Introduction
Breast cancer is the most prevalent type of cancer
affecting women worldwide and represents the leading cause of
cancer-related mortality after lung cancer (1). Three biomarkers are used in clinical
practice for tumor characterization and treatment: estrogen
receptor α, progesterone receptor and epidermal growth factor
receptor 2. Nevertheless, the benefits of therapies targeting these
mechanisms are often limited due to resistance. Indeed,
approximately one-third of patients with early-stage breast cancer
will develop resistance against tamoxifen over a 5-year treatment
period, and the majority of patients will become resistant against
trastuzumab over a 1-year period (2,3).
Moreover, no targeted therapies exist for the aggressive
triple-negative tumors, which express none of these three markers.
On the whole, epidemiological data reveal the urgent need for novel
therapeutic options.
Prostaglandins (PGs) are a family of biologically
active endogenous metabolites of arachidonic acid.
15-Deoxy-D12,14-prostaglandin J2 (15d-PGJ2) is a
dehydrated derivative of PGD2, that displays potent anticancer
properties (4–11). 15d-PGJ2 is a potent natural agonist
of peroxisome proliferator-activated receptor gamma (PPARγ)
(12,13). PPARγ belongs to a nuclear receptor
family that acts as ligand-activated transcription factor. The
activation of PPARγ requires ligand binding, heterodimerization
with retinoid X receptor and interaction with the PPAR response
element (PPRE) and co-activator recruitment (14–16).
Nevertheless, an increasing number of studies have demonstrated
that the antitumor effects of 15d-PGJ2 may also be mediated through
PPARγ-independent pathways (17–24).
In vivo, these effects have been confirmed in several mouse
tumor models (25–27). In breast cancer cell lines, the
anti-proliferative effects of 15d-PGJ2 remain modest compared to
several synthetic PPARγ ligands (20). Recent clinical trials relaunched
PPARγ agonist perspectives for the treatment of cancer. Indeed,
pioglitazone in combination with imatinib can erode the cancer stem
cell pool and efatutazone alone or in combination with folfiri has
been shown to provide some degree of control for metastatic cancer
(28–30).
In order to adapt 15d-PGJ2 for clinical use, more
potent derivatives are required. Several studies have demonstrated
that biotin-conjugated compounds are able to increase the uptake of
anticancer drugs in tumor cells (31–34).
Previously, we demonstrated that two biotinylated derivatives of
Δ2-troglitazone, PPARγ-inactive analogues of troglitazone (TGZ),
were more potent than the original compound in inhibiting cell
proliferation (35,36).
In the present study, we examined the
anti-proliferative activity of a biotin-conjugated 15d-PGJ2
(b-15d-PGJ2) on estrogen-dependent MCF-7 and triple-negative
MDA-MB-231 breast cancer cells. We demonstrate that b-15d-PGJ2 is
more efficient than 15d-PGJ2 in inhibiting cell proliferation and
inducing apoptosis. Molecular docking analysis indicated that
b-15d-PGJ2 was able to bind the ligand-binding domain of PPARγ, in
a similar manner as 15d-PGJ2. This compound also displayed an
improved PPARγ agonist activity, as measured by transactivation
experiments. RNA interference experiments revealed that a
PPARγ-dependent pathway was involved in b-15d-PGJ2-induced
apoptosis. This study provides a better understanding of the
anti-proliferative effects of PPARγ ligands and supports the
conception of more efficient derivatives, which may be used for the
development of novel therapeutic strategies for breast cancer.
Materials and methods
Reagents and cell lines
15d-PGJ2 and
15-deoxy-Δ12,14-prostaglandin J2-biotinamide
(b-15d-PGJ2) were purchased respectively, from Merk-Millipore
(Fontenay-sous-Bois, France) and Cayman Chemical Co. (Ann Arbor,
MI, USA). Efatutazone (RS5444; CS-7017) was obtained from
ChemScene, (Monmouth Junction, NJ, USA). The small-interfering RNA
(siRNA) duplexes targeting PPARγ and non-specific control siRNA
were purchased from Eurogentec (Angers, France) and PCR primers
from Euromedex (Souffelweyersheim, France).
The human breast cancer cell lines, MCF-7 and
MDA-MB-231, were purchased from the American Type Culture
Collection (ATCC, Manassas, VA, USA). Dulbecco’s modified Eagle’s
medium (DMEM), Leibovitz’s L-15 medium, trypsin-EDTA and PBS were
purchased from Life Technologies (Saint Aubin, France). Biotin,
ethanol (EtOH), dimethylsulfoxide (DMSO), fetal calf serum (FCS),
L-glutamine and all other chemicals were purchased from
Sigma-Aldrich (Lyon, France).
Cell culture and treatment
The MCF-7 and MDA-MB-231 cells were cultured at 37°C
under 5% CO2 in DMEM or without CO2 in L-15
medium, respectively. Both media were supplemented with 10% fetal
calf serum FCS and 2 mM L-glutamine. The cells were treated with
0.1% EtOH (vehicle) or various concentrations of 15d-PGJ2,
b-15d-PGJ2 and efatutazone in 1% FCS-containing medium. 15d-PGJ2
and efatutazone were dissolved at 50 mM in sterile DMSO. b-15d-PGJ2
was dissolved at 1.6 mM in EtOH. All these compounds were stored
frozen at −20°C.
Cell proliferation assays
The MCF-7 and MDA-MB-231 cells (0.8×105
cells/ml) were seeded in 12-well plates and treated with various
concentrations of 15d-PGJ2, b-15d-PGJ2, efatutazone (2 μM up
to 50 μM) and EtOH (control) for 24 h. For competition
experiments, various concentrations of free biotin (5 or 25
μM) were added together with 15d-PGJ2 or b-15d-PGJ2. Each
treatment was performed in triplicate. Cell proliferation was
measured using CellTiter-Glo™ Luminescent Cell Viability assay
(Promega, Charbonnières, France).
For the different compounds, the concentration
leading to a decrease of 50% in the number of viable cells
(IC50) was determined. Efatutazone was used as a gold
standard, since it is a PPARγ activator currently evaluated for
clinical trials (37).
Transient transfection assays
The MCF-7 cells (1.6×105 cells/ml) were
seeded in 24-well plates and transfected with
pPPRE3-tk-luc reporter (1 μg/well) and
pCMV-β-galactosidase (β-Gal) (0.6 μg/well), as an internal
control plasmid, in the presence of a human PPARγ expression vector
(2 μg/well). pPPRE3-tk-luc reporter comprises
three copies of PPRE from the promoter of the rat ACO gene. hPPARγ2
was cloned into the pcDNA3 vector. pCMV-β is a plasmid encoding
β-galactosidase under the control of the cytomegalovirus promotor.
The pPPRE3-tk-luc, the human PPARγ expression vector and
the pCMV-β-galactosidase construct were a gift from Professor P.
Becuwe, Dr L. Domenjoud and Professor O. Nusse, respectively. Cell
transfection was performed using Exgen 500 (Euromedex) according to
the manufacturer’s instructions. Following transfection, the cells
were allowed to grow for 24 h in DMEM supplemented with 10% FCS
stripped in dextran- coated charcoal and were treated with 10
μM of 15d-PGJ2, b-15d-PGJ2, efatutazone and ethanol as a
control for 24 h. Luciferase and β-Gal activities were measured as
previously described (35).
RNA interference
The MCF-7 and MDA-MB-231 cells (2×105
cells/ml) were seeded in 6-well culture plates and were transfected
with 100 nM of PPARγ siRNA duplex mix or control scRNAs [negative
control (OR-0030-neg05)] using Oligofectamine™ reagent (Life
Technologies) according to the manufacturer’s instructions. The
siRNA sequences against human PPARγ (PPARγ-siRNAs) were as follows:
5′-GUA-CCA-AAG-UGC-AAU-CAAATT-3′ and
5′-UUU-GAU-UGC-ACU-UUG-GUA-CTT-3′ for duplex no. 1,
5′-CAA-UCA-GAU-UGA-AGC-UUA-UTT-3′ and
5′-AUA-AGC-UUC-AAU-CUG-AUU-GTT-3′ for duplex no. 2. Twenty hours
later, the cells were exposed to 15d-PGJ2 (10 μM),
b-15d-PGJ2 (10 μM) or 0.1% EtOH (vehicle control) for 24 h
and harvested for RT-PCR or western blot analyses.
Semi-quantitative RT-PCR
The MCF-7 and MDA-MB-231 cells (2×105
cells/ml) were seeded overnight in 6-well plates and were exposed
to 15d-PGJ2 (10 μM), b-15d-PGJ2 (10 μM) or EtOH
(0.1%) for 24 h. RT-PCR was performed as previously described
(38). Briefly, cDNA was further
amplified by PCR with specific primers: 5′-GACCACTCCCACTCCTTT-3′
and 5′-CGACATTCAATTGCCATGAG-3′ for PPARγ,
5′-TACATGGGTGGGGTGTTGAA-3′ and 5′-AAGAGAGGCATCCTCACCC-3′ for
β-actin. Amplification was carried out under the following
conditions: i) initial denaturation 94°C for 2 min; ii) 94°C for 30
sec, 58°C for 30 sec and 72°C for 45 sec; iii) 10 min extension
step at 72°C. Subsequently, 20 μl of the PCR products were
mixed with loading buffer (5 μl) and submitted to
electrophoresis in a 1.5% agarose gel at 90 V for 35 min at room
temperature. The gel was stained with ethidium bromide, viewed and
photographed on a UV-transilluminator (Gel Doc 2000, Bio-Rad
Laboratories, Marnes-la-Coquette, France). The intensity of the
PPARγ signal was normalised to β-actin using Gel Doc 2000 and a
software package (Quantity One v.4·3·1) (both from Bio-Rad
Laboratories).
