Introduction
Prostate cancer is a malignancy that severely
affects the health of men worldwide. In the United States, an
estimated 161,360 cases of prostate cancer were newly diagnosed in
2017, leading to prostate cancer ranking first among cancers for
incidence rate and third for mortality rate (1). The incidence and mortality of
prostate cancer have increased in China in recent years due to
changes in lifestyle and the living environment. In China, prostate
cancer is one of the 10 most common cancers in men, and upward
trends in incidence and mortality rates have been observed
(2). The majority of prostate
cancer patients are at an advanced stage of cancer and have distant
metastasis when they are diagnosed, making the disease difficult to
treat (3). A good understanding of
the molecular mechanism of the progression of prostate cancer is
likely to aid its diagnosis and treatment.
Carboxyl terminus of Hsc-70-interacting protein
(CHIP), encoded by the STIP1 homology and U-box containing protein
1 (STUB1) gene, contains three domains: A three
tetratricopeptide repeat (TPR) domain at its N-terminus, a U-box
domain at its C-terminus, and a domain connecting the N-terminus
and C-terminus together. The TPR domain, which links to the
molecular chaperones Hsc70-Hsc70 and Hsc90, mediates
protein-protein interactions. The U-box domain displays an E3
ubiquitin ligase activity between the chaperone and proteasome
systems (4,5). As a new co-chaperone, CHIP serves an
important role in the folding, transport and degradation of
proteins (6). CHIP is an E3
ubiquitin ligase that induces the ubiquitination and degradation of
proteasomes and misfolded proteins, such as cystic fibrosis
transmembrane conductance regulator (7,8).
Substantial evidence indicates that CHIP is
associated with the development, migration and invasion of cancer.
Although the exact function of CHIP has not been elucidated, it is
considered to be a tumor suppressor in the majority of tumors. In
ovarian carcinoma, the overexpression of CHIP suppresses tumor
progression by inhibiting the aerobic glycolysis that provides
energy to tumor cells (9). In
addition, CHIP overexpression significantly inhibits the growth of
leukemia cells (10). Furthermore,
CHIP decreases the migration and invasion abilities of gastric
cancer cells to suppress the malignant transformation of the tumor
via the regulation of the nuclear factor κ-light-chain-enhancer of
activated B cells (NF-κB) signaling pathway (11). However, CHIP has been ascribed as
an oncogene in other tumors. A recent study revealed that CHIP
overexpression markedly increased the viability of thyroid cancer
cells and accelerated tumor growth in vivo (12). In another study, B-type hepatitis
virus-associated hepatocellular carcinoma, CHIP overexpression
exhibited a marked association with a larger tumor size and the
presence of portal vein invasion (13).
The specific role of CHIP in the progression of
prostate cancer remains unclear. In the present study, the
biological function and potential mechanism of CHIP in the
oncogenesis and progression of prostate cancer were explored. The
effects of the forced introduction of CHIP into DU145 human
prostatic cancer cells on various biological behaviors of the
cells, including proliferation, apoptosis, migration and invasion,
were investigated in vitro. The activation of the AKT (also
known as protein kinase B) signaling pathway and pro-apoptotic
protein levels in the CHIP-overexpressing cell lines were also
explored. In addition, a tissue microarray of human prostate cancer
was used to investigate the correlation of CHIP expression with
Gleason score.
Materials and methods
Cell culture
The DU145 cell line was obtained from American Type
Culture Collection (Manassas, VA, USA) and maintained in RPMI-1640
(cat. no. 1844368) with 10% fetal bovine serum (FBS, cat. no.
1428479) (both from Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), 100 U/ml penicillin, 100 µg/ml streptomycin and 2
mM glutamine. The cells were cultured in a humidified atmosphere
containing 5% CO2 at 37°C.
Transfection
The MSCV vector containing green fluorescent protein
(GFP; a gift from Professor Yong Zhao, Institute of Zoology,
Chinese Academy of Sciences) was used as the basis of a CHIP
overexpression vector. The full-length coding sequence of human
CHIP (hCHIP) was amplified by polymerase chain reaction (PCR) and
cloned into the MSCV-GFP vector to form MSCV-GFP-hCHIP. The primers
were designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
Two primer sequences of CHIP were selected as follows:
5′-AAAAAGATCTGGCGGCATGAAGGGCAAGG-3′ and
5′-GGGAACCTCAGTAGTCCTCCACCC-3′. A total of 3.2×105
phoenix A packaging cells (a gift from Professor Yong Zhao) were
seeded in a 35-mm dish and transfected with 2 µg
MSCV-GFP-hCHIP or control vector (MSCV-GFP) and 1 µg pCL-Amp
(gift from Professor Yong Zhao) using Lipofectamine 2000 (cat. no.
11668-019; Invitrogen; Thermo Fisher Scientific, Inc.). The
supernatants were collected after transfection for 48 h to infect
2×105 DU145 cells and provide DU145-control and
DU145-hCHIP cells. The individual clones were subsequently selected
by 5 ng/µl puromycin for 2 weeks.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was used to validate the effect of
STUB1 gene overexpression. Total RNA was extracted from the
cells using RNAiso Plus reagent (cat. no. 9109; Takara Bio, Inc.,
Otsu, Japan) and quantified. Then, 2 µg RNA was converted
into cDNA using Superscript M-MLV (cat. no. 28025-013; Invitrogen;
Thermo Fisher Scientific, Inc.). The conditions were as follows:
25°C for 10 min, 37°C for 50 min and 70°C for 15 min. qPCR was
performed in triplicate using SYBR Premix Ex Taq II (cat. no.
RR420L; Takara Bio, Inc.) with a LightCycler 480 System (Roche
Diagnostics, Basel, Switzerland). β-actin was used as an endogenous
control. Primer sequences were as follows: β-actin,
5′-GCTACGAGCTGCCTGACGG-3′ and 5′-TGTTGGCGTACAGGTCTTTGC-3′; STUB1,
5′-GCCAAGGAGCAGCGGCTGAA-3′ and 5′-CTCTCACGCTCCGCGGCAAT-3′. The
house-keeping gene (β-actin) and the target genes were reverse
transcribed together in a single run. The thermocycling conditions
were as follows: pre-incubation at 95°C for 30 sec, followed by 45
cycles of 95°C (30 sec), 58°C (15 sec) and 72°C (20 sec). The
relative expression levels of the target genes were quantified by
the 2−ΔΔCq method (14).
Western blot analysis
Whole-cell extracts, nuclear extracts (NEs) and
cytoplasmic extracts (CE) were prepared using suspension buffer (10
mM Tris-HCl, 0.1 M NaCl, 1 mM EDTA) and proteinase inhibitor (cat.
no. 11697498001; Roche Diagnostics, Mannheim, Germany) according to
standard procedures. The total protein was measured with a micro
BCA protein assay kit (cat. no. 23235; Thermo Fisher Scientific,
Inc.). Then 30 µg proteins were fractionated by 10% SDS-PAGE
and transferred to nitrocellulose membranes. After blocking with
milk for 1 h at 4°C, the membranes were probed with different
antibodies (Abs) with incubation overnight at 4°C. The membranes
were washed the next day and then incubated with appropriate
secondary Abs (1:10,000) at 4°C overnight. IRDye 680CW (cat. no.
926-32222) and IRDye 800CW secondary Abs (cat. no. 926-32213) were
obtained from LI-COR Biosciences (Lincoln, NE, USA). The bands were
scanned using an Odyssey system (LI-COR Biosciences). Band
densities were normalized to the β-actin or laminA/C loading
control. Primary Abs against transcription factor p65 (RelA;
sc-372), transcription factor RelB (RelB; sc-226), p105/p50
(sc-7178), p100/p52 (sc-298), proto-oncogene c-Rel (c-Rel; sc-70),
laminA/C (sc-20681), urokinase-type plasminogen activator (uPA;
sc-14019), TNF receptor-associated factor 2 (TRAF2; sc-876) and TNF
receptor-associated factor 3 (TRAF3; sc-4729) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Abs against
vimentin (CBL202) were purchased from EMD Millipore (Billerica, MA,
USA). Abs against uPA receptor (UPAR; #12713), B-cell lymphoma-2
(Bcl-2; #2870), Bcl-extra large (Bcl-xL; #2764), Bcl-2-like protein
11 (Bim; #2933), BH3 interacting-domain death agonist (BID; #2002),
Bcl-2-associated death promoter (Bad; #9239), p53 (#9919), CHIP
(#2080), matrix metal-loproteinase (MMP)2 (#13132), MMP9 (#13667),
integrin β1 (ITGB1; #9699), keratin8/18 (#4546), epithelial cell
adhesion molecule (EpCAM; #14452), N-cadherin (#13116), phosphatase
and tensin homolog (PTEN; #9559) and glycogen synthase kinase
(GSK)-3β (#9315), the Cell Cycle Regulation Sample kit (#9932) and
phospho-Akt Pathway Antibody Sampler kit (#9916) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Abs
against β-actin (AO1215a) were purchased from Abgent, Inc. (San
Diego, CA, USA).
Cell growth assay
The xCelligence RTCA instrument (ACEA Biosciences,
Inc., San Diego, CA, USA) was used to evaluate the growth of the
cells. In this assay, 8.0×103 cells were seeded in each
well in an E-plate and the impedance was continuously monitored for
72 h. For each well, a 'cell index' was generated, which was
determined by the number of cells seeded, the morphology and
overall size of the cells, and the degree to which the cells
interacted with the sensor surface. The cell index was continuously
monitored by the system and data were collected and analyzed using
RTCA 1.2 software (ACEA Biosciences, Inc.).
