Introduction
Cyclin-dependent kinases (CDKs) are present in all
known eukaryotes, and their regulatory functions during the cell
cycle are evolutionarily conserved. Cyclin-CDK complexes
phosphorylate specific substrates, according to the requirements of
a particular cell cycle phase. CDKs are regulated by cyclin
binding, phosphorylation and the binding of CDK inhibitors
(1). In addition to cell cycle
regulation, CDKs are involved in transcription, mRNA processing and
cellular differentiation (2–4).
In a number of human cancer types, CDKs are
overactive and CDK-inhibiting proteins are non-functional (5,6).
Therefore, CDKs are considered potential targets for anticancer
therapies, by interfering with CDK functions to selectively
interrupt cell cycle regulation in cancer cells (7). Flavopiridol (alvocidib) was the first
CDK inhibitor to be tested in clinical trials following its
identification in a screen for anticancer agents in 1992. It
competes for the ATP-binding site of CDKs (8). Although CDK inhibitors seem
therapeutically promising, their side-effects must be limited so
that only cancer cells are affected. AT7519, a pyrazole
3-carboxyamide compound, was developed by Astex and acts as an
inhibitor of CDK1, CDK2, CDK4, CDK6 and CDK9. Santo et al
showed demonstrated AT7519 exerts potent cytotoxic effects and
induces the apoptosis of multiple myeloma cells; AT7519 was also
found to be associated with the inhibition of in vivo tumor
growth and prolonged survival (9).
AT7519 has also been clinically evaluated in a phase I study of
patients with advanced refractory solid tumors or non-Hodgkin's
lymphoma (10), and in a phase II
clinical trial (NCT01183949). SNS-032 was developed by
Bristol-Myers Squibb; this compound exhibits potent and selective
inhibitory activity against CDK2, CDK7, and CDK9. Chen et al
demonstrated that SNS-032 effectively killed chronic lymphocytic
leukemia cells in vitro regardless of prognostic indicators
and treatment history (11). Two
phase I clinical studies of SNS-032 have been reported (12,13);
however, no further developments have been reported.
CDKs are not only required for proper cell cycle
progression, but are also involved in DNA damage repair,
particularly in the repair pathway choice between homologous
recombination (HR) and non-homologous end joining (NHEJ) (14). The phosphorylation of breast cancer
2 tumor suppressor (BRCA2) by CDK inhibits its interaction with
RAD51 and regulates the HR pathway for double-strand DNA repair
(15). In addition, eukaryotic
cells have been shown to respond to DNA lesions via the activation
of a complex signal transduction pathway, known as the DNA damage
checkpoint, which delays cell cycle progression, while stimulating
DNA repair (16). It has also been
reported that CDK1 inhibition abrogates S-phase cell cycle arrest
and the inefficient phosphorylation of ataxia telangiectasia
mutated (ATM)/ataxia telangiectasia and Rad3 related (ATR)
substrates, leading to the inhibition of BRCA1 recruitment to the
DNA damage foci (17). Although
the activation of DNA damage signals inhibits CDK complexes and
prevents cell cycle progression, CDK activity is also required for
checkpoint recovery through the activation of the Forkhead
transcription factor FoxM1 (18,19).
These studies suggest that the abrogation of HR repair and
checkpoint control by targeting CDKs can cause cellular
sensitization to DNA-damaging agents.
Therefore, in this study, we focused on the effects
of CDK inhibitors against solid tumors, such as cervical cancer,
that are prevalent in some developing countries (20). The most common cause of
refractoriness in cervical cancer treatment is resistance to
radiation, whereas the treatment outcomes of ovarian cancer are
dependent on the success of cytoreductive surgery and chemotherapy.
If a patient has a tumor refractory to radiation, salvage
chemotherapy can only elicit a response in a few cases. Currently,
the majority of ongoing clinical trials for cervical cancer
treatments investigate agents that target refractory disease.
Hence, we selected CDK inhibitors as candidate target agents for
refractory cervical cancer. In this study, we compared the
anticancer effects of several CDK inhibitors currently undergoing
clinical trials (data not shown). Among the inhibitors available,
the two most potent candidates, AT7519 and SNS-032, were selected
and further characterized in terms of cytotoxicity, senescence, and
metastasis using cervical cancer cell lines and a human xenograft
tumor model.
Materials and methods
Cells and reagents
The human cervical carcinoma cell lines, HeLa and
ME-180, were obtained from the Korean Cell Line Bank (Seoul,
Korea). The HeLa and ME-180 cells were maintained in Dulbecco's
modified Easgle's medium (DMEM) and Roswell Park Memorial Institute
(RPMI)-1640 medium (Welgene Inc., Korea), supplemented with 10%
fetal bovine serum (FBS) (Capricorn Scientific GmbH, Germany) and
100 units of penicillin and streptomycin (Welgene). The cells were
cultured in a humidified incubator containing 5% CO2 at
37°C. SNS-032 and AT7519 were purchased from Selleck Chemicals
(Houston, TX, USA).
Cell viability assay
The HeLa and ME-180 cells (2–5×103
cells/well) were plated in 96-well plates and allowed to attach for
24 h prior to treatment. The cells were exposed to 0.001, 0.01,
0.1, 0.5, 1, or 10 µM AT7519 and SNS-032 for 48 h, and cell
viability was measured using the resazurin reduction ratio
(21). Resazurin solution was
added to a final concentration of 50 µM, followed by
incubation for 2–4 h and spectroscopy at A600 (Epoch, BioTek
Instruments, Inc, Winooski, VT, USA). IC50 values were
calculated using the ED50 Plus v1.0 online (http://www.sciencegateway.org/protocols/cellbio/drug/data/ed50v10.xls).
Senescence-associated (SA)
β-galactosidase assay
The HeLa cells (1×104) were plated in
35-mm culture plates and treated with various concentrations of
0.05 or 0.1 µM of AT7519 and SNS-032 for 3 days. The cells
were fixed in 2% formaldehyde/0.2% glutaraldehyde for 15 min at
room temperature, and SA β-galactosidase staining was performed as
previously described (22).
Irradiation
Delivery of γ-radiation was achieved using a
dual-source 137Cs unit at a dose rate of 3.2 Gy/min with
a GC-3000 Elan irradiator (MDS Nordion, Ottawa, Canada).