Western blot analysis
The MCF-7 and MDA-MB-231 cells (2×105
cells/ml) were seeded in 6-well plates and were treated as
described below for 24 h and subjected to western blot analysis as
previously described (38). The
antibodies against cleaved PARP-1 (F21-852, BD Biosciences, Le
Pont-de-Claix, France) and caspase-7 (9494, Cell Signaling
Technology, Danvers, USA) were diluted at 1:1,000. The monoclonal
antibody against tubulin (EP1332Y, Epitomics, Burlingame, CA, USA)
and the polyclonal antibody against β-actin (SC-1615, Santa Cruz
Biotechnology, Dallas, TX, USA) were used diluted at 1:2,000.
Non-specific binding sites were blocked in TNT buffer (50 mM
Tris-HCl, 150 mM NaCl, 0.1% Tween-20) with 5% non-fat powder milk
and the membranes were incubated with the primary antibodies
diluted in blocking solution overnight at 4°C. The membranes were
probed with appropriate horseradish peroxidase-conjugated secondary
antibodies (SC-2005 for mouse antibody and SC-2004 for rabbit
antibody, Santa Cruz Biotechnology) for 1 h at room temperature. On
some western blots, the intensity of the bands corresponding to
cleaved PARP-1 and caspase-7 was normalized to β-actin or tubulin
by using Gel Doc 2000 and a software package (Quantity One v.4·3·1)
(both from Bio-Rad Laboratories).
Nuclear staining
The MCF-7 and MDA-MB-231 cells (2×105
cells/ml) were seeded in 6-well plates and were treated with
15d-PGJ2 or b-15d-PGJ2 (10 μM) for 24 h. Following
centrifugation (10 min, 1,200 rpm), the pellet was resuspended in
50 μl of media and the nuclei were stained with Hoechst dye
(Hoechst 33342; AAT Bioquest, Sunnyvale, CA, USA). Fluorescent
staining was observed under an Eclipse 80i microscope (Nikon,
Champigny-sur-Marne, France). Images were collected using LuciaG
software 4.81 (Laboratory imaging/Nikon).
Statistical analysis
The results of each experiment are expressed as the
mean ± standard error of the mean (SEM) of 3 to 5 different
experiments. Bars represent the means ± SEM. Statistical
differences were tested using analysis of variance (ANOVA) followed
by Bonferroni, Student-Newman-Keuls or Student’s t-test post hoc
comparisons (SPSS v11.0 Computer Software). Differences in which
the P-value was <0.05 were considered statistically
significant.
Molecular docking
Molecular docking of 15d-PGJ2 and b-15d-PGJ2 in the
ligand binding domain (LBD) of PPARγ was performed using the
Autodock software, version 4.2 (39). The target protein structure
corresponds to the Protein Data Bank (PDB) entry 3V9V (40). This structure was preferred over
PDB entry 2ZK1 (41), which
corresponds to 15d-PGJ2 bound to PPARγ as the former structure has
a better resolution (1.60 Å for 3V9V vs. 2.60 Å for 2ZK1,
respectively) and as its LBD pocket can accommodate larger ligands.
Ligands were manually built with the GaussView software (42). After a semiempirical PM6 (43) geometrical optimization with
Gaussian 09 (44), atomic charges
were computed using the AM1-BCC (45) scheme by the Antechamber program
from the AmberTools suite of programs (46). Atomic charges for the protein were
assigned according to the Amber ff12SB force field (47). A grid of 78×78×78 points centered
on the Cα of Cys285 (Helix3 of the LBD of PPARγ) was built with a
spacing of 0.375 Å using AutoGrid. The number of total docking runs
was set to 30, each with a population of 150 individuals, a maximum
number of generations set at 27,000 and a maximum number of energy
evaluations set at 108. All other parameters were given default
values.
Results
b-15d-PGJ2 markedly decreases cell
viability compared to the PPARγ agonists, 15d-PGJ2 and
efatutazone
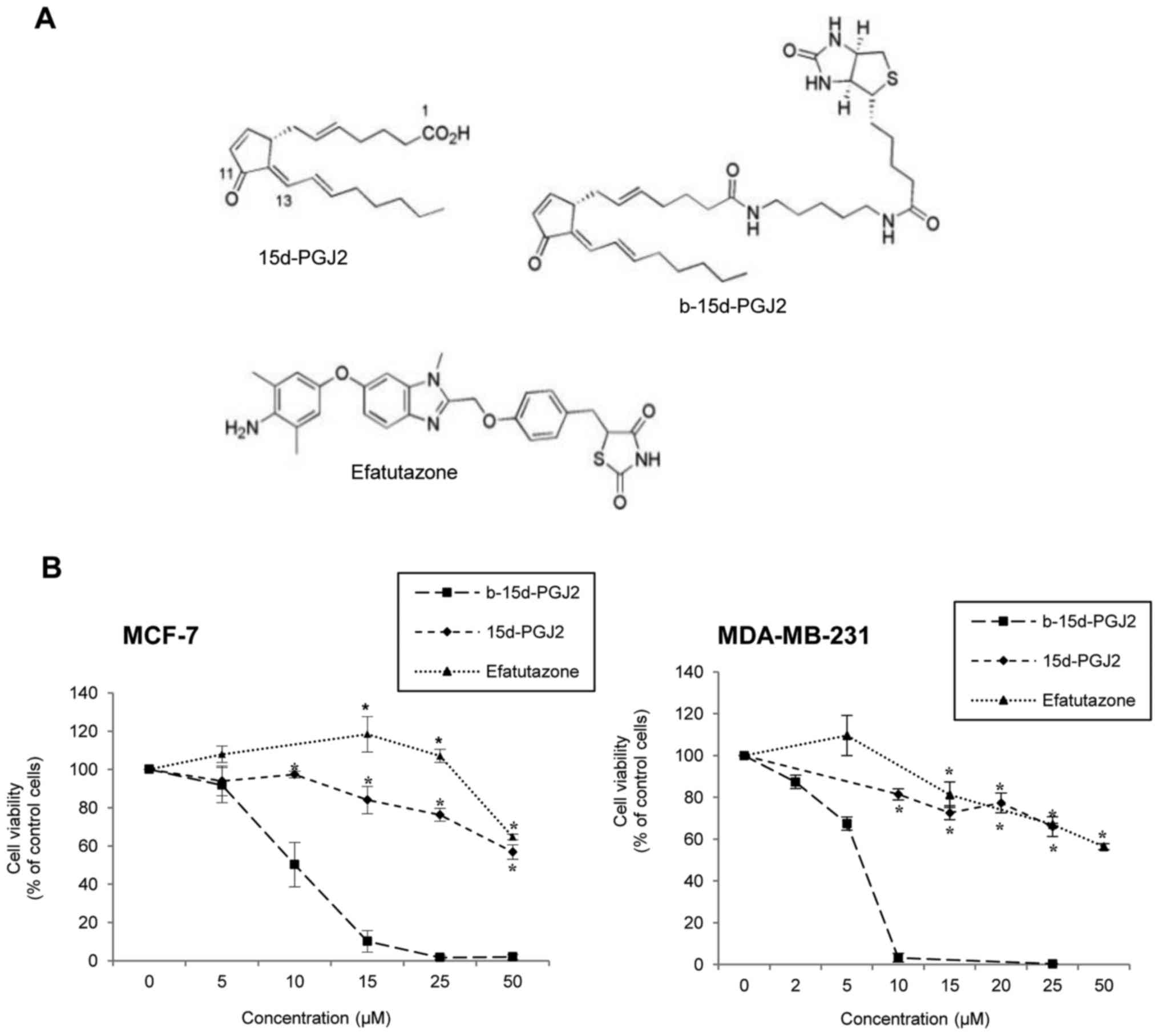
The anti-proliferative effects of 15d-PGJ2, its
biotinylated derivative, b-15d-PGJ2, and the PPARγ agonist,
efatutazone (Fig. 1A) were
measured on hormone-dependent MCF-7 and triple-negative MDA-MB-231
breast cancer cell lines. The cells were exposed to increasing
concentrations of each compound or corresponding ethanol
concentrations (control cells). Cell viability was determined after
24 h of treatment (Fig. 1B).
b-15d-PGJ2 induced a potent dose-dependent inhibition of MCF-7
(IC50, 9.8±1.2 μM) and MDA-MB-231
(IC50, 6.0±0.1 μM) cell proliferation, compared
to 15d-PGJ2 or efatutazone used as a positive control
(IC50 both >50 μM). The anti-proliferative
effect of b-15d-PGJ2 differed significantly from that of 15d-PGJ2
and efatutazone, from concentration as low as 15 and 10 μM
in the MCF-7 and MDA-MB-231 cells, respectively. Of note, the
MDA-MB-231 cells were significantly more sensitive than the MCF-7
cells to b-15d-PGJ2 treatment (IC50, P=0.016).