Bromodeoxyuridine (BrdU) assay
Cells (2.0×104 cells/well) were seeded in
a 96-well plate and cultured with fresh medium in a humidified
atmosphere containing 5% CO2 at 37°C for 24 h. BrdU from
a BrdU Cell Proliferation kit (cat. no. 2750; EMD Millipore) was
then added and the plate was incubated for 5 h at 37°C. The medium
was then removed and the cells were fixed at room temperature for
30 min. Following removal of the fixing solution, the cells were
washed twice with washing buffer, 100 µl/well diluted
anti-BrdU antibody was added, and the cells incubated for 1 h at
room temperature. The wells were then washed prior to the addition
of 100 µl/well peroxidase conjugate followed by 100
µl/well of TMB peroxidase substrate, with incubation for 30
min at room temperature following each addition. The reaction was
stopped by the addition of stop solution and the optical density
value at 450 nm (OD450) was measured.
Cell counting kit-8 (CCK-8) assay
Cell viability was measured using a CCK-8 assay
(cat. no. CK04; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). Cells (5.0×103 cells/well) were seeded into a
96-well plate and incubated at 37°C for 24, 48 and 72 h. RPMI-1640
medium was used as a blank control. Following incubation, 10
µl CCK-8 solution was added to each well and the plate was
continuously incubated at 37°C for 2 h. The OD450 of each well was
then monitored.
Cell cycle assay
Following culture for 48 h, cells were washed thrice
with ice-cold PBS and then fixed in cold 70% ethanol for ≥24 h at
4°C. Single cell suspensions were then prepared, in which the DNA
was stained using propidium iodide (PI; P4170; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) following the manufacturer's protocol.
Cell cycle analysis was conducted using a FACSCalibur flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with three
independent experiments performed. The stained cells were analysed
with BD CellQuest™ Pro software (BD Biosciences).
Cell apoptosis assay
Cells (2.0×105 cells/well) were incubated
in a 6-well plate and cultured with fresh medium for 24, 48 and 72
h. The cells were then washed thrice with ice-cold PBS and the
APC-Annexin V Binding Apoptosis assay kit (cat. no. 22837; AAT
Bioquest, Sunnyvale, CA, USA) was used to measure apoptosis
according to the manufacturer's protocol.
Cell migration assay
A cell migration assay was performed using the
xCelligence RTCA instrument with its CIM-plate. The upper chambers
of the CIM-plate were supplemented with 4×104 cells/well
in FBS-free RPMI-1640 medium. The lower chambers of CIM-plate were
supplemented with RPMI-1640 medium containing 10% FBS. Following
attachment, the impedance produced by cell migration towards the
lower chamber was continuously monitored for 24 h by the system.
The data were collected and analyzed by the RTCA 1.2 software.
A Transwell chamber (cat. no. 3422; Corning
Incorporated, Corning, NY, USA) was also used to measure cell
migration ability. The Transwell insert was supplemented with
4×104 cells/well in FBS-free RPMI-1640 medium, and
RPMI-1640 medium containing 10% FBS was added to the lower chamber.
After 48 h, the Transwell inserts were washed thrice with PBS and
fixed in methylethanol for ≥20 min. The Transwell inserts were then
stained with crystal violet for 10 min at room temperature, washed
and the remaining crystal violet-stained cells were gently removed
with a cotton swab tipped applicator. The cells were counted under
the light microscope (magnification, ×200) in ≥5 different fields
of view to calculate the mean number of migrated cells.
Scratch healing assay
When the cells reached 80–90% confluence, a pipette
tip was used to wound the cells and fresh RPMI-1640 medium with 10%
FBS was applied. Wound healing was observed with a light microscope
(System Microscope IX71) for 48 h. The gap closure was observed at
24 and 48 h, and compared with the width of the gap at 0 h.
Cell invasion assays
A cell invasion assay was performed with the
xCelligence RTCA instrument and CIM-plate as in the cell migration
assay. The upper chambers of the CIM-plate were pre-coated with
Matrigel (cat. no. 356234; BD Biosciences) and then supplemented
with 6×104 cells/well in FBS-free RPMI-1640 medium. The
lower chambers of the CIM-plate were supplemented with RPMI-1640
medium containing 10% FBS. Following attachment, the impedance
produced by cells invading through the Matrigel towards the lower
chamber was continuously monitored for 24 h by the system. The data
were collected and analyzed by the RTCA 1.2 software.
Transwell chambers were also used to measure cell
invasion. Matrigel was added to the Transwell insert and solidified
at 37°C for 4–6 h to form a thin gel layer. The Transwell insert
was supplemented with 6×104 cells/well in FBS-free
RPMI-1640 medium. The lower chamber was supplemented with RPMI-1640
medium containing 10% FBS. After 48 h, the Transwell inserts were
washed thrice with PBS and fixed in methylethanol for ≥20 min. The
Transwell insert was stained with crystal violet for 10 min at room
temperature. After that, the Transwell inserts were washed with PBS
and the remaining crystal violet-stained cells were gently removed
with a cotton swab tipped applicator. The cells were counted under
a light microscope (magnification, ×200) in ≥5 different fields of
view to calculate the mean number of invaded cells.
Tissue microarray
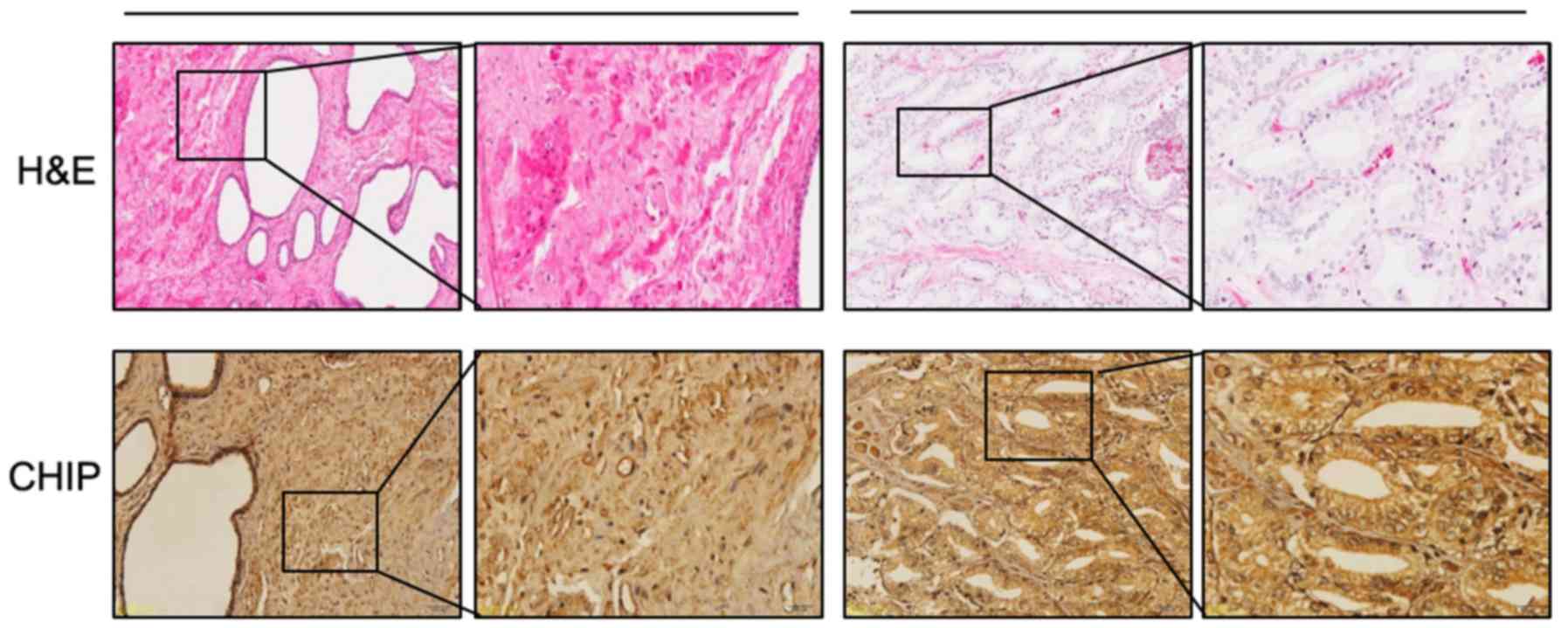
CHIP expression was examined by immunohistochemistry
(IHC), and hematoxylin and eosin (H&E) staining was also
examined. A tissue microarray of human prostate cancer
(HProA180PG04) was obtained from Shanghai Outdo Biotech Co., Ltd.
(Shanghai, China). All the tissue specimens in this microarray,
which included prostate cancer tissue and tissue adjacent to the
cancer, were obtained from 90 patients diagnosed with prostate
cancer. Tissue IHC was performed using anti-CHIP antibody (#2080;
Cell Signaling Technology, Inc.) according to the instructions of
the GTVision III Detection System/Mo & Rb IHC kit (GK500705;
Dako; Agilent Technologies, Inc., Santa Clara, CA, USA). CHIP
expression was graded on a scale of + to ++++ according to the
staining intensity of the cells. Positive CHIP staining in each
specimen was evaluated in the cytoplasm follows: +, weak staining,
light yellow; ++, moderate staining, light yellow brown; +++,
strong staining, brown; ++++, extreme staining, dark brown. The
grades were divided into two groups: low (+/++) and high
(+++/++++).
Statistical analysis
All experiments were repeated at least three times.
Data are expressed as the mean ± standard deviation. Statistical
comparisons were performed by Student's t-test. The clinical
characteristics of the subjects were expressed as the mean ±
standard deviation. Chi-square test was used to analyse the
associations between qualitative clinicopathological variables and
CHIP expression. P<0.05 was considered to indicate a
statistically significant difference. Data were analyzed with the
statistical analysis software SPSS 18.0 for Windows (SPSS Inc.,
Chicago, IL, USA).