Western blot analysis
The cells were lysed in radioimmunoprecipitation
assay (RIPA) buffer [50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.1% SDS,
0.5% deoxycholic acid, 1% NP-40] containing a protease inhibitor
and a phosphatase inhibitor cocktail and briefly sonicated. The
protein content was measured using the Coomassie (Bradford) Protein
assay kit (Thermo Fisher Scientific, Rockford, IL, USA). A total of
10–40 µg of cell lysates were separated on 8, 10 12, or 15%
SDS-polyacrylamide gels and transferred to nitrocellulose membranes
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were
immunoblotted with antibodies against poly(ADP-ribose) polymerase
(PARP)-1 (#SC-8007, 1:2,000), p53 (#SC-126, 1:2,000), Chk1
(#SC-8408, 1:2,000), cyclin A (#SC-239, 1:1,000), cyclin B1
(#SC-245, 1:1,000), cyclin D1 (#SC20044, 1:500), cyclin E (#SC-247,
1:500), β-actin (#SC-47778, 1:3,000) (all from Santa Cruz
Biotechnology, Santa Cruz, CA, USA), caspase 3 (#9662, 1:1,000),
cleaved caspase 3 (#4199, 1:1,000), phospho-p53 (ser15) (#9284,
1:2,000), phospho-Chk1 (ser345) (#2341, 1:1,000), phospho-Chk2
(thr68) (#2661, 1:2,000) (all from Cell Signaling Technology,
Danvers, MA, USA), γ-H2AX (ser139) (#05-636, 1:2,000), Chk2
(#07-057, 1:2,000), phospho-ATM (ser1981) (#05-740, 1:5,000)
(Millipore, USA) and ATM (#1549-1, 1:2,000) (Epitomics, Burlingame,
CA, USA). HRP-conjugated secondary antibodies were obtained from
Enzo Life Sciences (Farmingdale, NY, USA; ADI-SAB-100-J,
ADI-SAB-300-J, 1:20,000). Chemiluminescence was detected using
enhanced chemiluminescence detection reagents (Western Bright™ ECL
kit, Advansta CO, USA).
BrdU assay
The HeLa and ME-180 cells (1×106 cells)
were plated in 100-mm plates and exposed 1 µM of CDK
inhibitors for 16 h. For combination treatments with γ-irradiation,
1 µM AT7519 and 1 µM SNS-032 were added to the cells
1 h prior to irradiation, and the cells were further incubated for
4 h following irradiation. BrdU was pulsed for 30 min before cells
were harvested and fixed in 70% ethanol at −20°C for 16 h. The
fixed cells were rinsed with phosphate-buffered saline (PBS) 3
times and incubated in 1.5 M HCl for 30 min at room temperature.
After rinsing with PBS, the cells were resuspended in 100 µl
of 0.5% bovine serum albumin (BSA) in PBS and incubated for 10 min.
Fluorescein isothiocyanate (FITC)-conjugated BrdU antibody
(#11-5071-41, eBioscience/Thermo Fisher Scientific, Waltham, MA,
USA) was added to cells, followed by 1 h of incubation prior to the
analysis of the BrdU-positive cell profile using a flow cytometer
[BD FACSCalibur (SN. E97501075), BD Biosciences, San Jose, CA,
USA].
Migration and invasion assays
Eight-micrometer pore size Transwell filters
(Corning Inc., Corning, NY, USA) were placed into 24-well plates
and the upper chambers were covered with Matrigel for the invasion
assay (BD Biosciences). Following treatment with 0.5 µM
AT7519 or 0.2 µM SNS-032 for 24 h, HeLa (6×104
cells/well) and ME-180 (1.5×105 cells/well) cells were
suspended in 150 µl FBS-free medium and seeded onto the
filters. The lower chambers were filled with 500 µl medium
containing 10 or 20% FBS. At 16–48 h after seeding, the cells were
fixed with ice-cold methanol for 5 min, and stained with 0.5%
crystal violet (Sigma-Aldrich, St. Louis, MO, USA) in 20% methanol
for 10 min at room temperature. After washing with distilled water,
the cells on the top side of the filter were removed with a cotton
swab, then the number of migrated cells to the lower side were
counted.
Tube formation assay
A total of 50,000 human umbilical vein endothelial
cells (HUVECs; #C-12203, Promo Cell, Heidelberg, Germany) were
seeded into each well of a 24-well plate pre-coated with Matrigel;
and added CDK inhibitors simultaneously. The cells were incubated
overnight to allow the formation of tube-like structures.
Endothelial cell tube formation was assessed using an IN Cell
Analyzer imaging device (GE Healthcare Life Sciences, Pittsburgh,
PA, USA). Tubular structures were quantified by counting the number
of branches in each field 11 h after seeding.
Human xenograft tumor model
A total of 96 female BALB/c nude mice (5 weeks old,
weighing 15 g) were purchased from Orient Bio Co. (Seongnam, Korea)
and allowed to acclimate to the new environment for 1 week before
use. The room temperature and relative humidity were maintained at
22±3°C and 50±20%, respectively. The mice were allowed access to
water and food (Purina) ad libitum. For tumor generation,
exponentially growing 1×106 ME-180 cells were injected
subcutaneously into the right hind leg of each male BALB/c nude
mouse. Tumor diameters were measured using a caliper, and tumor
volumes were calculated with the following formula: V =
0.523×AxB2, where 'A' is the longest diameter and 'B' is
the shortest diameter of the tumors. All animal experiments were
conducted following a protocol approved by the Korea Institute of
Radiological and Medical Sciences (KIRAMS) Animal Care and Use
Committee (Reference no. KIRAMS 201400400). The weight of the mice
upon sacrifice was 25 g.
Tumor growth delay
The tumor-bearing mice were randomly divided into 6
groups of 8 mice per group and treated as follows: i) Control; ii)
irradiation; iii) AT7519; iv) irradiation and AT7519; v) SNS-032;
and vi) irradiation and SNS-032. When the tumors were 6–7 mm in
mean diameter, the mice were treated with AT7519 (15 mg/kg once a
day for 5 days for a 2-week duration) or SNS-032 (15 mg/kg injected
intraperitoneally every 2 days for a 2-week duration). For the
administration of radiation, the mice were lightly anesthetized
with 5 mg/kg tiletamine/zolazepam (Virbac ZoletilTM 50; Virbac
Lab., Carros, France), and the tumor-bearing legs were irradiated
with a 60Co irradiator (Thermatron 780, Atomic Energy of
Canada) at a dose rate of 1.3 Gy/min.
Assay of lung metastasis
The anti-metastatic potential in the 3 experimental
groups: [i) control; ii) AT7519; and iii) SNS-032 (16 mice per
group)] was tested using the spontaneous lung metastasis model.
Briefly, 1×106 ME-180 cells per mouse were administered
to the right thighs of 5-week-old male BALB/c nude mice. When the
tumors reached a diameter of 6–7 mm, the mice were randomly
assigned to one of the three groups. Mouse lungs were removed on
days 45 and 60 following treatment and fixed with Bouin's solution
to count lung nodules under a polarizing light microscope with a 4X
objective lens.
Statistical analyses
Data were analyzed using the Kruskal-Wallis
non-parametric statistical test followed by the Mann-Whitney U test
using Bonferroni correction to adjust the probability. Statistical
analyses were performed using IBM SPSS Statistics version 20.
