Introduction
Cholangiocarcinoma (CCA) is one of the leading types
of cancer in Thailand, with patients having a poor prognosis and
treatment outcome. As there is no effective chemotherapeutic
treatment at present, additional therapeutic approaches with
specific and effective targets are required for CCA treatment. In
Thailand, the incidence rate of CCA is associated with the
prevalence of Opisthorchis viverrini infection (1). Chronic inflammation, often coupled
with injury of the bile duct epithelium, is a common and important
contributor to the malignant transformation of cholangiocytes.
Nitric oxide, activated by inflammatory cytokines, is implicated in
inflammation and the carcinogenesis of CCA (2). Interleukin 6 (IL6) is among the
inflammatory cytokines that have been associated with the
pathogenesis of CCA. The levels of IL6 have been observed to be
elevated in patients with CCA, compared with those in healthy
controls (3). Through the AKT
signaling pathway, IL6 upregulates myeloid leukemia 1, resulting in
resistance to apoptosis in CCA (4). The involvement of other signaling
molecules has also been investigated in CCA cell growth and
survival.
Several studies have reported the involvement of
phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of
rapamycin (mTOR) signaling in CCA. It has been shown that the
expression and activation of AKT protein increase during the
development and progression of CCA (5,6), and
that AKT is a promising target for CCA treatment (7). In addition, an activated PI3K/AKT
signaling pathway is frequently found in CCA cells resistant to
chemotherapy (8).
Epidemiological investigations have revealed gender
disparities in CCA, for example, the incidence rate is higher in
men (9). Liver biopsies from
patients with intrahepatic CCA are positive for the estrogen
receptor (ER), both ERα and ERβ subtypes, whereas normal liver
cholangiocytes are ER-negative (10). The growth-stimulating effects of
estrogen (E2) via ER signaling or crosstalk with the epidermal
growth factor receptor (EGFR) and AKT pathways are well established
in breast cancer and endometrial cancer (11–14).
E2 has also been shown to be involved in the proliferation
(10,15) and invasion (16) of CCA. Additionally, E2 stimulates
the production of IL6 in biliary epithelial cells (17). These observations suggest that the
disruption of E2 may be useful for CCA therapy.
Genistein (GE), an isoflavone found in soybeans, has
multi-targeted biological effects in cancer cells, leading to the
inhibition of cell growth and induction of apoptotic cell death in
several cancer cell lines, including breast (18), lung (19) and colon cancer (20). In addition to being a
phytoestrogen, the biological effects of GE have been reported to
be associated with tyrosine kinase inhibition, anti-inflammatory
effects (21,22) and the inhibition of AKT (23,24).
The present study investigated the effects of GE and
the involvement of E2 in human CCA cell lines by targeting AKT,
EGFR receptor tyrosine kinase, IL6, and ER. The results showed that
GE exhibited cytotoxicity in human HuCCA-1 and RMCCA-1 cell lines.
GE reduced the protein levels of p-AKT and p-EGFR, and the
production of IL6. Additionally, GE induced downregulation of the
expression of ERα at the mRNA and protein levels. The present study
demonstrated for the first time, to the best of our knowledge, that
E2 deprivation potentiated the GE-induced reduction in the
production of IL6, and protein levels of total AKT, p-AKT (Ser473)
and p-EGFR (Tyr1173) in CCA.
Materials and methods
Chemicals and reagents
GE, 17-β estradiol (E2) and triciribine were
purchased from Sigma-Aldrich; EMD Millipore (Billerica, MA, USA).
LY294002 was obtained from Cayman Chemical Company (Ann Arbor, MI,
USA). AG1478 was acquired from Merck Millipore (Darmstadt,
Germany). Ham's F12 medium, phenol red-free RPMI-1640 medium, and
supplements were purchased from Gibco; Thermo Fisher Scientific,
Inc. (Waltham, MA, USA). Fetal bovine serum (FBS) treated with
dextran-charcoal (CSS) was purchased from HyClone; GE Healthcare
Life Sciences (Logan, UT, USA). Antibodies were obtained as
follows: Anti-human p-AKT (Ser473) cat. no. 13038, AKT cat. no.
9272, p-p38 (Thr180/Tyr182) cat. no. 9215, p38 cat. no. 9217,
p-ERK1/2 (Thr202/Tyr204) cat. no. 9106 and ERK1/2 cat. no. 9102
from Cell Signaling Technology, Inc. (Danvers, MA, USA); ERα cat.
no. sc-8002, p-ERα (Ser118) cat. no. sc-12915, p-EGFR (Tyr1173)
cat. no. sc-12351 and EGFR cat. no. sc-03 from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA); ERβ cat. no. 05-824 and
inducible nitric oxide synthase (iNOS) cat. no. ABN26 from Merck
Millipore, and β-actin cat. no. A5316 from Sigma-Aldrich; EMD
Millipore (Billerica, MA, USA). HRP-conjugated secondary antibodies
obtained were as follows: Anti-rabbit IgG (cat. no. 5127) from Cell
Signaling Technology, Inc.; anti-goat IgG (cat. no. HAF109) from
R&D System Inc. (Minneapolis, MN, USA); and anti-mouse IgG
(cat. no. 1706516) from Bio-Rad Laboratories, Inc. (Hercules, CA,
USA).
Cell lines and cell culture
The HuCCA-1 and RMCCA-1 human intrahepatic CCA cell
lines were established and kindly provided by Professor Stitaya
Sirisinha and Dr Kawin Leelawat, respectively (25,26).
The cells were cultured in complete medium (FBS condition)
containing Ham's F12 culture medium supplemented with 10% FBS, 2 mM
L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin.
For the E2-deprived condition (CSS condition), cells were switched
to a culture of phenol red-free RPMI-1640 medium supplemented with
10% CSS, 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin for 24 h prior to treatment. The cells were maintained
at 37°C in a humidified atmosphere of 5% CO2.
The difference between the FBS and CSS cultures was
the concentration of steroid hormone E2. The E2 concentration in
FBS is reported to be >100 pg/ml (27), whereas the concentration in CSS is
in the range of 15–20 pg/ml (from COA of product). This model has
been commonly used by others and by our group at the Laboratory of
Pharmacology, Chulabhorn Research Institute (Bangkok, Thailand) as
in vitro E2-deprived cell culture condition (28,29).
Cell viability assay
The cells maintained in the FBS condition were
plated into 96-well plates at 1×104 cells/well.
Following incubation for 24 h, the medium containing various
concentrations of GE (50–200 µM) or vehicle (0.1% DMSO) were
added in triplicate wells and incubated at 37°C for 24 or 48 h.
Following the manufacturer's protocol, following this period of
incubation, 10 µl of PrestoBlue™ cell viability reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to detect
cell viability. The reaction was measured by spectrofluorometry to
detect fluorescence at 560 nm for excitation and 590 nm for
emission using a SpectraMax microplate reader (Molecular Devices
LLC, Sunnyvale, CA, USA).
Western blot analysis
The HuCCA-1 or RMCCA-1 cells were plated on a 100-mm
culture dish at 4×106 cells with 10 ml per plate and
treated with GE (50, 100 and 200 µM for 0.5, 6 or 24 h),
LY294002 (20 µM for 0.5, 6 or 24 h), triciribine (20
µM for 0.5, 6 or 24 h), or AG1478 (0.01 µM for 6 h)
in either FBS or CSS conditions, according to each experimental
design. Following the exposure period, the cells were collected and
centrifuged 500 × g for 10 min at 4°C. The pellets were then
re-suspended in RIPA buffer containing 50 mM Tris (pH 7.4), 150 mM
NaCl, 1 mM EDTA, 0.1% SDS, 1% sodium deoxycholate, 1% NP40 and
protease inhibitors (Calbiochem; EMD Millipore) mixed with 100 mM
PMSF and phosphatase inhibitors (1 mM Na3VO4,
100 mM NaF and 1 nM okadaic acid). The protein concentration was
determined using Bradford reagent according to the manufacturer's
protocol (Bio-Rad Laboratories, Inc.). For immunoblotting, 50
µg of total protein was separated using 7.5% SDS-PAGE and
then electro-transferred onto a nitrocellulose membrane (Amersham
Biosciences, Buckinghamshire, UK). The membrane was incubated in
blocking buffer [5% non-fat dry milk in TBST buffer (10 mM Tris-HCl
pH 8.0, 150 mM NaCl, and 0.05% Tween-20)] for 1 h at room
temperature. The membrane was probed overnight with primary
antibody p-AKT (Ser473) (1:1,000), AKT (1:2,000), p-p38
(Thr180/Tyr182) (1:2,000), p38 (1:2,000), p-ERK1/2 (Thr202/Tyr204)
(1:2,000), ERK1/2 (1:2,000), ERα (1:1,000), p-ERα (Ser118)
(1:1,000), p-EGFR (Tyr1173) (1:1,000), EGFR (1:1,000), ERβ
(1:2,000) or iNOS (1:1,000) at 4°C, followed by incubation with
appropriate HRP-conjugated secondary antibody (1:3,000) at room
temperature for 2 h. Subsequently, the blot was reacted with a
chemiluminescent substrate (Amersham; GE Healthcare Life Sciences,
Chalfont, UK), and exposed to X-ray film (GE Healthcare Life
Sciences). The protein levels were quantified by densitometry using
ImageQuant TL 7.0 software (GE Healthcare Life Sciences). The
expression level of each protein was normalized to the
corresponding protein level of β-actin derived from the same
blot.
Enzyme-link immunosorbent assay
(ELISA)
The culture medium collected following treatment was
analyzed for levels of IL6 using the DuoSet ELISA Development kit
(R&D System Inc.). The absorbance was immediately measured at
450 nm, with the wavelength correction at 570 nm, on a SpectraMax
microplate reader (Molecular Devices LLC., San Jose, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total mRNA was isolated from 4×106
HuCCA-1 cells using an RNA purification cell kit (5 PRIME GmbH,
Hamburg, Germany) according to the manufacturer's recommendations.