Results
Establishing a CHIP-overexpressing cell
line
To understand the role of CHIP in DU145 prostate
cancer cells, a plasmid overexpressing CHIP was constructed and
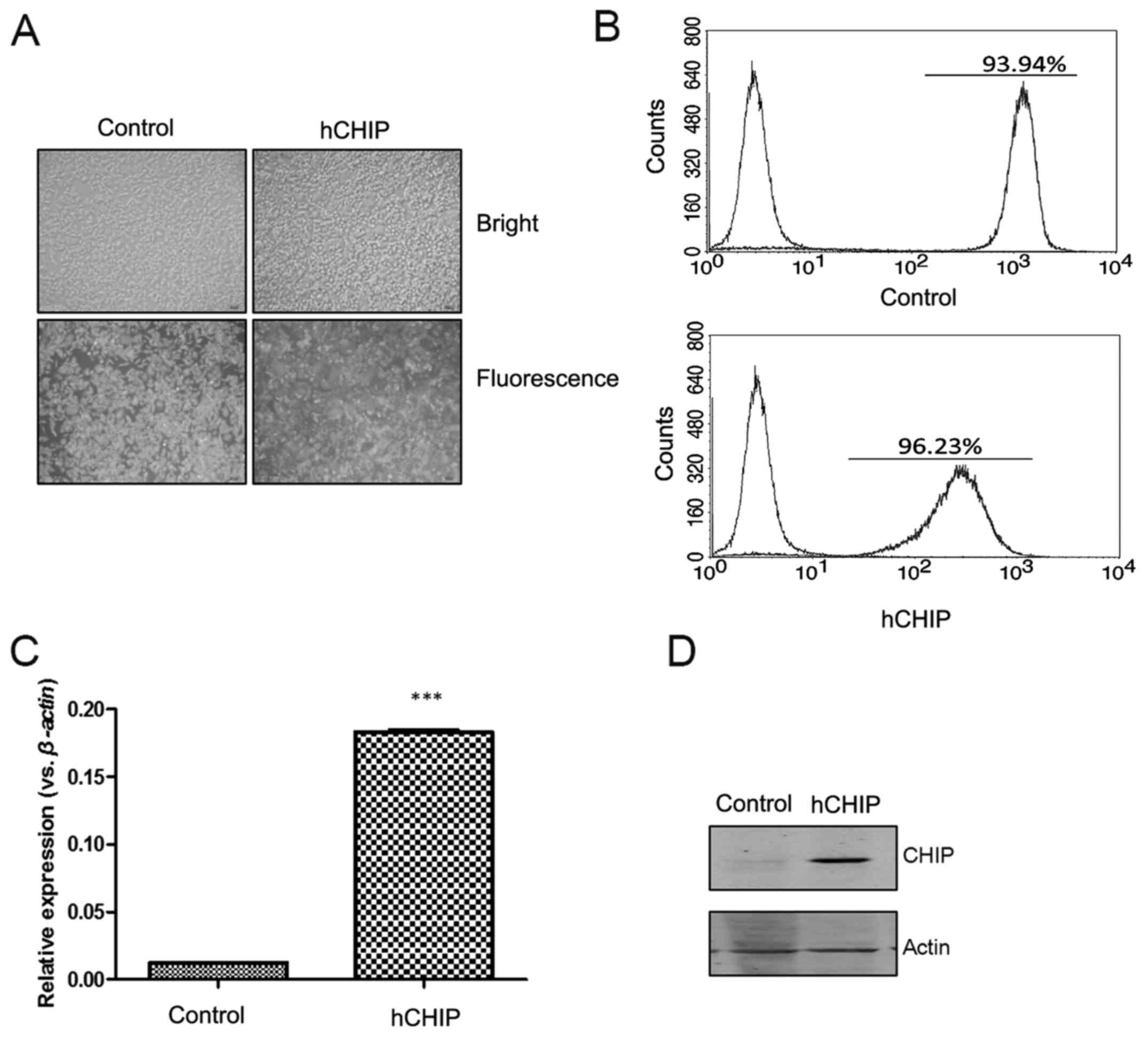
transfected into DU145 cells. When observed using fluorescence
microscopy, the DU145-control and DU145-hCHIP cells presented
strong GFP signals (Fig. 1A). The
percentages of GFP-positive cells, detected by flow cytometry, in
the DU145-control and DU145-hCHIP cells were 93.94 and 96.23%,
respectively (Fig. 1B). As shown
in Fig. 1C, the CHIP mRNA
expression of selected clones, which were expanded for 2 weeks, was
examined by RT-qPCR. The expression of CHIP mRNA was significantly
increased in the DU145-hCHIP cells compared with the DU145-control
cells. The protein levels of CHIP in the DU145-hCHIP cells were
also observed to be markedly increased compared with those in the
DU145-control cells when evaluated using western blotting (Fig. 1D). The results indicated that the
DU145 prostate cancer cell line over-expressing CHIP was
established successfully.
CHIP overexpression promotes DU145 cell
growth
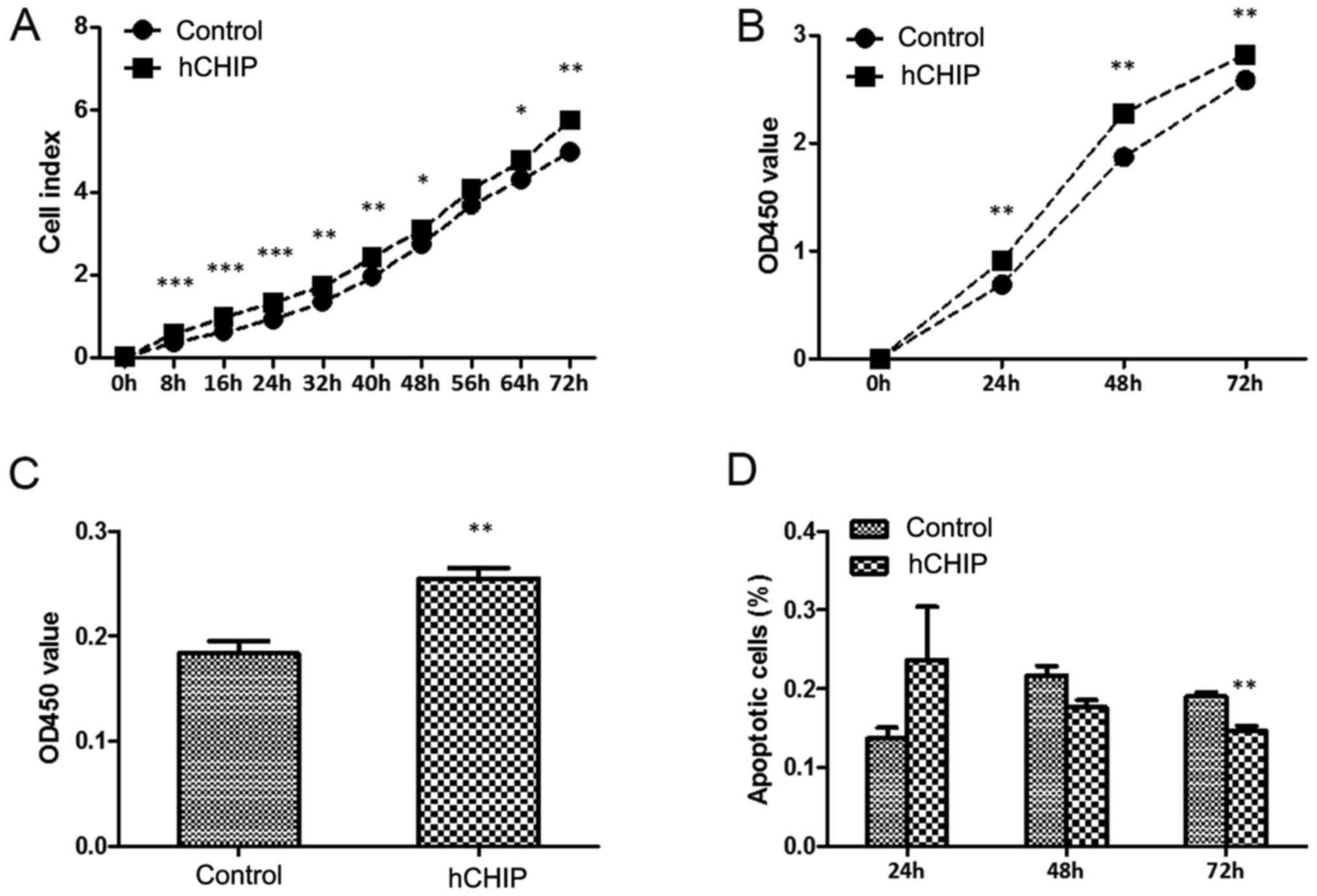
To investigate the effect of CHIP overexpression on
cell growth, the real-time xCelligence system was used. The DU145
cells overexpressing CHIP grew faster than the DU145-control cells,
and a significant difference was observed between the two
established cell lines during the 72-h continuous monitoring
(Fig. 2A). A CCK8 assay and BrdU
proliferation assay were performed to investigate the proliferation
capability of the DU145 cells. In the CCK8 assay, the OD450 values
of the DU145-control group were 0.690±0.068, 1.872±0.174 and
2.584±0.099 at 24, 48 and 72 h, respectively compared with
0.980±0.076, 2.276±0.127 and 2.821±0.047, respectively in the
DU145-hCHIP group (Fig. 2B). The
proliferation of the DU145-hCHIP cells was much greater than that
of the DU145-control cells and statistically significant
differences were detected between the two established cell lines
during the 72-h continuous monitoring. Similar results were
obtained in the BrdU proliferation assay. The OD450 value of the
DU145-control cells was 0.184±0.023 and that of the DU145-hCHIP
cells was 0.255±0.020 (Fig. 2C).
The proliferation of the DU145 cells with ectopic CHIP expression
was significantly increased. Cellular apoptosis was measured by
flow cytometry and the results are shown in Fig. 2D. The DU145-control cells comprised
0.203±0.025, 0.210±0.020 and 0.200±0.010% apoptotic cells at 24, 48
and 72 h, respectively, whereas the DU145-hCHIP cells comprised
0.173±0.025, 0.177±0.015 and 0.143±0.015% apoptotic cells at 24, 48
and 72 h. Apoptosis was reduced in the DU145-hCHIP cells compared
with the DU145-control cells, and a statistically significant
difference in the frequencies of apoptotic cells was detected
between the two types of cells at 72 h. Considered together, these
results indicate that CHIP overexpression accelerated the growth of
DU145 cells, likely due to the promotion of cellular proliferation
and inhibition of cellular apoptosis.