Results
AT7519 and SNS-032 inhibit cervical
cancer cell growth via the induction of apoptosis, senescence, and
cytostasis
To determine whether the CDK inhibitors, AT7519 and
SNS-032, exert anticancer effects, cell viability assays were
performed in two cervical cancer cell lines, HeLa and ME-180. As
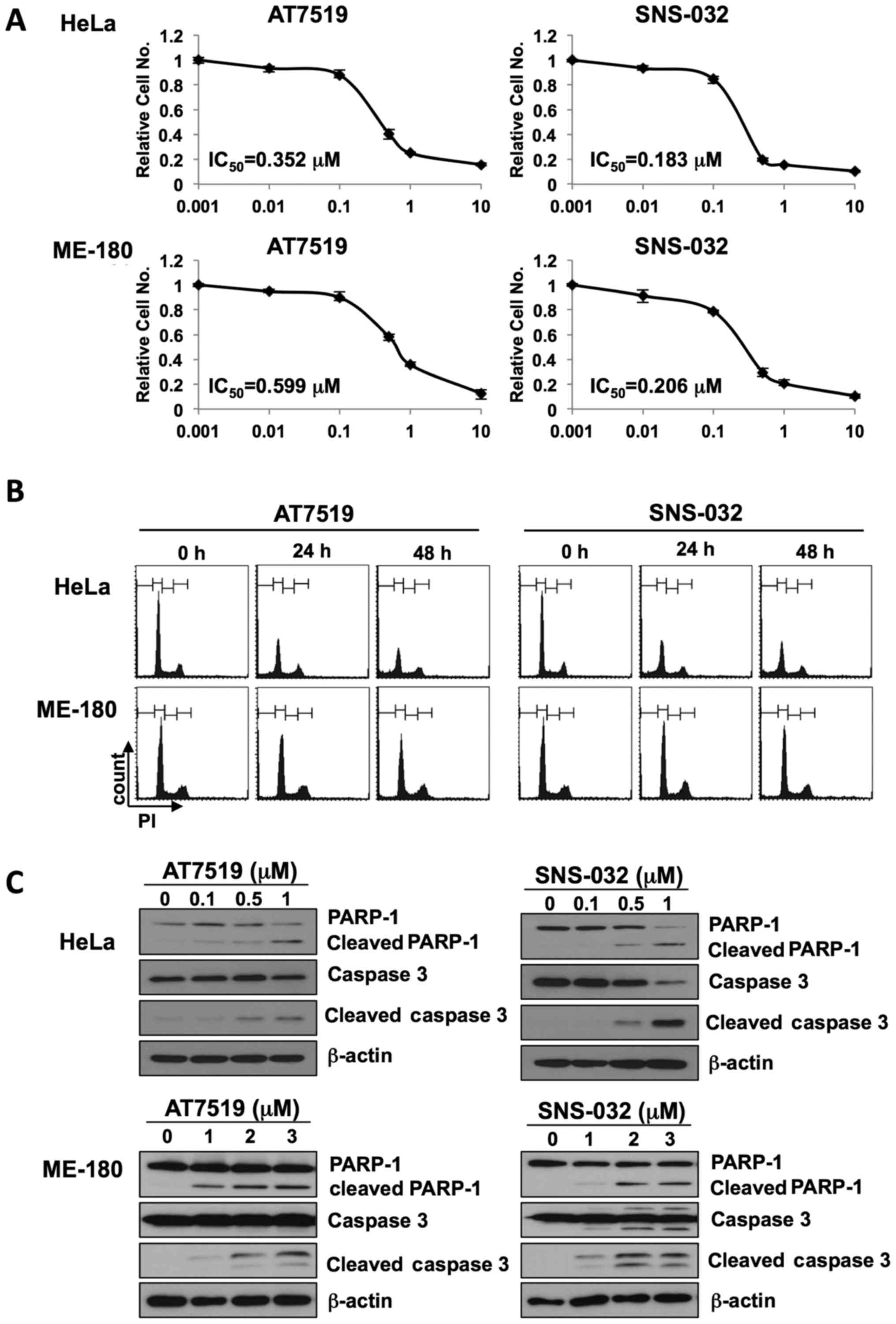
shown in Fig. 1A, cell growth was
markedly inhibited in a dose-dependent manner. AT7519 inhibited
HeLa and ME-180 cell growth with IC50 values of 0.352
and 0.599 µM, respectively, whereas SNS-032 inhibited HeLa
and ME-180 cell growth with IC50 values of 0.183 and
0.206 µM, respectively. To determine the anticancer
mechanisms of action of AT7519 and SNS-032, we first analyzed the
cell cycle following treatment with either AT7519 or SNS-032. As
shown in Fig. 1B, these CDK
inhibitors dysregulated cell cycle progression and increased the
sub-G1 cell population, particularly in the HeLa cells. Thus, we
next examined whether these drugs induced the apop-tosis of
cervical cancer cells. As shown Fig.
1C, the cleavage of PARP-1 and caspase 3, classical markers of
apoptosis, was increased following treatment with either AT7519 or
SNS-032, in a dose-dependent manner. The HeLa cells were more
susceptible to apoptosis by these CDK inhibitors. As CDK inhibition
disrupts cell cycle progression, we investigated whether prolonged
exposure to AT7519 or SNS-032 induces premature cellular
senescence. The results of an SA β-galactosidase assay revealed
that the numbers of senescent cells increased 2.5- and 3.5-fold
following treatment with 0.05 or 0.1 µM AT7519,
respectively, for 3 days. Similar to AT7519, the number of SA
β-galactosidase-stained cells increased by 2.4- and 3.7-fold
following treatment with 0.05 or 0.1 µM SNS-032,
respectively (Fig. 1D). SA β-gal
positivity was also increased in the ME-180 cells following
treatment with the CDK inhibitors. Finally, we measured the
actively proliferating cell populations by BrdU incorporation
assay. As shown in Fig. 1E, the
population of BrdU-positive cells decreased from 18–4% following
treatment with 1 µM AT7519 and to 3% with 1 µM
SNS-032, respectively. The inhibition of cellular proliferation was
dependent on the dose of the inhibitor applied (data not shown). As
cyclins are the crucial cofactors of CDKs and seem to be
deregulated in various cases of cervical cancer, we examined the
levels of several key cyclins by western blot analysis. The levels
of all the cyclins, (namely cyclin D1, E, A and B1) investigated
were decreased by AT7519 and SNS-032 treatment (Fig. 1F). These results suggest that
AT7519 and SNS-032 inhibited the growth of cervical cancer cells by
inducing cell cycle deregulation, apoptosis, cellular senescence
and cytostasis.