Contaminating DNA was removed using the Turbo DNA-free™ DNase kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.). A master
mixture was created in a 50 µl total reaction volume
containing 1X RNA-direct™ SYBR®-Green Realtime
PCR Master Mix (Toyobo Co., Ltd., Osaka, Japan), 0.2 µM
forward primer, 0.2 µM reverse primer and 250 ng/µl
RNA sample. The RT-qPCR analysis was performed on an
ABI-StepOnePlus thermocycler (Applied Biosystems; Thermo Fisher
Scientific, Inc.), starting with the RT reaction (10 min at 50°C),
followed by 40 cycles of two-step PCR (95°C for 10 sec and 60°C for
30 sec). Standard melting curve analysis was performed. The
housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase, was
used for normalization. The relative quantification of the target
genes was performed using StepOnePlus™ software version 2.1
(Applied Biosystems; Thermo Fisher Scientific, Inc.), based on the
2−ΔΔCq method, and resulted in the relative
transcription level of the target genes in treated cells, compared
with those in control cells. The primer sequences are listed in
Table I.
 | Table IPrimers pairs for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primers pairs for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Primer
sequence | Product size
(bp) | Accession no. |
|---|
| ERα
forward |
5′-CGTGGTGCCCCTCTATGA-3′ | 165 | NM_000125.3 |
| ERα
reverse |
5′-TCCCCCGTGATGTAATAC-3′ | | |
| ERβ1
forward |
5′-GAATGCCCACGTGCTTCGCG-3′ | 150 | NM_001271877.1 |
| ERβ1
reverse |
5′-GCCTGTGACCTCTGTGGGCC-3′ | | |
| ERβ2
forward |
5′-ACCCTCTAAATCAACTCGGTGGCCT-3′ | 240 | NM_001291723.1 |
| ERβ2
reverse |
5′-CCCTGGGCAGTTAAGGAGACCT-3′ | | |
| GAPDH
forward |
5′-GAAGGTGAAGGTCGGAGTC-3′ | 226 | NM_002046.5 |
| GAPDH
reward |
5′-GAAGATGGTGATGGGATTTC-3′ | | |
Statistical analysis
Data are expressed as the mean ± standard error of
the mean calculated from at least three independent experiments.
Multiple comparisons were performed using one-way analysis of
variance with Dunett's multiple comparisons test by Stata™ program
version 10.1 (StataCorp, College Station, TX, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
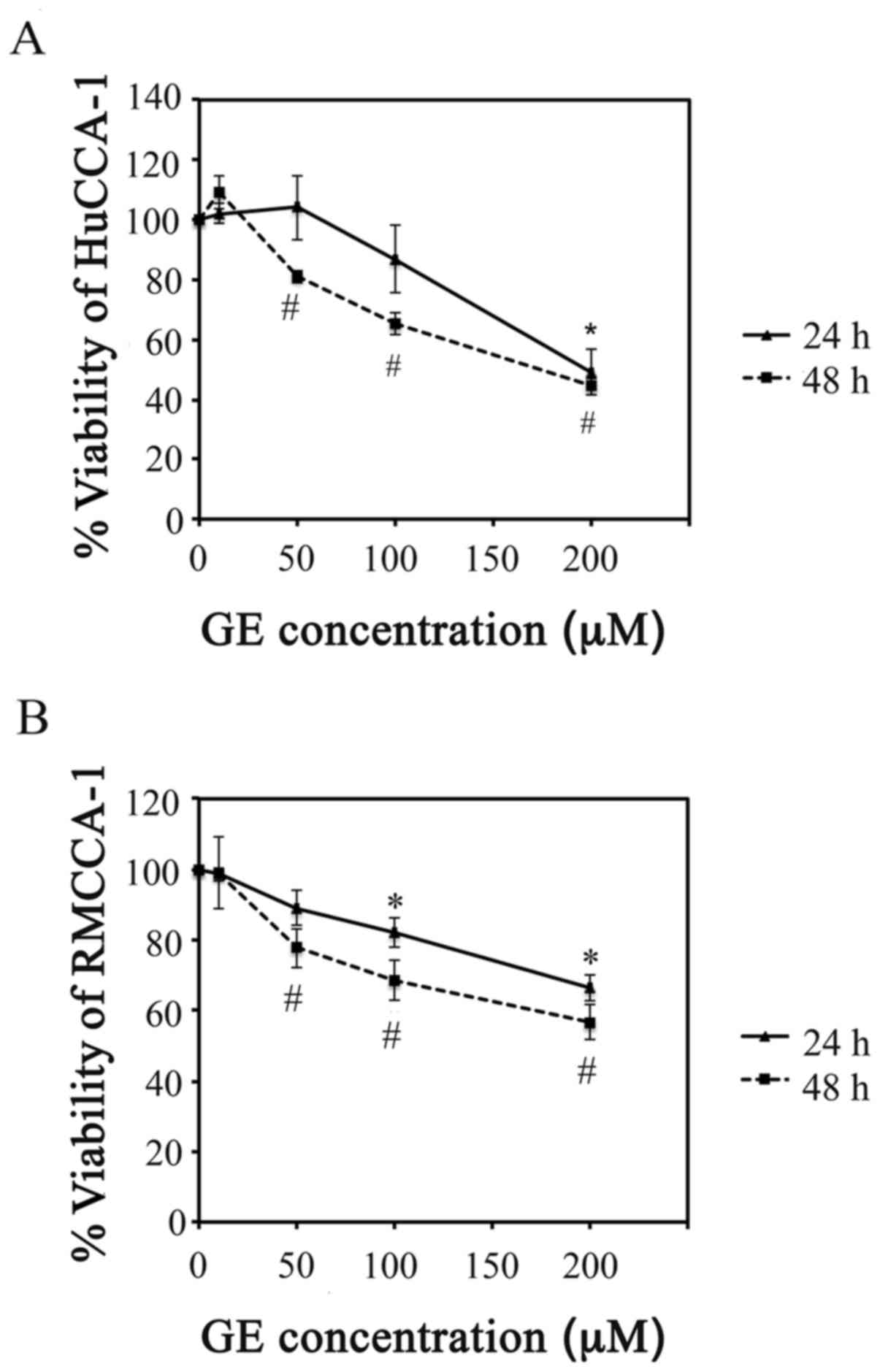
GE reduces HuCCA-1 and RMCCA-1 cell
viability
Two human CCA cell lines, HuCCA-1 and RMCCA-1, were
used to investigate the cytotoxic effect of GE. As shown in
Fig. 1, GE concentrations ranging
between 10 and 200 µM exhibited a dose- and time-dependent
induction of cytotoxic effects in the HuCCA-1 and RMCCA-1 cells. A
statistically significant cytotoxic effect (P<0.05) of GE was
observed, starting at a GE concentration of 50 µM in the
HuCCA-1 cells (Fig. 1A) and 100
µM in the RmCCA-1 cells (Fig.
1B) when treated for 48 h.
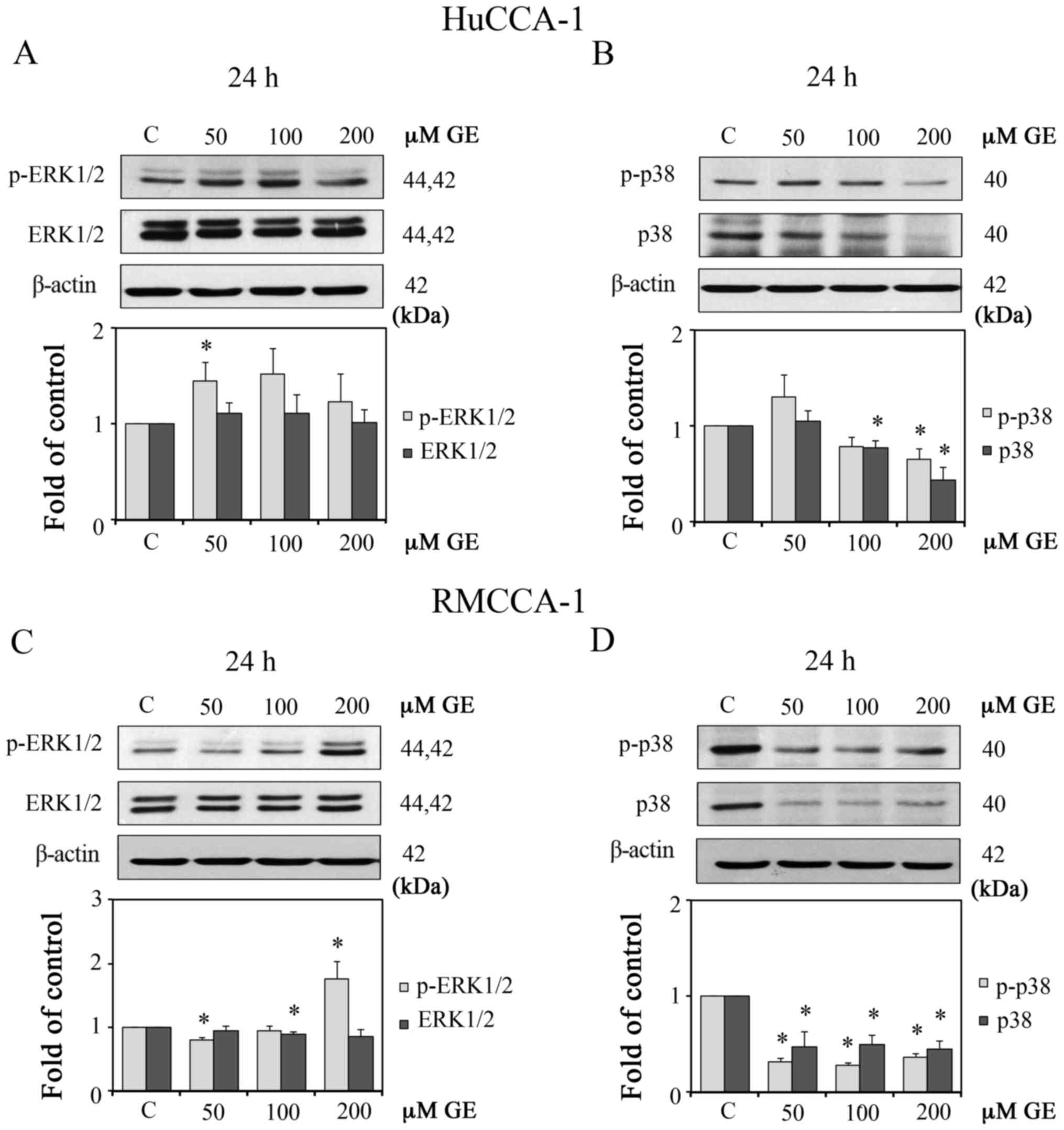
GE reduces the protein expression of
p-AKT in HuCCA-1 and RMCCA-1 cells
AKT protein is a promising target for CCA treatment
as its expression and activation increase during the development
and progression of CCA (5–7). Studies in colorectal cancer cells
have demonstrated that GE can induce apoptosis by inhibiting the
activation of AKT (30).
Therefore, the present study examined whether GE attenuated the
activity of AKT in the CCA cell lines. LY294002 (PI3K inhibitor)
and triciribine (AKT inhibitor) were used as positive controls. As
shown in Fig. 2A–F, GE
downregulated the protein expression of p-AKT (Ser473) in the two
cell lines.