CHIP regulates proliferation- and
apoptosis-associated proteins
Western blotting results for the cells are presented
in Fig. 3. The levels of proteins
associated with the NF-κB signaling pathway were examined. As shown
in Fig. 3B, there were no evident
changes in the expression of RelA, RelB, p50, p52 and c-Rel in NE
and CE portions of the DU145-hCHIP cells compared with the
DU145-control cells. The protein levels of TRAF2 and TRAF3, which
are upstream molecules of the NF-κB signaling pathway, were also
examined. TRAF2 positively regulates canonical and non-canonical
NF-κB activity and TRAF3 negatively regulates non-canonical NF-κB
activity (15). In the present
study, there were no clear differences in the expression of TRAF2
and TRAF3 between the DU145-hCHIP cells and the DU145-control
cells.
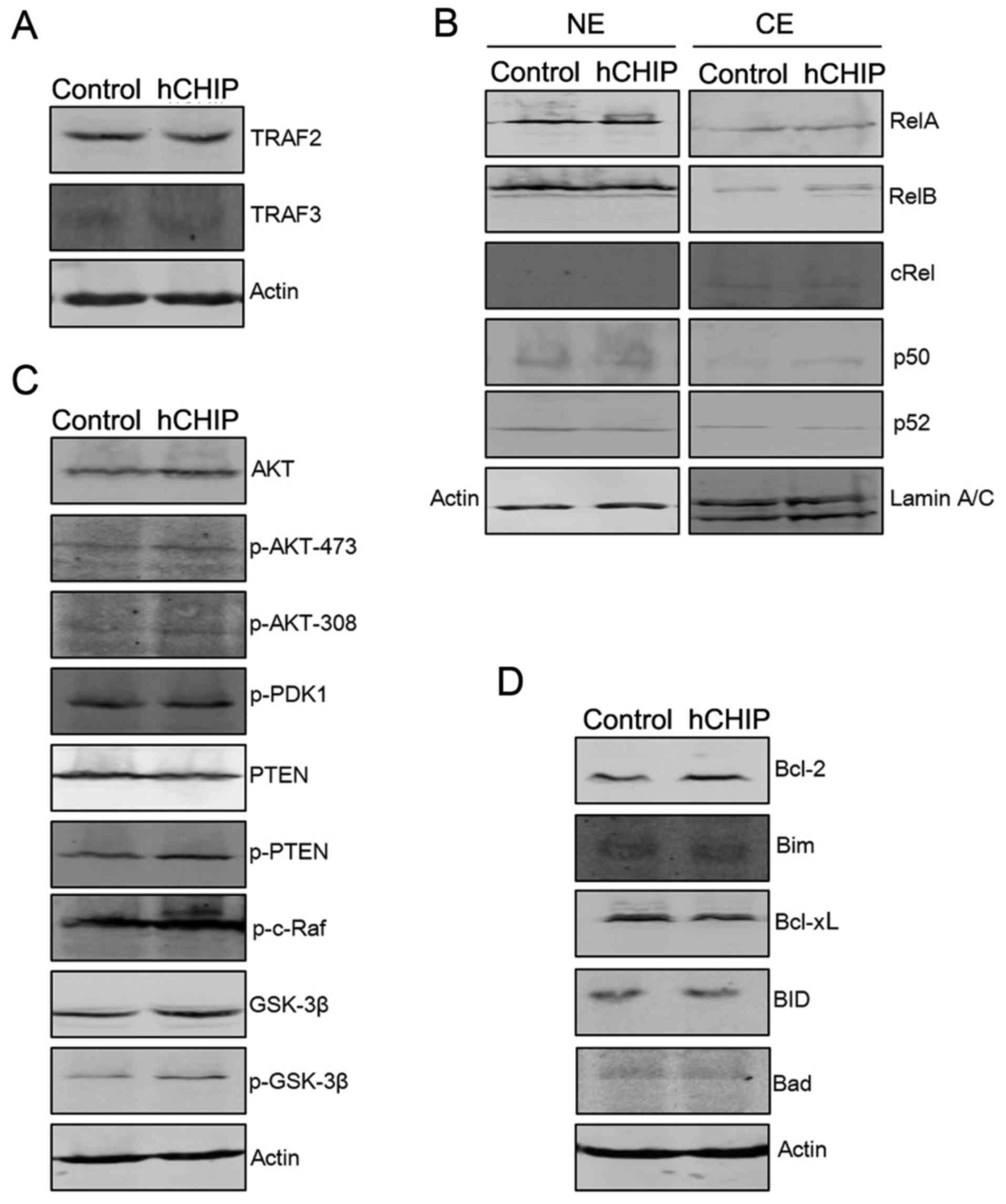 | Figure 3CHIP overexpression regulates
proliferation- and apoptosis-associated proteins. (A) Protein
expression levels of TRAF2 and TRAF3 were analyzed by western
blotting. The level of each protein was normalized against actin.
(B) The protein expression levels of nuclear factor-κB subunits
were analyzed by western blotting. Protein expression in the NE and
CE portions were normalized against actin and laminA/C,
respectively. (C) The expression levels of proteins associated with
the AKT signaling pathway were analyzed by western blotting. The
level of each protein was normalized against actin. (D) Expression
levels of pro- and anti-apoptotic proteins were analyzed by western
blotting. The level of each protein was normalized against actin.
CHIP, carboxyl terminus of Hsc-70-interacting protein; hCHIP, human
CHIP; NE, nuclear extract; CE, cytoplasmic extract; TRAF, TNF
receptor-associated factor; RelA, transcription factor p65; RelB,
transcription factor RelB; cRel, proto-oncogene c-Rel; AKT, protein
kinase B; p, phosphorylated; PDK, 3-phosphoinositide-dependent
protein kinase; PTEN, phosphatase and tensin homolog; c-Raf,
proto-oncogene c-Raf; GSK, glycogen synthase kinase; Bcl-2, B-cell
lymphoma-2; Bim, Bcl-2-like protein 11; Bcl-xL, Bcl-extra large;
BID, BH3 interacting-domain death agonist; Bad, Bcl-2-associated
death promoter. |
AKT signaling molecules were also examined by
western blotting to investigate whether they are involved in the
altered cellular proliferation induced by the expression of CHIP.
As shown in Fig. 3C, the
expression levels of total AKT, p-AKT-473, p-AKT-308, p-PTEN,
p-c-Raf, GSK-3β and p-GSK-3β were higher in the DU145-hCHIP cells
compared with the DU145-control cells. The overexpression of CHIP
led to a reduction in the expression level of PTEN. However, the
phosphorylation levels of 3-phosphoinositide-dependent protein
kinase 1 were similar in the two types of DU145 cells (Fig. 3C). The protein levels of Bcl-2, an
anti-apoptotic protein, were markedly increased in the DU145-hCHIP
cells compared with the DU145-control cells. By contrast, the
protein levels of Bim, a pro-apoptotic protein were reduced in the
DU145-hCHIP cells (Fig. 3D).
Together, these results indicate that CHIP over-expression led to
activation of the AKT signaling pathway. In addition, the promoted
expression of anti-apoptotic proteins, such as Bcl-2, and
suppressed expression of pro-apoptotic proteins, such as Bim, may
have contributed to the reduction of apoptosis in the DU145-hCHIP
cells.
CHIP overexpression promotes the G0-G1
phase of the cell cycle
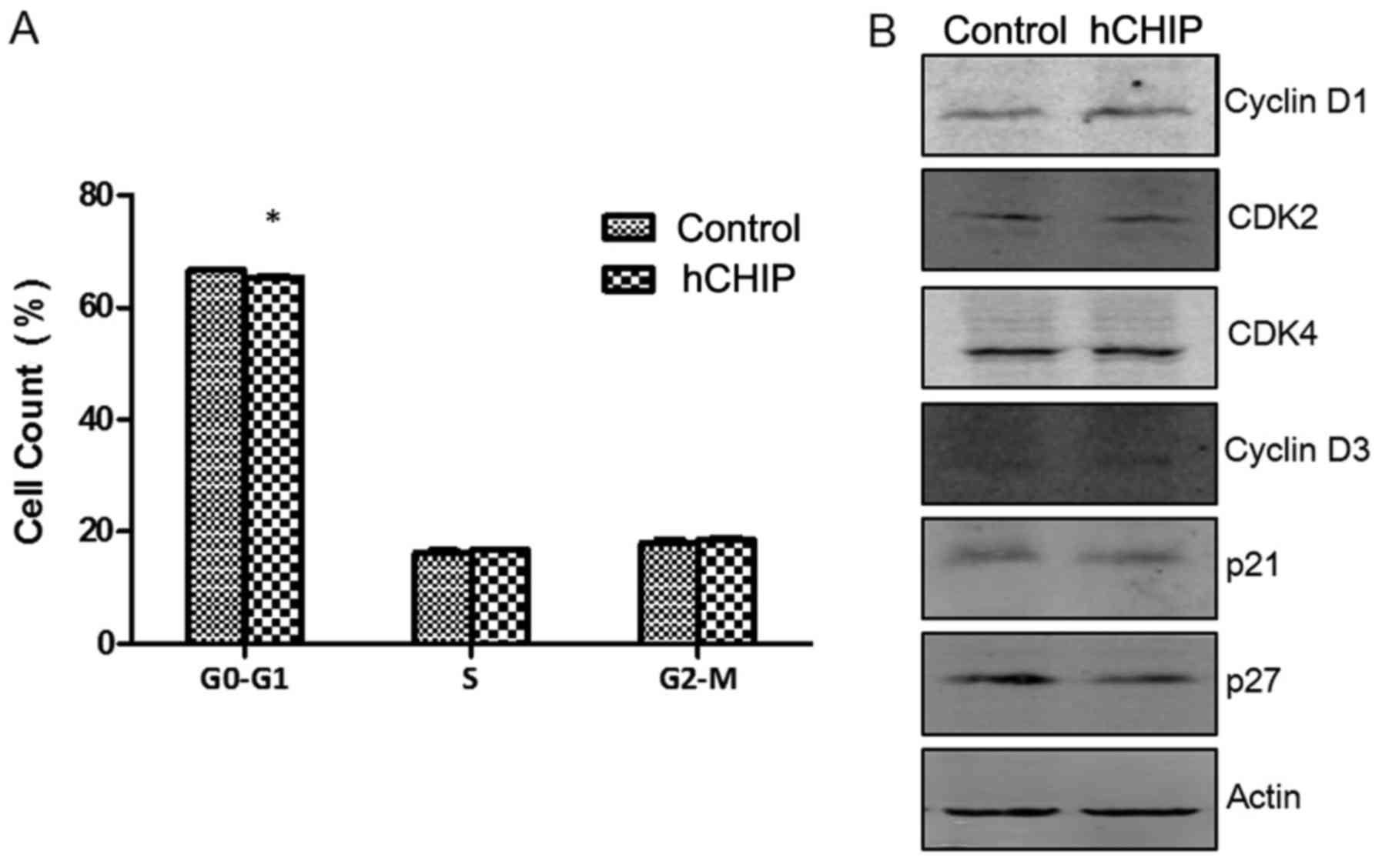
Cell-cycle analyses and cellular DNA content
measurements were performed using PI staining. The percentages of
cells in the G0-G1, S and G2-M cell-cycle phases were 66.56±0.41,
16.15±0.826 and 18.02±0.91% respectively in the DU145-control cells
and 65.34±0.47, 16.78±0.19 and 18.53±0.64% in the DU145-hCHIP
cells. No significant differences were observed between the
DU145-hCHIP and DU145-control cells with regard to the proportions
in the S and G2-M phases. However, a significant difference was
detected in the proportion of cells in the G0-G1 phase between the
two cell lines (P<0.05) (Fig.