AT7519 and SNS-032 inhibit in vivo tumor
growth and sensitize cervical cancer cells to radiation
To confirm the anticancer effects of these
inhibitors in vivo and to examine their radiosensitizing
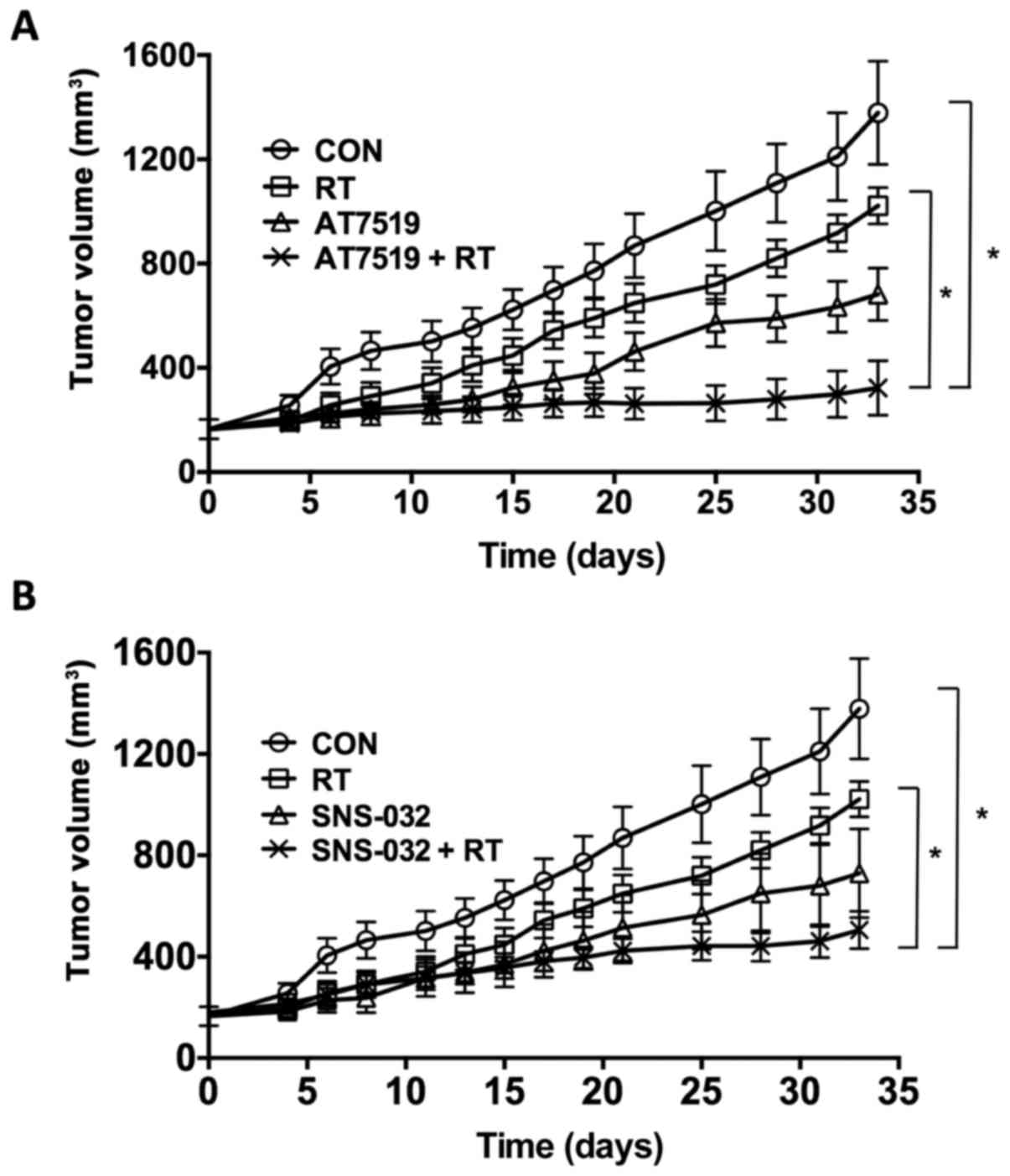
effects, a human xenograft tumor was established. As shown in
Fig. 2, the growth of subcutaneous
ME-180 xenograft tumors in the legs of BALB/c nude mice were
examined following exposure to various CDK inhibitors. The
treatments commenced when the tumor volume reached 164–180
mm3. The volume of the control tumors (CON)
progressively increased after reaching 400–500 mm3. The
irradiation of the tumors of mice treated with a single exposure of
9 Gy (RT) suppressed tumor growth. The growth of the tumors of the
mice treated with AT7519 or SNS-032 was slower than that of tumors
of the controls or the tumors from the mice treated only with
irradiation. The growth of the tumors of the mice that were both
irradiated and treated with AT7519 or SNS-032 was significantly
slower than that of the tumors of the mice treated with irradiation
alone (P<0.05). Consequently, while the volume of the tumors of
the untreated controls increased 2-fold in approximately 5 days,
the volume of the tumors from the mice treated with irradiation
doubled in 10 days. When irradiation was combined with treatment
with AT7519 or SNS-032, the tumor-doubling time was delayed to 33
or 15 days, respectively.
AT7519 and SNS-032 modulate DNA damage
response signaling and sensitize cells to radiation in vitro
As AT7519 and SNS-032 were shown to sensitize ME-180
xenograft tumors to radiation in vivo, we examined the
mechanisms through which CDK inhibitors radiosensitize ME-180
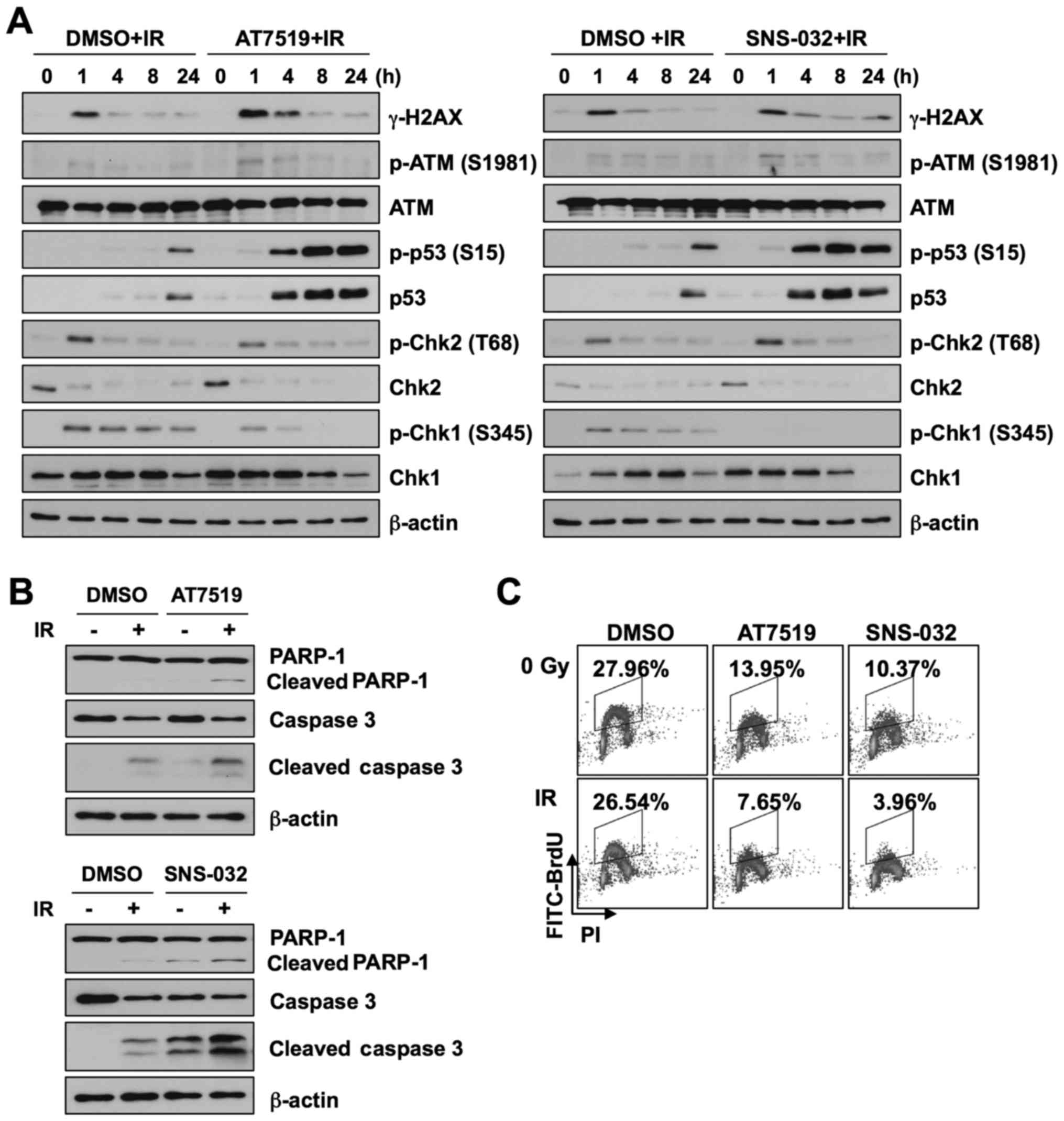
cells. We first assessed the activation of the DNA damage signaling
pathway. The ME-180 cells were treated with 1 µM AT7519 or 1
µM SNS-032 for 1 h, followed by γ-irradiation. We found that
the levels of γ-H2AX, a DNA double-strand break marker, were
slightly increased in the CDK inhibitor-treated cells than in the
cells treated with radiation alone (Fig. 3A). Although the phosphorylation
levels of ATM and Chk2 were similar, p53 was markedly activated 4 h
following both irradiation and treatment with AT7519 or SNS-032. In
addition, AT7519 and SNS-032 completely abolished Chk1
phosphorylation induced by IR. These results suggest that AT7519
and SNS0-032 enhance cellular radiosensitivity via p53 activation
and Chk1 inhibition. To determine whether AT7519 or SNS-032
accelerate apoptosis induced by radiation, the levels of apoptotic
markers were examined by western blot analyses (Fig. 3B). The combination of AT7519 or
SNS-032 with radiation increased the population of apoptotic cells.
