Introduction
Osteosarcoma (OS) is the most common malignant
primary bone tumor, particularly in children and adolescents in
both sexes (1). It has been
reported that, from the epidemiological point of view, OS has a
predilection for the metaphyseal portions of the long bone, as well
as the distal femur and proximal tibia, which account for
approximately 50% of all cases of OS (2). Furthermore, OS is a highly aggressive
disease and the lungs are the primary target tissue for the
metastasis of OS. Up to 25% of patients diagnosed with OS also
suffer from lung metastases (3).
OS mostly affects adolescents. In approximately three quarters of
the cases, the age of patients with OS is between 15–25 years. The
median age is 17 years, with a trend in the predominance of the
male sex. There may be some correlation between OS and bone
development, as during adolescence growth spurt, bone also grows
rapidly (4). A relatively greater
number of patients with OS have germline mutations in either the
retinoblastoma or p53 genes (5). This discovery indicates that these
genes may be involved in the occurrence of the OS and provide us
with some clues for future OS therapy. The clinical characteristics
of OS include pain and swelling of the soft tissues, which are the
most common symptoms of patients with OS (6). Currently, inpatient treatment for OS
involves chemotherapy such as cisplatin, doxorubicin and
methotrexate (6,7). However, the 5-year survival rate for
patients treated with this chemotherapeutic regimen is only 70%. In
addition, 20% of patients with metastatic or recurrent disease
exhibit chemoresistance, which limits the effectiveness of
chemotherapy in malignant tumors, particularly OS (5–7).
Thereby, it is urgent to investigate new pathways or to explore
novel targets for the treatment of OS, particularly chemoresistant
OS.
MicroRNAs (miRNAs or miRs) are single-stranded small
RNAs which are approximately 22–24 nucleotides in length (8). They play an important role in the
post-transcriptional regulation of gene expression. As miRNAs can
bind to the complementary site of the 3′UTR sequence within the
targeting mRNAs, they can mediate target RNA degradation or
suppress translation (9–12). miR-18a is a member of the miR-17-92
family cluster, which includes 6 individual miRNAs: miR-17,
miR-18a, miR-19a, miR-20a, miR-19b1 and miR-92a1, which are
amplified in lymphomas and other cancer cells (13–15).
This cluster and its paralogues play important roles in cancer
development due to their ability to suppress the expression of a
number of tumor-associated proteins, such as p53 and Akt (13–15),
the overexpression of which promotes cell viability and reduces
apoptosis by regulating cell cycle progression (16–19).
The miR-17-92 cluster also plays an important role in normal growth
and skeletal development. The deletion or duplication miR-17-92 or
that of its paralogues can interrupt skeletal development,
resulting in smaller embryos and post-natal death due to
ventricular septal defects (20).
Recently, much attention has been paid to miRNAs,
such as the miR-17 family, which has been reported to regulate
tumor growth in various types of cancer. miR-18a has been reported
to function as an onco-miRNA, promoting cell viability and
facilitating tumor progression (21,22).
However, it has also been demonstrated that the abnormally high
expression of miR-18a in gastric cancer tissues inhibits the
expression of interferon regulatory factor 2 to modulate tumor
protein p53 (TP53) expression (16). miR-18a promotes the viability of
esophageal squamous cell carcinoma cells by increasing cyclin D1
expression (22). miR-18a
expression has also been shown to be ele vated in prostate cancer
and to promote tumorigenesis through the suppression of
serine/threonine-protein kinase 4 (STK4) in vitro and in
vivo (23). In addition,
miR-18a has been shown to be upregulated in glioblastoma tissues,
enhancing cell viability and facilitating the exit of cells from
cell cycle arrest (24).
Recently, it has been reported that miR-1, miR-9,
miR-18a, miR-18b, miR-126, miR-133b, miR-144, miR-195 and miR-451
expression levels are consistently decreased in both cell lines and
clinical samples of OS compared with normal bone tissues (25–27).
According the study by Namløs et al in 2012, miR-18a
expression in patients with OS was significantly decreased compared
with that in normal adjacent tissue (25). However, the exact role of miR-18a
in the development of OS remains to be determined. Therefore, in
this study, we aimed to investigate the role of miR-18a in OS. We
found miR-18a induced OS cell (MG63 and Saos-2 cells) apoptosis and
suppressed cell migration and invasion. The results form our in
vivo experiments using mice also demonstrated that miR-18a
significantly inhibited tumor growth. Immunohistochemistry also
confirmed that Ki67 and p-AKT expression was increased by miR-18a.
Furthermore, mediator complex subunit 27 (MED27) was identified as
a direct target of miR-18a with bioinformatics tools and validated
in both OS cell lines.
Materials and methods
Cell culture
The human osteosarcoma cell lines, MG63 and Saos-2,
were purchased from the Stem Cell Bank, Chinese Academy of Sciences
(Shanghai, China). The MG63 cells were propagated in Dulbecco's
modified Eagle's medium (DMEM; Thermo Fisher Scientific, Waltham,
MA, USA) and the Saos-2 cells were cultured in RPMI-1640 medium
(Sigma, St. Louis, MO, USA). Both culture media were supplemented
with 10% fetal bovine serum (Thermo Fisher Scientific) and
gentamicin (40 μg/ml, Sigma). All the cells were cultured at
37°C with 5% CO2 in a humidified cell culture incubator
(Sanyo, Tokyo, Japan).
Transient cell transfection
A total of 3×105 MG63 cells and
3×105 Saos-2 cells were plated in 6-well plates and
transfected with 100 nM miR-18a mimics, 100 nM miR-18a inhibitor or
the scramble control, which were synthesized by Shanghai GenePharma
Co. (Shanghai, China), using Lipofectamine® 2000 reagent
(Invitrogen/Thermo Fisher Scientific) according to the
manufacturer's instructions. At 48 h following transfection, cell
viability, and protein and mRNA expression levels were analyzed in
each group of cells.
Cell viability assay
The Cell Counting kit-8 (#C0038, Beyotime, Shanghai,
China) was used to determine cell viability. According to the
instructions of the manufacturer, the procedure was as follows: The
MG63 or Saos-2 cells at the logarithmic growth phase were plated in
a 96-well plate at a density of 3,000 cells/well. The cells were
then transfected with 100 nM miR-18a mimics, miR-18a inhibitor or
the control and cultured for a 72 h. At indicated time-points, 20
μl cell counting kit solution was added to each wall, and
the cells were further incubated for 0.5 h in a cell incubator.
Moreover, the cells were transfected with 10, 30, 50, 100 or 150 nM
miR-18a mimics, the inhibitor or the control (mock) for 72 h. The
number of living cells was measured using a microplate reader
(#168-1130, Bio-Rad Laboratories, Hercules, CA, USA), at a 450 nm
wavelength. The inhibitory rate = 1 - the OD value of the miR-18a
mimic transfected/OD value of the control group. The half maximal
inhibitory concentration (IC50) was then calculated.
Wound healing assay
The MG63 or Saos-2 cells were plated into 6-well
plates (2×105 cells/well) and transfected with 30 nM
miR-18a mimics, miR-18a inhibitor or the control (mock). When the
cells grew to 100% confluence, a vertical wound was made down
through the cell monolayer using 1,000 μl pipette tip to
press firmly and swiftly against the top of the tissue culture
plate. The media and cell debris were carefully aspirated prior to
further culture. The wound was captured under a microscope (Leica
Microsystems, Inc., Buffalo Grove, IL, USA) at each indicated
time-point. The snapshot image was used to analyze the distance of
one side of the wound to the other side using a scale bar.