4A). As shown in Fig. 4B, the
levels of proteins regulated in the cell cycle were detected by
western blotting. The results suggested that CHIP overexpression
promoted the expression of cyclin-dependent kinase 2, cyclin D1 and
cyclin D3, and inhibited the expression of the cell cycle
inhibitory proteins p21 and p27. Therefore, it may be concluded
that CHIP overexpression promoted the proliferation and G0-G1
transition of prostate cancer cells.
CHIP affects the migration ability of
DU145 prostate cancer cells
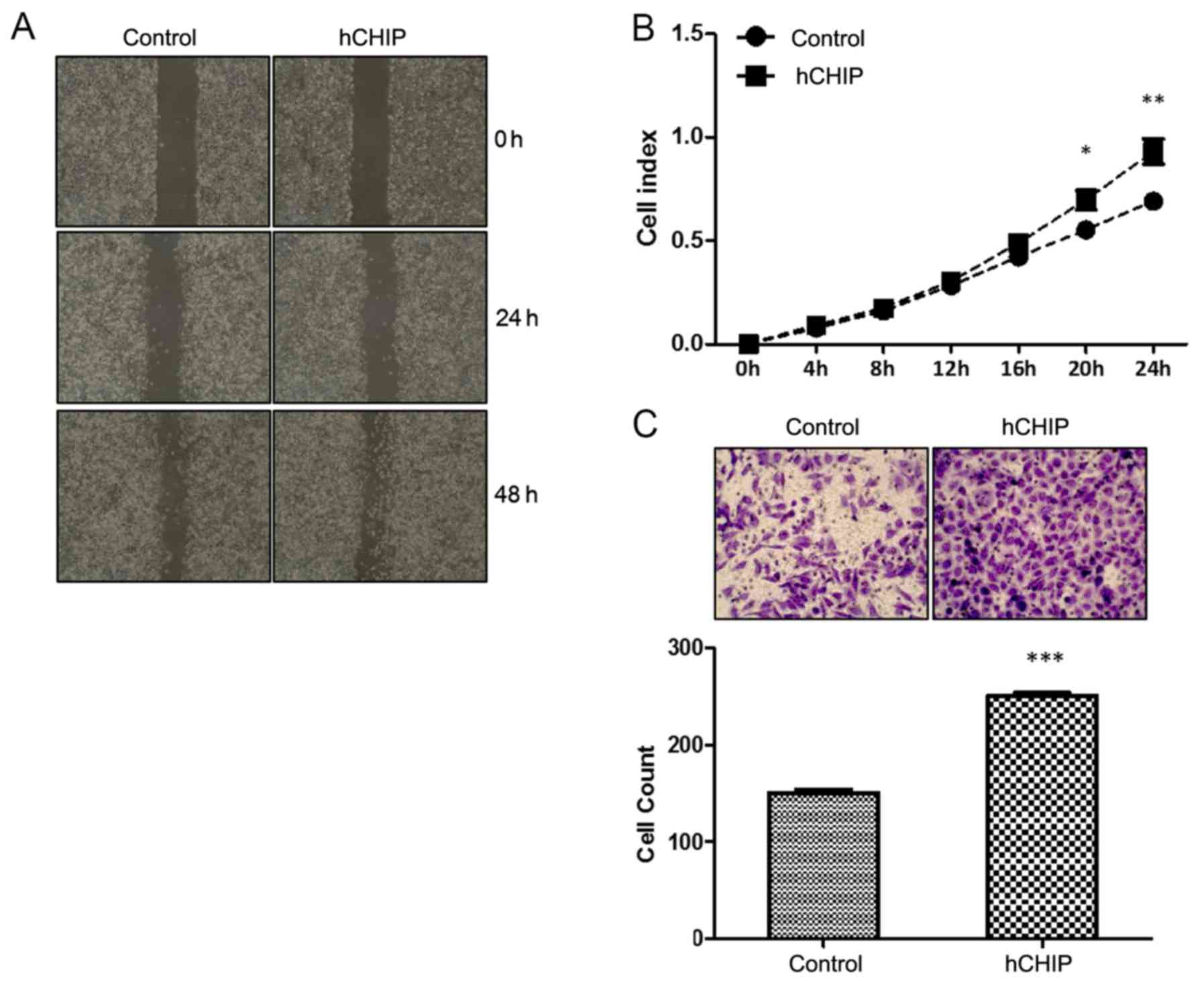
The migration ability of the cells was examined
in vitro using three different methods, namely a scratch
healing assay, the real-time xCelligence system and a Transwell
assay. In the scratch healing assay, the wound area was reduced to
a greater extent by the CHIP-overexpressing cells than by the
control cells (Fig. 5A). The
real-time xCelligence system assay indicated that the DU145-hCHIP
cells migrated faster than the DU145-control cells during the 24-h
continuous monitoring, and statistically significant differences
were detected between the two cell lines at 20 and 24 h (Fig. 5B). The Transwell cell migration
assay also demonstrated that the overexpression of CHIP
significantly increased the migration ability of the cells, as
shown in Fig. 5C. Migratory cell
numbers were increased ~1.5-fold (P<0.0001) for the DU145-hCHIP
cells compared with the DU145-control cells (Fig. 5C). Therefore, it may be concluded
that the overexpression of CHIP increased the migration ability of
the DU145 prostate cancer cells.
CHIP affects the invasion ability of
DU145 prostate cancer cells
To examine the effect of CHIP on the invasion
ability of DU145 cells, the real-time xCelligence system was used
and Transwell cell invasion assays were performed. As shown in
Fig. 6A, the DU145 cells
overexpressing CHIP invaded through the Matrigel faster than the
control cells did, and there was a statistically significant
difference between the cell index for the two cell lines during the
24-h continuous monitoring. In the Transwell assay, the invasive
cells numbers were significantly increased ~6-fold (P<0.0001)
for the DU145-hCHIP cells compared with the DU145-control cells
(Fig. 6B).