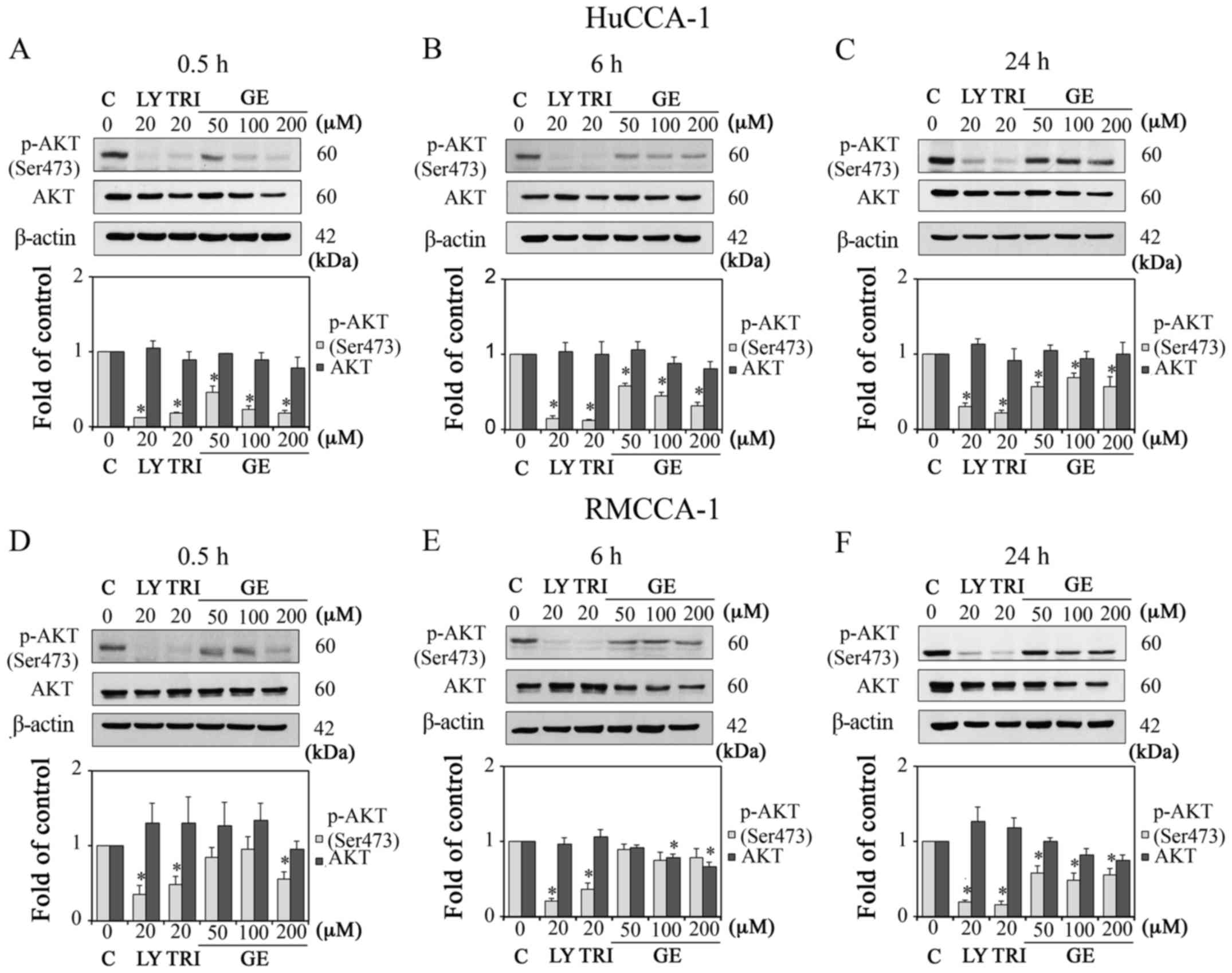 | Figure 2Effect of GE on AKT proteins in
HuCCA-1 and RMCCA-1 cells. Western blot analysis and quantification
of the expression levels of p-AKT (Ser473) and AKT in cells treated
with 10, 50, 100 and 200 µM GE, 20 µM LY and 20
µM TRI, in complete medium. Results in HuCCA-1 cells at (A)
0.5 h, (B) 6 h and (C) 24 h post-treatment, and RMCCA-1 cells at
(D) 0.5 h, (E) 6 h and (F) 24 h post-treatment. Data are presented
as the mean ± standard error of the mean of at least three
independent experiments. *P<0.05, vs. C. GE,
genistein; LY, LY294002; TRI, triciribine; C, control (0.1% DMSO
vehicle); p-, phosphorylated. |
GE alters EGFR signaling in HuCCA-1 and
RMCCA-1 cells
As AKT is one of the downstream molecules of EGFR,
exhibiting prolonged activation in CCA (31), and GE is a known tyrosine kinase
inhibitor (32), the present study
aimed to determine the effect of GE on the protein expression of
p-EGFR (Tyr1173) in the CCA cell lines. As shown in Fig. 3A–F, GE suppressed the protein
expression levels of p-EGFR (Tyr1173) in the HuCCA-1 and RMCCA-1
cell lines.
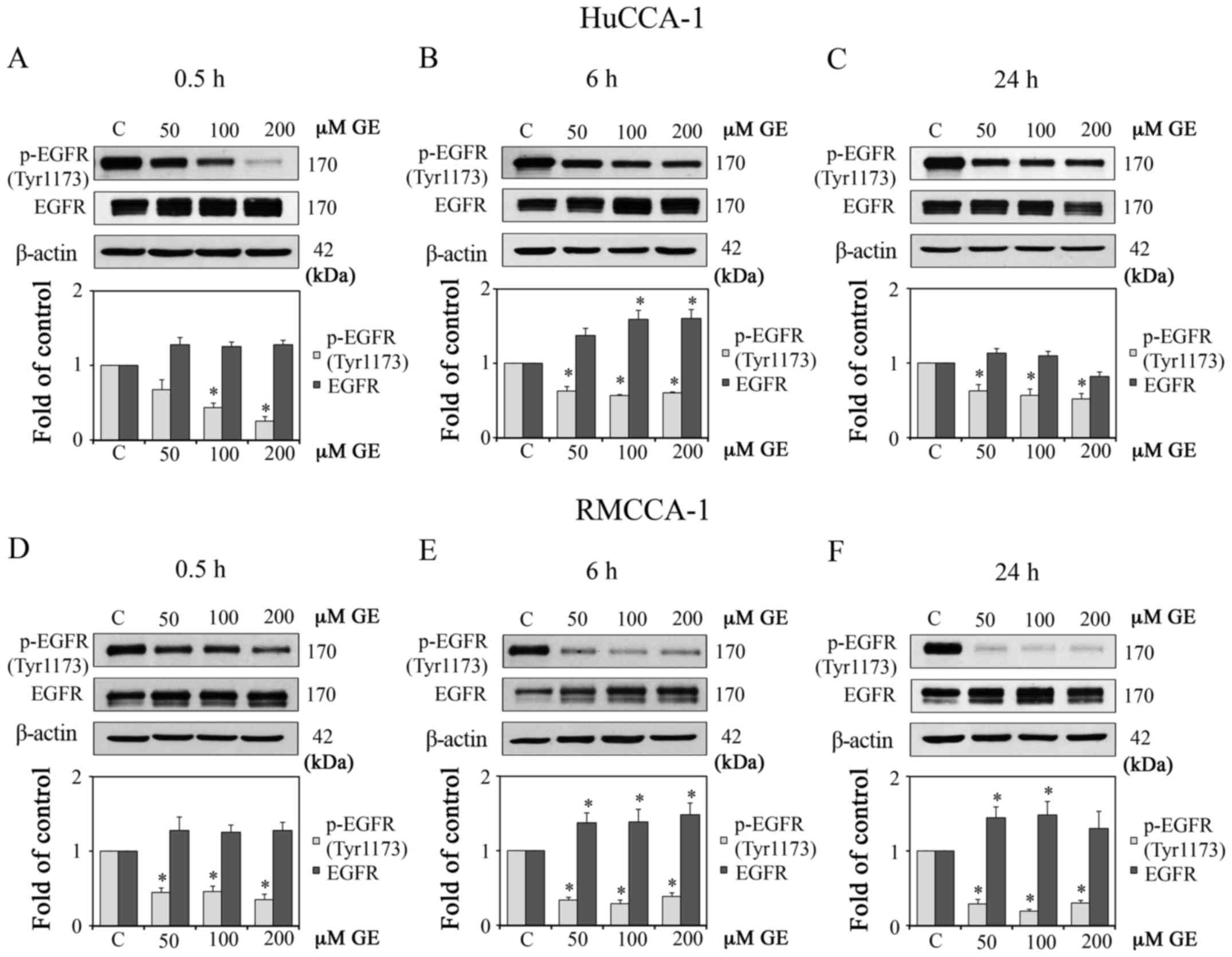 | Figure 3Effect of GE on EGFR proteins in
HuCCA-1 and RMCCA-1 cells. Western blot analysis and quantification
of the expression levels of p-EGFR (Tyr1173) and EGFR in cells
treated with 10, 50, 100 and 200 µM GE, in complete medium.
Results of HuCCA-1 cells at (A) 0.5 h, (B) 6 h and (C) 24 h
post-treatment, and (D-F) RMCCA-1 cells at (D) 0.5 h, (E) 6 h and
(F) 24 h post-treatment. Data are presented as the mean ± standard
error of the mean of at least three independent experiments.
*P<0.05, vs. C. GE, genistein; EGFR, epidermal growth
factor receptor; C, control (0.1% DMSO vehicle); p-,
phosphorylated. |
The mitogen-activated protein kinases (MAPKs), both
extracellular signal-regulated kinase (ERK)1/2 and p38, are
involved in cell proliferation of KMCH-1 malignant cholangiocyte
cells (33). Therefore, the
present study investigated the effect of GE on ERK1/2 and p38. The
results showed that GE decreased the protein expression levels of
p-p38 (Thr180/Tyr182) but enhanced the protein levels of p-ERK1/2
in the two CCA cell lines, as shown in Fig. 4A–D.
Activation of AKT in HuCCA-1 cells is
mediated by EGFR and other upstream molecules
The previous experiments demonstrated that GE
induced a reduction of active p-EGFR and p-AKT. The activation of
AKT can be mediated by upstream molecules other than EGFR. It has
been shown that non-genomic ER signaling can crosstalk with EGFR,
which leads to the activation of ERK1/2 and consequently induces
cell growth (29). The present
study examined whether the activation of AKT in CCA cells is
mediated only by EGFR, or involves other molecules, including ER.
The preliminary results revealed that HuCCA-1 cells expressed ERα
and ERβ proteins, whereas RMCCA-1 cells expressed only ERβ protein.