We then assessed the effects of the combination of radiation and
CDK inhibitor treatment on S-phase cell cycle progression by BrdU
incorporation assay (Fig. 3C).
Although there was no inhibition of proliferation following
treatment with radiation alone under our experimental conditions,
the combination of AT7519 or SNS-032 and IR significantly decreased
the population of cells in the S phase by 50%.
AT7519 and SNS-032 inhibit cell migration
and invasion
To evaluate the effects of AT7519 and SNS-032 on
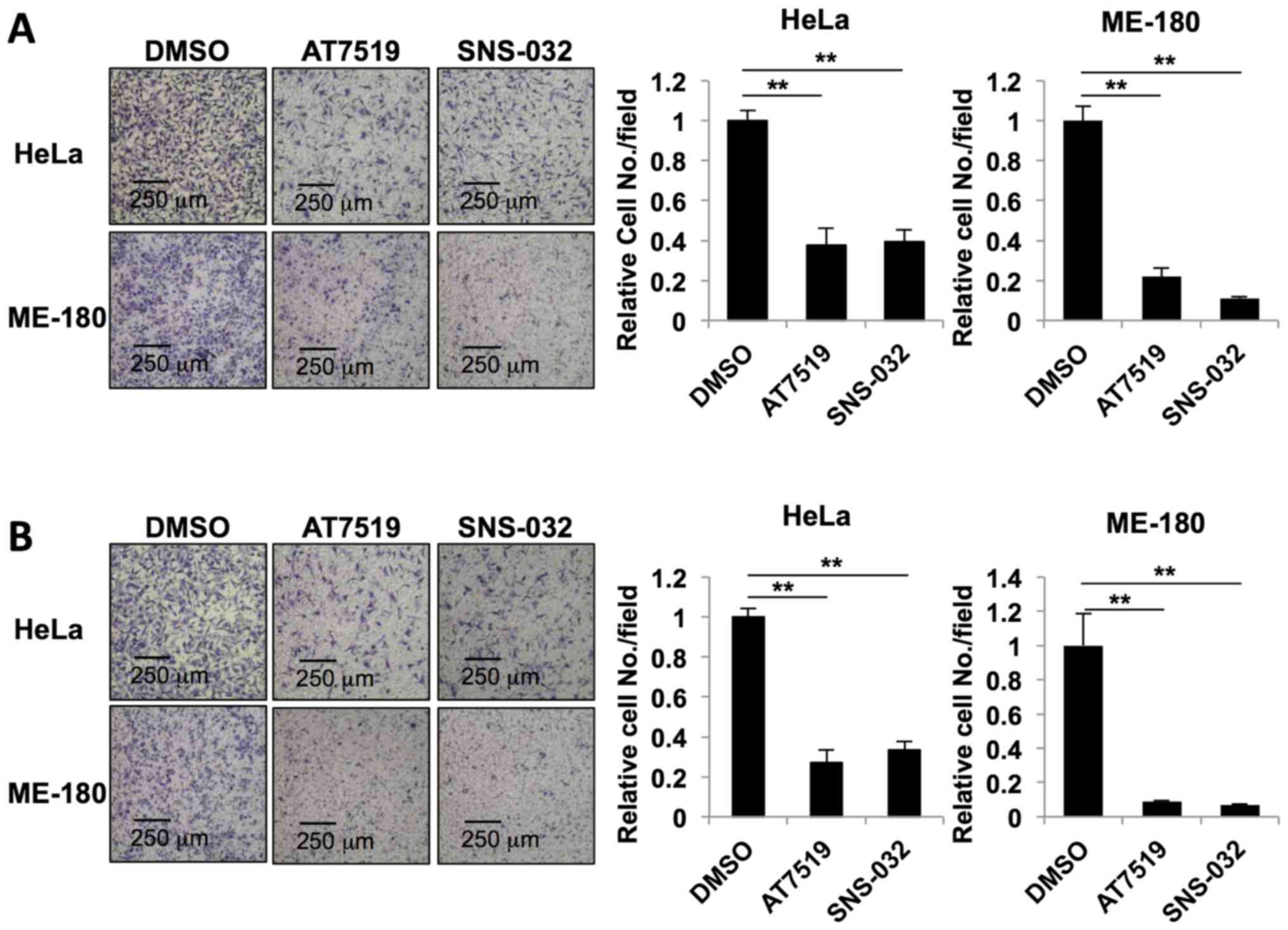
cancer metastasis, we performed cell migration and invasion assays.
For the migration assay, the HeLa and ME-180 cells were seeded in
Transwell® chambers and treated with 0.5 µM
AT7519 or 0.2 µM SNS-032 for 24 h. Both AT7519 and SNS-032
inhibited cervical cancer cell migration (Fig. 4A). The number of migratory HeLa
cells was significantly reduced following treatment with AT7519 and
SNS-032 to 0.38±0.08 and 0.393±0.059, respectively. The number of
migratory ME-180 cells were also reduced following treatment with
AT7519 (0.220±0.045) and SNS-032 (0.111±0.010). The inhibitory
effects of AT7519 and SNS-032 on cell invasion were also confirmed
(Fig. 4B). The number of invasive
HeLa cells was reduced following treatment with 0.5 µM
AT7519 (0.273±0.063) and 0.2 µM SNS-032 (0.334±0.045). The
number of invasive ME-180 cells was also reduced following
treatment with AT7519 (0.087±0.063) and SNS-032 (0.070±0.004).
