Introduction
Colon carcinogenesis is a multistep and complex
process, involving the accumulation of a string of genetic and
epigenetic alterations in normal colonic epithelial cells (1). Some of the frequent alterations found
in colon cancer involve mutations in the tumor suppressors p53 and
adenomatous polyposis coli (APC), as well as those in the
oncogenes, BRAF, phosphoinositide 3-kinase (PI3K),
K-ras and β-catenin, some of which are closely
associated with the control of cell proliferation (1). The overexpression of
proliferation-associated proteins, such as leucine-rich
repeat-containing G-protein coupled receptor 5 (Lgr5), EPH receptor
B3 (EphB3) and Sox9 is also common in cancer (2–4).
Overproliferation in the intestinal epithelium is a crucial
mechanism deregulating the control of the turnover of intestinal
epithelial cells, and is progressively incontrollable in colon
carcinogenesis (2). Excessive
proliferation is a hallmark of cancer, leading to an enhanced risk
of cell self-clonal expansion, which in turn, increases the
possibility of the occurrence of tumorigenic, including the
accumulation of genetic mutations, angiogenesis, growth under
stress conditions and an extended life span (5). Studies directly investigating the
regulatory mechanisms of malignant cell proliferation are still
largely and urgently required.
Cystatin 1 (CST1), a secretory peptide and a member
of the type 2 cystatin superfamily, has been reported to be closely
associated with cell proliferation and metastasis in several types
of cancer, such as pancreatic malignant neoplasm, gastric cancer,
esophageal squamous cell carcinoma and colorectal cancer (6–9). Our
previous study using microarray analysis identified CST1 as
one of the most highly expressed genes on the array in pancreatic
cancer. Further experiments demonstrated that CST1 is a useful
biomarker for pancreatic cancer cell proliferation (6). Moreover, it has been demonstrated
that the upregulation of CST1 contributes to cell proliferation and
the inhibition of cathepsin in gastric cancer; cathepsin is widely
expressed in tissues and plays a role in cell proliferation, the
immune response and tissue remolding (7). CST1 was believed to be an independent
predictor of the 5-year survival rate of patients with surgically
resected esophageal squamous cell carcinoma (8). Moreover, CST1 has been reported to be
a novel biomarker of colorectal cancer (9). However, the detailed mechanisms
responsible for the regulatory effects of CST1 on colon cancer cell
proliferation remain to be elucidated.
MicroRNAs (miRNAs or miRs) play an important role in
tumor cell proliferation. With the dysregulation of miRNAs, the
risk of carcinogenesis increases after Dicer 1, a critical enzyme
needed for miRNA processing, is genetically manipulated (10,11).
Let-7d miRNA is an important family member of the let-7 miRNAs,
which suppresses a multitude of oncofetal mRNAs and other
pro-proliferative and/or pro-metastatic targets, such as
high-mobility group AT-hook 2 (HMGA2), insulin-like growth
factor 2 mRNA-binding protein 1 (IGF2BP1), IGF2BP2
and nuclear receptor subfamily 6 group A member 1 (NR6A1)
(12–15). A number of studies have
demonstrated that let-7d plays pivotal role in the initiation and
development of several malignant neoplasms. Let-7d has been proven
to be negatively associated with pancreatic ductal adenocarcinoma,
breast cancer, oral cancer, prostate cancer, and head and neck
cancer (16–20). Moreover, some researchers
demonstrated that let-7d inhibited glioblastoma multiforme cell
proliferation and accelerated cell apoptosis in melanoma,
colorectal, and ovarian cancer (21,22).
However, the association between let-7d and colorectal cancer has
been rarely reported.
In the current study, we used microarrays to
identify that CST1 is one of the most highly expressed mRNAs
in colon cancer samples, compared with normal colon tissues, and we
then further verified CST1 expression in more clinical
samples by reverse transcription-quantitative PCR (RT-qPCR). We
then detected 5 miRNAs (miR15b, miR-145, miR-126, miR-21 and
let-7d), which are known as tumor-associated genes in cancer,
including colon cancer and normal tissues (23–25).
We found that let-7d expression was significantly decreased
in colon cancer tissues and it was the only miRNA which was related
to CST1 in colorectal cancer cell lines following transfection with
miRNA mimics. We verified that CST1 and let-7d have an effect on
colorectal cancer cell proliferation. We further identified the
CST1/p65 pathway as the downstream pathway of let-7d in modulating
colorectal cancer cell proliferation via the overexpression of
let-7d or the silencing of CST1. The results of the current study
suggest that the let-7d/CST1/p65 pathway is a novel transduction
pathway found in colorectal cancer cell proliferation and that it
may prove to be useful in the prevention and treatment of
colorectal cancer.