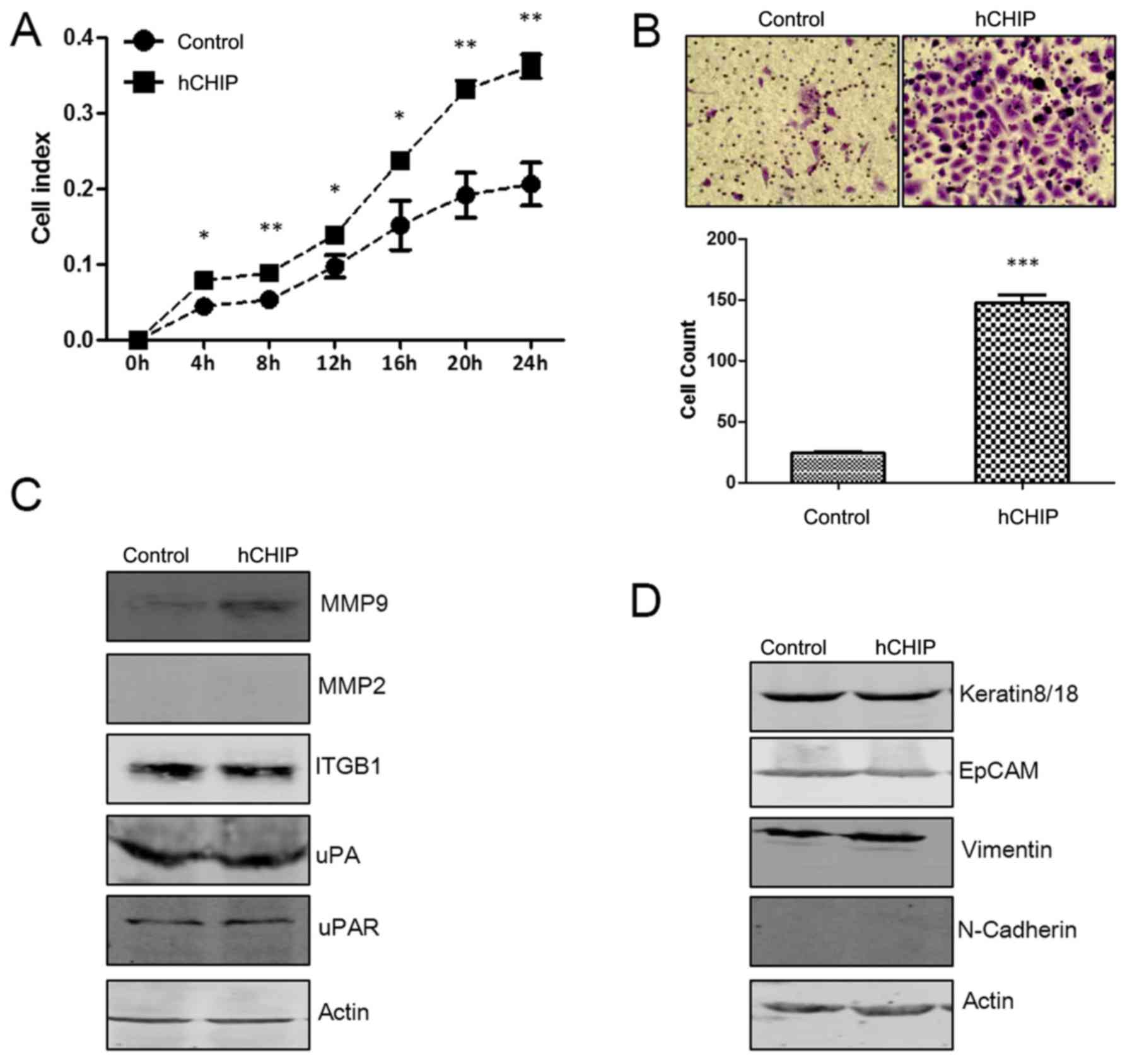 | Figure 6CHIP overexpression promotes the
invasive ability of DU145 cells. (A) The invasive ability of cells
was detected using a real-time xCelligence system. Each plate was
inoculated with 6×104 cells and invasion was examined
during a 24-h monitoring (*P<0.05 and
**P<0.01; Student's t-test). (B) Transwell invasion
assay of the two established cell lines. The cells were cultured in
Transwell chambers for 48 h and fixed and stained with crystal
violet. The invaded cells from five 20× fields were counted under a
microscope and quantified results are presented
(***P<0.001; Student's t-test). (C) Protein
expression levels of MMP9, MMP2, ITGB1, uPA and uPAR were analyzed
by western blotting. The level of each protein was normalized
against actin. (D) Protein expression levels of keratin8/18, EpCAM,
vimentin and N-cadherin were analyzed by western blotting. The
level of each protein was normalized against actin. CHIP, car-boxyl
terminus of Hsc-70-interacting protein; hCHIP, human CHIP; MMP,
matrix metalloproteinase; ITGB1, integrin β1; uPA, urokinase-type
plasminogen activator; uPAR, uPA receptor; EpCAM, epithelial cell
adhesion molecule. |
MMP and uPA systems serve an important role in the
invasion of tumor cells (16). In
the present study, it was observed that the protein levels of MMP9
and uPAR were increased in the DU145-hCHIP cells compared with the
DU145-control cells. However, no marked differences in the
expression of MMP2 and uPA were detected. ITGB1 is associated with
the function of invasion (17),
and no difference in ITGB1 protein levels was evident between the
two cell lines (Fig. 6C).
Epithelial mesenchymal transition (EMT) enables cancer cells to
invade and migrate, finally resulting in metastasis (18). Biomarkers of the EMT process were
investigated in the present study. The results demonstrated that
CHIP overexpression upregulated the protein level of vimentin,
which is a mesenchymal marker (19). As epithelial markers, the protein
levels of N-cadherin, EpCAM and keratin8/18 (20–23)
were not affected by the overexpression of CHIP (Fig. 6D). These results suggest that the
overexpression of CHIP promoted the invasive ability of prostate
cancer by inducing the EMT process.
CHIP expression in human prostate cancer
tissues
The expression levels of CHIP in 90 pairs of
prostate cancer tissues and adjacent tissues were detected by IHC.
The expression of CHIP in the prostate cancer cells was compared
with that in the cells adjacent to the cancer. Representative cases
of CHIP expression in cancer tissues and adjacent tissues are shown
in Fig. 7. Positive
immunohistochemical staining for CHIP was present mainly in the
cytoplasm and on the membrane of the prostate cancer cells and the
expression of CHIP in the prostate cancer tissues was higher than
that in the adjacent tissues. Overall, high staining of CHIP was
detected in 53/90 (58.89%) prostate cancer tissues, while
relatively low staining of CHIP was detected in 37/90 (41.11%)
cancer samples. The association between CHIP expression and the
clinical pathological characteristics of the prostate cancer
patients is presented in Table I.
The CHIP expression level exhibited a positive association with the
Gleason scores of the prostate cancer patients (P<0.005), but
did not differ according to the age of the patients (P=0.097). The
results indicate that CHIP expression was increased in the prostate
cancer tissue and that CHIP overexpression is an indicator of the
cancer grade, and hence a poor prognosis.
 | Table IAssociation between CHIP expression
as determined by immunohistochemistry and the clinical pathological
characteristics of patients with prostate cancer. |
Table I
Association between CHIP expression
as determined by immunohistochemistry and the clinical pathological
characteristics of patients with prostate cancer.
|
Characteristics | Patients, n
(%) | CHIP expression, n
(%)
| P-value |
|---|
| High | Low |
|---|
| All patients | 90 | 53 (58.89) | 37 (41.11) | |
| Age (years)a | | | | 0.970 |
| ≤70 | 46 (51.11) | 27 | 19 | |
| >70 | 44 (48.89) | 26 | 18 | |
| Gleason score | | | | 0.005 |
| 5–6 | 29 (32.22) | 11 | 18 | |
| 7–10 | 61 (67.78) | 42 | 19 | |
Discussion
In the present study, CHIP was demonstrated to be
important in the progression of prostate cancer. As an
androgen-dependent cancer, prostate cancer is effectively treated
by androgen withdrawal in the early stages. However, with
progression of the disease, prostate cancer may become
androgen-independent and resistant to hormone therapy (24). As androgen-independent cells, DU145
cells have a very low degree of differentiation and powerful
migration and invasion abilities (25). In the present study, the
overexpression of CHIP promoted cell proliferation and suppressed
apoptosis in the DU145 prostate cancer cells, which was associated
with activation of the AKT signaling pathway and the regulation of
pro- and anti-apoptotic proteins. The migration and invasion of
DU145 cells were also accelerated by the overexpression of CHIP,
which was associated with the upregulation of MMP9 and uPAR.
Furthermore, the present study revealed that overexpression of CHIP
increased the protein level of vimentin, which is a mesenchymal
marker, but not the protein levels of epithelial markers in the EMT
process. In addition, evidence that increased CHIP expression is
positively associated with a high histological grade of prostate
cancer was obtained. Collectively, these results indicate that CHIP
serves an oncogenic role in human prostate cancer.
CHIP is an E3 ubiquitin ligase and an HSP70 and
HSP90 interacting co-chaperone. CHIP functions as a tumor
suppressor in numerous types of tumors, including colorectal
cancer, gastric cancer and breast cancer (26–28).
However, there is also evidence to suggest that CHIP is a powerful
oncogene in other types of tumors (12,13).
In the present study, the results indicated that CHIP functions as
an oncogenic driver of prostate cancer cell proliferation. The
forced expression of CHIP in DU145 cells increased cell
proliferation by activation of the AKT signaling pathway,
upregulation of p-PTEN and downregulation of PTEN. The AKT
signaling pathway promotes the proliferation, migration and
invasion of cancer cells and PTEN is a well-known suppressor of
this pathway; however, PTEN can lose its tumor suppressor function
by phosphorylation (29). It has
previously been demonstrated that CHIP overexpression is associated
with the phosphorylation of PTEN, which activates the AKT signaling
pathway (30). In MCF7 and MCF10A
human breast cancer cells, CHIP activates the AKT/forkhead box O3
(FoxO)/Bim signaling pathway to induce proliferation and apoptosis
resistance by downregulating the level of PTEN (31). In human prostate cancer cells, the
overexpression of CHIP leads to the accelerated degradation and
elevated ubiquitination of endogenous PTEN, while the depletion of
endogenous CHIP stabilizes PTEN (32). In thyroid cancer tissue and cell
lines, the upregulation of CHIP increases the cell viability and
growth via activation of AKT and MAPK pathways, while the
downregulation of CHIP induces the opposite results (12). The present study demonstrated that
the expression of PTEN was downregulated in DU145-hCHIP cells;
however, the mechanism requires further exploration.
Previous evidence suggests that CHIP modulates
mitotic arrest to suppress the proliferation of prostate cancer
cells by degrading the androgen receptor in androgen-dependent
LNCaP prostate cancer cells (33).
In gastric cancer, CHIP overexpression promotes the ubiquitination
and degradation of p65, which are canonical NF-κB signaling pathway
activities, to inhibit the growth of xenografts and blood vessel
formation in nude mice (27). In
breast cancer cells, CHIP induces the downregulation of TRAF2 to
inhibit NF-κB-mediated cell invasion (34). Unlike the previous findings, the
present study indicated that CHIP had no association with the
canonical or non-canonical NF-κB signaling pathways.
In the present study, CHIP-overexpression attenuated
the apoptosis of the DU145 prostate cancer cells. The data revealed
that Bcl-2, an anti-apoptotic protein, was markedly upregulated in
the CHIP-overexpressing DU145 cells, whereas Bim, a pro-apoptotic
protein, was downregulated. A previous study revealed that
excessive CHIP expression promoted abnormal AKT activation,
inactivated the FoxO3 signaling pathway and decreased the level of
Bim (31). AKT phosphorylation
releases Bcl-2 from the polymer to serve an anti-apoptotic function
and decreases the proteins level of Bax and Bad (35,36).