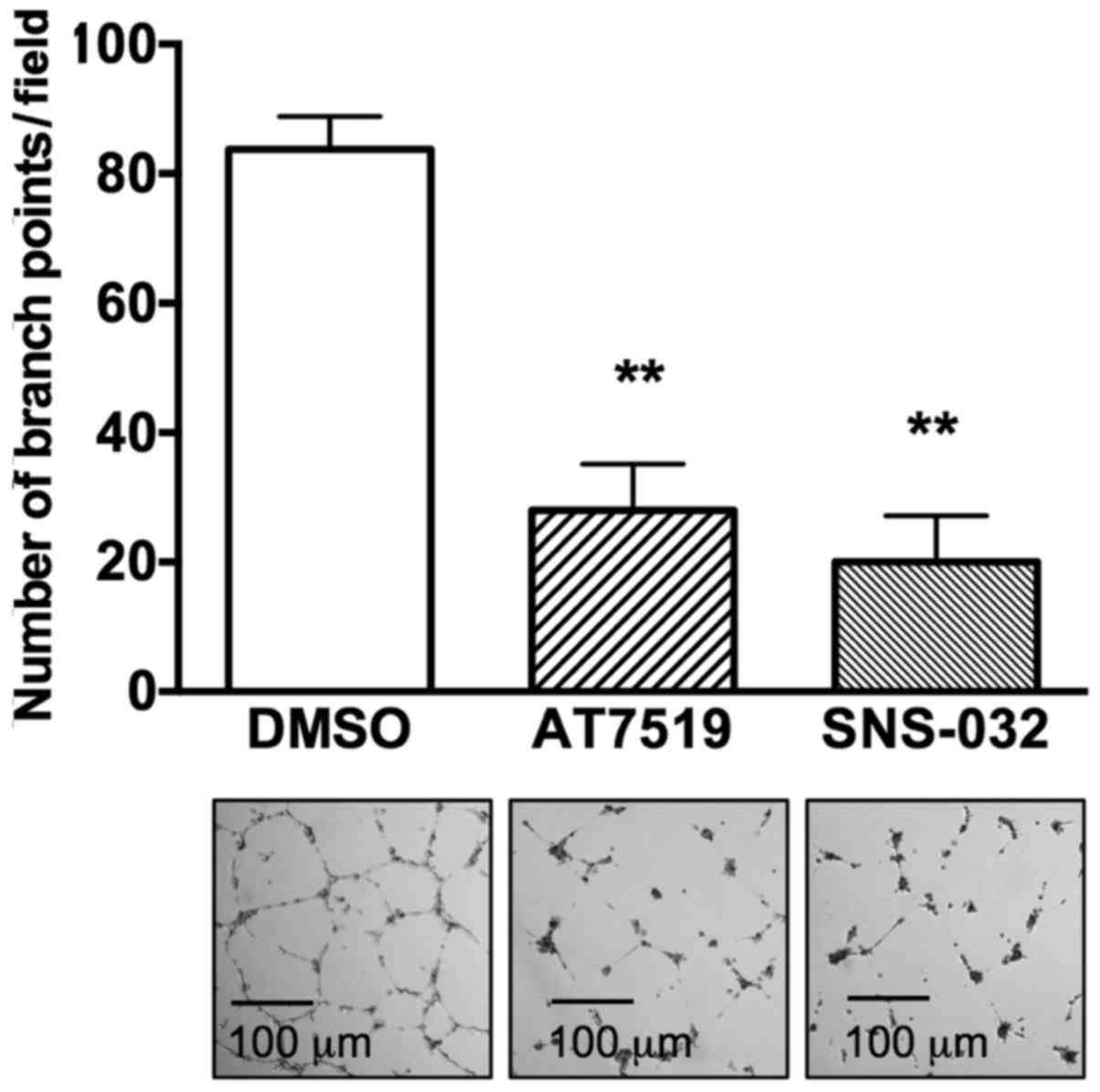
Anti-angiogenic effects of AT7519 and
SNS-032
As angio-genesis accelerates tumor metastasis, the
anti-angiogenic activities of the CDK inhibitors were measured. As
shown in Fig. 5, HUVECs formed a
well-organized tubular structure on Matrigel®,
representing functional activity of endothelial cells. However,
both AT7519 and SNS-032 inhibited tube formation. Compared to
83.75±2.56 branch points in the control group, the number of branch
points per field decreased to 28±3.56 (P<0.001) and 20±3.58
(P<0.001) in the presence of AT7519 and SNS-032, respectively.
From this result, CDK inhibitors appeared to suppress tumor
angiogenesis, in addition to suppressing the invasion and migration
of cervical cancer cells.
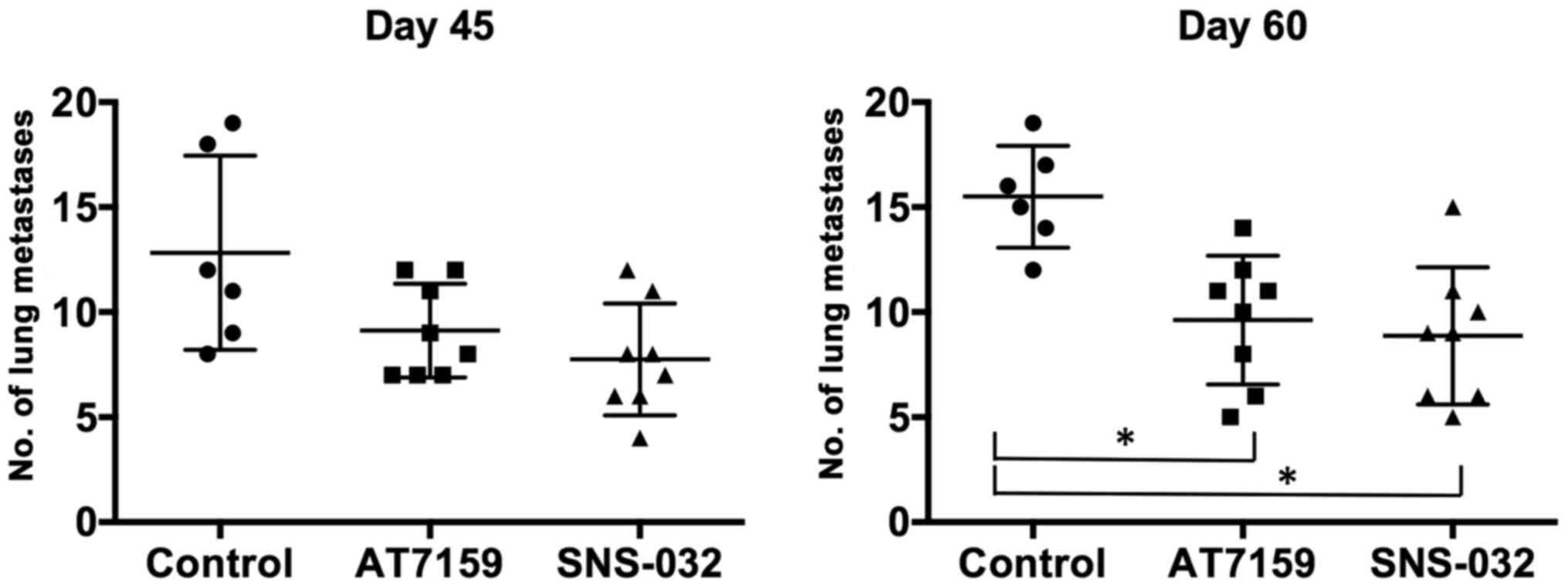
Lung metastasis
To confirm the anti-metastatic activity of CDK
inhibitors in vivo, a spontaneous lung metastasis model was
employed using an ME-180 xenograft tumor model. Forty-five days
after treatment, the average number of lung nodules of the control
group was 12.83±1.89. The number of lung nodules decreased to
7.75±0.94 in the AT7519 group and to 9.12±0.79 in the SNS-032
group. The anti-metastatic effects were more prominent after 60
days. Compared to 15.5±0.99 in the control group, the number of
lung nodules was 8.88±1.16 (P<0.05) and 9.63±1.08 (P<0.05) in
the AT7519 and SNS-032 groups, respectively (Fig. 6). These results confirm that both
AT7519 and SNS-032 were able to suppress tumor metastasis to the
lungs in vivo.