Therefore, only HuCCA-1 cells were utilized in this and the
following experiments in order to investigate the role of ER in the
effects of GE. To examine whether the GE-inhibited activation of
AKT is mediated by EGFR or other molecules, the EGFR inhibitor
(AG1478) was utilized as a positive control. As shown in Fig. 5A and B, GE markedly reduced the
protein expression levels of p-EGFR (Tyr1173) and p-AKT (Ser473),
whereas AG1478 (10 nM) inhibited the protein expression of p-EGFR
(Tyr1173) only. These results suggested that, in addition to EGFR,
other upstream molecules may be involved in the phosphorylation of
AKT in HuCCA-1 cells.
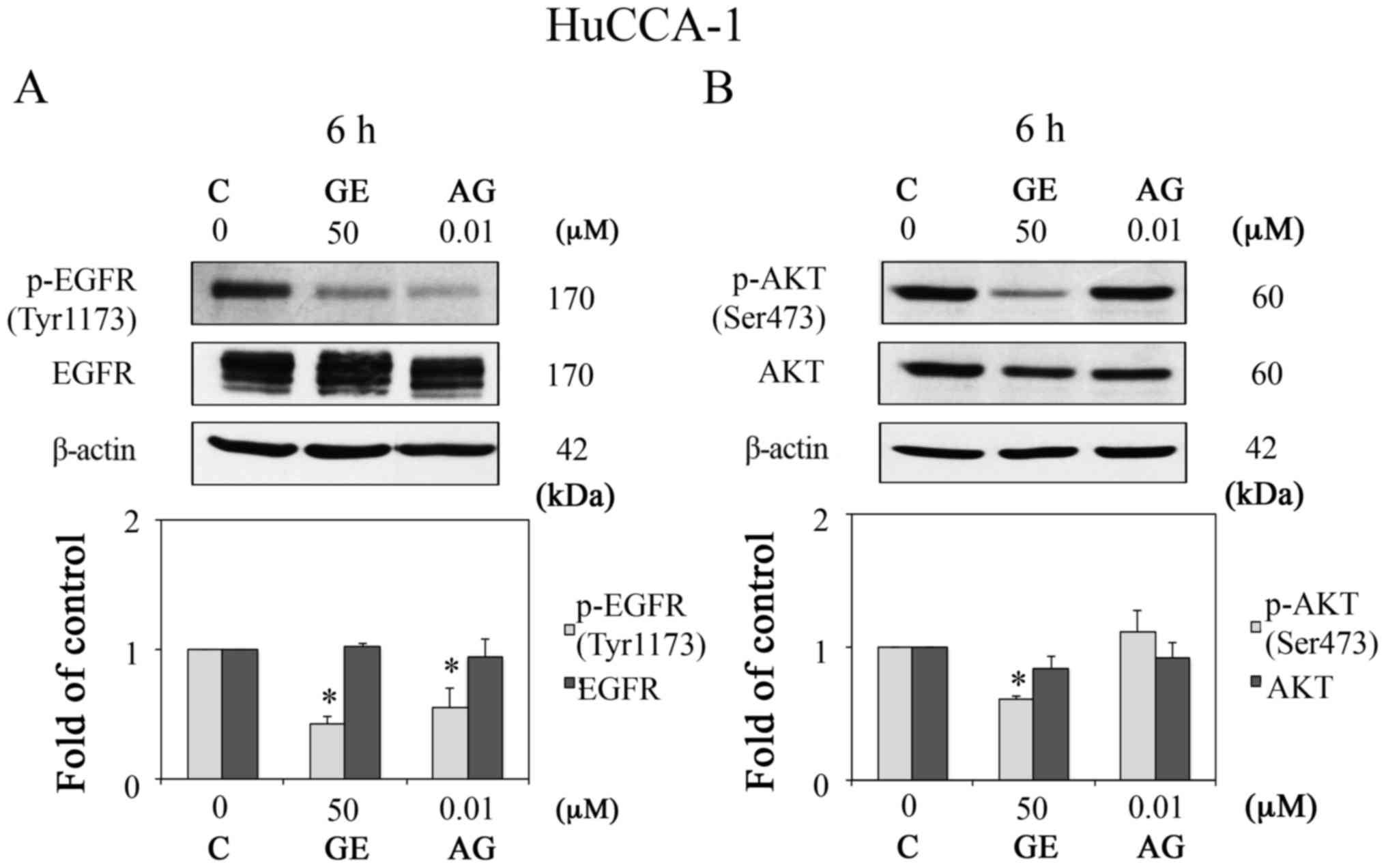 | Figure 5Activation of AKT in HuCCA-1 cells is
not only mediated by EGFR. Western blot analysis of (A) p-EGFR
(Tyr1173) and EGFR, and (B) p-AKT (Ser473) and AKT, in HuCCA-1
cells treated with 0.1% DMSO vehicle control, 10 nM AG, and 50
µM GE for 6 h in complete medium (upper panels), and
quantification of the western blot data (lower panels). Data are
presented as the mean ± standard error of the mean of at least five
independent experiments. *P<0.05, vs. C. GE,
genistein; AG, AG1478; C, control; EGFR, epidermal growth factor
receptor; p-, phosphorylated. |
Reduction of total AKT protein and
inhibition of the activation of EGFR by GE are enhanced under
conditions of E2 deprivation
The two CCA cell lines used in the present study are
intrahepatic CCA cell lines. HuCCA-1 cells were originated from
O. viverrini-infected patients, whereas RMCCA-1 cells were
derived from a patient, whose parasitic infection information was
not reported (25,26). Therefore, the two cell lines were
used in the present study. E2 is involved in CCA cancer
proliferation (10,15) and crosstalk exists between ER and
EGFR signaling in breast cancer cell lines (29). Therefore, it was hypothesized that
E2 may be partly involved in the activation of EGFR in CCA cells.
In order to investigate the role of E2 in the effects of GE,
experiments designed in CSS conditions were compared with those in
FBS conditions. To confirm whether E2 is involved in the GE-induced
activation of EGFR, HUCCA-1 cells were cultured and treated with GE
under CSS conditions. As expected, GE markedly suppressed the
protein expression level of p-EGFR (0.61±0.11-, 0.57±0.10-and
0.55±0.05-fold of control at GE concentrations of 50, 100, and 200
µM, respectively) under E2 deprivation (Fig. 6A). However, in FBS conditions
(Fig. 3), GE reduced the protein
expression levels of p-EGFR to a lesser degree (0.78±0.11-,
0.66±0.10- and 0.64±0.08-fold of control for GE concentrations of
50, 100, and 200 µM, respectively). Additionally, the
down-regulation of total protein levels of AKT induced by GE under
E2-deprivation (0.93±0.05-, 0.88±0.10- and 0.66±0.06-fold of
controls for GE concentrations of 50, 100, and 200 µM,
respectively; Fig. 6B) was higher,
compared with the corresponding effect in FBS conditions
(1.07±0.10-, 0.88±0.09- and 0.81±0.09-fold of control for GE
concentrations of 50, 100, and 200 µM, respectively;
Fig. 2). The inhibitory effects of
GE on p-EGFR (Tyr1173) and total AKT were enhanced in the CSS
condition, compared with those in the FBS condition. These findings
supported the hypothesis that E2 and its receptors may be involved
in the activation of EGFR, and may interfere with the reduced
phosphorylation of EGFR induced by GE.
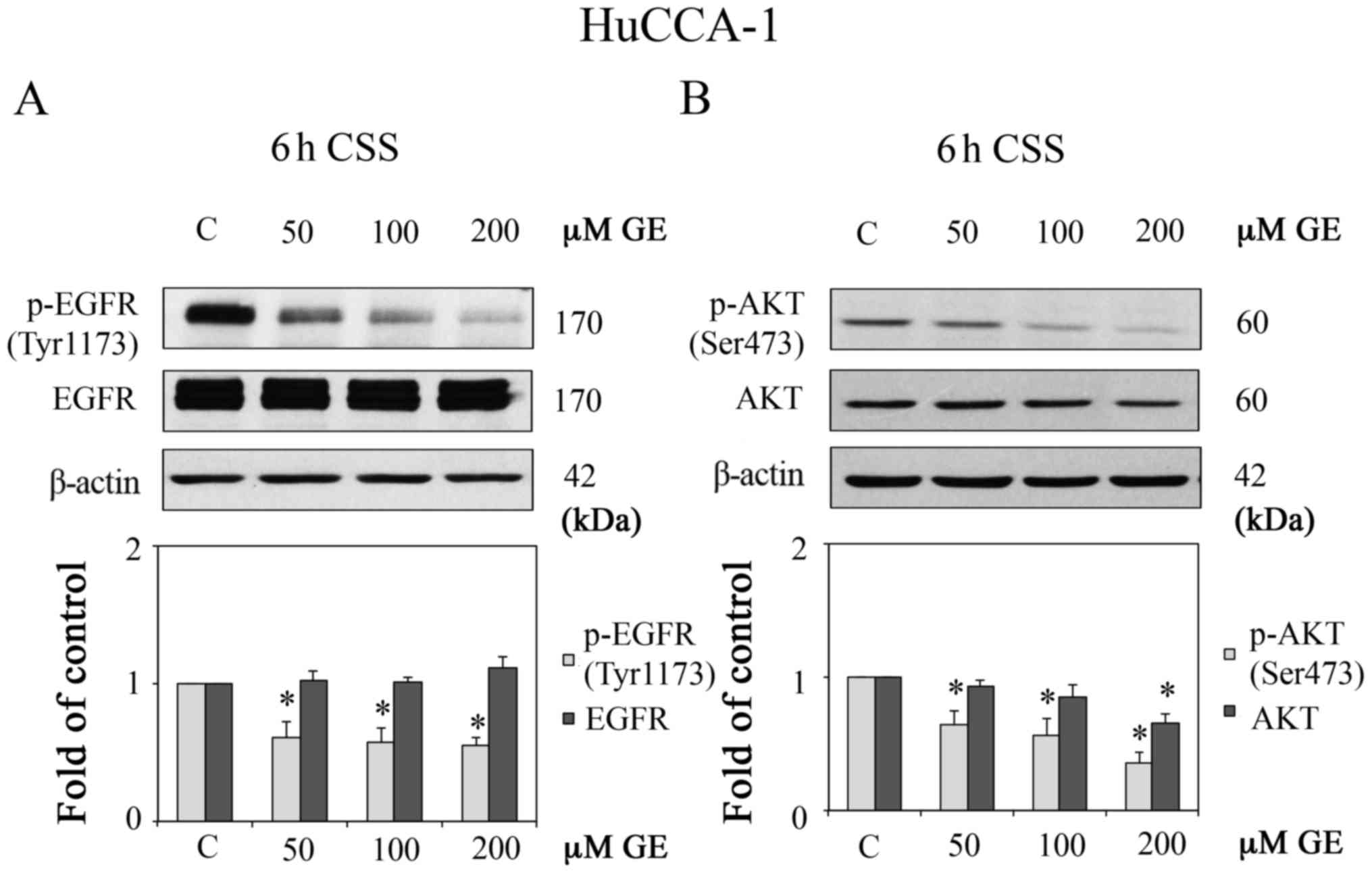 | Figure 6Effect of GE on AKT and EGFR under E2
deprivation. Western blot analysis of (A) p-EGFR (Tyr1173) and
EGFR, and (B) p-AKT (Ser473) and AKT, in HuCCA-1 cells treated with
50, 100 and 200 µM GE for 6 h in CSS conditions (upper
panels), and quantification of the western blot data (lower
panels). In CCS conditions, cells in culture plates were maintained
in CCS condition 1 day prior to continual treatment with E2 or GE
for 6 h. Data are presented as the mean ± standard error of the
mean of at least five independent experiments.