Transwell cell invasion assay
A total of 100 μl of the MG63 or Saos-2 cells
(1×106 cells/ml) transfected with 30 nM miR-18a, miR-18a
inhibitor or the control (mock) were plated on the top of the
membrane in a Transwell insert into a 24-well plate. When the cells
had settled down, 600 μl 30% fetal bovine serum was added to
the bottom of the lower chamber in a 24-well plate. The cells that
did not migrate from the top of the upper membrane and remaining
culturing media were removed carefully using cotton swabs, while
the migrated cells into the bottom of the lower chamber in the well
were fixed and stained with crystal violet (#R40052, Thermo Fisher
Scientific) at room temperature for 48 h. The cells in different
fields of view were counted under a microscope (#CKX41, Olympus
America, Inc., Center Valley, MA, USA) and using the average sum of
cells to analyze the invasion rate.
Flow cytometric analysis of cell
apoptosis
Cell apoptosis was measured using Annexin V-FITC and
propidium iodide (PI) (Thermo Fisher Scientific) according to the
manufacturer's instructions. Briefly, 1×106 MIA PaCa-2
cells were plated into 100-mm dishes (Corning, New York, NY, USA)
and then transfected with miR-128 mimics or the mock control (100
nM). After 72 h, the cells were lysis, and stained with
Annexin-FITC and PI prior to being subjected to flow cytometry
(#660344 flow cytometer, BD Biosciences, Franklin Lakes, NJ, USA).
This assay can sort and identify the early apoptotic (Annexin
V-FITC+/PI−) cells, the primary necrotic
(Annexin V-FITC−/PI+) cells, and late
apoptotic (Annexin V-FITC+/PI+) cells. For
each group, the samples were examined in triplicate.
Predicted target analysis of miR-18a
miRecords (http://c1.accurascience.com/miRecords/) is an online
database which can be used to predict the binding site of miRNA to
its targets. It integrates the results of several online miRNA
target prediction tools, including DIANAmicroT, miRanda, PicTar and
TargetScan (28). This online
software was used to predict the potential targets of miR-18a.
3′UTR-luciferase reporter gene assay
All vectors were purchased from Genewiz (Beijing,
China), which carried MED27 containing the predicted miR-18a
binding sites with the wild-type or mutant 3′UTR. The MG63 or
Saos-2 cells were plated into a 24-well plate and transfected with
MED27 vector or mutant vector using Lipofectamine® 2000
transfection reagent (Thermo Fisher Scientific), according to the
manufacturer's instructions. After 4 h, 100 nM of miR-18a mimics or
the control (mock) were transfected into the cells, respectively.
After a further 48 h of culture, the cells were lysed and the
luciferase activities were analyzed by a dual luciferase assay kit
(Promega, Madison, WI, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the MG63 or Saos-2 cells
using the PicoPure™ RNA Isolation kit (Arcturus, Sunnyvale, CA,
USA) according to the manufacturer's instructions. Subsequently, 2
μg of RNA were utilized for cDNA synthesis using SuperScript
III RNase H Reverse Transcriptase (Thermo Fisher Scientific).
Following the reverse transcription reactions, the miR-18a
expression level was detected by microRNA assay (Thermo Fisher
Scientific). The primer for GAPDH was as follows: forward,
5′-AATGCATCCTGCACCACCAA′ and reverse, 5′-GTAGCCATATTCATTGTCATA′.
The qPCR procedure was performed as follows: for the holding stage:
Step 1, heating from 25°C to 95°C at the rate of 1.6°C/sec and
holding for 2 min at 50°C; step 2, heating from 50°C to 95°C at the
rate of 1.6°C/sec and then holding for 10 min at 95°C. For the PCR
stage: Step 1, initial denaturation at 95°C for 15 sec; step 2,
annealing extension at 60°C for 1 min. The temperature was cooled
down from 95°C to 60°C at the rate of 1.6°C/sec and the
denaturation and extension were then repeated for 40 cycles. Ct was
measured at the PCR stage with the by ViiTM 7 system (#4458571,
Bio-Rad Laboratories) Based on the real-time PCR results of Ct
number, the expression of mRNA levels was calculated using the
2−∆∆Cq method (29) and
normalized to the internal reference control, GAPDH.
Western blot analysis
The MG63 or Saos-2 cells were collected at 72 h
following transfection with miR-18a mimics or the mock control.
Tumor tissues were collected at the end of the experiment (5 weeks)
and then total proteins were extracted by using 1X loading buffer.
The protein concentration was quantified using the BCA Protein
Assay kit (Thermo Fisher Scientific). A total of 30 μg of
protein were loaded per lane per group, and electrophoresis was
performed on a 10% Tris-SDS gel. Following electrophoresis, the gel
was blotted onto polyvinylidene fluoride membranes (Thermo Fisher
Scientific). For the following locking and antibody incubation, the
iBind kit was used according the manufacturer's instructions. The
primary antibodies used were as follows: MED27 (1:500; #SAB1411657;
Sigma), Bax (1:500; #5023), Bcl-2 (1:500; #2872), p-Akt (1:500;
#4060), Akt (1:500; #4691), matrix metalloproteinase (MMP)2 (1:500;
#40994) and MMP9 (1:500; #13667) (all from Cell Signaling
Technology, Danvers, MA, USA). Cleaved caspase-3 (1:1,000;
#sc-98785) and GAPDH (1:1,500; #sc-66163) were purchased from Santa
Cruz Biotechnology, Inc., (Santa Cruz, CA, USA). The secondary
antibody (horseradish peroxidase conjugated anti-rabbit) was
provided from Cell Signaling Technology, Inc. (#7074, 1:3,000). The
signal was detected with super sensitive regent (Thermo Fisher
Scientific), and the specific proteins were detected with the
ChemDoc™ imaging system (Bio-Rad Laboratories). Quantification of
the protein data was carried out using the density in the blots
with Image Lab software version 4.0 (Bio-Rad Laboratories).
In vivo mouse tumor xenograft model
A total of 30 (5–6 weeks old, weighing 18–21 g)
female BALB/c nude mice were purchased from Vital River
Laboratories (Beijing, China) and housed in an SPF environment with
a 12-h/12-h light/dark cycle at the Animal Center in Xinhua
Hospital. These mice were supplied with free water and food and the
temperature was maintained at 22±2°C with 40–70% relative humidity.
Some of the MG63 cells were transfected with the lentivirus with
the miR-18a sequence, while the other cells were only transfected
with the lentivirus with mock sequence (Shanghai GenePharma Co.).
The mice were subcutaneously implanted with these two modified MG63
cells and the tumor size and mouse body weights were monitored once
a week for successive 5 weeks. The mice were allowed free access to
food and water. The MG63 osteosarcoma cells transfected with the
miR-18a mimics or the mock control lentivirus (5×107 in
100 μl in PBS) were subcutaneously injected into the left
flank of each mouse. It should be noted that a total of 30 mice
were subcutaneously implanted with these the modified MG63 cells.
