsky N: Identification and characterization of circular RNAs as
a new class of putative biomarkers in human blood. PLoS One.
10:e01412142015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kroll KW, Mokaram NE, Pelletier AR,
Frankhouser DE, Westphal MS, Stump PA, Stump CL, Bundschuh R,
Blachly JS and Yan P: Quality control for RNA-Seq (QuaCRS): An
integrated quality control pipeline. Cancer Inform. 13(Suppl 3):
7–14. 2014.PubMed/NCBI
|
|
40
|
Chen C, Khaleel SS, Huang H and Wu CH:
Software for pre-processing Illumina next-generation sequencing
short read sequences. Source Code Biol Med. 9:82014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pertea M, Kim D, Pertea GM, Leek JT and
Salzberg SL: Transcript-level expression analysis of RNA-seq
experiments with HISAT, StringTie and Ballgown. Nat Protoc.
11:1650–1667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The Sequence Alignment/Map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Anders S, Pyl PT and Huber W: HTSeq - a
Python framework to work with high-throughput sequencing data.
Bioinformatics. 31:166–169. 2015. View Article : Google Scholar
|
|
44
|
Pertea M, Pertea GM, Antonescu CM, Chang
TC, Mendell JT and Salzberg SL: StringTie enables improved
reconstruction of a transcriptome from RNA-seq reads. Nat
Biotechnol. 33:290–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Huang W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar :
|
|
46
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar
|
|
47
|
Kadota PO, Hajjiri Z, Finn PW and Perkins
DL: Precision subtypes of T cell-mediated rejection identified by
molecular profiles. Front Immunol. 6:5362015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang XO, Dong R, Zhang Y, Zhang JL, Luo
Z, Zhang J, Chen LL and Yang L: Diverse alternative back-splicing
and alternative splicing landscape of circular RNAs. Genome Res.
26:1277–1287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Salzman J, Chen RE, Olsen MN, Wang PL and
Brown PO: Cell-type specific features of circular RNA expression.
PLoS Genet. 9:e10037772013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li Z, Huang C, Bao C, Chen L, Lin M, Wang
X, Zhong G, Yu B, Hu W, Dai L, et al: Exon-intron circular RNAs
regulate transcription in the nucleus. Nat Struct Mol Biol.
22:256–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: circRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liang D and Wilusz JE: Short intronic
repeat sequences facilitate circular RNA production. Genes Dev.
28:2233–2247. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Alhasan AA, Izuogu OG, Al-Balool HH, Steyn
JS, Evans A, Colzani M, Ghevaert C, Mountford JC, Marenah L,
Elliott DJ, et al: Circular RNA enrichment in platelets is a
signature of transcriptome degradation. Blood. 127:e1–e11. 2016.
View Article : Google Scholar :
|
|
54
|
Thomas LF and Sætrom P: Circular RNAs are
depleted of polymorphisms at microRNA binding sites.
Bioinformatics. 30:2243–2246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo JU, Agarwal V, Guo H and Bartel DP:
Expanded identification and characterization of mammalian circular
RNAs. Genome Biol. 15:4092014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gao Y, Wang J, Zheng Y, Zhang J, Chen S
and Zhao F: Comprehensive identification of internal structure and
alternative splicing events in circular RNAs. Nat Commun.
7:120602016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Caiment F, Gaj S, Claessen S and Kleinjans
J: High-throughput data integration of RNA-miRNA-circRNA reveals
novel insights into mechanisms of benzo[a]pyrene-induced
carcinogenicity. Nucleic Acids Res. 43:2525–2534. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ghosal S, Das S, Sen R, Basak P and
Chakrabarti J: Circ2Traits: A comprehensive database for circular
RNA potentially associated with disease and traits. Front Genet.
4:2832013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu YC, Li JR, Sun CH, Andrews E, Chao RF,
Lin FM, Weng SL, Hsu SD, Huang CC, Cheng C, et al: CircNet: A
database of circular RNAs derived from transcriptome sequencing
data. Nucleic Acids Res. 44D:D209–D215. 2016. View Article : Google Scholar
|
|
60
|
Dudekula DB, Panda AC, Grammatikakis I, De
S, Abdelmohsen K and Gorospe M: CircInteractome: A web tool for
exploring circular RNAs and their interacting proteins and
microRNAs. RNA Biol. 13:34–42. 2016. View Article : Google Scholar :
|
|
61
|
Huang M, Zhong Z, Lv M, Shu J, Tian Q and
Chen J: Comprehensive analysis of differentially expressed profiles
of lncRNAs and circRNAs with associated co-expression and ceRNA
networks in bladder carcinoma. Oncotarget. 7:47186–47200. 2016.
View Article : Google Scholar : PubMed/NCBI
|