b-15d-PGJ2 is a more potent inducer of
apoptosis
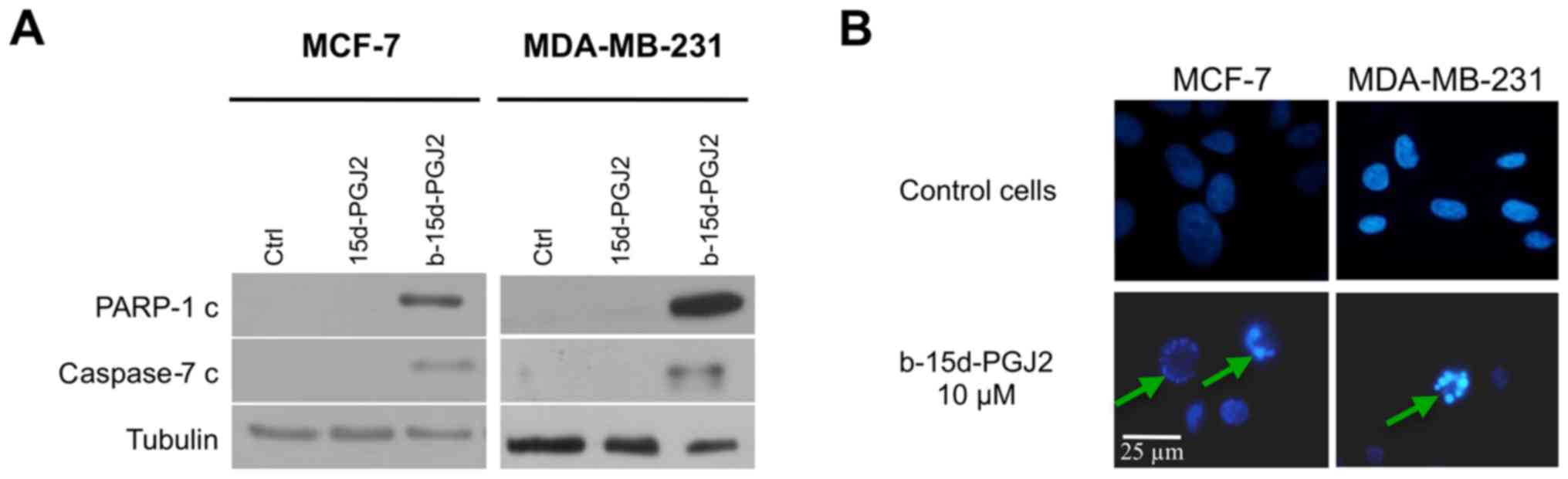
First, we investigated the activation of the
executioner caspase, caspase-7 (since caspase-3 is not expressed in
MCF-7 cells), as well as PARP-1 cleavage, since it is a well-known
caspase substrate (Fig. 2A). At
the concentration of 10 μM b-15d-PGJ2, western blot analysis
revealed the cleavage of caspase-7 and the cleavage of PARP-1 after
24 h of treatment compared with the 15d-PGJ2-treated or the control
cells. To ascertain our results, we examined the effects of
b-15d-PGJ2 on nuclear morphology in Hoechst-stained cells. Both
cell lines were treated for 24 h with b-15d-PGJ2 (10 μM). As
depicted in Fig. 2B, the treated
cells exhibited characteristics of apoptosis, such as cell
shrinkage, nuclear condensation and fragmentation compared to the
untreated controls. Taken together, these results provide insight
into the induction of apoptosis in both cell lines following
treatment with 10 μM b-15d-PGJ2.
Biotin group is not responsible for the
enhanced anti-proliferative effects of b-15d-PGJ2
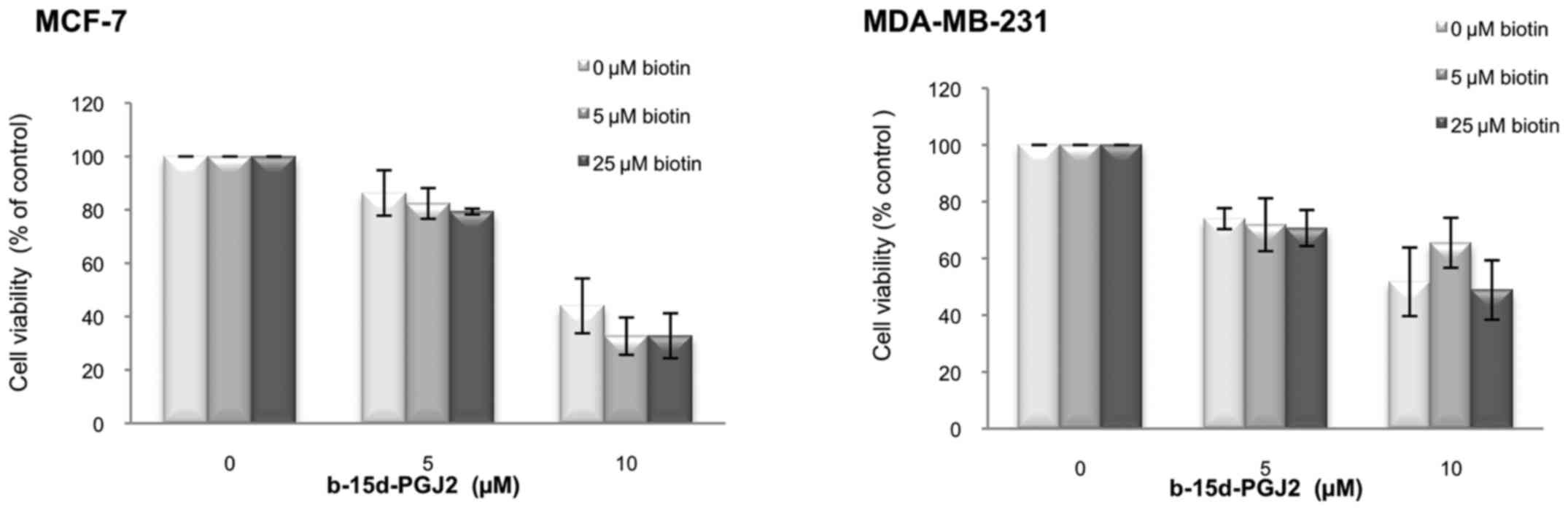
We then performed competition experiments with free
biotin to determine whether the enhanced anti-proliferative effect
of b-15d-PGJ2 could be the result of an increased internalization
mediated by a biotin membrane receptor (Fig. 3). We examined viability of the
MCF-7 and MDA-MB-231 cells following 24 h of treatment with
b-15d-PGJ2 (0, 5 and 10 μM) in the presence of increasing
concentrations of biotin (0, 5 and 25 μM). In both cell
lines, free biotin up to 25 μM in the culture medium did not
significantly modify the effects of b-15d-PGJ2 on cell viability.
This result thus excluded biotin receptors as a cause for the
enhanced anti-proliferative effects of b-15d-PGJ2.
b-15d-PGJ2 is a potent PPARγ agonist
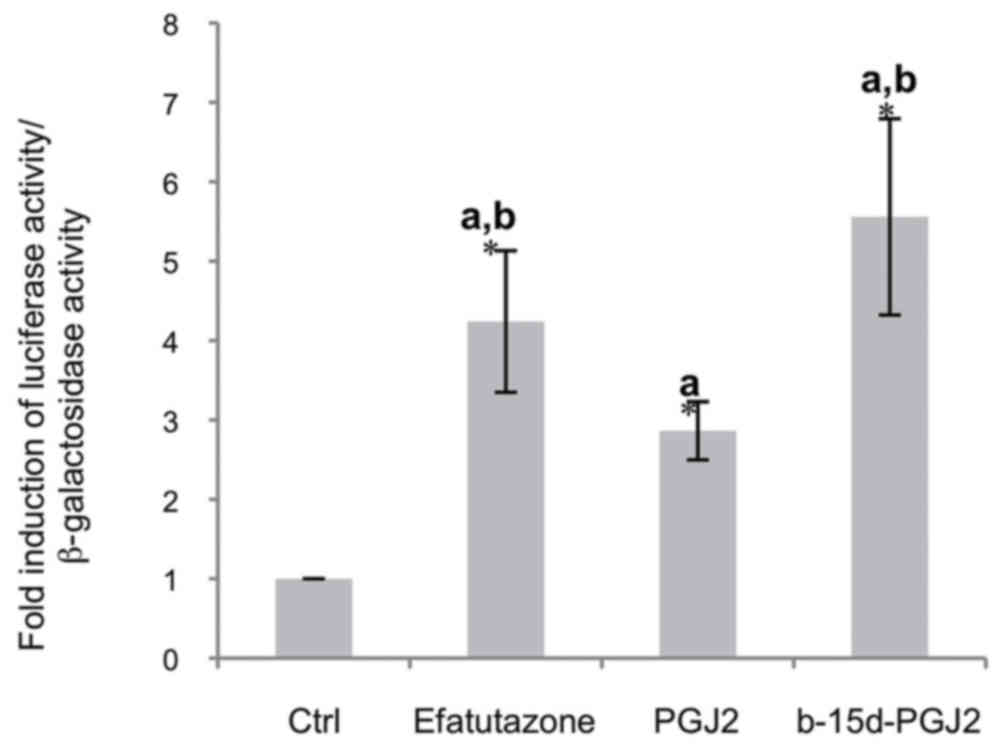
In order to examine whether the biotin moiety could
affect PPARγ activation, we compared the effects of b-15d-PGJ2 to
the PPARγ agonists, efatutazone and 15d-PGJ2. The MCF-7 cells were
transiently transfected with a pPPRE3-tk-luc vector in
the presence of a human PPARγ expression vector. Following 24 h of
treatment with 10 μM efatutazone, 15d-PGJ2 or b-15d-PGJ2,
luciferase activity was stimulated 4.2-fold by efatutazone and
5.5-fold by b-15d-PGJ2, whereas 15d-PGJ2 induced only a 3.2-fold
stimulation (Fig. 4). This result
clearly indicated that b-15d-PGJ2 is a potent PPARγ agonist.