Discussion
In this study, we demonstrated that the CDK
inhibitors, AT7519 and SNS-032, suppressed the growth of cancer
cells in a dose-dependent manner (Fig.
1A). SNS-032 was more potent than AT7519, with a lower
IC50 value. The mechanisms of growth inhibition can be
summarized as cell cycle dysregulation, apoptosis, premature
senescence and cytostasis. Although there was no a shift to a
specific cell cycle phase, it seems likely that regulated cell
cycle progression was inhibited by these CDK inhibitors (Fig. 1B). Quantitative measurements of the
cleavage of PARP-1 and caspase 3, and SA β-galactosidase staining
revealed that AT7519 and SNS-032 treatment induced the apoptosis
and premature senescence of both HeLa and ME-180 cells (Fig. 1C and D). Although AT7519 and
SNS-032 induced the apoptosis of HeLa cells, apoptosis did not seem
to be a major mechanism of action in the ME-180 cells. However,
there is a possibility that apoptosis could be induced in the
ME-180 cells at higher concentration. It has been reported that CDK
inhibitors exert cytostatic effects at lower concentrations, but
induce apoptosis at higher concentrations. However, their
mechanistic actions have not yet been clarified. Roscovitine has
been reported to induce apoptosis at moderate cytotoxic
concentrations by decreasing mitochondria membrane potential
(23) or by reducing the amounts
of the caspase inhibitor, XIAP (24). In addition, we investigated other
possible anticancer mechanisms shown in Fig. 1D–F rather than focusing on the
apoptosis of HeLa cells. As CDKs are known to be master regulators
of cell cycle progression, we hypothesized that cytostatic growth
arrest may contribute to the anticancer effects of AT7519 and
SNS-032, as reported for other anticancer agents (25). From the results of the BrdU
incorporation assay, in the ME-180 cells treated with CDK
inhibitors, the number of cells in the S phase decreased in a
dose-dependent manner (Fig. 1E),
suggesting that these CDK inhibitors exert cytostatic rather than
cytotoxic effects on ME-180 cells. The depletion of cyclin D1, E, A
and B1 (Fig. 1F) may be one of the
crucial mechanisms of action of AT7519 and SNS-032, leading to cell
cycle dysregulation and cytostasis. Growth arrest at various points
of the cell cycle is known to eventually trigger cell death.
Whether a drug is cytostatic or cytotoxic depends on the dose, the
schedule of administration, the phase of the cell cycle during
which the drug acts and in which the cell resides, and the cellular
context (26). On the whole, it is
suggested AT7519 and SNS-032 exert anticancer effects through cell
cycle deregulation, premature senescence and cytostasis in both
cervical cancer cells.
We then examined the synergistic effects of
radiation and CDK inhibitors in vivo on tumor growth rate
using a human xenograft tumor model (Fig. 2). Hence, we suggest that CDK
inhibitors may be beneficial additions to standard
chemoradiotherapy regimens for patients with cervical cancer.
Initially, we hypothesized that the modulation of DNA double-strand
break repair through the inhibition of HR and prolonged G(2)-M arrest may be a major sensitizing
mechanism of AT7519 and SNS-032, as shown in other studies
(27–29). However, although the γ-H2AX levels
were slightly elevated, DNA damage was efficiently repaired in the
ME-180 cells following irradiation (Fig. 3A). By contrast, AT7519 or SNS-032
treatment induced p53 activation and inhibited the phosphorylation
of Chk1 at Ser345 following irradiation (Fig. 3A). From these results, it can be
concluded that the DNA damage-independent activation of p53 and
cell cycle checkpoint deregulation occurs through the inhibition of
Chk1 and contributes to radiaosensitization.
Metastasis is the most life-threatening event in
patients with cancer. We assessed the effects of AT7519 and SNS-032
on the aggressiveness of cervical cancer cells in terms of
invasion, angiogenesis and metastasis. The biological behavior of
tumors is very important to consider during treatment, as although
current modalities of cancer therapy have improved, they are
insufficient to adequately treat aggressive tumors. The
aggressiveness of tumors is usually defined by rapid invasion,
accelerated angiogenesis and early metastasis. As shown in Fig. 4, CDK inhibitor treatment reduced
the migration and invasion of HeLa and ME-180 cells.
Anti-angiogenic therapy has been extensively utilized since
bevacizumab was introduced as a treatment for patients with
recurrent cervical cancer during the GOG 240 trial (30,31).
Both AT7519 and SNS-032 inhibited tube formation in HUVECs, which
represent functional endothelial cells (Fig. 5). This finding suggests that CDK
inhibitors may be possible candidates for use in a combination
regimen of chemotherapeutics and irradiation for patients with
recurrent cervical cancer.
Overall survival is often determined by the presence
of distant organ metastases, such as of the liver, lungs and brain.
Li et al suggested that lymph node-only metastases are
better than organ metastases in patients with cervical cancer
(32). Therefore, we assessed the
effects of AT7519 and SNS-032 on organ metastases using a
spontaneous metastasis model. The AT7519- and SNS-032-treated
groups exhibited a statistically significant decrease in lung
metastases from xenografted cervical cancer cells (Fig. 6). These findings are well in
agreement with the results of the in vitro experiments
(Figs. 4 and 5). Based on these findings, the CDK
inhibitors, AT7519 and SNS-032, can enhance the efficacy of a
combination of chemo- and radiotherapies, thus impeding tumor cell
progression for the treatment of advanced and metastatic cases of
cervical cancer.
Abbreviations:
|
CDK
|
cyclin-dependent kinase
|
|
SA
|
senescence-associated
|
|
IR
|
ionizing radiation
|
Acknowledgments
Not applicable.