Materials and methods
Tissue samples
Colon malignant neoplasm and matched non-tumor
tissues from 10 patients were harvested during surgery from The
Third Affiliated Hospital of Sun Yat-sen University (Guangzhou,
China). The information of the patients is presented in Table I. Their diagnosis was dependent on
pathological analysis. Matched non-tumor tissues were normal colon
mucosa with biopsy tissues from the suspected patients examined
using a colonoscope, which were confirmed pathological normality.
The specimens were instantly frozen following excision and reserved
at −80°C for microarray, RT-qPCR, western blot analysis and
immunohistochemical analysis. This study was approved by the
Institutional Review Board at The Third Affiliated Hospital of Sun
Yat-sen University. Written informed consent was obtained from each
patient.
 | Table IThe clinical characteristics of the
patients. |
Table I
The clinical characteristics of the
patients.
| Clinicopathology
and characteristic | Total |
|---|
| Total, n | 10 |
| Age (mean ±
SD) | 58±8.5 |
| Age, n (%) | |
| <65 | 2 |
| ≥65 | 8 |
| Sex | |
| Male | 4 |
| Female | 6 |
| Tumor location | |
| Right colon | 7 |
| Left colon | 3 |
| T category | |
| pT1 | 1 |
| pT2 | 2 |
| pT3 | 4 |
| pT4 | 3 |
| N category | |
| 0 | 3 |
| 1 | 4 |
| 2 | 3 |
| Distant
metastasis | |
| Absent | 6 |
| Present | 4 |
|
Differentiation | |
| Well/moderate | 6 |
| Poor | 4 |
Microarray experiment
The four normal samples were normal colon mucosa
with biopsy tissues from the suspected patients examined using a
colonoscope. Six colon malignant neoplasm tissues were harvested
during surgery. The samples were subsequently verified by histology
assay. The microarray analysis was carried out by Capital Bio Corp.
(Beijing, China). The array data were analyzed for data
summarization, normalization and quality control using the
GeneSpring software v12 (Agilent). The data were Log2 transformed
and median centred by genes using the Adjust Data function of
Cluster 3.0 software and then further analyzed with hierarchical
clustering with average linkage.
Cell culture and transfection
The human colorectal cancer cell lines, including
HCT116 (colon cancer), SW480 (colon adenocarcinoma), HT-29
(rectosigmoid adenocarcinoma) were purchased from the American Type
Culture Collection (ATCC, Manassas, VA, USA). The cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL,
New York, NY, USA) supplemented with 10% heat-inactivated fetal
bovine serum and incubated in a humidified condition at 37°C with
5% CO2. For overexpression or knock down experiments, 3
siRNAs targeting CST1 (20 µM CST1 siRNA; oligo
kit; GenePharma, Shanghai, China), the let-7d mimic and
inhibitor kit (GenePharma) and miR15b, miR-145, miR-126, miR-21
mimic kit (GenePharma) were transfected into the colorectal cancer
cell lines using Lipofectamine 3000 (1901443, Invitrogen, Carlsbad,
CA, USA) according to the manufacturer's instructions; the
sequences of siRNAs and mimics are listed in Table II. The most effective siRNA
sequence (number 3, Table II; the
results of the most effective siRNA are not shown) was screened to
attain a transfection efficiency of >90% and was selected to be
used in the experiments. Following 1 day of incubation, the
transfection medium with Lipofectamine 3000 (Invitrogen) was
discarded and regular culture medium was added for incubation in a
humidified atmoshpere at 37°C with 5% CO2. At 48 h after
transfection, the human colorectal cancer cell lines were used in
further experiments. Subsequently, Bay117082 (B5556; Sigma, St.
Louis, MO, USA), an NNF-κB inhibitor was added at an optimal dose
of 30 µmol/l into the medium of HCT116 cells for 2 h before
further experiments.
| Table IISequences of siRNAs and mimics
used. |
Table II
Sequences of siRNAs and mimics
used.
| Gene | Sequence |
|---|
| CST1 siRNA | 1.
5′-GGUACUAAGAGCCAGGCAATT-3′ |
|
5′-UUGCCUGGCUCUUAGUACCTT-3′ |
| 2.