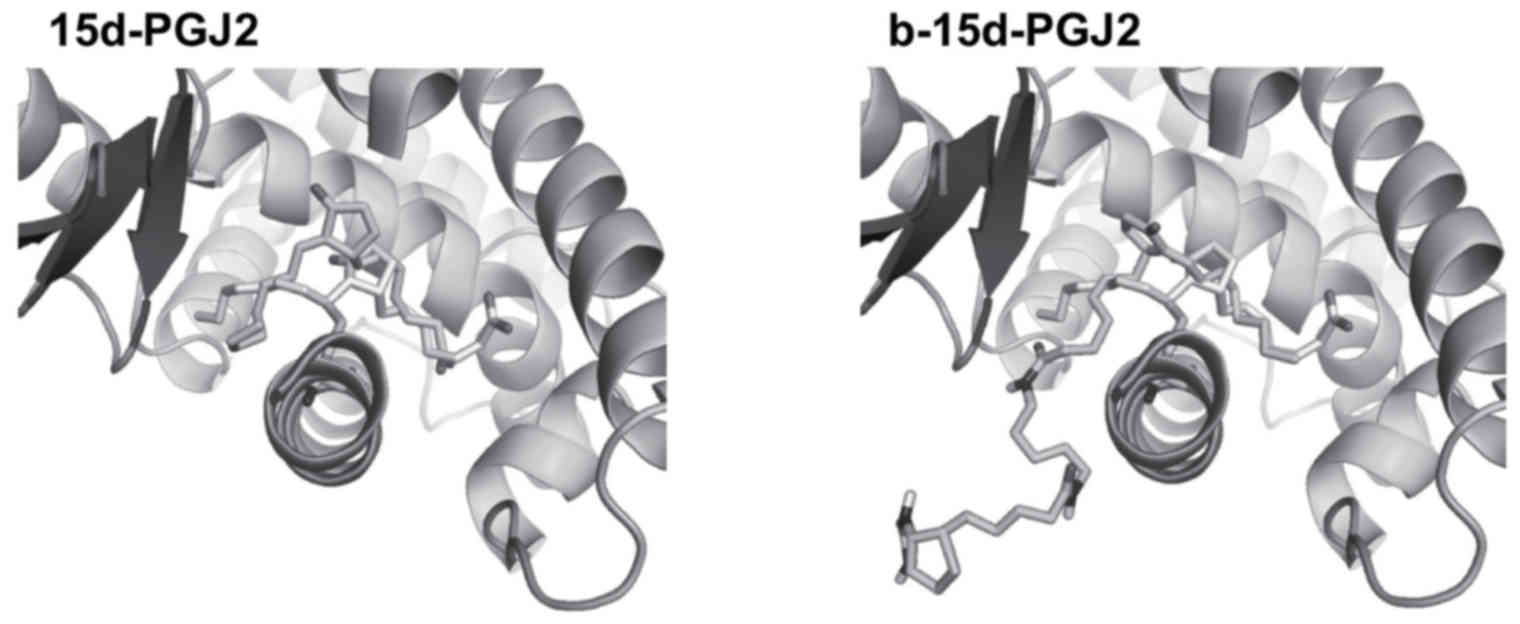
b-15d-PGJ2 docks into the LBD of PPARγ,
in a similar manner as 15d-PGJ2
In order to determine the mechanisms through which
b-15d-PGJ2 activates PPARγ, we performed docking of both ligands
into the ligand binding pocket of the receptor. The docking of
15d-PGJ2 in the LBD of PPARγ yielded a pose in the 3V9V structure
that was very similar to the X-ray pose (PDB entry code 2ZK1). It
is important to note that in the 2ZK1 entry, a covalent bond
between 15d-PGJ2 and Cys285 was present, which cannot be reproduced
by Autodock. However, in the best pose, the distance that we
obtained between the sulfur atom of Cys285 and C-13 of 15d-PGJ2
(the exocyclic Michael acceptor) remained small and compatible with
a subsequent chemical bonding (Fig.
5).
Our docking protocol being able to reproduce in the
3V9V structure a correct pose for 15d-PGJ2, we docked b-15d-PGJ2
into the same target protein using the same protocol. In solution,
each of the two amide bonds of b-15d-PGJ2 can adopt configurations
Z or E. Therefore, we considered in our docking study, 4 different
types of structures for b-15d-PGJ2 (i.e., ZZ, ZE, EZ, or EE). Our
docking results were very similar for all 4 isomers. In all cases,
the biotin moiety lies out of the PPARγ ligand-binding pocket. The
only significant interaction of this molecular fragment was the
interaction of the first amide bond (i.e., the amide bond linked to
the 15d-PGJ2 moiety) and Ser342. Regardless of its configuration,
the amide bond formed a hydrogen bond with either the backbone of
Ser342 (N-H of Ser342 donates to the C=O of the amide bond) and/or
the side chain of Ser342 (the amide bond N-H donates to the
hydroxyl oxygen of Ser342). The ‘15d-PGJ2’ moiety adopted a similar
conformation than the best 15d-PGJ2 docking pose with a closer
distance between Cys285 and C-13 of 15d-PGJ2 (3.1 vs. 3.8 Å between
the two atoms for the b-15d-PGJ2 pose vs. the 15d-PGJ2 pose,
respectively). Taken together, our results indicated that
b-15d-PGJ2 activated PPARγ via a covalent bond with Cys285 in a
very similar manner to what has been reported for 15d-PGJ2
(41).
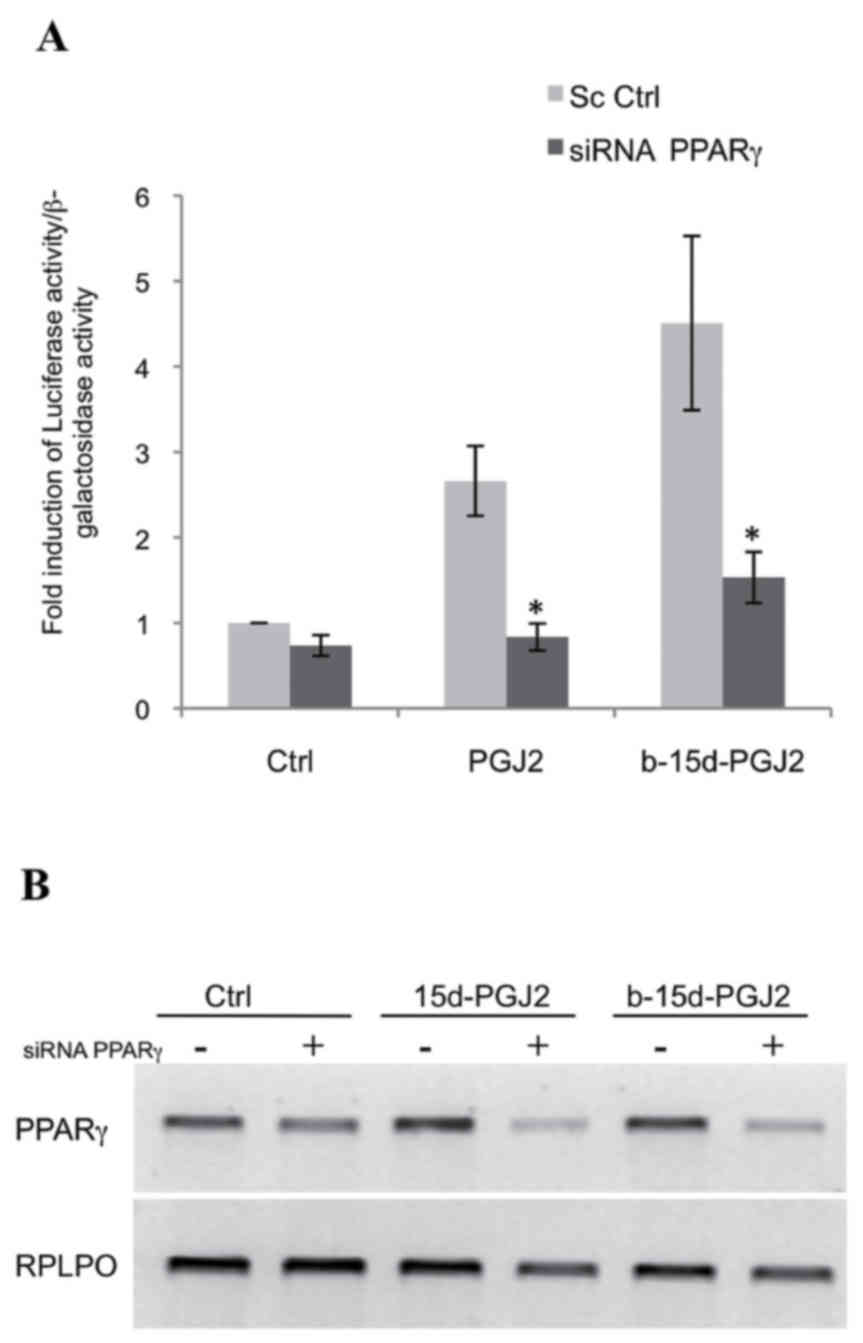
b-15d-PGJ2-induced apoptosis is
attenuated with PPARγ silencing
To determine whether b-15d-PGJ2-induced apoptosis
was mediated by PPARγ, we used siRNA directed against this receptor
in the MCF-7 cells. First, we verified the silencing of PPARγ by
performing a functional assay, in which the MCF-7 cells were
co-transfected with the reporter construct,
pPPRE3-tk-luc, a human PPARγ-expressing vector and PPARγ
siRNA or a scramble sequence. The cells were then treated with
15d-PGJ2, b-15d-PGJ2 (10 μM, 24 h) or ethanol as a control.