However as the tumor formatting rate is approximately 70%, we
selected 20 mice with good tumor formatting for the experiment. The
other 10 mice with no or very small tumors were not used. Thus, in
the experiments, there were 10 mice per group. The tumor volumes
(TV) were calculated using calipers once per week, which were
calculated as follows: TV = (width2 × length)/2. Both
tumor width and length are presented in mm. At the end of the
experiment, the mice were sacrificed. The tumors were then excised
from the mice and weighed. A portion of tumors was fixed into 10%
PFA as soon as possible for further use. Another portion of tumors
was lysed for western blot analysis. The maximum diameter of a
single tumor was 15.12 mm and the maximum tumor volume was 727.54
mm3 in our study. No mouse developed multiple tumors.
All the above-mentioned procedures using these nude mice were
approved by the Xin Hua Hospital Animal Experimental Ethics
Committee (Approval no. 201703678).
Immunohistochemistry (IHC) assay
Ki67 as a proliferation protein was used to detect
the cell proliferation in the tumor tissues isolated from the mice.
All tissues were fixed with formalin and paraffin-embedded in
advance means immediately when the tissue samples were isolated
from the mouse bodies. They were then stored for use at -80°C or
deparaffinized, rehydrated and then immersed in a target retrieval
solution (pH 6.0), and boiled at medium baking temperature 3 times
for 10 min once in a microwave. They were then incubated with 3%
BSA for 1 h and then incubated with primary antibody against Ki67
(1:500; #701198, Thermo Fisher Scientific) for 1 h. After washing
with PBS 3 times, the samples were then incubated with biotinylated
secondary antibody (1:8,000; #65-6140, Thermo Fisher Scientific)
followed by the addition of horseradish peroxidase-conjugated
streptavidin (#N100, Thermo Fisher Scientific). For TUNEL assay,
the procedure was carried out according to the instructions of the
manufacturer (Roche Diagnostics, Indianapolis, IN, USA). The
samples were counterstained with hematoxylin and the
target-positive cells were counted in 3–4 different fields and
photographed using the EVOS™ FL Auto Imaging System (Thermo Fisher
Scientific).
Statistical analysis
Statistical analyses between 2 groups and multiple
groups were carried out using a two-tailed Student's t-test and
one-way ANOVA followed by Dunnett's test, respectively (GraphPad
Prism7, La Jolla, CA, USA). In all the assays, a P-value <0.01
was considered to indicate a statistically significant difference.
All experiments were performed in triplicate, except for the tissue
samples from the tumor xenograft model for IHC and all values are
presented as the means ± SD.
Results
miR-18a inhibits MG63 and Saos-2 cell
viability
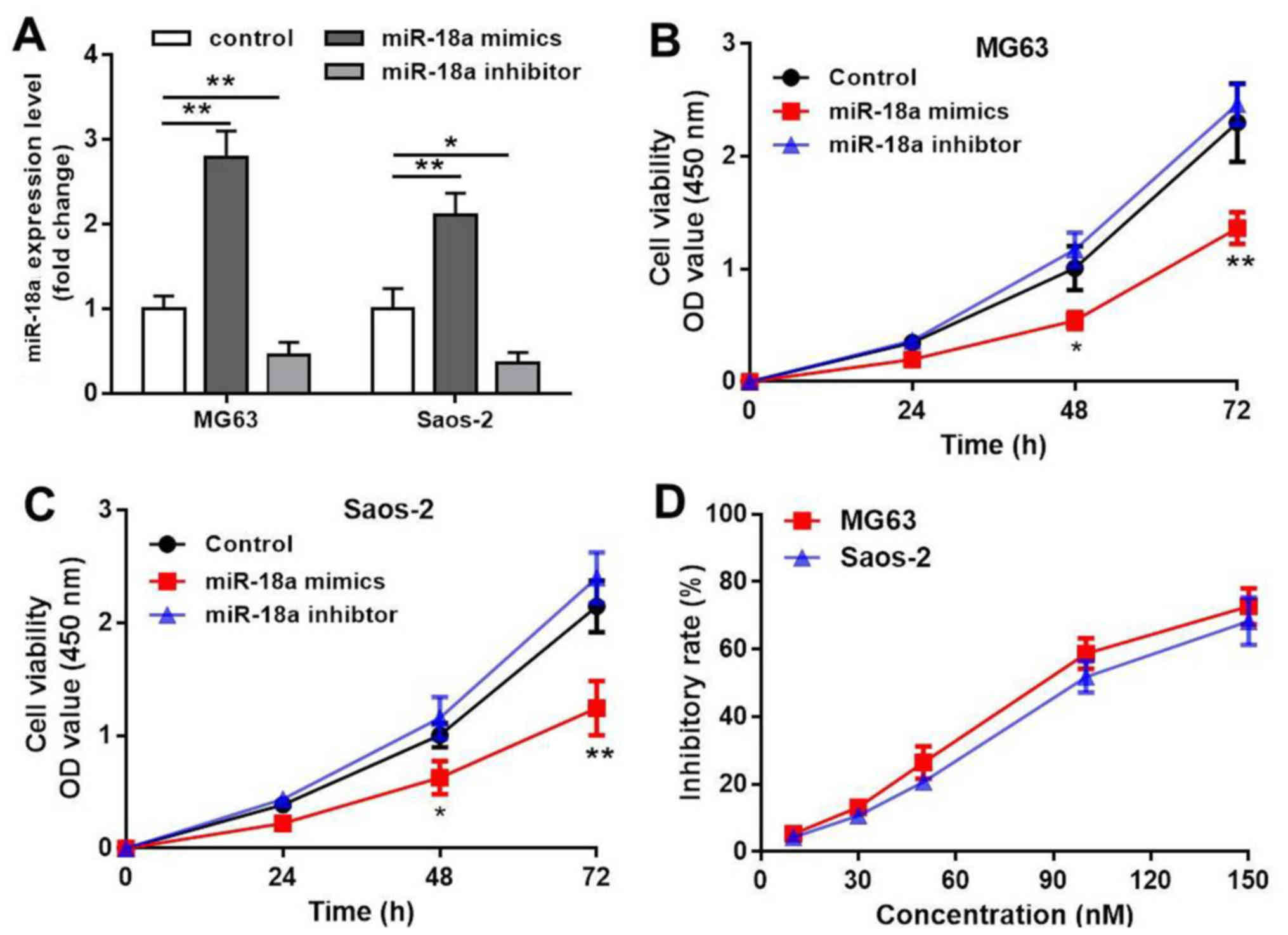
Firstly, we transfected the MG63 and Saos-2 cells
with miR-18a mimics or miR-18a inhibitor and confirmed that the
miR-18a levels were significantly upregulated or downregulated,
respectively (P<0.01, Fig. 1A).
Subsequently, the effects of miR-18a on OS cell viability were
measured by CCK8 assay. The results revealed that transfection with
100 nM miR-18a mimic inhibited MG63 and Saos-2 cell growth in a
time-dependent manner (P<0.01, Fig.
1B and C). In addition, the IC50 values of the
miR-18a-transfected MG63 and Saos-2 cells were 88.6 and 95.8 nM,
respectively (Fig. 1D), which
indicated that 100 nM of miR-18a appeared to be cytotoxic.