*P<0.05, vs. C. GE, genistein; E2, estrogen; CSS,
E2-deprived conditions; C, 0.1% DMSO vehicle control in CSS; EGFR,
epidermal growth factor receptor; p-, phosphorylated. |
GE reduces the production of IL6 and
alters the expression of iNOS in HuCCA-1 cells
The inflammatory cytokine IL6 is one of the
therapeutic targets for CCA (34)
and GE has been reported to inhibit the IL6 produced by macrophages
(21). Previous experiments have
revealed that GE reduces the activation of AKT, which is also a
downstream molecule of the IL6 pathway. Therefore, the present
study investigated the effect of GE treatment in HuCCA-1 cells on
the production of IL6 and the inflammation-associated protein,
iNOS. As shown in Fig. 7A,
treatment with GE for 6 h decreased the level of IL6 in a
dose-dependent manner in the FBS and CSS conditions. Of note, with
the same concentration of GE, treatment in the CSS condition led to
a more marked reduction in IL6, compared with that in the FBS
condition. The fold changes from the controls in the FBS condition
were 0.10±0.04, 0.25±0.02 and 0.41±0.07, whereas those in the CSS
condition were 0.33±0.04, 0.54±0.04 and 0.66±0.05 at GE
concentrations of 50, 100 and 200 µM, respectively. In the
CSS condition, GE reduced the protein level of iNOS, whereas GE
significantly increased the protein level of iNOS in the FBS
condition (Fig. 7B).
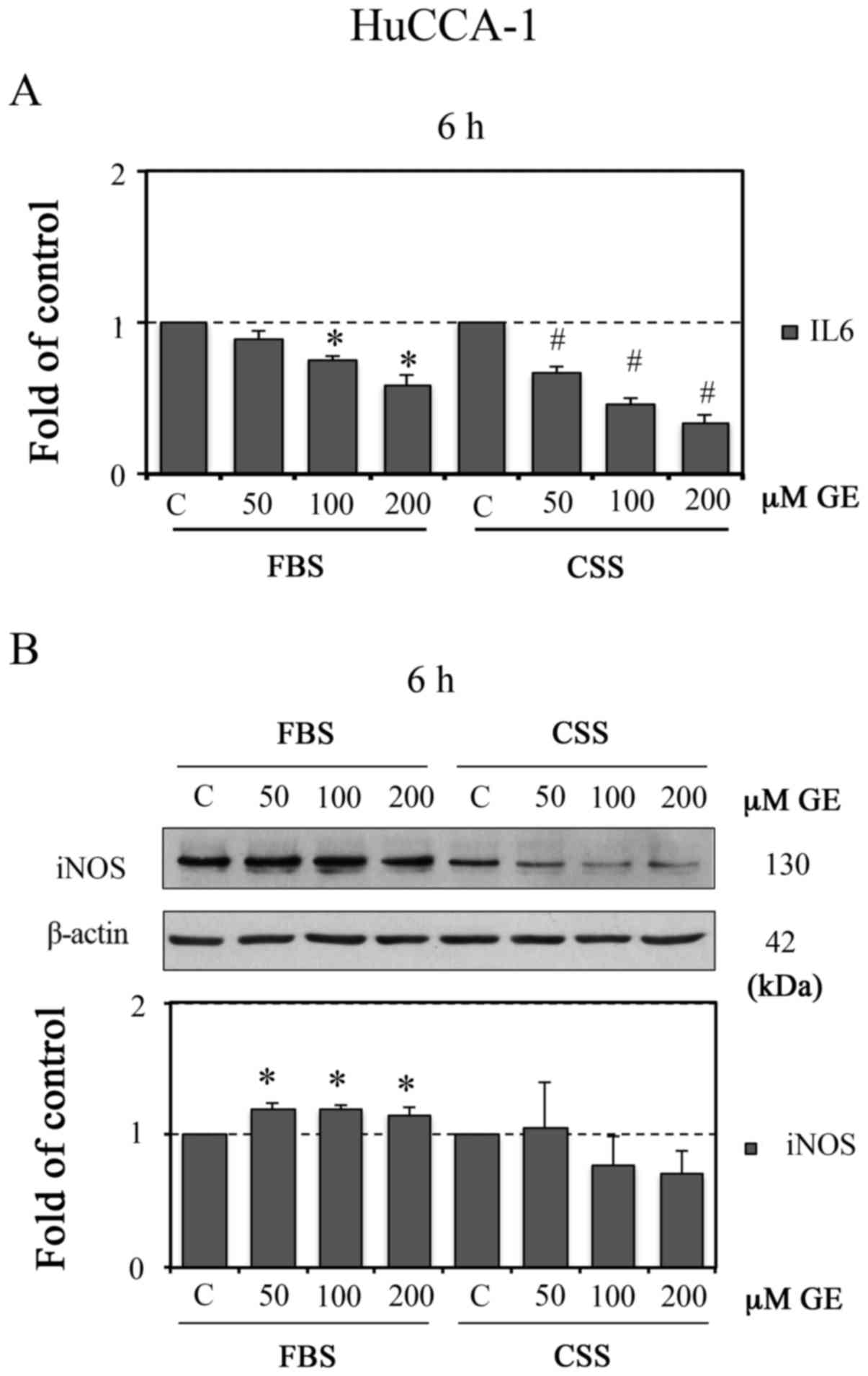 | Figure 7GE reduces induction of IL6 in
HuCCA-1 cells. HuCCA-1 cells treated with 50, 100 and 200 µM
GE for 6 h in FBS or CSS conditions. Following treatment, the
cultured media were collected to determine the (A) protein level of
IL6 by enzyme-link immunosorbent assay analysis. The level of IL6
(pg/ml) was normalized to the total protein of each sample
(µg). The cell lysates were harvested to evaluate the
expression level of (B) iNOS using western blot analysis (upper
panel), with quantification of the western blot data (lower panel).
Data are presented as the mean ± standard error of the mean of at
least four independent experiments. *P<0.05, vs. C in
FBS; #P<0.05, vs. C in CSS. GE, genistein; FBS,
complete medium; CSS, E2-deprived conditions; C, 0.1% DMSO vehicle
control; IL6, interleukin 6; iNOS, inducible nitric oxide
synthase. |
GE alters the protein and gene expression
of ER in HuCCA-1 cells
The results of the present study showed that GE
effectively downregulated the phosphorylation of EGFR and induction
of IL6 in CSS, compared with FBS conditions, suggesting that ER may
be involved in those effects. In addition, GE has been reported to
reduce the expression of ER in rat prostates (35). Therefore, the present study further
investigated the effects of GE treatment on the expression of ER in
HuCCA-1 cells. As shown in Fig.
8A, GE dose-dependently downregulated the protein expression of
ERα at 24 h in FBS and CSS conditions. In addition, the results
demonstrated that treatment with GE (100 and 200 µM) for 24
h in the CSS condition significantly reduced the level of p-ERα
(Ser118), whereas GE at a concentration of 50 µM increased
the level of p-ERα (Ser118). Additionally, the GE-induced
downregulation of ERβ was observed following 6 h of treatment in
the FBS condition and following 24 h of treatment in CSS condition
(Fig. 8B).
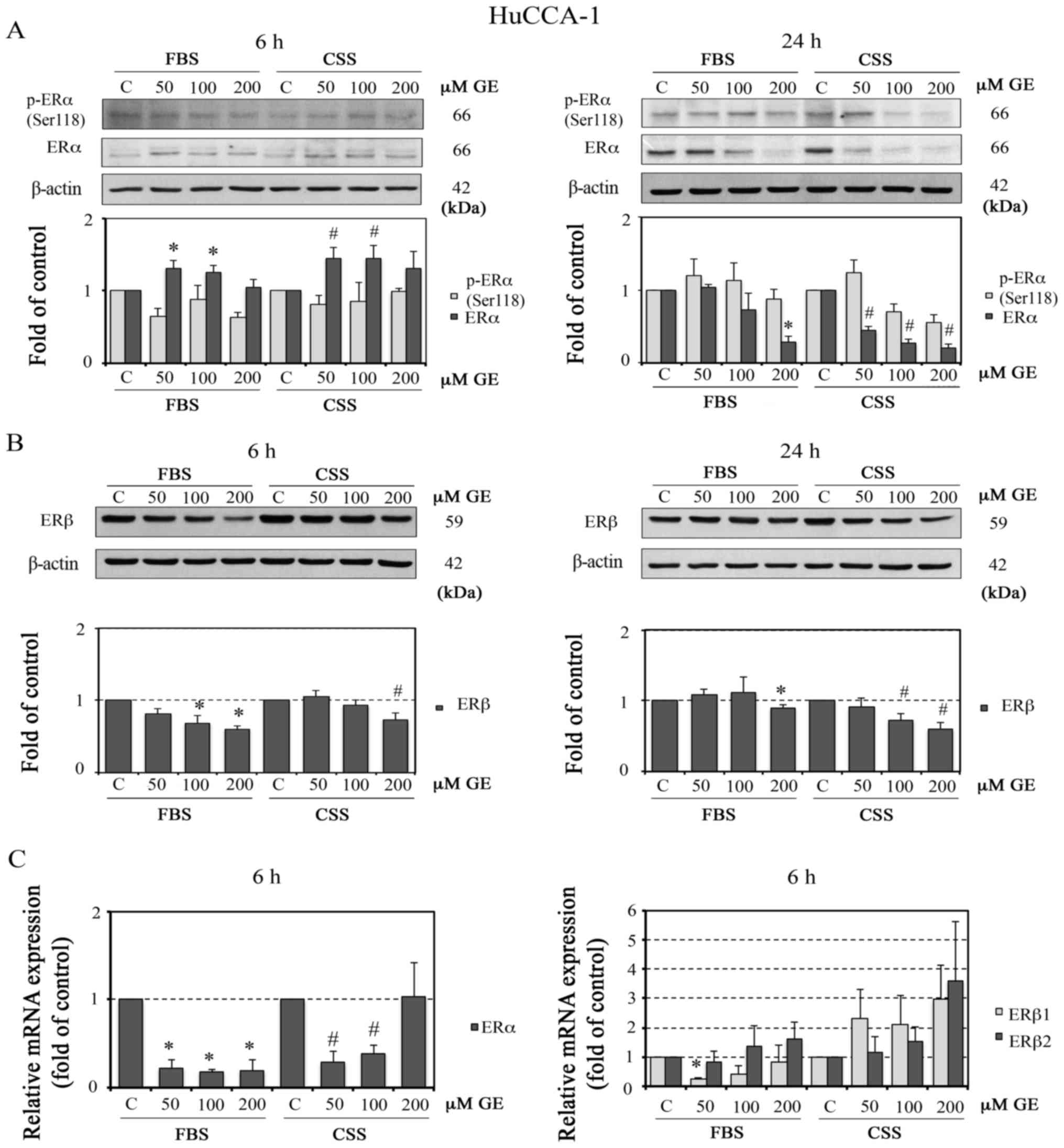 | Figure 8Effect of GE on protein and mRNA
expression of ER in HuCCA-1 cells. Western blot analysis of protein
expression of (A) p-ERα (Ser118) and ERα and (B) ERβ in HuCCA-1
cells treated with 50, 100 and 200 µM GE for 6 h (left) and
24 h (right) in FBS or in CSS (upper panels), with quantification
of the western blot data (lower panels). Relative mRNA expression
of (C) ERα (left), and of ERβ1 and ERβ2
(right) in HUCCA-1 cells treated with or 50, 100 and 200 µM
GE for 6 h in FBS or in CSS, analyzed using reverse
transcription-quantitative polymerase chain reaction analysis. Data
are presented as the mean ± standard error of the mean of at least
three independent experiments. *P<0.05, vs. C in FBS;
#P<0.05, vs. C in CSS. GE, genistein; FBS, complete
medium; CSS, E2-deprived conditions; C, 0.1% DMSO vehicle control
in FBS or CSS; ER, estrogen receptor; p-, phosphorylated. |
To obtain further insight into the effect of GE on
ERα and ERβ in HuCCA-1 cells, RT-qPCR analysis of the mRNA
expression of ERα and ERβ (ERβ1 and
ERβ2) was performed. ERβ1 was selected as ERβ1
is translated to ERβ protein with a MW of 59 kDa, corresponding to
the ERβ protein expressed in HuCCA-1 cells. In addition, the mRNA
expression of ERβ2 was investigated as this gene is widely
used in investigations of several types of cancer and has been
shown to inhibit ERα (36). The
results demonstrated that treatment with GE for 6 h, with the
exception of 200 µM in CSS conditions, reduced the mRNA
levels of ERα. However, despite mediating the downregulation
of ERβ at the protein level, GE induced an upregulation in the gene
expression levels of ERβ1 and ERβ2, particularly in
CSS conditions (Fig. 8C).