In the presence of PPARγ siRNA, we observed a potent and
significant decrease in luciferase activity following treatment
with 15d-PGJ2 (3.2-fold) and following treatment with b-15d-PGJ2
(2.9-fold) compared to scramble sequence (Fig. 6A). These data were then confirmed
by the analysis of the PPARγ mRNA levels. (Fig. 6B). We then also examined the effect
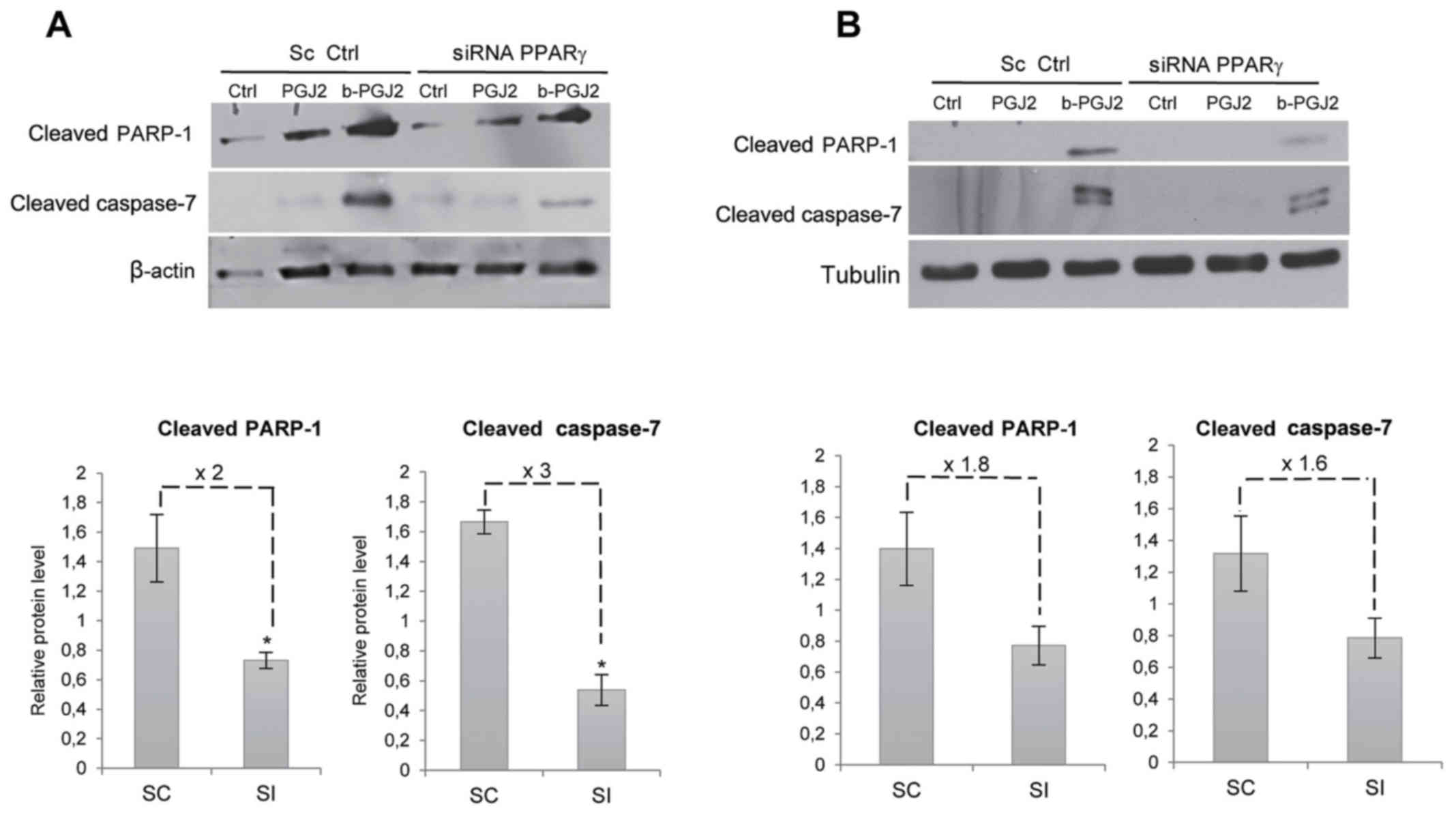
of PPARγ silencing on the b-15d-PGJ2-induced apoptosis of both cell
lines. After PPARγ silencing, the pro-apoptotic signals usually
induced by b-15d-PGJ2 were significantly reduced in the MCF-7
cells: the level of cleaved PARP-1 and cleaved caspase-7 exhibited
a 2- and 3-fold decrease, respectively (Fig. 7A). PPARγ silencing led to a similar
trend in the MDA-MB-231 cells, with a decrease of cleaved PARP-1
and caspase-7 of 1.8- and 1.6-fold, respectively; however, no
significant difference was observed (Fig. 7B).
Discussion
De novo or acquired resistance to current
therapies constitutes a main obstacle in breast cancer therapy. In
this context, the development of alternative treatments remains
essential. The PPARγ agonist, 15d-PGJ2, has been broadly studied
for its anticancer properties (4–11,48,49).
Recently, we described that biotinylation enhanced the
anti-proliferative activity of TGZ, a synthetic agonist of PPARγ,
in breast cancer cells (36). The
aim of the present study was to determine whether the biotinylation
of the natural PPARγ agonist, 15d-PGJ2, could further enhance its
activity on breast cancer cells.
In this context, we examined the effects of
b-15d-PGJ2 on two breast cancer cell lines: MCF-7
(estrogen-dependent) and MDA-MB-231 (triple-negative) cells.
According to previous studies, 15d-PGJ2 inhibited the proliferation
of various cell lines with an IC50 value >50
μM (20,50). By contrast, the linkage of a biotin
group to the terminal carboxylic acid of 15d-PGJ2 greatly enhanced
its anti-proliferative effects on both cell lines. b-15d-PGJ2
significantly reduced MCF-7 cell viability with an IC50
value of 9.8±1.2 μM. b-15d-PGJ2 appeared more potent on the
MDA-MB-231 cells, with an IC50 value of 6.0±0.1
μM. No viable cells remained in the presence of b-15d-PGJ2
(10 μM), whereas 80% of the cells were viable in presence of
15d-PGJ2 (10 μM). In previous studies, we observed that the
MDA-MB-231 cells were more sensitive than the MCF-7 cells to the
biotinylated derivatives of TGZ (35,36).
Of note, the b-15d-PGJ2 inhibition efficiency was significantly
higher than that of efatutazone, a recent PPARγ agonist undergoing
clinical investigation, and whose activity has been demonstrated
in vitro and in xenograft models (51,52).
Moreover, it has been described that PPARγ ligands and 15d-PGJ2
induce apoptosis (53,54). In the present study, we observed
strong apoptotic signals in response to a 24 h treatment with
b-15d-PGJ2 (10 μM), while no signal was detectable in
response to treatment with 15d-PGJ2 (10 μM) in both cell
lines.
Subsequenlty, we investigated the cause of this
improved efficiency. One might suggest that biotin can improve the
cellular intake of b-15d-PGJ2 through receptor-mediated
internalization (31–34,55).
Biotin competition experiments were performed to test this
hypothesis. However, the presence of free biotin did not decrease
the anti-proliferative activity. We also investigated whether
biotin could improve the general cellular uptake. To shed light
into this matter, we performed cooperation experiments between
biotin and 15d-PGJ2. In the MCF-7 and MDA-MB-231 cells, no
significant differences were observed in the anti-proliferative
effects of 15d-PGJ2 in presence of 5 or 10 μM of biotin
(data not shown). Thus, the improved activity was not the result of
an increased uptake of the molecule mediated by the biotin group.
Most likely, the structural modification of 15d-PGJ2 elicited a
better activity of the biotinylated compound as we observed
previously using biotinylated derivatives of TGZ (35,36).