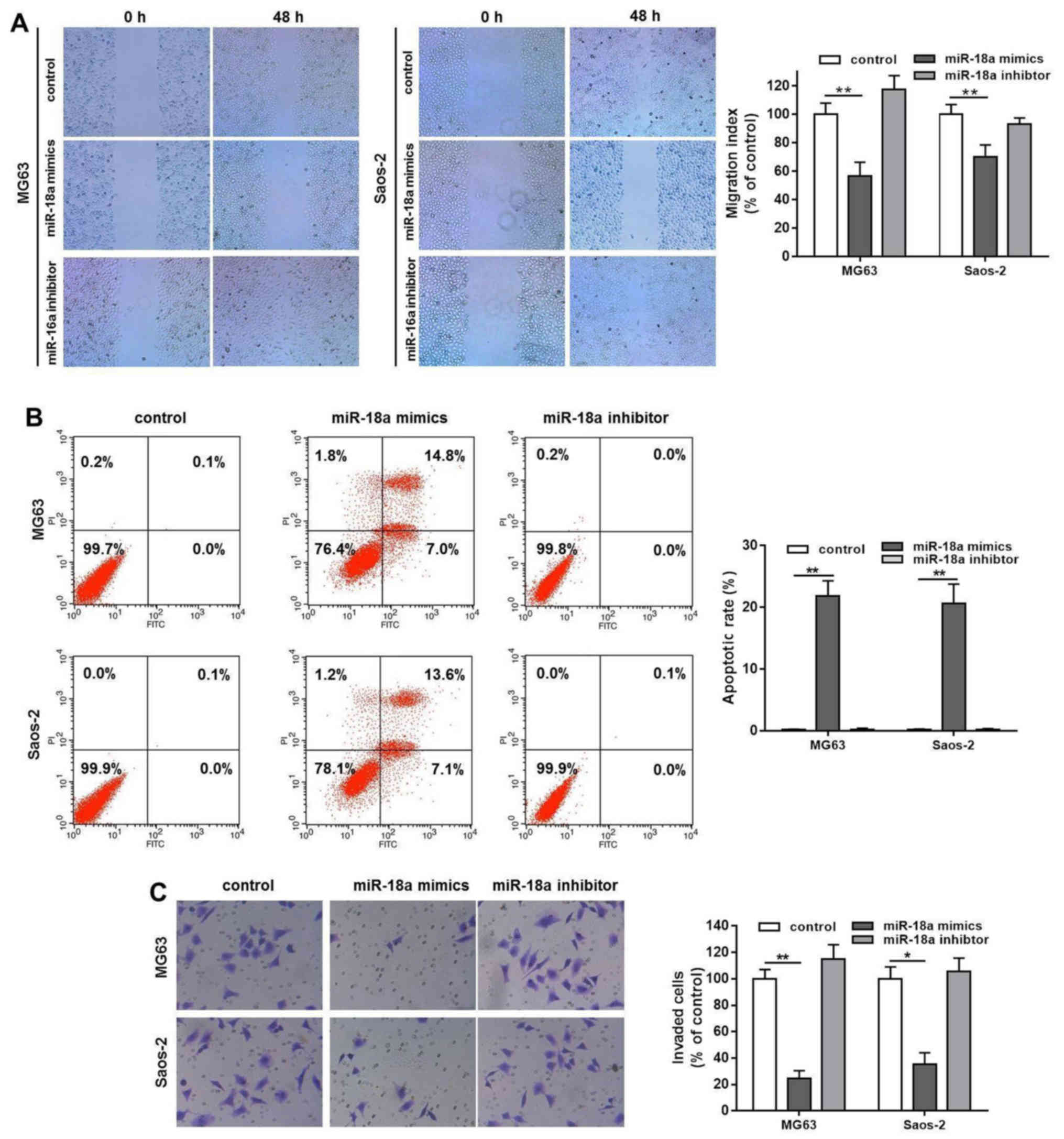
miR-18a mimics inhibits OS cell migration
and invasion, and induces cellular apoptosis
Wound healing assay was used to examine the effects
of miR-18a on OS cell migration. As shown in Fig. 2A, transfection with miR-18a mimics
markedly inhibited MG63 cell migration and invasion, whereas
transfection with miR-18a inhibitor had no effect and the
inhibitory effect of miR-18a mimcs on Saos-2 cell migration was
relatively weak, but compared with the control there was still a
difference. This might due to the rapid proliferative ability of
the Saos-2 cells. In addition, the results of flow cytometry assay
indicated that transfection with miR-18a mimics increased Annexin
V-FITC/PI double staining positive rate in both the MG63 and Saos-2
cells (Fig. 2B), which indicated
that miR-18a significantly induced MG63 and Saos-2 cell apoptosis
compared with the control (Fig.
2B). Similar to the results of wound healing assay, the results
of Transwell assay demonstrated that transfection with miR-18a
mimics blocked the invasive ability of the MG63 and Saos-2 cells
(Fig. 2C). The invasion rate in
the control group was considered as 100%, while this rate was
decreased to only 20 or 30% following transfection of the cells
with miR-18a mimics (Fig. 2C).
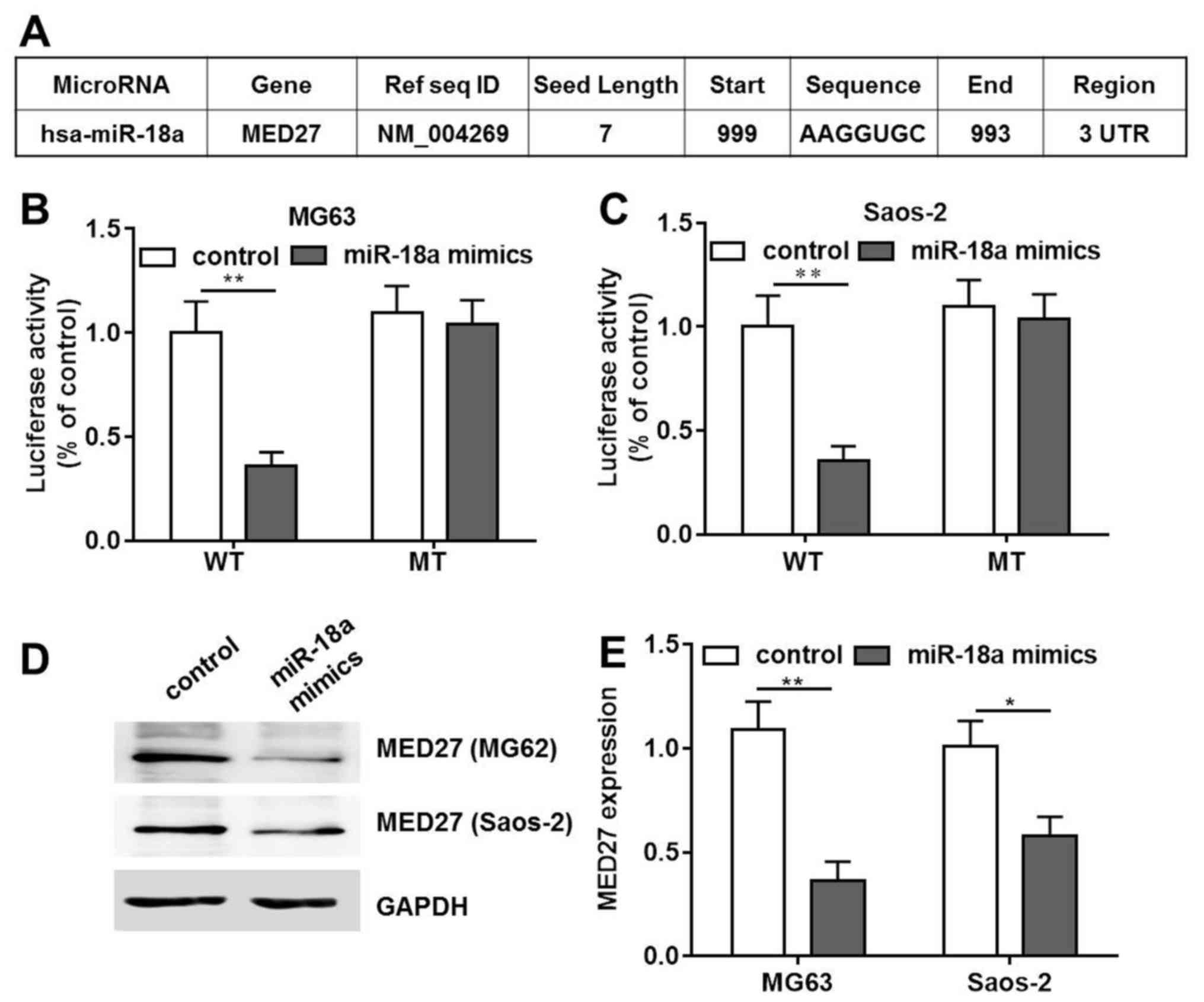
miR-18a mimics induce OS cell apoptosis
by targeting MED27
Bioinformatics tools were used in order to
investigate the antitumor mechanisms of miR-18a in the MG63 and
Saos-2 cells. The use of the online bioinformatics tools,
TargetScan (http://www.targetscan.org/vert_61/) and miRanda
(http://www.microrna.org/microrna/)