Bcl-2 and Bcl-xL are anti-apoptotic members of the Bcl-2 family
that regulate cell apoptosis. The results of the present study
revealed no marked differences in the protein expression levels of
Bcl-xL, but clear differences in the protein expression levels of
Bcl-2. Although Bcl-2 and Bcl-xL have similar structural features
(37), they undergo different
changes in certain situations. It has been shown that the
expression of Bcl-xL is high in some hemopoietic tumors, while the
expression of Bcl-2 is low (38).
In present study, proliferation was significantly promoted and
apoptosis was inhibited in the CHIP-overexpressing DU145 cells.
These results are consistent with those reported in the previous
studies.
Observation of cell cycle revealed that DU145-hCHIP
cells went through the G0-G1 phase faster than DU145-control cells.
The western blotting results indicated that CHIP overexpression
triggered the upregulation of cyclin D1 and the downregulation of
p21 and p27. Cyclin D1, p21 and p27 are cell cycle-dependent
regulatory factors that act as the downstream molecules of the AKT
signaling pathway (39). Previous
studies have indicated that the activated AKT pathway induces the
expression of cyclin D1 to promote the cell cycle by the
phosphorylation of GSK-3β (40,41).
The AKT signaling pathway inhibits apoptosis and promotes cellular
proliferation via the regulation of p27 and GSK-3β (42–44).
CHIP is able to induce the ubiquitylation and degradation of p21,
which leads to transient cell cycle arrest in the G1 phase
(45). Previous studies suggested
that the inhibition of CHIP upregulates the level of
TGF-β-responsive p21 and p15 to affect cell cycle transition
(46,47). Cyclin D1 and p27 are directly
modulated by the AKT signaling pathway at the transcriptional level
to modulate cell proliferation (48–50).
These observations lead to the conclusion that CHIP promoted the
proliferation of prostate cancer cells via the promotion of cell
cycle progression.
The high mortality rate of prostate cancer is
associated with the high metastatic ability and invasive nature of
this cancer (3). Degradation of
the basement membrane barriers and the extracellular matrix (ECM)
are crucial steps in the pathogenesis of prostate cancer (51). The functions of MMPs in the
progression of tumor invasion and tissue remodeling have been
confirmed (52). MMP2 and MMP9 are
gelatinases and the two enzymes are relevant to the degradation of
ECM through strong proteolytic activity (53). Previous studies indicated that high
expression levels of CHIP are associated with the suppression of
MMP9, which inhibits breast cancer invasion (7,34).
In a similar manner to MMPs, uPA and uPAR are vital determinants in
the degradation of the EMC; studies indicate that uPAR acts as a
single transducer that associates with certain cell surface
molecules, such as integrin, in cell migration and adhesion
(54–56). In DU145 and PC3 cell lines, the
downregulation of MMP9 and uPAR inhibited angiogenesis, Matrigel
invasion and wound healing ability, and induced apoptosis (57). Consistently with the previous
findings, the present study indicated that the upregulation of MMP9
and uPAR was associated with the increased migration and invasion
ability of CHIP-overexpressing prostate cancer cell lines. However,
CHIP did not appear to affect the expression levels of MMP2, ITGB1
and uPA.
EMT is a crucial process that changes epithelial
cells to the migratory mesenchymal phenotype (19). During tumori-genesis, normal cancer
cells acquire the ability to metastasize via EMT. In this
procession, the acquisition of a mesenchymal phenotype by
epithelial cells is accompanied by the upregulation of mesenchymal
markers, including vimentin and N-cadherin, and the downregulation
of epithelial markers, including N-cadherin, EpCAM and keratin8/18
(51,58). The present study demonstrated that
CHIP overexpression upregulated the protein level of vimentin in
DU145 prostate cancer cells, but did change the expression levels
of N-cadherin, EpCAM and keratin8/18. Therefore, it may be
concluded that CHIP overexpression increases the expression of
vimentin and thereby induces EMT.
In the present study, the CHIP expression level was
observed to be significantly elevated in prostate cancer tissues
compared with normal tissue, and the expression level of CHIP was
associated with the Gleason score, but not with age. These findings
are in line with previous findings in gliomas and esophageal
squamous cell carcinoma (59,60).
In view of the observation that CHIP is able to distinguish between
different stages in the progression of prostate cancer, it may have
potential as a clinical marker of prostate cancer.
In conclusion, CHIP overexpression significantly
promoted the growth, migration and invasion of prostate cancer
cells. The results of the present study suggest that CHIP effected
the progression of proliferation in prostate cancer cells via
downregulation of the expression of PTEN, which activated the AKT
signaling pathway. Activation of the AKT signaling pathway
significantly induced cell proliferation by facilitating cell
growth, suppressing apoptosis and promoting G0-G1 transition. CHIP
expression was positively associated with the ability of tumor
cells to invade and migrate. CHIP possibly induced the EMT process
in the prostate cancer cells to confer migratory and invasive
properties to the cells. However, the molecular mechanism of CHIP
in the process of migration and invasion required further
elucidation. CHIP expression is associated with the Gleason scores
of prostate cancer patients. High expression levels of CHIP were
detected in prostate cancer tissues and were positively associated
with high Gleason scores. These findings establish a
tumor-promoting role for CHIP in the tumorigenesis of prostate
cancer and have important implications for the diagnosis and
therapy of this disease.
Acknowledgments
The authors greatly acknowledge Jingjing Xu and Jun
Zhou for excellent technical assistance and suggestions.
Funding
The present study was supported by the Natural
Science Foundation of Jiangsu Provincial (grant no.
BK20151211).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding authors on reasonable
request.
Authors' contributions
LC performed research, collected and analyzed data.
JZ performed research. HJD and FL collected clinical information.
FG designed and performed research, collected, analyzed and
interpreted data, and wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of
interest.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Litwin MS and Tan HJ: The diagnosis and
treatment of prostate cancer: A review. JAMA. 317:2532–2542. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Paul I and Ghosh MK: The E3 ligase CHIP:
Insights into its structure and regulation. BioMed Res Int.
2014:9181832014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Murata S, Minami Y, Minami M, Chiba T and
Tanaka K: CHIP is a chaperone-dependent E3 ligase that
ubiquitylates unfolded protein. EMBO Rep. 2:1133–1138. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paul I and Ghosh MK: A CHIPotle in
physiology and disease. Int J Biochem Cell Biol. 58:37–52. 2015.
View Article : Google Scholar
|
|
7
|
McDonough H and Patterson C: CHIP: A link
between the chaperone and proteasome systems. Cell Stress
Chaperones. 8:303–308. 2003. View Article : Google Scholar
|
|
8
|
Connell P, Ballinger CA, Jiang J, Wu Y,
Thompson LJ, Höhfeld J and Patterson C: The co-chaperone CHIP
regulates protein triage decisions mediated by heat-shock proteins.
Nat Cell Biol. 3:93–96. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shang Y, He J, Wang Y, Feng Q, Zhang Y,
Guo J, Li J, Li S, Wang Y, Yan G, et al: CHIP/Stub1 regulates the
Warburg effect by promoting degradation of PKM2 in ovarian
carcinoma. Oncogene. 36:4191–4200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yonezawa T, Takahashi H, Shikata S, Liu X,
Tamura M, Asada S, Fukushima T, Fukuyama T, Tanaka Y, Sawasaki T,
et al: The ubiquitin ligase STUB1 regulates stability and activity
of RUNX1 and RUNX1-RUNX1T1. J Biol Chem. 292:12528–12541. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
1Liu F, Zhou J, Zhou P, Chen W and Guo F:
The ubiquitin ligase CHIP inactivates NF-κB signaling and impairs
the ability of migration and invasion in gastric cancer cells. Int
J Oncol. 46:2096–2106. 2015. View Article : Google Scholar
|
|
12
|
Zhang L, Liu L, He X, Shen Y, Liu X, Wei
J, Yu F and Tian J: CHIP promotes thyroid cancer proliferation via
activation of the MAPK and AKT pathways. Biochem Biophys Res
Commun. 477:356–362. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jin Y, Zhou L, Liang ZY, Jin KM, Zhou WX
and Xing BC: Clinicopathologic and prognostic significance of
carboxyl terminus of Hsp70-interacting protein in HBV-related
hepatocellular carcinoma. Asian Pac J Cancer Prev. 16:3709–3713.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) Method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
15
|
Tang X, Zhang L and Wei W: Roles of TRAFs
in NF-κB signaling pathways mediated by BAFF. Immunol Lett.
196:113–118. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mali AV, Joshi AA, Hegde MV and Kadam ShS:
Enterolactone suppresses proliferation, migration and metastasis of
MDA-MB-231 breast cancer cells through inhibition of uPA induced
plasmin activation and MMPs-mediated ECM remodeling. Asian Pac J
Cancer Prev. 18:905–915. 2017.PubMed/NCBI
|
|
17
|
Kurozumi A, Goto Y, Matsushita R, Fukumoto
I, Kato M, Nishikawa R, Sakamoto S, Enokida H, Nakagawa M, Ichikawa
T, et al: Tumor-suppressive microRNA-223 inhibits cancer cell
migration and invasion by targeting ITGA3/ITGB1 signaling in
prostate cancer. Cancer Sci. 107:84–94. 2016. View Article : Google Scholar
|
|
18
|
Grant CM and Kyprianou N: Epithelial
mesenchymal transition (EMT) in prostate growth and tumor
progression. Transl Androl Urol. 2:202–211. 2013.
|
|
19
|
Nakazawa M and Kyprianou N:
Epithelial-mesenchymal-transition regulators in prostate cancer:
Androgens and beyond. J Steroid Biochem Mol Biol. 166:84–90. 2017.