One might suggest that biotinylation could alter the lipophilicity
of 15d-PGJ2 and increase its entry into the cell. We then
calculated the logP of both molecules. The introduction of the
biotin group induces a slight decrease of the ClogP of 0.21, which
is not so significant. However, the non-biotinylated molecule bears
a carboxylic acid group, which is ionized at the pH of the culture
medium. This may lead to a lower diffusion of 15d-PGJ2 through the
cellular membrane, and could explain a lower activity than the
biotinylated compound.
Subsequently, we determined whether biotin could
modify the 15d-PGJ2 PPARγ agonist activity. Co-transfection
experiments demonstrated a clear improvement of PPARγ stimulation.
Indeed, b-15d-PGJ2 induced a 5.5-fold stimulation compared to the
solvent control. This activation was not statistically different
from those induced by efatutazone. Moreover, in this condition,
both ligands were significantly more efficient than 15d-PGJ2.
The question of whether the biotinylation of 15dPGJ2
could improve its binding to PPARγ still remained unanswered. Thus,
to assess this question, we performed a docking analysis of
b-15d-PGJ2 into PPARγ. It appeared that b-15d-PGJ2 potentially
docked into the LBD of PPARγ in a very similar manner as 15d-PGJ2.
However, there were two differences: i) the distance between Cys285
of the receptor and C-13 of the prostaglandin derivative is shorter
(3.1 Å) in the case of the biotinylated derivative than for the
non-biotinylated molecule (3.8 Å). Since this distance may affect
the formation of a beneficial covalent bond between the two
species, this could lead to a different transactivation ability of
both molecules; ii) in the case of b-15d-PGJ2, a new H-bond
interaction was seen with the receptor, compared to 15d-PGJ2. This
interaction is related to the amide bond between 15d-PGJ2 and the
1,5-diaminopentane linker used for the biotinylation. Indeed, it
may establish a hydrogen bond with Ser342 and this could also lead
to a different transactivation ability of both molecules. As a
perspective, these two issues could be addressed as follows. First,
as previously reported by Schopfer et al (56), a covalent binding of b-15d-PGJ2 to
PPARγ LBD could be checked by means of HPLC-MS/MS after incubation.
If this linkage was confirmed, a co-crystallization experiment
could permit X-ray diffraction analysis, enabling the study of
tight interactions between both partners. Second, a new and
non-commercial analog of b-15d-PGJ2 could be chemically
synthetized, with a structure which would not permit the hydrogen
bond with Ser342. For example, this could be obtained by replacing
both amide bonds of the linker by ester bonds. If this interaction
was significant, such a new compound should be less active.
It may also be interesting to investigate whether
the presence of b-15d-PGJ2 may improve interaction with
co-activators or allow recruitment of additional activators.
Moreover, we questioned the exact role played by PPARγ in the
pro-apoptotic activity of b-15d-PGJ2. We observed a PPARγ-dependent
effect, since the pro-apoptotic response was significantly reduced
following PPARγ silencing in MCF-7 cells. Despite the similar
tendency, this decrease was not significant in the MDA-MB-231
cells, suggesting the possibility of a different mechanism of
action. PPARγ-independent mechanisms are probably also involved, as
illustrated in other cell lines for 15d-PGJ2. For instance, as
previously demonstrated, 15d-PGJ2 induced apoptosis through both
PPARγ-dependent and PPARγ-independent pathways in Jurkat T
lymphocytes, in spite of a specific role of PPARγ in cell death
(57). The level of these two
pathways may differ between the MCF-7 and MDA-MB-231 cells.
Nevertheless, both pathways can result from the covalent binding of
15d-PGJ2 by Michael’s addition to nucleophilic components targeting
proteins of intracellular signaling pathways (17,20,58)
or the PPARγ LBD (59,60). In conclusion, the biotinylation of
15d-PGJ2 markedly enhanced the anti-proliferative and pro-apoptotic
activities in breast cancer cells, with a higher efficiency toward
the triple-negative MDA-MB-231 cells compared to the
estrogen-sensitive MCF-7 cells.
The present study contributes to the understanding
of the mechanisms of action of b-15d-PGJ2 and suggests that its
biotinylation may be a promising tool for the further development
of novel therapeutic agents for triple-negative breast cancer. As a
perspective, this study could be extended by in vivo
experiments using mouse tumor xenografts derived from breast cancer
cells. A number of studies have demonstrated the in vivo
therapeutic effects of 15d-PGJ2 in inflammatory diseases, as well
as in cancer (26,27,61);
however, no in vivo data obtained with b-15d-PGJ2 are
available to date, at least to the best of our knowledge.
Nevertheless, the in vivo administration of other
biotinylated compounds has already been described. For instance,
biotinylated nanoparticles containing doxorubicin appeared safer
than doxorubicin solution in terms of cardio-compatibility and
overall effect on morphology of vital organs (62). Besides, when administrated once
weekly to patients with deep veinous thrombosis, idrabiotaparinux,
the biotinylated form of the anticoagulant idraparinux, appeared to
be at least as effective and safe as idraparinux during the 6-month
treatment period (63). These data
may encourage the commencement of in vivo studies with
b-15d-PGJ2. The future of this compound will depend on the
demonstration of its safety and efficacy.
Glossary
Abbreviations
Abbreviations:
|
DMSO
|
dimethylsulfoxide
|
|
EtOH
|
ethanol
|
|
FCS
|
fetal calf serum
|
|
LBD
|
ligand binding domain
|
|
PDB
|
protein data bank
|
|
PG
|
prostaglandin
|
|
15d-PGJ2
|
15-deoxy-∆12,14-prostaglandin J2
|
|
b-15d-PGJ2
|
biotin-conjugated
15-deoxy-∆12,14-prostaglandin J2
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
PPRE
|
PPAR response element
|
|
TGZ
|
troglitazone
|
Acknowledgments
The authors would like to thank Mrs. Marine Geoffroy
for statistical analyses. The pPPRE3-tk-luc, the human
PPARγ expression vector and the pCMV-β-galactosidase (β-Gal)
construct were a generous gift from Professor Philippe Becuwe, Dr
Lionel Domenjoud and Professor Oliver Nusse, respectively.
Funding
This study was supported by grants from the ‘Ligue
Contre le Cancer’, the ‘Conseil régional de Lorraine’ the
Université de Lorraine and CNRS. Christelle Colin was recipient of
a PhD fellowship from the ‘Ministère de l’enseignement supérieur et
de la recherche’. Claudia Cerella was supported by a Waxweiler
grant for cancer prevention research from the Action Lions ‘Vaincre
le Cancer’. Research at LBMCC is financially supported by the
Fondation de Recherche Cancer et Sang, the Recherches Scientifiques
Luxembourg Association, the Een Haerz fir kriibskrank Kanner
Association, the Action Lions Vaincre le Cancer Association and the
Télévie Luxembourg. MD was supported by the National Research
Foundation (NRF), by the MEST of Korea for Tumor Microenvironment
Global Core Research Center (GCRC) grant (grant number
2011-0030001), by the Seoul National University Research grant.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors’ contributions
CCo provided substantial contributions to the design
of the study and acquisition, analysis, and interpretation of data
for the study, and also revised the draft critically for important
intellectual content. MM and AK provided substantial contributions
to data acquisition and interpretation of the data, and also
revised the draft critically for important intellectual content.
CCe, MD, SF, MB and GM provided substantial contributions to the
design of the study and interpretation of the data, and also
revised the draft critically for important intellectual content.
IGV and SK provided substantial contributions to the conception and
design of the study and the acquisition of data, and also drafted
the manuscript. All authors gave final approval of the version be
published, and agree to be accountable for all aspects of the study
in ensuring that questions related to the accuracy or integrity of
any part of the study are appropriately investigated and
resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Referevnces
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Early Breast Cancer Trialists’
Collaborative Group (EBCTCG): Effects of chemotherapy and hormonal
therapy for early breast cancer on recurrence and 15-year survival:
An overview of the randomised trials. Lancet. 365:1687–1717. 2005.
View Article : Google Scholar
|
|
3
|
Nahta R and Esteva FJ: HER2 therapy:
Molecular mechanisms of trastuzumab resistance. Breast Cancer Res.
8:2152006. View
Article : Google Scholar
|
|
4
|
Diers AR, Dranka BP, Ricart KC, Oh JY,
Johnson MS, Zhou F, Pallero MA, Bodenstine TM, Murphy-Ullrich JE,
Welch DR, et al: Modulation of mammary cancer cell migration by
15-deoxy-delta(12,14)-prostaglandin J(2): Implications for
anti-metastatic therapy. Biochem J. 430:69–78. 2010. View Article : Google Scholar
|
|
5
|
Chen YC, Shen SC and Tsai SH:
Prostaglandin D(2) and J(2) induce apoptosis in human leukemia
cells via activation of the caspase 3 cascade and production of
reactive oxygen species. Biochim Biophys Acta. 1743:291–304. 2005.