predicted MED27 as a target of miR-18a with experiments were
carried out to confirm whether MED27 was a direct target of miR-18a
(data not shown). Two plasmids was constructed as follows: One was
wild-type MED27 and the other was the mutant with the MED27-3′UTR
mutation. Both plasmids were fused with a luciferase reporter gene
(Fig. 3A). The results of
luciferase assay using the OS cells (MG63 and Saos-2) indicated
that transfection with miR-18a mimics significantly inhibited the
luciferase activity of the wild-type construct. However, no
difference was observed between the control and the
miR-18a-trasnfected group with the MED27 mutant. Moreover, the
results of both cell lines revealed comparable activity (Fig. 3B and C). The results of western
blot analyses also confirmed that transfection with miR-18a mimics
downregulated MED27 protein expression in both the MG63 and Saos-2
cells (Fig. 3D and E). These
findings indicated that MED27 was the direct target of miR-18a in
the OS cells.
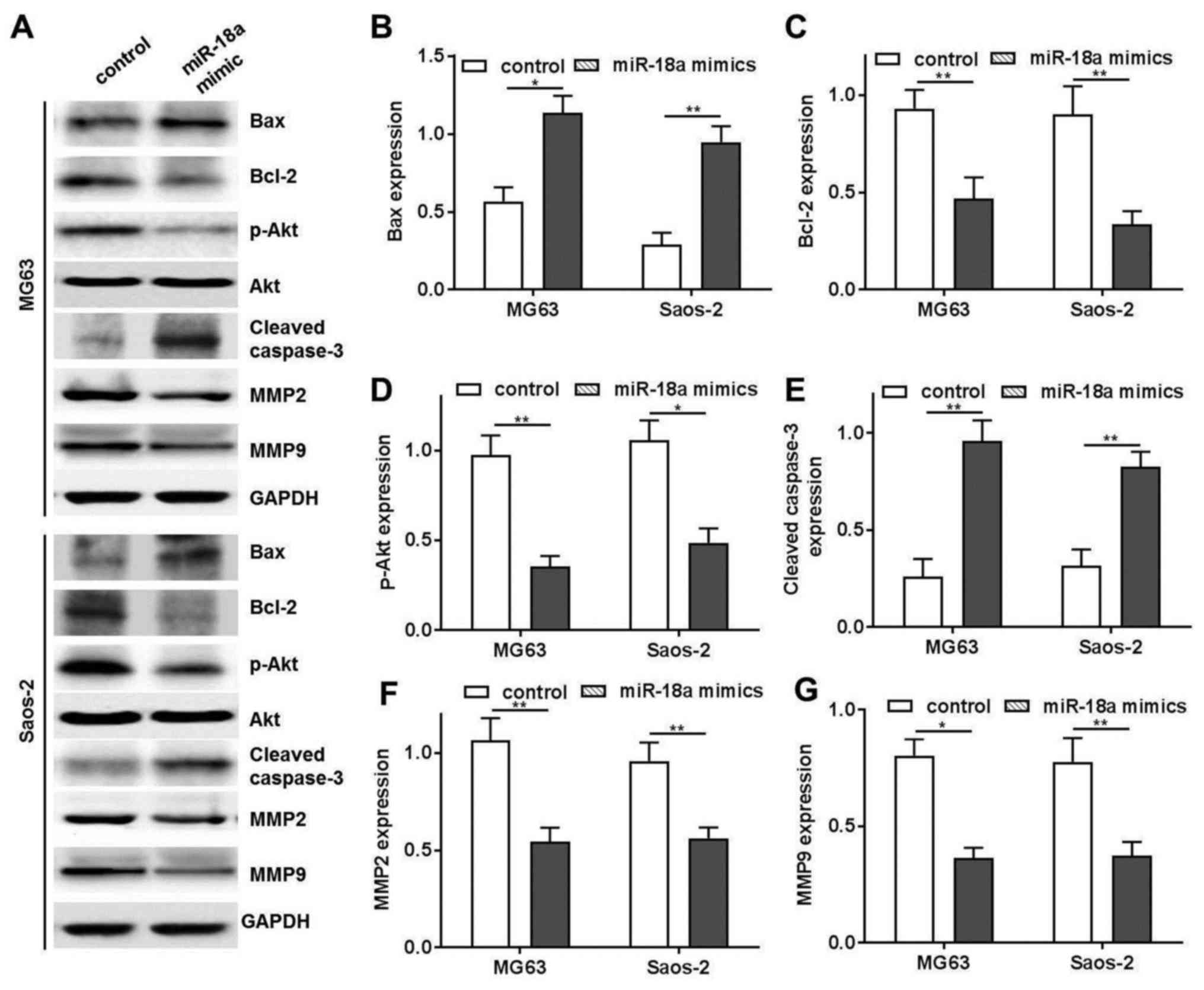
miR-18a mimics induce OS cell apoptosis
via the caspase-3 dependent pathway
In order to further investigate the antitumor
mechanisms of miR-18a, we examined the expression of several
proteins, including Bax, Bcl-2, p-Akt and cleaved caspse-3 in the
MG63 and Saos-2 cells. The results indicated that transfection with
miR-18a mimics downregulated Bcl-2 and p-Akt expression (Fig. 4A–C), which are the negative
regulators of cellular apoptosis. We also found that transfection
with miR-18a upregulated Bax and cleaved caspse-3 expression, which
are the promoters of cellular apoptosis (Fig. 4A, D and E). In addition,
transfection with miR-18a mimics inhibited OS cell migration and