Discussion
In the present study, GE concentrations of 10, 50,
100 and 200 µM were selected for examining its cytotoxicity
in CCA cell lines (Fig. 1).
Treatment for 24 and 48 h with GE at 50, 100 and 200 µM
reduced cell viability ~0–60% of that of the control. Previous
studies have reported that GE (60 µM, 72 h) caused a 70%
reduction in cell viability in HT-29 cells (20), and GE (50 µM, 72 h) induced
40% cell death in MDA-MB231 cells (23). Therefore, the range of GE
concentrations used in the present study included non-toxic to
cytotoxic concentrations and were in line with concentrations used
in previous studies. Based on its effects on CCA cells, GE
concentrations of 50–200 µM were selected for further
experiments.
The results of the present study suggested that the
CCA cells were not particularly sensitive to GE. CCA is known to be
resistant to chemotherapy, and this type of cancer has a poor
prognosis with a high mortality rate (37). Therefore, identifying novel drugs
is required. Despite showing that high concentrations are required
to induce cytotoxicity, GE may be of translational value in CCA. In
addition, cancer cells exhibit higher sensitivity to the cytotoxic
effect of GE than normal cells. In human leukemic HL-60 and MOLT-4
cells, 185 µM of GE induced cytotoxicity, whereas no
cytotoxic effect was found in normal human lymphocytes (38). In addition, this natural-occurring
compound is found in soybean products, which are usually consumed
by the Asian population. The daily intake of GE is 49.88
µMol per day (13.48 mg per day) in the Japanese population
and higher in the vegan Asian population (39). Therefore, the GE concentrations of
50 and 100 µM used in the present study are possible to
achieve in physiological conditions. However, the in vitro
concentrations of GE used in the present study require further
consideration and conversion prior to utilization in in vivo
investigations.
The expression and activation of AKT protein
increases during the development and progression of CCA (6). Furthermore, an activated PI3K/AKT
signaling pathway is frequently found in CCA cells resistant to
chemotherapy (8). The present
study investigated the effects of GE in CCA cells. The results
demonstrated that GE reduced CCA cell viability and inhibited the
phosphorylation of AKT. The resulting data are in agreement with
previous reports that showed GE inhibits cell growth in several
types of cancer, including HCT-116 and LoVo colorectal cancer cells
by reducing the activation of AKT (30), and inhibiting the activation of
PI3K/AKT enhances cytotoxicity of oxaliplatin in CCA cell lines
(40).
The present study showed that GE significantly
inhibited the activity of EGFR, as evidenced by a decrease in
active p-EGFR (Tyr1173). Previous reports have shown that EGFR is
involved in the proliferation and progression of CCA (31,41,42).
It has been shown that GE suppresses the proliferation of
proliferative cholangitis in rat models, where the expression of
EGFR is high (43). Furthermore,
GE conjugated with recombinant human EGF has the capability to
inhibit EGFR tyrosine kinase and to trigger rapid apoptotic cell
death in EGFR-positive MDA-MB231 and BT-20 breast cancer cells
(44). These reports support the
results of the present study that GE inhibited the activities of
AKT and EGFR, and this was involved in the reduced viability
observed in the HuCCA-1 and RMCCA-1 cells.
It is well established that MAPK signaling can be
activated upon the activation of EGFR. ERK1/2 and p38 are members
of the MAPK family of cell signaling proteins. The signaling
cascades of ERK1/2 and p38 and their upstream components, including
ras, Raf, MEK and EGFR are associated with the transformation and
progression of several types of cancer (45). A study investigating the KMCH-1
malignant cholangiocyte cell line indicated that mitogens can
stimulate cell growth via the activation of ERK1/2 and p38.
However, only p38 was involved in the anchorage-independent growth
of this cell line (33). The
present study revealed that GE inhibited the activation of p38, as
evidenced by the reduction in the protein expression level of p-p38
in HuCCA-1 and RMCCA-1 cells. This finding is in line with a
previous report in human prostate cancer cell lines that GE
inhibited the activation of p38, leading to the inhibition of cell
invasion (46). Therefore, it is
possible that GE may also inhibit anchorage-independent transformed
cell growth through the inactivation of p38 in CCA. Despite the
reduced activation of p38, the results of the present study
demonstrated that GE increased the protein expression levels of
p-ERK1/2 in the two CCA cell lines. The activation of ERK1/2 is
known to be involved in cell proliferation. However, several
studies have demonstrated enhancement in the protein levels of
p-ERK1/2 in cells undergoing apoptosis. For example, cisplatin
increased the protein levels of p-ERK1/2 in a dose- and
time-dependent manner, which was involved in the induction of
apoptosis in HeLa cells (47). The
GE-induced induction of G2/M cell cycle arrest in MDA-MB231 human
breast cancer cells was also mediated via the activation of ERK1/2
(48). These reports are in line
with the results of the present study, which showed that GE induced
the phosphorylation of ERK1/2 in CCA cells. Taken together, the
present study suggested that ERK1/2 and p38 were involved in the
growth inhibitory effect of GE in CCA cells, but through a
different mechanism of action. GE affects the inhibition of cell
invasion and induction of cell cycle arrest through MAPK signaling.
However, the present study did not focus on the effects of GE on
cell cycle machinery. The effects of GE on MAPK signaling in CCA
require further investigation.
AKT is the intermediate molecule of various
pathways, including EGFR. Upon inhibiting EGFR kinase activity with
AG1478, the activity of AKT is reduced, as demonstrated by a
reduction in the phosphorylation of AKT in colorectal
adenocarcinoma (49),
nasopharyngeal carcinoma (50) and
breast cancer cells (51). In the
present study, although GE treatment led to a reduction in the
phosphorylation of EGFR and AKT, treatment with AG1478, an EGFR
kinase inhibitor, in the HuCCA-1 cells reduced the phosphorylation
of EGFR only, and not its downstream molecule AKT. These results
suggested that, at least in HuCCA-1 cells, activation of the
phosphorylation of AKT is induced not only by EGFR. Human epidermal
growth factor receptor 2, another member of the EGFR family, has
been shown to be involved in HuCCA-1 cell proliferation and
invasion through the AKT pathway (52). In addition to the EGFR family of
proteins, the activation of AKT by IL6 has also been reported. It
has been shown that the growth of CCA cell lines stimulated by IL6
occurred via an AKT-dependent mechanism (53). Of note, in the present study, the
suppression of p-AKT by GE was more prominent following a short
(0.5 h) duration of treatment, compared with a longer (6–24 h)
duration of treatment. This may be due to cell compensation for the
reduced activity of AKT, which is a crucial molecule in several
pathways, particularly those involved in cell survival and
chemoresistance in CCA (5–8). Additionally, a previous study
suggested that E2 enhances the protein expression levels of p-AKT,
p-ERK1/2 and ERα, leading to the stimulation of CCA cell
proliferation (10).
Previous studies have indicated that there is
crosstalk between ERα and EGFR in breast cancer, leading to
activation of the downstream molecules of EGFR signaling pathways,
including MAPK and AKT pathways (12,29,31,54,55).
Additionally, it has been reported that serum levels of E2 in
patients with CCA increase in concentrations corresponding to an
optimal dose for stimulating the proliferation and invasion of CCA
cell lines in vitro (16).
Based on the above information, and as GE is also an agonist of
ERβ, the present study investigated whether E2 and ER were involved
in the inhibitory effect of GE observed in the CCA cell lines. The
results showed that the GE-induced inhibition of the activities of
AKT and EGFR was enhanced in the absence of E2. This suggested the
involvement of E2 in the inhibitory effect of GE and the growth of
HuCCA-1 cells. In the presence of E2, GE competes with E2 for ER
binding and, as it has a higher affinity towards ERβ, causes a
growth inhibitory effect, which is a predominant effect of the
activation of ERβ (56). In E2
deprivation, GE binds to ERβ, causing increased growth inhibition.