View Article : Google Scholar
|
|
6
|
Kaikkonen S, Paakinaho V, Sutinen P,
Levonen AL and Palvimo JJ: Prostaglandin 15d-PGJ(2) inhibits
androgen receptor signaling in prostate cancer cells. Mol
Endocrinol. 27:212–223. 2013. View Article : Google Scholar
|
|
7
|
Kim KR, Kim HJ, Lee SK, Ma GT, Park KK and
Chung WY: 15-deoxy-Δ12,14-prostaglandin J2 inhibits
osteolytic breast cancer bone metastasis and estrogen
deficiency-induced bone loss. PLoS One. 10:e01227642015. View Article : Google Scholar
|
|
8
|
Trindade-da-Silva CA, Reis CF, Vecchi L,
Napimoga MH, Sperandio M, Matias Colombo BF, Alves PT, Ward LS,
Ueira-Vieira C and Goulart LR: 15-Deoxy-Δ(12,14)-prostaglandin J2
induces apoptosis and upregulates SOCS3 in human thyroid cancer
cells. PPAR Res. 2016:41062972016. View Article : Google Scholar
|
|
9
|
Wu YC, Jhao YT, Cheng YC and Chen Y:
15-Deoxy-Δ12,14-prostaglandin J2inhibits migration of
human thyroid carcinoma cells by disrupting focal adhesion complex
and adherens junction. Oncol Lett. 13:2569–2576. 2017. View Article : Google Scholar
|
|
10
|
Yaacob NS, Nasir R and Norazmi MN:
Influence of 17β-estradiol on 15-deoxy-Δ12,14
prostaglandin J2 -induced apoptosis in MCF-7 and MDA-MB-231 cells.
Asian Pac J Cancer Prev. 14:6761–6767. 2013. View Article : Google Scholar
|
|
11
|
Muhammad SN, Mokhtar NF and Yaacob NS:
15d-PGJ2 induces apoptosis of MCF-7 and MDA-MB-231 cells via
increased intracellular calcium and activation of caspases,
independent of ERα and ERβ. Asian Pac J Cancer Prev. 17:3223–3228.
2016.
|
|
12
|
Forman BM, Tontonoz P, Chen J, Brun RP,
Spiegelman BM and Evans RM: 15-Deoxy-delta 12, 14-prostaglandin J2
is a ligand for the adipocyte determination factor PPAR gamma.
Cell. 83:803–812. 1995. View Article : Google Scholar
|
|
13
|
Kliewer SA, Lenhard JM, Willson TM, Patel
I, Morris DC and Lehmann JM: A prostaglandin J2 metabolite binds
peroxisome proliferator-activated receptor gamma and promotes
adipocyte differentiation. Cell. 83:813–819. 1995. View Article : Google Scholar
|
|
14
|
Issemann I and Green S: Activation of a
member of the steroid hormone receptor superfamily by peroxisome
proliferators. Nature. 347:645–650. 1990. View Article : Google Scholar
|
|
15
|
Mangelsdorf DJ and Evans RM: The RXR
heterodimers and orphan receptors. Cell. 83:841–850. 1995.
View Article : Google Scholar
|
|
16
|
Yang W, Rachez C and Freedman LP: Discrete
roles for peroxisome proliferator-activated receptor gamma and
retinoid X receptor in recruiting nuclear receptor coactivators.
Mol Cell Biol. 20:8008–8017. 2000. View Article : Google Scholar
|
|
17
|
Kim HJ, Kim JY, Meng Z, Wang LH, Liu F,
Conrads TP, Burke TR, Veenstra TD and Farrar WL:
15-deoxy-Delta12,14-prostaglandin J2 inhibits
transcriptional activity of estrogen receptor-alpha via covalent
modification of DNA-binding domain. Cancer Res. 67:2595–2602. 2007.
View Article : Google Scholar
|
|
18
|
Wang JJ and Mak OT: Induction of apoptosis
by 15d-PGJ2 via ROS formation: An alternative pathway without PPARγ
activation in non-small cell lung carcinoma A549 cells.
Prostaglandins Other Lipid Mediat. 94:104–111. 2011. View Article : Google Scholar
|
|
19
|
Kar R, Singha PK, Venkatachalam MA and
Saikumar P: A novel role for MAP1 LC3 in nonautophagic cytoplasmic
vacuolation death of cancer cells. Oncogene. 28:2556–2568. 2009.
View Article : Google Scholar
|
|
20
|
Lecomte J, Flament S, Salamone S, Boisbrun
M, Mazerbourg S, Chapleur Y and Grillier-Vuissoz I: Disruption of
ERalpha signalling pathway by PPARgamma agonists: Evidences of
PPARgamma-independent events in two hormone-dependent breast cancer
cell lines. Breast Cancer Res Treat. 112:437–451. 2008. View Article : Google Scholar
|
|
21
|
Qin C, Burghardt R, Smith R, Wormke M,
Stewart J and Safe S: Peroxisome proliferator-activated receptor
gamma agonists induce proteasome-dependent degradation of cyclin D1
and estrogen receptor alpha in MCF-7 breast cancer cells. Cancer
Res. 63:958–964. 2003.
|
|
22
|
Uchida K and Shibata T:
15-Deoxy-Delta(12,14)-prostaglandin J2: An electrophilic trigger of
cellular responses. Chem Res Toxicol. 21:138–144. 2008. View Article : Google Scholar
|
|
23
|
Malaviya A and Sylvester PW: Synergistic
antiproliferative effects of combined γ -tocotrienol and PPAR γ
antagonist treatment are mediated through PPAR γ -independent
mechanisms in breast cancer cells. PPAR Res. 2014:4391462014.
View Article : Google Scholar
|
|
24
|
de Jong E, Winkel P, Poelstra K and
Prakash J: Anticancer effects of 15d-prostaglandin-J2 in wild-type
and doxorubicin-resistant ovarian cancer cells: Novel actions on
SIRT1 and HDAC. PLoS One. 6:e251922011. View Article : Google Scholar
|
|
25
|
Koyama M, Izutani Y, Goda AE, Matsui TA,
Horinaka M, Tomosugi M, Fujiwara J, Nakamura Y, Wakada M, Yogosawa
S, et al: Histone deacetylase inhibitors and
15-deoxy-Delta12,14-prostaglandin J2 synergistically
induce apoptosis. Clin Cancer Res. 16:2320–2332. 2010. View Article : Google Scholar
|
|
26
|
Shin SW, Seo CY, Han H, Han JY, Jeong JS,
Kwak JY and Park JI: 15d-PGJ2 induces apoptosis by reactive oxygen
species-mediated inactivation of Akt in leukemia and colorectal
cancer cells and shows in vivo antitumor activity. Clin Cancer Res.
15:5414–5425. 2009. View Article : Google Scholar
|
|
27
|
Prakash J, Bansal R, Post E, de
Jager-Krikken A, Lub-de Hooge MN and Poelstra K: Albumin-binding
and tumor vasculature determine the antitumor effect of
15-deoxy-Delta-(12,14)-prostaglandin-J(2) in vivo. Neoplasia.
11:1348–1358. 2009. View Article : Google Scholar
|
|
28
|
Prost S, Relouzat F, Spentchian M,
Ouzegdouh Y, Saliba J, Massonnet G, Beressi JP, Verhoeyen E,
Raggueneau V, Maneglier B, et al: Erosion of the chronic myeloid
leukaemia stem cell pool by PPARγ agonists. Nature. 525:380–383.
2015. View Article : Google Scholar
|
|
29
|
Komatsu Y, Yoshino T, Yamazaki K, Yuki S,
Machida N, Sasaki T, Hyodo I, Yachi Y, Onuma H and Ohtsu A: Phase 1
study of efatutazone, a novel oral peroxisome
proliferator-activated receptor gamma agonist, in combination with
FOLFIRI as second-line therapy in patients with metastatic
colorectal cancer. Invest New Drugs. 32:473–480. 2014. View Article : Google Scholar
|
|
30
|
Murakami H, Ono A, Takahashi T, Onozawa Y,
Tsushima T, Yamazaki K, Jikoh T, Boku N and Yamamoto N: Phase I
study of Efatutazone, an oral PPARγ agonist, in patients with
metastatic solid tumors. Anticancer Res. 34:5133–5141. 2014.
|
|
31
|
Russell-Jones G, McTavish K, McEwan J,
Rice J and Nowotnik D: Vitamin-mediated targeting as a potential
mechanism to increase drug uptake by tumours. J Inorg Biochem.
98:1625–1633. 2004. View Article : Google Scholar
|
|
32
|
Shi JF, Wu P, Jiang ZH and Wei XY:
Synthesis and tumor cell growth inhibitory activity of biotinylated
annonaceous aceto-genins. Eur J Med Chem. 71:219–228. 2014.