invasion via the downregulation of MMP2 and MMP9 (Fig. 4A, F and G)
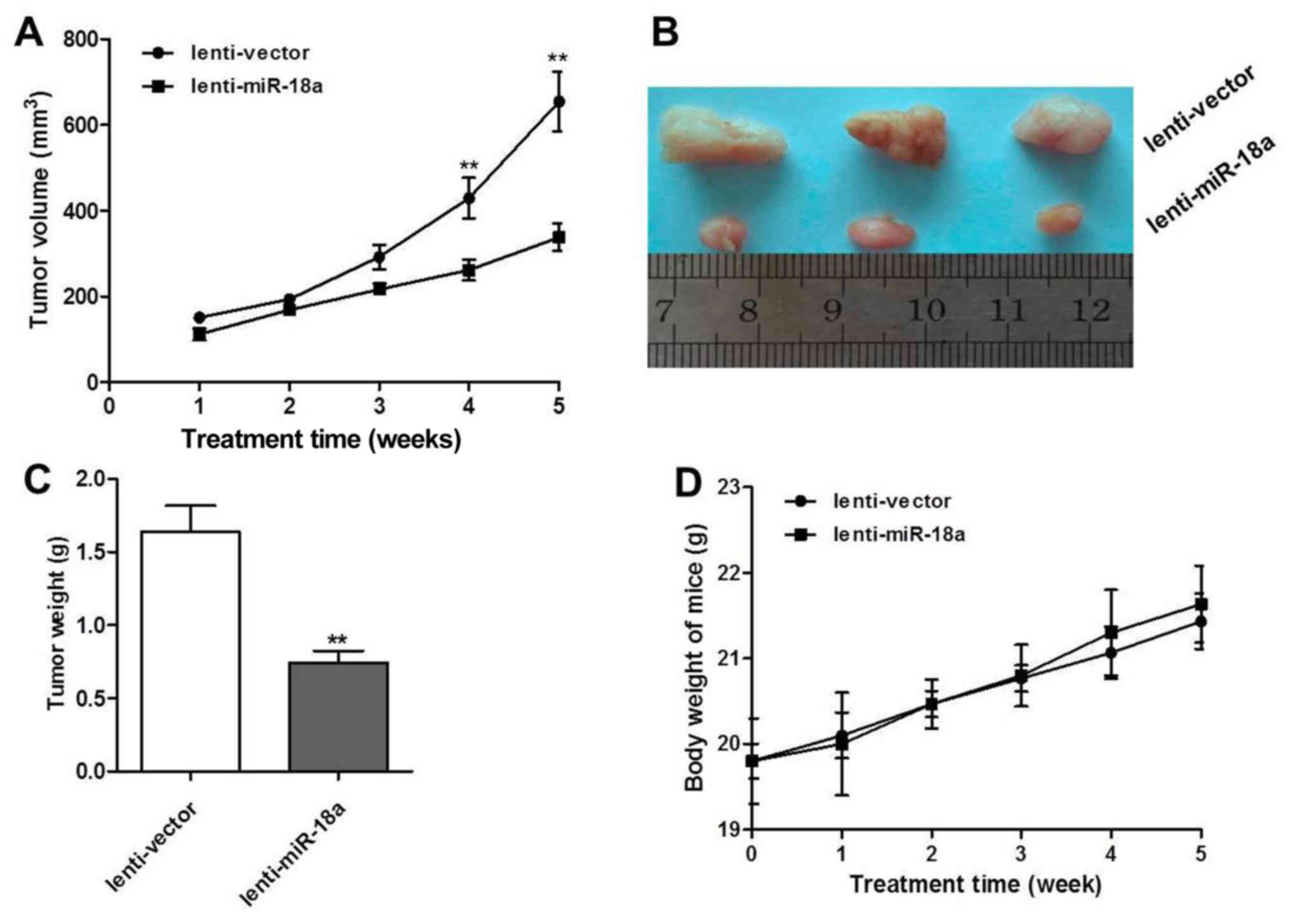
miR-18a mimics inhibit tumor growth in a
MG63 tumor xenograft model
In order to determine whether miR-18a inhibits OS
tumor growth activity in vivo, lentivirus-infected MG63
cells were established and injected into nude mice. From the
results, it was found that the tumors derived from the cells
transfected with the miR-18a mimics grew at a significantly slower
rate compared with the lentivirus control cells over a period of 4
weeks. The tumor size in the miR-18a lentivirus group ranged from
280 to 320 mm3 compared with that in the control group,
which ranged from 500 to 700 mm3 over a period of 5
weeks (Fig. 5A). At the end of the
experiment, the tumor weight in the lentivirus miR-18a group was
only 0.7 g compared with that in the control group which was
approximately 1.6 g (Fig. 5B and
C). Moreover, no obvious changes in body weight were observed
in the 2 groups (Fig. 5D).
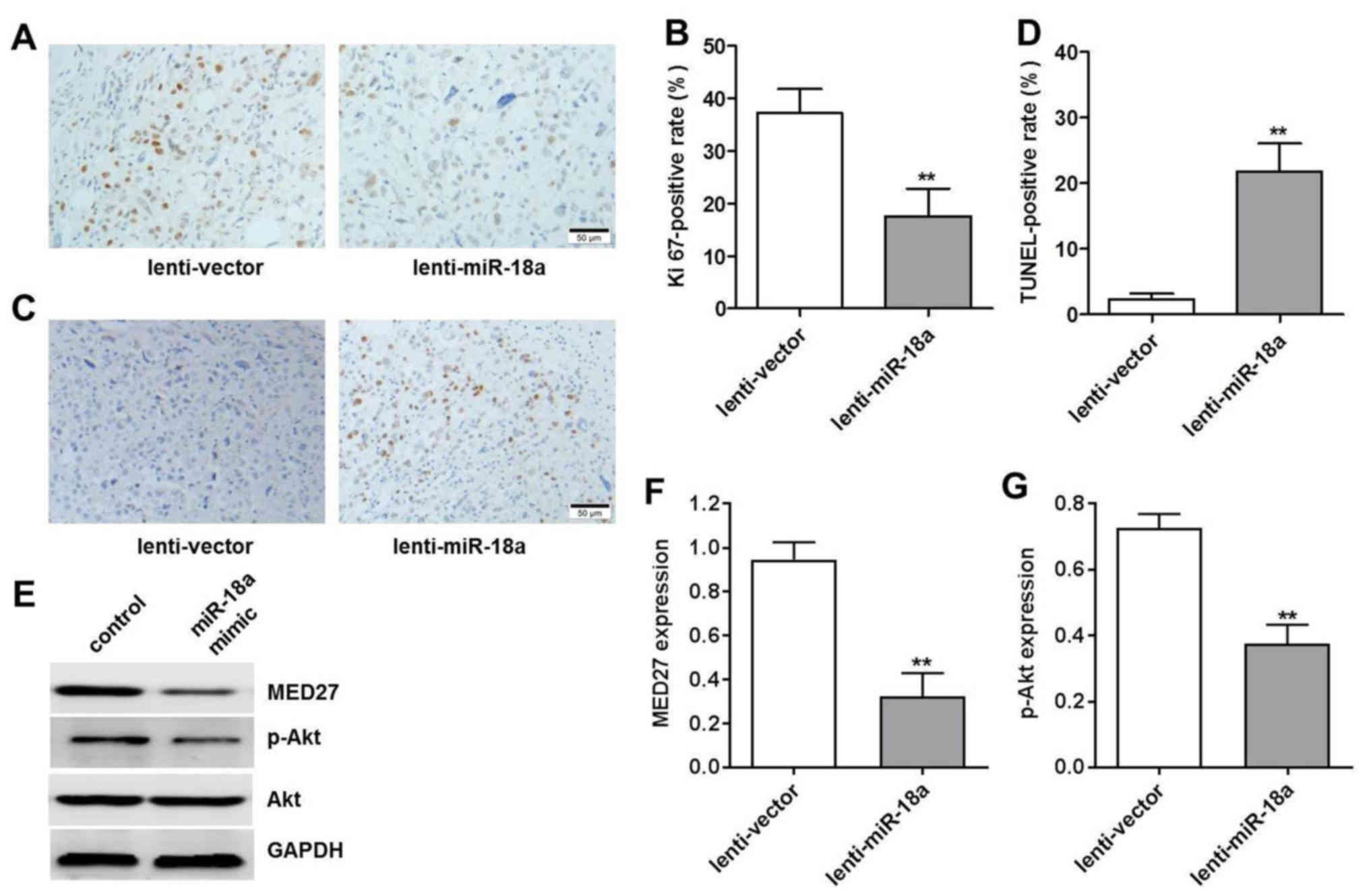
Effects of miR-18a mimics on Ki67, TUNEL
and MED27/p-Akt expression levels in tumor xenografts
To further validate the antitumor activity of
miR-18a on OS tumor xenogafts, we examined the Ki67 and TUNEL
positive cell rate by IHC. The results revealed that the number of
Ki67-postive cells decreased by 20% and that of TUNEL-positive
cells increased by approximately 20% in the lentivirus miR-18a
group compared with the lentivirus control (Fig. 6A–D). We also confirmed that miR-18a
significantly inhibited MED27 and p-Akt expression in vivo,
which was consistent with the findings in vitro (Fig. 6E–G).