However, the growth inhibitory effect of GE may be independent of
the ER-mediated pathway. As reported in other cell lines and
evidenced in CCA cells in the present study, GE possesses a
tyrosine kinase-inhibitory effect. The GE-induced inhibition of the
activities of EGFR and AKT may lead to a reduction of CCA cell
growth. Therefore, the inhibition of EGFR and AKT evidenced
following GE treatment may be mediated via its tyrosine kinase
inhibiting effects and/or via interfering with the crosstalk
between non-genomic ER and EGFR signaling.
CCA is associated with chronic inflammation of the
bile duct (57). Upon binding to
its receptor, E2, has been shown to act as an anti-inflammatory and
anticarcinogenic by inhibiting the production of IL6 in DEN-induced
liver cancer in animal models (58). The cytokine IL6 is important in CCA
cell growth. Its signaling involves a complex network of different
pathways, including the AKT pathways (4,59)
and MAPK (60) pathways. The data
presented in the present study showed that GE reduced the
production of IL6 in HuCCA-1 cells. This result is in line with
previous reports that GE possesses anti-inflammatory properties
(22) and can inhibit the
production of IL6 by macrophages stimulated with lipopolysaccharide
(21). Additionally, the present
study found that the GE-induced decrease in the production of IL6
in CSS conditions was more marked, compared with that in FBS
conditions. These results suggested that E2 was involved in the
phosphorylation of AKT, and also in the induction of IL6 in HuCCA-1
cells. This finding is in agreement with a previous report that E2
stimulated the production of IL6 in non-malignant biliary
epithelial cells and malignant biliary epithelial cells expressing
ERα (17). Therefore, in addition
to EGFR, the results suggested that E2, ER and IL6 may also be
involved in the inhibitory effect of GE and the growth of CCA
cells.
Although it has been reported that GE can attenuate
the nitric oxide released via iNOS in activated macrophages
(61), the results of the present
study demonstrated that GE increased the protein expression levels
of iNOS in HuCCA-1 cells, despite decreased levels of IL6. However,
a report by Nakaya et al demonstrated that GE and E2
upregulated the protein expression and activity of iNOS through ER
pathways in RAW246.7 macrophage cells (62). This finding is in accordance with
the results of the present study, which showed that GE induced the
expression of iNOS in the presence of E2. The discrepancy in these
findings may be due to differences in cell type and inflammatory
stages. The HuCCA-1 cell line was established from CCA tissue
fragments surgically removed from a male patient from Thailand
diagnosed with chronic infection due to liver fluke (25).
The results of the present study suggested the
involvement of E2 on the growth-inhibiting effects of GE in CCA
cells. This suggests that its receptor, ER, is likely to be
involved. GE has been identified as an agonist of ERβ; however,
this compound can bind ERα and ERβ, of which binding affinity to
ERβ is 20–30-fold higher than that to ERα (63,64).
The data in the present study showed that, at 24 h, GE (100 and 200
µM) in CSS conditions reduced the protein expression levels
of p-ERα (Ser118), ERα and ERβ, whereas the mRNA expression of ERα
was downregulated at 6 h. This result is consistent with a previous
report that showed that GE downregulated the expression of ER in
rat prostates (35). In the
present study, although GE reduced the protein expression of ERβ,
there were no significant changes in the mRNA expression levels of
ERβ1 and ERβ2 in the CSS conditions. These results
suggested that the GE-induced reduction in ERβ protein was not at
the transcriptional level. However, the effects of GE on p-ERβ were
not measured in the present study, as information on the function
of p-ERβ remains limited. As the ER pathway has been shown to be
associated with the AKT (12) and
IL6 (17) signaling cascades, the
downregulation of ER induced by GE observed in the present study
may be involved in the GE-induced inhibition of IL6 and AKT in
CCA.
In conclusion, the present study is the first, to
the best of our knowledge, to demonstrate that GE can reduce the
growth of CCA cell lines via inhibition of the activities of EGFR,
AKT and IL6. The mechanism was, in part, associated with
downregulation of the protein and mRNA expression levels of ER. The
present study provided preliminary results for further
investigations to generate further translational value for GE in
the treatment of CCA, for example, modifying GE structure to
improve its activity, using GE to sensitize CCA to conventional
treatments, and potentially using GE for chemoprevention as a
therapeutic supplement in CCA.
Acknowledgments
Not applicable.
Funding
This study was supported in part by a grant (no.
EHT-R-4/2556) from The Center of Excellence on Environmental Health
and Toxicology, Science and Technology Postgraduate Education and
Research Development Office (PERDO), Ministry of Education,
Thailand.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
PT performed the experiments, data analysis and
drafted the manuscript. AT initiated and monitored the research,
supervised the study design, data interpretation and the drafting
and revision of manuscript. PW and JS performed data interpretation
and also assisted in the drafting of the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Songserm N, Promthet S, Wiangnon S and
Sithithaworn P: Prevalence and co-infection of intestinal parasites
among thai rural residents at high-risk of developing
cholangiocarcinoma: A cross-sectional study in a prospective cohort
study. Asian Pac J Cancer Prev. 13:6175–6179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jaiswal M, LaRusso NF, Burgart LJ and
Gores GJ: Inflammatory cytokines induce Dells by a nitric
oxide-dependent mechanism. Cancer Res. 60:184–190. 2000.PubMed/NCBI
|
|
3
|
Sripa B, Thinkhamrop B, Mairiang E, Laha
T, Kaewkes S, Sithithaworn P, Periago MV, Bhudhisawasdi V,
Yonglitthipagon P, Mulvenna J, et al: Elevated plasma IL-6
associates with increased risk of advanced fibrosis and
cholangiocarcinoma in individuals infected by Opisthorchis
viverrini. PLoS Negl Trop Dis. 6:e16542012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kobayashi S, Werneburg NW, Bronk SF,
Kaufmann SH and Gores GJ: Interleukin-6 contributes to Mcl-1
up-regulation and TRAIL resistance via an Akt-signaling pathway in
cholangiocarcinoma cells. Gastroenterology. 128:2054–2065. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meng F, Henson R, Lang M, Wehbe H,
Maheshwari S, Mendell JT, Jiang J, Schmittgen TD and Patel T:
Involvement of human micro-RNA in growth and response to
chemotherapy in human cholangiocarcinoma cell lines.
Gastroenterology. 130:2113–2129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yothaisong S, Thanee M, Namwat N,
Yongvanit P, Boonmars T, Puapairoj A and Loilome W: Opisthorchis
viverrini infection activates the PI3K/AKT/PTEN and Wnt/β-catenin
signaling pathways in a Cholangiocarcinogenesis model. Asian Pac J
Cancer Prev. 15:10463–10468. 2014. View Article : Google Scholar
|
|
7
|
Ewald F, Nörz D, Grottke A, Hofmann BT,
Nashan B and Jücker M: Dual Inhibition of PI3K-AKT-mTOR- and
RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and
reverses acquired resistance to MEK-inhibitors. Invest New Drugs.
32:1144–1154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoon H, Min JK, Lee JW, Kim DG and Hong
HJ: Acquisition of chemoresistance in intrahepatic
cholangiocarcinoma cells by activation of AKT and extracellular
signal-regulated kinase (ERK)1/2. Biochem Biophys Res Commun.
405:333–337. 2011. View Article : Google Scholar
|
|
9
|
Kamsa-ard S, Wiangnon S, Suwanrungruang K,
Promthet S, Khuntikeo N, Kamsa-ard S and Mahaweerawat S: Trends in
liver cancer incidence between 1985 and 2009, Khon Kaen, Thailand:
Cholangiocarcinoma. Asian Pac J Cancer Prev. 12:2209–2213.
2011.
|
|
10
|
Alvaro D, Barbaro B, Franchitto A, Onori
P, Glaser SS, Alpini G, Francis H, Marucci L, Sterpetti P,
Ginanni-Corradini S, et al: Estrogens and insulin-like growth
factor 1 modulate neoplastic cell growth in human
cholangiocarcinoma. Am J Pathol. 169:877–888. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shao W and Brown M: Advances in estrogen
receptor biology: Prospects for improvements in targeted breast
cancer therapy. Breast Cancer Res. 6:39–52. 2004. View Article : Google Scholar :
|
|
12
|
Guo RX, Wei LH, Tu Z, Sun PM, Wang JL,
Zhao D, Li XP and Tang JM: 17 β-estradiol activates PI3K/Akt
signaling pathway by estrogen receptor (ER)-dependent and
ER-independent mechanisms in endometrial cancer cells. J Steroid
Biochem Mol Biol. 99:9–18. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nicholson RI, McClelland RA, Robertson JF
and Gee JM: Involvement of steroid hormone and growth factor
cross-talk in endocrine response in breast cancer. Endocr Relat
Cancer. 6:373–387. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osborne CK, Shou J, Massarweh S and Schiff
R: Crosstalk between estrogen receptor and growth factor receptor
pathways as a cause for endocrine therapy resistance in breast
cancer. Clin Cancer Res. 11:865s–870s. 2005.PubMed/NCBI
|
|
15
|
Mancino A, Mancino MG, Glaser SS, Alpini
G, Bolognese A, Izzo L, Francis H, Onori P, Franchitto A,
Ginanni-Corradini S, et al: Estrogens stimulate the proliferation
of human cholangiocarcinoma by inducing the expression and
secretion of vascular endothelial growth factor. Dig Liver Dis.
41:156–163. 2009. View Article : Google Scholar :
|
|
16
|
Hunsawong T, Singsuksawat E, In-chon N,
Chawengrattanachot W, Thuwajit C, Sripa B, Paupairoj A, Chau-in S
and Thuwajit P: Estrogen is increased in male cholangiocarcinoma
patients' serum and stimulates invasion in cholangiocarcinoma cell
lines in vitro. J Cancer Res Clin Oncol. 138:1311–1320. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Isse K, Specht SM, Lunz JG III, Kang LI,
Mizuguchi Y and Demetris AJ: Estrogen stimulates female biliary
epithelial cell interleukin-6 expression in mice and humans.
Hepatology. 51:869–880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakla MS, Shenouda NS, Ansell PJ,
Macdonald RS and Lubahn DB: Genistein affects HER2 protein
concentration, activation, and promoter regulation in BT-474 human
breast cancer cells. Endocrine. 32:69–78. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu D, Yan L, Wang L, Tai W, Wang W and
Yang C: Genistein enhances the effect of cisplatin on the
inhibition of non-small cell lung cancer A549 cell growth in vitro
and in vivo. Oncol Lett. 8:2806–2810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu Z, Li W and Liu F: Inhibition of
proliferation and induction of apoptosis by genistein in colon
cancer HT-29 cells. Cancer Lett. 215:159–166. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ji G, Zhang Y, Yang Q, Cheng S, Hao J,
Zhao X and Jiang Z: Genistein suppresses LPS-induced inflammatory
response through inhibiting NF-κB following AMP kinase activation
in RAW 264.7 macrophages. PLoS One. 7:e531012012. View Article : Google Scholar
|
|
22
|
Verdrengh M, Jonsson IM, Holmdahl R and
Tarkowski A: Genistein as an anti-inflammatory agent. Inflamm Res.