View Article : Google Scholar
|
|
33
|
Taheri A, Dinarvand R, Atyabi F, Nouri F,
Ahadi F, Ghahremani MH, Ostad SN, Borougeni AT and Mansoori P:
Targeted delivery of methotrexate to tumor cells using biotin
functionalized metho-trexate-human serum albumin conjugated
nanoparticles. J Biomed Nanotechnol. 7:743–753. 2011. View Article : Google Scholar
|
|
34
|
Yang W, Cheng Y, Xu T, Wang X and Wen LP:
Targeting cancer cells with biotin-dendrimer conjugates. Eur J Med
Chem. 44:862–868. 2009. View Article : Google Scholar
|
|
35
|
Colin C, Salamone S, Grillier-Vuissoz I,
Boisbrun M, Kuntz S, Lecomte J, Chapleur Y and Flament S: New
troglitazone derivatives devoid of PPARγ agonist activity display
an increased antiproliferative effect in both hormone-dependent and
hormone-independent breast cancer cell lines. Breast Cancer Res
Treat. 124:101–110. 2010. View Article : Google Scholar
|
|
36
|
Salamone S, Colin C, Grillier-Vuissoz I,
Kuntz S, Mazerbourg S, Flament S, Martin H, Richert L, Chapleur Y
and Boisbrun M: Synthesis of new troglitazone derivatives:
Anti-proliferative activity in breast cancer cell lines and
preliminary toxicological study. Eur J Med Chem. 51:206–215. 2012.
View Article : Google Scholar
|
|
37
|
Pishvaian MJ, Marshall JL, Wagner AJ,
Hwang JJ, Malik S, Cotarla I, Deeken JF, He AR, Daniel H, Halim AB,
et al: A phase 1 study of efatutazone, an oral peroxisome
proliferator-activated receptor gamma agonist, administered to
patients with advanced malignancies. Cancer. 118:5403–5413. 2012.
View Article : Google Scholar
|
|
38
|
Colin-Cassin C, Yao X, Cerella C, Chbicheb
S, Kuntz S, Mazerbourg S, Boisbrun M, Chapleur Y, Diederich M,
Flament S, et al: PPARγ-inactive Δ2-troglitazone independently
triggers ER stress and apoptosis in breast cancer cells. Mol
Carcinog. 54:393–404. 2015. View Article : Google Scholar
|
|
39
|
Morris GM, Huey R, Lindstrom W, Sanner MF,
Belew RK, Goodsell DS and Olson AJ: AutoDock4 and AutoDockTools4:
Automated docking with selective receptor flexibility. J Comput
Chem. 30:2785–2791. 2009. View Article : Google Scholar
|
|
40
|
Furukawa A, Arita T, Fukuzaki T, Satoh S,
Mori M, Honda T, Matsui Y, Wakabayashi K, Hayashi S, Araki K, et
al: Substituents at the naphthalene C3 position of
(−)-Cercosporamide derivatives significantly affect the maximal
efficacy as PPARγ partial agonists. Bioorg Med Chem Lett.
22:1348–1351. 2012. View Article : Google Scholar
|
|
41
|
Waku T, Shiraki T, Oyama T, Fujimoto Y,
Maebara K, Kamiya N, Jingami H and Morikawa K: Structural insight
into PPARgamma activation through covalent modification with
endogenous fatty acids. J Mol Biol. 385:188–199. 2009. View Article : Google Scholar
|
|
42
|
Dennington R, Keith TA and Millam JM:
GaussView, version 5.0. Semichem, Inc.; Shawnee Mission, KS:
2008
|
|
43
|
Stewart JJ: Optimization of parameters for
semiempirical methods V: Modification of NDDO approximations and
application to 70 elements. J Mol Model. 13:1173–1213. 2007.
View Article : Google Scholar
|
|
44
|
Frisch MJ, Trucks GW, Schlegel HB,
Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci
B, Petersson GA, et al: Gaussian 09, revision D.01. Gaussian, Inc;
Wallingford, CT: 2013
|
|
45
|
Jakalian A, Jack DB and Bayly CI: Fast,
efficient generation of high-quality atomic charges. AM1-BCC model:
II. Parameterization and validation. J Comput Chem. 23:1623–1641.
2002. View Article : Google Scholar
|
|
46
|
Case DA, Berryman JT, Betz RM, Cerutti DS,
Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW,
et al: AMBER 2015. University of California; San Francisco, CA:
2015
|
|
47
|
Maier JA, Martinez C, Kasavajhala K,
Wickstrom L, Hauser KE and Simmerling C: ff14SB: Improving the
accuracy of protein side chain and backbone parameters from ff99SB.
J Chem Theory Comput. 11:3696–3713. 2015. View Article : Google Scholar
|
|
48
|
Qiao L, Dai Y, Gu Q, Chan KW, Ma J, Lan
HY, Zou B, Rocken C, Ebert MP and Wong BC: Loss of XIAP sensitizes
colon cancer cells to PPARgamma independent antitumor effects of
troglitazone and 15-PGJ2. Cancer Lett. 268:260–271. 2008.
View Article : Google Scholar
|
|
49
|
Ray DM, Akbiyik F and Phipps RP: The
peroxisome proliferator-activated receptor gamma (PPARgamma)
ligands 15-deoxy-Delta12,14-prostaglandin J2 and
ciglitazone induce human B lymphocyte and B cell lymphoma apoptosis
by PPARgamma-independent mechanisms. J Immunol. 177:5068–5076.
2006. View Article : Google Scholar
|
|
50
|
Shen D, Deng C and Zhang M: Peroxisome
proliferator-activated receptor gamma agonists inhibit the
proliferation and invasion of human colon cancer cells. Postgrad
Med J. 83:414–419. 2007. View Article : Google Scholar
|
|
51
|
Pirat C, Farce A, Lebègue N, Renault N,
Furman C, Millet R, Yous S, Speca S, Berthelot P, Desreumaux P, et
al: Targeting peroxisome proliferator-activated receptors (PPARs):
Development of modulators. J Med Chem. 55:4027–4061. 2012.
View Article : Google Scholar
|
|
52
|
Shimazaki N, Togashi N, Hanai M, Isoyama
T, Wada K, Fujita T, Fujiwara K and Kurakata S: Anti-tumour
activity of CS-7017, a selective peroxisome proliferator-activated
receptor gamma agonist of thiazolidinedione class, in human tumour
xenografts and a syngeneic tumour implant model. Eur J Cancer.
44:1734–1743. 2008. View Article : Google Scholar
|
|
53
|
Ishihara S, Rumi MA, Okuyama T and
Kinoshita Y: Effect of prostaglandins on the regulation of tumor
growth. Curr Med Chem Anticancer Agents. 4:379–387. 2004.
View Article : Google Scholar
|
|
54
|
Zhang Z, Xu Y, Xu Q and Hou Y: PPARγ
against tumors by different signaling pathways. Onkologie.
36:598–601. 2013. View Article : Google Scholar
|
|
55
|
Kansara V, Luo S, Balasubrahmanyam B, Pal
D and Mitra AK: Biotin uptake and cellular translocation in human
derived retinoblastoma cell line (Y-79): A role of hSMVT system.
Int J Pharm. 312:43–52. 2006. View Article : Google Scholar
|
|
56
|
Schopfer FJ, Cole MP, Groeger AL, Chen CS,
Khoo NK, Woodcock SR, Golin-Bisello F, Motanya UN, Li Y, Zhang J,
et al: Covalent peroxisome proliferator-activated receptor gamma
adduction by nitro-fatty acids: Selective ligand activity and
anti-diabetic signaling actions. J Biol Chem. 285:12321–12333.
2010. View Article : Google Scholar
|
|
57
|
Ferreira-Silva V, Rodrigues AC, Hirata TD,
Hirabara SM and Curi R: Effects of 15-deoxy-Delta12, 14
prostaglandin J2 and ciglitazone on human cancer cell cycle
progression and death: The role of PPARgamma. Eur J Pharmacol.
580:80–86. 2008. View Article : Google Scholar
|
|
58
|
Marcone S and Fitzgerald DJ: Proteomic
identification of the candidate target proteins of
15-deoxy-delta12,14-prostaglandin J2. Proteomics.
13:2135–2139. 2013. View Article : Google Scholar
|
|
59
|
Itoh T, Fairall L, Amin K, Inaba Y, Szanto
A, Balint BL, Nagy L, Yamamoto K and Schwabe JW: Structural basis
for the activation of PPARgamma by oxidized fatty acids. Nat Struct
Mol Biol. 15:924–931. 2008. View Article : Google Scholar
|
|
60
|
Shiraki T, Kamiya N, Shiki S, Kodama TS,
Kakizuka A and Jingami H: Alpha,beta-unsaturated ketone is a core
moiety of natural ligands for covalent binding to peroxisome
proliferator-activated receptor gamma. J Biol Chem.
280:14145–14153. 2005. View Article : Google Scholar
|
|
61
|
Surh YJ, Na HK, Park JM, Lee HN, Kim W,
Yoon IS and Kim DD: 15-Deoxy-Δ12,14-prostaglandin
J2, an electrophilic lipid mediator of anti-inflammatory
and pro-resolving signaling. Biochem Pharmacol. 82:1335–1351. 2011.
View Article : Google Scholar
|
|
62
|
Singh Y, Durga Rao Viswanadham KK, Kumar
Jajoriya A, Meher JG, Raval K, Jaiswal S, Dewangan J, Bora HK, Rath
SK, Lal J, et al: Click biotinylation of PLGA template for biotin
receptor oriented delivery of doxorubicin hydrochloride in 4T1
cell-induced breast cancer. Mol Pharm. 14:2749–2765. 2017.
View Article : Google Scholar
|
|
63
|
Equinox Investigators: Efficacy and safety
of once weekly subcutaneous idrabiotaparinux in the treatment of
patients with symptomatic deep venous thrombosis. J Thromb Haemost.
9:92–99. 2011. View Article : Google Scholar
|