Discussion
Cancer is not only one of the main causes of death
worldwide, but is also one of the most rapidly growing causes of
death (2,12). OS mainly affects adolescents and
70% of patients with OS succumb to the disease due to OS metastasis
and chemoresistance (6,7). Thus, it is mandatory to develop novel
targets or markers for the efficient treatment of OS. Over the past
decades, miRNAs have been identified by researchers as tumor
markers; however, the journey from bench to bedside is still a long
one, and it may take time for miRNAs to be used as targets in
clinical practice (28–30). miRNAs are a group of small
non-coding RNAs (ncRNAs) that post-transcriptionally regulate gene
expression by targeting the 3′UTR of their corresponding mRNAs.
Dysregulated miRNAs have been regarded as a novel type of
'onco-miRNAs' or 'tumor suppressors', which may play essential
roles in cancer initiation, progression and metastasis (12,31–33).
miR-18a belongs to a large miRNA cluster known as
the miR-17-92 cluster, which encodes a total of 5 miRNAs, including
miR-17, miR-19a, miR-20a, miR-19b and miR-92a. A number of studies
have focused on the regulation of miR-17-92 in multiple types of
tumors from lymphomas to solid tumors (20–24,32).
Moreover, it has been reported that the miR-17-92 gene cluster
transcript can be activated by the c-myc, N-myc and E2F families
(34,35). Of noted, the expression levels of
each miRNA in the cluster are not exactly parallel with each other,
suggesting that the processing or stability of the miRNAs is
differentially regulated (33). It
has been previously reported that miR-17-92 plays an essential role
in the progression and development of breast cancer and that the
overexpression of miR-17 promotes human breast cancer cell
migration and invasion through the downregulation of HMG-box
transcription factor 1 (HBP1), which is the regulator of miR-17
stability (36,37). Furthermore, it has been
demonstrated that miR-18a reduces DNA damage repair signaling and
increases cellular radiosensitivity (37).
In the present study, we demonstrated that miR-18a
inhibited the viability, migration, and invasion, and induced the
apoptosis of the MG63 and Saos-2 cells. In vivo experiments
also confirmed that miR-18a suppressed tumor growth and that this
was accompanied by a decrease in the Ki67-positive cell rate, a
decrease in Bcl-2 and p-Akt expression, and by the upregulation of
Bax. The above-mentioned findings support the notion that miR-18a
functions as a tumor suppressor. In addition, the results of
bioinformatics analysis and luciferase assay validated MED27 as the
direct target of miR-18a.
Given the demonstration of the mutations or
overexpression of some MED proteins in various human cancers
(38–40), MED proteins have been recognized to
play an increasingly essential role in tumorigenesis and
development. Although MED subunits have recently been reported to
be involved in tumor growth (41–45),
these findings were limited to MED1, 12, 14, 15, 19, 24 and 28, and
little was known about the functional role of other subunits of the
MED complex in carcinogenesis, including MED27 (46). In this study, we investigated and
demonstrated the functional significance of MED27 in osteosarcoma
progression. Of note, we found that MED27 protein expression was
decreased by transfection with miR-18a mimics along with the
downregulation of p-AKT expression in OS cells. Taken together,
these results may aid to in the elucidation of the role miR-18a,
particularly its role in the regulation of OS cell apoptosis and
suggest that MED27 may be a novel potential target in the treatment
of OS.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD and JL were responsible for sample collection,
and the experiment design and execution. LS and PS analyzed and
interpreted the experimental data. JD, MH and QC drafted the
manuscript, interpreted the experimental data and were involved in
the conception of the study. JL reviewed and approved the final
draft of this manuscript prior to submission. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All the procedures using nude mice were approved by
the Xin Hua Hospital Animal Experimental Ethics Committee (Approval
no. 201703678).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
2
|
Whelan J, McTiernan A, Cooper N, Wong YK,
Francis M, Vernon S and Strauss SJ: Incidence and survival of
malignant bone sarcomas in England 1979–2007. Int J Cancer.
131:E508–E517. 2012. View Article : Google Scholar
|
|
3
|
Longhi A, Errani C, De Paolis M, Mercuri M
and Bacci G: Primary bone osteosarcoma in the pediatric age: State
of the art. Cancer Treat Rev. 32:423–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eilber FR and Rosen G: Adjuvant
chemotherapy for osteosarcoma. Semin Oncol. 16:312–322.
1989.PubMed/NCBI
|
|
5
|
Pápai Z, Féja CN, Hanna EN, Sztán M, Oláh
E and Szendrôi M: P53 overexpression as an indicator of overall
survival and response to treatment in osteosarcomas. Pathol Oncol
Res. 3:15–19. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sakamoto A and Iwamoto Y: Current status
and perspectives regarding the treatment of osteosarcoma:
Chemotherapy. Rev Recent Clin Trials. 3:228–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He H, Ni J and Huang J: Molecular
mechanisms of chemoresistance in osteosarcoma (Review). Oncol Lett.
7:1352–1362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lim LP, Glasner ME, Yekta S, Burge CB and
Bartel DP: Vertebrate microRNA genes. Science. 299:15402003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar
|
|
11
|
Tagawa H and Seto M: A microRNA cluster as
a target of genomic amplification in malignant lymphoma. Leukemia.
19:2013–2016. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
He L, Thomson JM, Hemann MT,
Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe
SW, Hannon GJ, et al: A microRNA polycistron as a potential human
oncogene. Nature. 435:828–833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hayashita Y, Osada H, Tatematsu Y, Yamada
H, Yanagisawa K, Tomida S, Yatabe Y, Kawahara K, Sekido Y and
Takahashi T: A polycistronic microRNA cluster, miR-17-92, is
overexpressed in human lung cancers and enhances cell
proliferation. Cancer Res. 65:9628–9632. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–222. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olive V, Bennett MJ, Walker JC, Ma C,
Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ and He L: miR-19
is a key oncogenic component of mir-17-92. Genes Dev. 23:2839–2849.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen YJ, Wu H, Zhu JM, Li XD, Luo SW, Dong
L, Liu TT and Shen XZ: MicroRNA-18a modulates P53 expression by
targeting IRF2 in gastric cancer patients. J Gastroenterol Hepatol.