View Article : Google Scholar
|
|
20
|
Zhang T, Zhao G, Yang C, Dong P, Watari H,
Zeng L, Pfeffer LM and Yue J: Lentiviral vector mediated-ASAP1
expression promotes epithelial to mesenchymal transition in ovarian
cancer cells. Oncol Lett. 15:4432–4438. 2018.PubMed/NCBI
|
|
21
|
Chaudhari PR, Charles SE, D'Souza ZC and
Vaidya MM: Hemidesmosomal linker proteins regulate cell motility,
invasion and tumorigenicity in oral squamous cell carcinoma derived
cells. Exp Cell Res. 360:125–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nassour M, Idoux-Gillet Y, Selmi A, Côme
C, Faraldo ML, Deugnier MA and Savagner P: Slug controls
stem/progenitor cell growth dynamics during mammary gland
morphogenesis. PLoS One. 7:e534982012. View Article : Google Scholar
|
|
23
|
Wu F, Zhu J, Mao Y, Li X, Hu B and Zhang
D: Associations between the epithelial-mesenchymal transition
phenotypes of circulating tumor cells and the clinicopathological
features of patients with colorectal cancer. Dis Markers.
2017:94745322017. View Article : Google Scholar
|
|
24
|
Szostak MJ and Kyprianou N:
Radiation-induced apoptosis: Predictive and therapeutic
significance in radiotherapy of prostate cancer (Review). Oncol
Rep. 7:699–706. 2000.PubMed/NCBI
|
|
25
|
Motta M, Dondi D, Moretti RM, Montagnani
Marelli M, Pimpinelli F, Maggi R and Limonta P: Role of growth
factors, steroid and peptide hormones in the regulation of human
prostatic tumor growth. J Steroid Biochem Mol Biol. 56:107–111.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Ren F, Wang Y, Feng Y, Wang D, Jia
B, Qiu Y, Wang S, Yu J, Sung JJ, et al: CHIP/Stub1 functions as a
tumor suppressor and represses NF-κB-mediated signaling in
colorectal cancer. Carcinogenesis. 35:983–991. 2014. View Article : Google Scholar
|
|
27
|
Wang S, Wu X, Zhang J, Chen Y, Xu J, Xia
X, He S, Qiang F, Li A, Shu Y, et al: CHIP functions as a novel
suppressor of tumour angiogenesis with prognostic significance in
human gastric cancer. Gut. 62:496–508. 2013. View Article : Google Scholar
|
|
28
|
Tsuchiya M, Nakajima Y, Hirata N,
Morishita T, Kishimoto H, Kanda Y and Kimura K: Ubiquitin ligase
CHIP suppresses cancer stem cell properties in a population of
breast cancer cells. Biochem Biophys Res Commun. 452:928–932. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Torres J and Pulido R: The tumor
suppressor PTEN is phosphorylated by the protein kinase CK2 at its
C terminus. Implications for PTEN stability to proteasome-mediated
degradation. J Biol Chem. 276:993–998. 2001. View Article : Google Scholar
|
|
30
|
Brazil DP, Yang ZZ and Hemmings BA:
Advances in protein kinase B signalling: AKTion on multiple fronts.
Trends Biochem Sci. 29:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lv Y, Song S, Zhang K, Gao H and Ma R:
CHIP regulates AKT/FoxO/Bim signaling in MCF7 and MCF10A cells.
PLoS One. 8:e833122013. View Article : Google Scholar :
|
|
32
|
Ahmed SF, Deb S, Paul I, Chatterjee A,
Mandal T, Chatterjee U and Ghosh MK: The chaperone-assisted E3
ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN
for proteasomal degradation. J Biol Chem. 287:15996–16006. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sarkar S, Brautigan DL, Parsons SJ and
Larner JM: Androgen receptor degradation by the E3 ligase CHIP
modulates mitotic arrest in prostate cancer cells. Oncogene.
33:26–33. 2014. View Article : Google Scholar :
|
|
34
|
Jang KW, Lee KH, Kim SH, Jin T, Choi EY,
Jeon HJ, Kim E, Han YS and Chung JH: Ubiquitin ligase CHIP induces
TRAF2 proteasomal degradation and NF-κB inactivation to regulate
breast cancer cell invasion. J Cell Biochem. 112:3612–3620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Henshall DC, Araki T, Schindler CK, Lan
JQ, Tiekoter KL, Taki W and Simon RP: Activation of
Bcl-2-associated death protein and counter-response of Akt within
cell populations during seizure-induced neuronal death. J Neurosci.
22:8458–8465. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roy MJ, Vom A, Czabotar PE and Lessene G:
Cell death and the mitochondria: Therapeutic targeting of the BCL-2
family-driven pathway. Br J Pharmacol. 171:1973–1987. 2014.
View Article : Google Scholar :
|
|
38
|
Swanson PJ, Kuslak SL, Fang W, Tze L,
Gaffney P, Selby S, Hippen KL, Nunez G, Sidman CL and Behrens TW:
Fatal acute lymphoblastic leukemia in mice transgenic for B
cell-restricted Bcl-xL and c-myc. J Immunol. 172:6684–6691. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee JT, Lehmann BD, Terrian DM, Chappell
WH, Stivala F, Libra M, Martelli AM, Steelman LS and McCubrey JA:
Targeting prostate cancer based on signal transduction and cell
cycle pathways. Cell Cycle. 7:1745–1762. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Alan Diehl J, Cheng M and Martine F:
Roussel and Sherr CJ: Glycogen synthase kinase-3b regulates cyclin
D1 proteolysis and subcellular localization. Genes Dev.
12:3499–3511. 1998. View Article : Google Scholar
|
|
41
|
Shaw M, Cohen P and Alessi DR: The
activation of protein kinase B by H2O2 or
heat shock is mediated by phosphoinositide 3-kinase and not by
mitogen-activated protein kinase-activated protein kinase-2.
Biochem J. 336:241–246. 1998. View Article : Google Scholar
|
|
42
|
Zhang BG, Li JF, Yu BQ, Zhu ZG, Liu BY and
Yan M: microRNA-21 promotes tumor proliferation and invasion in
gastric cancer by targeting PTEN. Oncol Rep. 27:1019–1026. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vazquez F, Ramaswamy S, Nakamura N and
Sellers WR: Phosphorylation of the PTEN tail regulates protein
stability and function. Mol Cell Biol. 20:5010–5018. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hill R and Wu H: PTEN, stem cells, and
cancer stem cells. J Biol Chem. 284:11755–11759. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Biswas K, Sarkar S, Du K, Brautigan DL,
Abbas T and Larner JM: The E3 ligase CHIP mediates p21 degradation
to maintain radio-resistance. Mol Cancer Res. 15:651–659. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Canaff L, Vanbellinghen JF, Kanazawa I,
Kwak H, Garfield N, Vautour L and Hendy GN: Menin missense mutants
encoded by the MEN1 gene that are targeted to the proteasome:
Restoration of expression and activity by CHIP siRNA. J Clin
Endocrinol Metab. 97:E282–E291. 2012. View Article : Google Scholar
|
|
47
|
Massagué J and Gomis RR: The logic of
TGFbeta signaling. FEBS Lett. 580:2811–2820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou W, He MR, Jiao HL, He LQ, Deng DL,
Cai JJ, Xiao ZY, Ye YP, Ding YQ, Liao WT, et al: The
tumor-suppressor gene LZTS1 suppresses colorectal cancer
proliferation through inhibition of the AKT-mTOR signaling pathway.
Cancer Lett. 360:68–75. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cui YM, Jiang D, Zhang SH, Wu P, Ye YP,
Chen CM, Tang N, Liang L, Li TT, Qi L, et al: FOXC2 promotes
colorectal cancer proliferation through inhibition of FOXO3a and
activation of MAPK and AKT signaling pathways. Cancer Lett.
353:87–94. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Singh M, Yelle N, Venugopal C and Singh
SK: EMT: Mechanisms and therapeutic implications. Pharmacol Ther.
182:80–94. 2018. View Article : Google Scholar
|
|
52
|
Werb Z: ECM and cell surface proteolysis:
Regulating cellular ecology. Cell. 91:439–442. 1997. View Article : Google Scholar
|
|
53
|
Lukaszewicz-Zając M, Mroczko B and
Szmitkowski M: Gastric cancer - The role of matrix
metalloproteinases in tumor progression. Clin Chim Acta.
412:1725–1730. 2011. View Article : Google Scholar
|
|
54
|
Rao JS: Molecular mechanisms of glioma
invasiveness: The role of proteases. Nat Rev Cancer. 3:489–501.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dass K, Ahmad A, Azmi AS, Sarkar SH and
Sarkar FH: Evolving role of uPA/uPAR system in human cancers.
Cancer Treat Rev. 34:122–136. 2008. View Article : Google Scholar
|
|
56
|
Yuan ZL, Guan YJ, Chatterjee D and Chin
YE: Stat3 dimerization regulated by reversible acetylation of a
single lysine residue. Science. 307:269–273. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nalla AK, Gorantla B, Gondi CS, Lakka SS
and Rao JS: Targeting MMP-9, uPAR, and cathepsin B inhibits
invasion, migration and activates apoptosis in prostate cancer
cells. Cancer Gene Ther. 17:599–613. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gurzu S, Turdean S, Kovecsi A, Contac AO
and Jung I: Epithelial-mesenchymal, mesenchymal-epithelial, and
endothelial-mesenchymal transitions in malignant tumors: An update.
World J Clin Cases. 3:393–404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xu T, Zhou Q, Zhou J, Huang Y, Yan Y, Li
W, Wang C, Hu G, Lu Y and Chen J: Carboxyl terminus of
Hsp70-interacting protein (CHIP) contributes to human glioma
oncogenesis. Cancer Sci. 102:959–966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wen J, Luo KJ, Hu Y, Yang H and Fu JH:
Metastatic lymph node CHIP expression is a potential prognostic
marker for resected esophageal squamous cell carcinoma patients.
Ann Surg Oncol. 20:1668–1675. 2013. View Article : Google Scholar : PubMed/NCBI
|