52:341–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong L, Li Y, Nedeljkovic-Kurepa A and
Sarkar FH: Inactivation of NF-kappaB by genistein is mediated via
Akt signaling pathway in breast cancer cells. Oncogene.
22:4702–4709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gossner G, Choi M, Tan L, Fogoros S,
Griffith KA, Kuenker M and Liu JR: Genistein-induced apoptosis and
autophagocytosis in ovarian cancer cells. Gynecol Oncol. 105:23–30.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sirisinha S, Tengchaisri T, Boonpucknavig
S, Prempracha N, Ratanarapee S and Pausawasdi A: Establishment and
charac-terization of a cholangiocarcinoma cell line from a Thai
patient with intrahepatic bile duct cancer. Asian Pac J Allergy
Immunol. 9:153–157. 1991.PubMed/NCBI
|
|
26
|
Rattanasinganchan P, Leelawat K,
Treepongkaruna SA, Tocharoentanaphol C, Subwongcharoen S,
Suthiphongchai T and Tohtong R: Establishment and characterization
of a cholangiocarcinoma cell line (RMCCA-1) from a Thai patient.
World J Gastroenterol. 12:6500–6506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Milo GE, Malarkey WB, Powell JE, Blakeslee
JR and Yohn DS: Effects of steroid hormones in fetal bovine serum
on plating ang cloning of human cells in vitro. In Vitro. 12:23–30.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wilson VS, Bobseine K and Gray LE Jr:
Development and characterization of a cell line that stably
expresses an estrogen-responsive luciferase reporter for the
detection of estrogen receptor agonist and antagonists. Toxicol
Sci. 81:69–77. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakareangrit W, Thiantanawat A,
Visitnonthachai D, Watcharasit P and Satayavivad J: Sodium arsenite
inhibited genomic estrogen signaling but induced pERα (Ser118) via
MAPK pathway in breast cancer cells. Environ Toxicol. 31:1133–1146.
2016. View Article : Google Scholar
|
|
30
|
Qin J, Teng J, Zhu Z, Chen J and Huang WJ:
Genistein induces activation of the mitochondrial apoptosis pathway
by inhibiting phosphorylation of Akt in colorectal cancer cells.
Pharm Biol. 54:74–79. 2016. View Article : Google Scholar
|
|
31
|
Yoon JH, Gwak GY, Lee HS, Bronk SF,
Werneburg NW and Gores GJ: Enhanced epidermal growth factor
receptor activation in human cholangiocarcinoma cells. J Hepatol.
41:808–814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akiyama T, Ishida J, Nakagawa S, Ogawara
H, Watanabe S, Itoh N, Shibuya M and Fukami Y: Genistein, a
specific inhibitor of tyrosine-specific protein kinases. J Biol
Chem. 262:5592–5595. 1987.PubMed/NCBI
|
|
33
|
Tadlock L and Patel T: Involvement of p38
mitogen-activated protein kinase signaling in transformed growth of
a cholangiocarcinoma cell line. Hepatology. 33:43–51. 2001.
View Article : Google Scholar
|
|
34
|
Braconi C, Swenson E, Kogure T, Huang N
and Patel T: Targeting the IL-6 dependent phenotype can identify
novel therapies for cholangiocarcinoma. PLoS One. 5:e151952010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fritz WA, Wang J, Eltoum IE and
Lamartiniere CA: Dietary genistein down-regulates androgen and
estrogen receptor expression in the rat prostate. Mol Cell
Endocrinol. 186:89–99. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haldosén LA, Zhao C and Dahlman-Wright K:
Estrogen receptor β in breast cancer. Mol Cell Endocrinol.
382:665–672. 2014. View Article : Google Scholar
|
|
37
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Traganos F, Ardelt B, Halko N, Bruno S and
Darzynkiewicz Z: Effects of genistein on the growth and cell cycle
progression of normal human lymphocytes and human leukemic MOLT-4
and HL-60 cells. Cancer Res. 52:6200–6208. 1992.PubMed/NCBI
|
|
39
|
Nakamura Y, Tsuji S and Tonogai Y:
Determination of the levels of isoflavonoids in soybeans and
soy-derived foods and estimation of isoflavonoids in the Japanese
daily intake. J AOAC Int. 83:635–650. 2000.PubMed/NCBI
|
|
40
|
Leelawat K, Narong S, Udomchaiprasertkul
W, Leelawat S and Tungpradubkul S: Inhibition of PI3K increases
oxaliplatin sensitivity in cholangiocarcinoma cells. Cancer Cell
Int. 9:32009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshikawa D, Ojima H, Iwasaki M, Hiraoka
N, Kosuge T, Kasai S, Hirohashi S and Shibata T:
Clinicopathological and prognostic significance of EGFR, VEGF, and
HER2 expression in cholangiocarcinoma. Br J Cancer. 98:418–425.
2008. View Article : Google Scholar
|
|
42
|
Yoshikawa D, Ojima H, Kokubu A, Ochiya T,
Kasai S, Hirohashi S and Shibata T: Vandetanib (ZD6474), an
inhibitor of VEGFR and EGFR signalling, as a novel
molecular-targeted therapy against cholangiocarcinoma. Br J Cancer.
100:1257–1266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang L, Jiang LS, Yan LN, Li FY, Wang W,
Cheng NS and Wen TF: Effects of epidermal growth factor receptor
inhibitor genistein on proliferative cholangitis in rats. J Surg
Res. 162:59–67. 2010. View Article : Google Scholar
|
|
44
|
Uckun FM, Narla RK, Jun X, Zeren T,
Venkatachalam T, Waddick KG, Rostostev A and Myers DE: Cytotoxic
activity of epidermal growth factor-genistein against breast cancer
cells. Clin Cancer Res. 4:901–912. 1998.PubMed/NCBI
|
|
45
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang X, Chen S, Xu L, Liu Y, Deb DK,
Platanias LC and Bergan RC: Genistein inhibits p38 map kinase
activation, matrix metalloproteinase type 2, and cell invasion in
human prostate epithelial cells. Cancer Res. 65:3470–3478. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li Z, Li J, Mo B, Hu C, Liu H, Qi H, Wang
X and Xu J: Genistein induces G2/M cell cycle arrest via stable
activation of ERK1/2 pathway in MDA-MB-231 breast cancer cells.
Cell Biol Toxicol. 24:401–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bishnupuri KS, Luo Q, Murmu N, Houchen CW,
Anant S and Dieckgraefe BK: Reg IV activates the epidermal growth
factor receptor/Akt/AP-1 signaling pathway in colon
adenocarcinomas. Gastroenterology. 130:137–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhu XF, Liu ZC, Xie BF, Li ZM, Feng GK,
Yang D and Zeng YX: EGFR tyrosine kinase inhibitor AG1478 inhibits
cell proliferation and arrests cell cycle in nasopharyngeal
carcinoma cells. Cancer Lett. 169:27–32. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wei Z, Song X and Shaikh ZA: Cadmium
promotes the proliferation of triple-negative breast cancer cells
through EGFR-mediated cell cycle regulation. Toxicol Appl
Pharmacol. 289:98–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Treekitkarnmongkol W and Suthiphongchai T:
High expression of ErbB2 contributes to cholangiocarcinoma cell
invasion and proliferation through AKT/p70S6K. World J
Gastroenterol. 16:4047–4054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Frampton G, Invernizzi P, Bernuzzi F, Pae
HY, Quinn M, Horvat D, Galindo C, Huang L, McMillin M, Cooper B, et
al: Interleukin-6-driven progranulin expression increases
cholangiocarcinoma growth by an Akt-dependent mechanism. Gut.
61:268–277. 2012. View Article : Google Scholar
|
|
54
|
Shou J, Massarweh S, Osborne CK, Wakeling
AE, Ali S, Weiss H and Schiff R: Mechanisms of tamoxifen
resistance: Increased estrogen receptor-HER2/neu cross-talk in
ER/HER2-positive breast cancer. J Natl Cancer Inst. 96:926–935.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Levin ER: Bidirectional signaling between
the estrogen receptor and the epidermal growth factor receptor. Mol
Endocrinol. 17:309–317. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Marzioni M, Torrice A, Saccomanno S,
Rychlicki C, Agostinelli L, Pierantonelli I, Rhönnstad P, Trozzi L,
Apelqvist T, Gentile R, et al: An oestrogen receptor β-selective
agonist exerts anti-neoplastic effects in experimental intrahepatic
cholangiocarcinoma. Dig Liver Dis. 44:134–142. 2012. View Article : Google Scholar
|
|
57
|
Gores GJ: Cholangiocarcinoma: Current
concepts and insights. Hepatology. 37:961–969. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Naugler WE, Sakurai T, Kim S, Maeda S, Kim
K, Elsharkawy AM and Karin M: Gender disparity in liver cancer due
to sex differences in MyD88-dependent IL-6 production. Science.
317:121–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Johnson C, Han Y, Hughart N, McCarra J,
Alpini G and Meng F: Interleukin-6 and its receptor, key players in
hepatobiliary inflammation and cancer. Transl Gastrointest Cancer.
1:58–70. 2012.PubMed/NCBI
|
|
60
|
Park J, Tadlock L, Gores GJ and Patel T:
Inhibition of interleukin 6-mediated mitogen-activated protein
kinase activation attenuates growth of a cholangiocarcinoma cell
line. Hepatology. 30:1128–1133. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hämäläinen M, Nieminen R, Vuorela P,
Heinonen M and Moilanen E: Anti-inflammatory effects of flavonoids:
Genistein, kaempferol, quercetin, and daidzein inhibit STAT-1 and
NF-kappaB activations, whereas flavone, isorhamnetin, naringenin,
and pelargonidin inhibit only NF-kappaB activation along with their
inhibitory effect on iNOS expression and NO production in activated
macrophages. Mediators Inflamm. 2007:456732007. View Article : Google Scholar
|
|
62
|
Nakaya M, Tachibana H and Yamada K:
Isoflavone genistein and daidzein up-regulate LPS-induced inducible
nitric oxide synthase activity through estrogen receptor pathway in
RAW264.7 cells. Biochem Pharmacol. 71:108–114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kuiper GG, Lemmen JG, Carlsson B, Corton
JC, Safe SH, van der Saag PT, van der Burg B and Gustafsson JA:
Interaction of estrogenic chemicals and phytoestrogens with
estrogen receptor β. Endocrinology. 139:4252–4263. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Manas ES, Xu ZB, Unwalla RJ and Somers WS:
Understanding the selectivity of genistein for human estrogen
receptor-β using X-ray crystallography and computational methods.
Structure. 12:2197–2207. 2004. View Article : Google Scholar : PubMed/NCBI
|