31:155–163. 2016. View Article : Google Scholar
|
|
17
|
Speidel D: Transcription-independent p53
apoptosis: An alternative route to death. Trends Cell Biol.
20:14–24. 2010. View Article : Google Scholar
|
|
18
|
Chen H, Zhou L, Wu X, Li R, Wen J, Sha J
and Wen X: The PI3K/AKT pathway in the pathogenesis of prostate
cancer. Front Biosci. 21:1084–1091. 2016. View Article : Google Scholar
|
|
19
|
Wang KC, Botting KJ, Zhang S, McMillen IC,
Brooks DA and Morrison JL: Akt signaling as a mediator of cardiac
adaptation to low birth weight. J Endocrinol. 233:R81–R94. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu Y, Thomson JM, Wong HY, Hammond SM and
Hogan BL: Transgenic overexpression of the microRNA miR-17-92
cluster promotes proliferation and inhibits differentiation of lung
epithelial progenitor cells. Dev Biol. 310:442–453. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo Z, Dai Y, Zhang L, Jiang C, Li Z, Yang
J, McCarthy JB, She X, Zhang W, Ma J, et al: miR-18a promotes
malignant progression by impairing microRNA biogenesis in
nasopharyngeal carcinoma. Carcinogenesis. 34:415–425. 2013.
View Article : Google Scholar
|
|
22
|
Zhang W, Lei C, Fan J and Wang J: miR-18a
promotes cell proliferation of esophageal squamous cell carcinoma
cells by increasing cylin D1 via regulating PTEN-PI3K-AKT-mTOR
signaling axis. Biochem Biophys Res Commun. 477:144–149. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hsu TI, Hsu CH, Lee KH, Lin JT, Chen CS,
Chang KC, Su CY, Hsiao M and Lu PJ: MicroRNA-18a is elevated in
prostate cancer and promotes tumorigenesis through suppressing STK4
in vitro and in vivo. Oncogenesis. 3:e992014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu W, Zhou X, Yu T, Bao Z, Zhi T, Jiang K,
Nie E, Wang Y, Zhang J and You Y: The malignancy of miR-18a in
human glioblastoma via directly targeting CBX7. Am J Cancer Res.
7:64–76. 2017.PubMed/NCBI
|
|
25
|
Namløs HM, Meza-Zepeda LA, Barøy T,
Østensen IH, Kresse SH, Kuijjer ML, Serra M, Bürger H,
Cleton-Jansen AM and Myklebost O: Modulation of the osteosarcoma
expression phenotype by microRNAs. PLoS One. 7:e480862012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fenger JM, Roberts RD, Iwenofu OH, Bear
MD, Zhang X, Couto JI, Modiano JF, Kisseberth WC and London CA:
miR-9 is overexpressed in spontaneous canine osteosarcoma and
promotes a metastatic phenotype including invasion and migration in
osteoblasts and osteosarcoma cell lines. BMC Cancer. 16:7842016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baumhoer D: Molecular characterization of
osteosarcomas. Pathologe. 34(Suppl 2): 260–263. 2013.In German.
View Article : Google Scholar
|
|
28
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37(Database): D105–D110. 2009.
View Article : Google Scholar
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Ji J and Wang XW: New kids on the block:
Diagnostic and prognostic microRNAs in hepatocellular carcinoma.
Cancer Biol Ther. 8:1686–1693. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Giráldez MD, Lozano JJ, Ramírez G, Hijona
E, Bujanda L, Castells A and Gironella M: Circulating microRNAs as
biomarkers of colorectal cancer: Results from a genome-wide
profiling and validation study. Clin Gastroenterol Hepatol.
11:681–8.e3. 2013. View Article : Google Scholar
|
|
32
|
Lawrie CH: MicroRNA expression in
lymphoma. Expert Opin Biol Ther. 7:1363–1374. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schulte JH, Horn S, Otto T, Samans B,
Heukamp LC, Eilers UC, Krause M, Astrahantseff K, Klein-Hitpass L,
Buettner R, et al: MYCN regulates oncogenic microRNAs in
neuroblastoma. Int J Cancer. 122:699–704. 2008. View Article : Google Scholar
|
|
35
|
Woods K, Thomson JM and Hammond SM: Direct
regulation of an oncogenic micro-RNA cluster by E2F transcription
factors. J Biol Chem. 282:2130–2134. 2007. View Article : Google Scholar
|
|
36
|
Guil S and Cáceres JF: The multifunctional
RNA-binding protein hnRNP A1 is required for processing of miR-18a.
Nat Struct Mol Biol. 14:591–596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li H, Bian C, Liao L, Li J and Zhao RC:
miR-17-5p promotes human breast cancer cell migration and invasion
through suppression of HBP1. Breast Cancer Res Treat. 126:565–575.
2011. View Article : Google Scholar
|
|
38
|
Nagasawa S, Maeda I, Fukuda T, Wu W,
Hayami R, Kojima Y, Tsugawa K and Ohta T: MED12 exon 2 mutations in
phyllodes tumors of the breast. Cancer Med. 4:1117–1121. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shaikhibrahim Z, Menon R, Braun M,
Offermann A, Queisser A, Boehm D, Vogel W, Rüenauver K, Ruiz C,
Zellweger T, et al: MED15, encoding a subunit of the mediator
complex, is overexpressed at high frequency in castration-resistant
prostate cancer. Int J Cancer. 135:19–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kämpjärvi K, Mäkinen N, Kilpivaara O,
Arola J, Heinonen HR, Böhm J, Abdel-Wahab O, Lehtonen HJ, Pelttari
LM, Mehine M, et al: Somatic MED12 mutations in uterine
leiomyosarcoma and colorectal cancer. Br J Cancer. 107:1761–1765.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao M, Yang X, Fu Y, Wang H, Ning Y, Yan
J, Chen YG and Wang G: Mediator MED15 modulates transforming growth
factor beta (TGFβ)/Smad signaling and breast cancer cell
metastasis. J Mol Cell Biol. 5:57–60. 2013. View Article : Google Scholar
|
|
42
|
Jin F, Irshad S, Yu W, Belakavadi M,
Chekmareva M, Ittmann MM, Abate-Shen C and Fondell JD: ERK and AKT
signaling drive MED1 overexpression in prostate cancer in
association with elevated proliferation and tumorigenicity. Mol
Cancer Res. 11:736–747. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li XH, Fang DN and Zeng CM: Knockdown of
MED19 by short hairpin RNA-mediated gene silencing inhibits
pancreatic cancer cell proliferation. Cancer Biother Radiopharm.
26:495–501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen W, Rogatsky I and Garabedian MJ:
MED14 and MED1 differentially regulate target-specific gene
activation by the glucocorticoid receptor. Mol Endocrinol.
20:560–572. 2006. View Article : Google Scholar
|
|
45
|
Huang CY, Chou YH, Hsieh NT, Chen HH and
Lee MF: MED28 regulates MEK1-dependent cellular migration in human
breast cancer cells. J Cell Physiol. 227:3820–3827. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Conaway RC and Conaway JW: Function and
regulation of the Mediator complex. Curr Opin Genet Dev.
21:225–230. 2011. View Article : Google Scholar : PubMed/NCBI
|