Funding
This study was supported by a grant from the Korea
Institute of Radiological and Medical Sciences (KIRAMS), funded by
Ministry of Science and ICT (MSICT), Republic of Korea (50531-2018;
50458-2014).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MHK and JHJ conceived and designed the experiments;
MAK, WK and HRJ performed the experiments; MAK, WK, HRJ, YJS, MK,
and JJJ curated and analyzed the data; MAK, WK, HRJ, YJS, MHK, and
JHJ wrote and edited manuscript. All authors have read and approved
the nal version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were conducted following a
protocol approved by the KIRAMS Animal Care and Use Committee
(Reference no. KIRAMS 201400400).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Malumbres M and Barbacid M: Mammalian
cyclin-dependent kinases. Trends Biochem Sci. 30:630–641. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bertoli C, Skotheim JM and de Bruin RA:
Control of cell cycle transcription during G1 and S phases. Nat Rev
Mol Cell Biol. 14:518–528. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Swaffer MP, Jones AW, Flynn HR, Snijders
AP and Nurse P: CDK substrate phosphorylation and ordering the cell
cycle. Cell. 167:1750–1761. e1716. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malumbres M and Barbacid M: To cycle or
not to cycle: A critical decision in cancer. Nat Rev Cancer.
1:222–231. 2001. View
Article : Google Scholar
|
|
6
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Senderowicz AM: Flavopiridol: The first
cyclin-dependent kinase inhibitor in human clinical trials. Invest
New Drugs. 17:313–320. 1999. View Article : Google Scholar
|
|
9
|
Santo L, Vallet S, Hideshima T, Cirstea D,
Ikeda H, Pozzi S, Patel K, Okawa Y, Gorgun G, Perrone G, et al:
AT7519, A novel small molecule multi-cyclin-dependent kinase
inhibitor, induces apoptosis in multiple myeloma via GSK-3beta
activation and RNA polymerase II inhibition. Oncogene.
29:2325–2336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen EX, Hotte S, Hirte H, Siu LL, Lyons
J, Squires M, Lovell S, Turner S, McIntosh L and Seymour L: A Phase
I study of cyclin-dependent kinase inhibitor, AT7519, in patients
with advanced cancer: NCIC Clinical Trials Group IND 177. Br J
Cancer. 111:2262–2267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen R, Wierda WG, Chubb S, Hawtin RE, Fox
JA, Keating MJ, Gandhi V and Plunkett W: Mechanism of action of
SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic
lymphocytic leukemia. Blood. 113:4637–4645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heath EI, Bible K, Martell RE, Adelman DC
and Lorusso PM: A phase 1 study of SNS-032 (formerly BMS-387032), a
potent inhibitor of cyclin-dependent kinases 2, 7 and 9
administered as a single oral dose and weekly infusion in patients
with metastatic refractory solid tumors. Invest New Drugs.
26:59–65. 2008. View Article : Google Scholar
|
|
13
|
Tong WG, Chen R, Plunkett W, Siegel D,
Sinha R, Harvey RD, Badros AZ, Popplewell L, Coutre S, Fox JA, et
al: Phase I and pharmacologic study of SNS-032, a potent and
selective Cdk2, 7, and 9 inhibitor, in patients with advanced
chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol.
28:3015–3022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Trovesi C, Manfrini N, Falcettoni M and
Longhese MP: Regulation of the DNA damage response by
cyclin-dependent kinases. J Mol Biol. 425:4756–4766. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Esashi F, Christ N, Gannon J, Liu Y, Hunt
T, Jasin M and West SC: CDK-dependent phosphorylation of BRCA2 as a
regulatory mechanism for recombinational repair. Nature.
434:598–604. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Finn K, Lowndes NF and Grenon M:
Eukaryotic DNA damage checkpoint activation in response to
double-strand breaks. Cell Mol Life Sci. 69:1447–1473. 2012.
View Article : Google Scholar
|
|
17
|
Johnson N, Cai D, Kennedy RD, Pathania S,
Arora M, Li YC, D'Andrea AD, Parvin JD and Shapiro GI: Cdk1
participates in BRCA1-dependent S phase checkpoint control in
response to DNA damage. Mol Cell. 35:327–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Laoukili J, Alvarez M, Meijer LA, Stahl M,
Mohammed S, Kleij L, Heck AJ and Medema RH: Activation of FoxM1
during G2 requires cyclin A/Cdk-dependent relief of autorepression
by the FoxM1 N-terminal domain. Mol Cell Biol. 28:3076–3087. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alvarez-Fernández M, Halim VA, Krenning L,
Aprelia M, Mohammed S, Heck AJ and Medema RH: Recovery from a
DNA-damage-induced G2 arrest requires Cdk-dependent activation of
FoxM1. EMBO Rep. 11:452–458. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
21
|
O'Brien J, Wilson I, Orton T and Pognan F:
Investigation of the Alamar Blue (resazurin) fluorescent dye for
the assessment of mammalian cell cytotoxicity. Eur J Biochem.
267:5421–5426. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, Medrano EE, Linskens M, Rubelj I and Pereira-Smith
O: A biomarker that identifies senescent human cells in culture and
in aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Arisan ED, Obakan P, Coker-Gurkan A,
Calcabrini A, Agostinelli E and Unsal NP: CDK inhibitors induce
mitochondria-mediated apoptosis through the activation of polyamine
catabolic pathway in LNCaP, DU145 and PC3 prostate cancer cells.
Curr Pharm Des. 20:180–188. 2014. View Article : Google Scholar
|
|
24
|
Mohapatra S, Chu B, Zhao X, Djeu J, Cheng
JQ and Pledger WJ: Apoptosis of metastatic prostate cancer cells by
a combination of cyclin-dependent kinase and AKT inhibitors. Int J
Biochem Cell Biol. 41:595–602. 2009. View Article : Google Scholar
|
|
25
|
Sparreboom A, de Jonge MJ and Verweij J:
The use of oral cytotoxic and cytostatic drugs in cancer treatment.
Eur J Cancer. 38:18–22. 2002. View Article : Google Scholar
|
|
26
|
Rixe O and Fojo T: Is cell death a
critical end point for anticancer therapies or is cytostasis
sufficient? Clin Cancer Res. 13:7280–7287. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kodym E, Kodym R, Reis AE, Habib AA, Story
MD and Saha D: The small-molecule CDK inhibitor, SNS-032, enhances
cellular radiosensitivity in quiescent and hypoxic non-small cell
lung cancer cells. Lung Cancer. 66:37–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Raghavan P, Tumati V, Yu L, Chan N,
Tomimatsu N, Burma S, Bristow RG and Saha D: AZD5438, an inhibitor
of Cdk1, 2, and 9, enhances the radiosensitivity of non-small cell
lung carcinoma cells. Int J Radiat Oncol Biol Phys. 84:e507–e514.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Storch K and Cordes N: The impact of CDK9
on radiosensitivity, DNA damage repair and cell cycling of HNSCC
cancer cells. Int J Oncol. 48:191–198. 2016. View Article : Google Scholar
|
|
30
|
Tewari KS, Sill MW, Long HJ III, Penson
RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao
MM, et al: Improved survival with bevacizumab in advanced cervical
cancer. N Engl J Med. 370:734–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Monk BJ and Tewari KS: Evidence-based
therapy for recurrent cervical cancer. J Clin Oncol. 32:2687–2690.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li H, Wu X and Cheng X: Advances in
diagnosis and treatment of metastatic cervical cancer. J Gynecol
Oncol. 27:e432016. View Article : Google Scholar : PubMed/NCBI
|