5′-GGUGGCAUCUAUAACGCAGTT-3′ |
|
5′-CUGCGUUAUAGAUGCCACCTT-3′ |
| 3.
5′-GCCAUCAGCGAGUAUAACATT-3′ |
|
5′-UGUUAUACUCGCUGAUGGCTT-3′ |
| Let-7d mimic |
5′-AGAGGUAGUAGGUUGCAUAGUU-3′ |
|
5′-AACUAUGCAACCUACUACCUCU-3′ |
| miRNA15b |
5′-UAGCAGCACAUCAUGGUUUACA-3′ |
|
5′-UGUAAACCAUGAUGUGCUGCUA-3′ |
| miR-145 |
5′-GUCCAGUUUUCCCAGGAAUCCCU-3′ |
|
5′-AGGGAUUCCUGGGAAAACUGGAC-3′ |
| miR-126 |
5′-CAUUAUUACUUUUGGUACGCG-3′ |
|
5′-CGCGUACCAAAAGUAAUAAUG-3′ |
| miR-21 |
5′-UAGCUUAUCAGACUGAUGUUGA-3′ |
|
5′-UCAACAUCAGUCUGAUAAGCUA-3′ |
RNA extraction and RT-qPCR
With the RNAgents Total RNA Isolation system
(Promega, Madison, WI, USA), total RNA was extracted from the
cancer tissues, matched normal tissues and cell lines according to
the manufacturer's instructions. For miRNA quantification, with
primer sets (GenePharma), total RNA was reverse transcribed into
cDNA for quantitative PCR. U6 was used to normalize the expression
of let-7d. First-strand cDNA was synthesized using Superscript
Reverse Transcriptase (Invitrogen) following the manufacturer's
instructions. cDNA was diluted with diethyl pyrocarbonate-treated
water at a 1:10 ratio. Quantitative PCR was executed using specific
primers and the QuantiTest SYBR-Green PCR kit (Cat. no.
04707516001) according to the manufacturer's instructions (Roche,
Basel, Switzerland) on an Applied Biosystems 7500 real-time PCR
machine (Applied Biosystems, Foster City, CA, USA). The mRNA
primers used and the sequences were as follows: CST1 sense,
5′-GGTACAGCGTGCCCTTCA-3′ and antisense, 5′-TTGGGCTGGGACTTGGTA-3′,
171-bp product; proliferating cell nuclear antigen (PCNA)
sense, 5′-CCTGCTGGGATATTAGCTCCA-3′ and antisense,
5′-CAGCGGTAGGTGTCGAAGC-3′, 109-bp product; cyclin D1 sense,
5′-GCTGCGAAGTGGAAACCATC-3′ and antisense,
5′-CCTCCTTCTGCACACATTTGAA-3′, 135-bp product; cyclin E
sense, 5′-CCTGCGCGAGAAGGAACTG-3′ and antisense,
5′-CGTTGTAGCGATCCATGAAGTG-3′, 173-bp product; and β-actin
sense, 5′-GTCTTCCCCTCCATCGTG-3′ and antisense, 5′-AGGGTGAGGATGC
CTCTCTT-3′, 113-bp product. The reaction conditions were as
follows: 95°C, 5 min; (95°C, 15 sec; 60°C, 30 sec) ×40 cycles. mRNA
expression was normalized to β-actin.
Immunohistochemistry
Formalin-fixed tissues were embedded in paraffin and
sliced. The 4-mm-thick sections were then incubated with monoclonal
antibodies against CST1 (1:100, Sigma) overnight at 4°C and then
incubated with goat anti-rabbit secondary antibodies (sc-2006,
Santa Cruz Biotechnology, Santa Cruz, CA, USA).
3,3′-Diaminobenzidine was used as the chromogen. The sections were
then dyed using hematoxylin and mounted. CST1 expression was
evaluated qualitatively by two independent pathological experts
unaware of the clinical and pathological information.
Western blot analysis
Total protein was extracted from the tissues and
cell lines using tissue extraction reagent (FNN0071, Invitrogen)
and cell extraction buffer (FNN0011, Invitrogen), purified and
assessed qualitatively by western blot analysis. Total protein
extracts (40 µg) were shifted to 10% gradient SDS-PAGE gels
to separate the proteins by different molecular weight and
transferred onto nitrocellulose membranes for antigen-antibody
reaction. The membranes were then blocked with 5% skimmed milk in
PBS for 2 h and mixed with primary antibodies against CST1
(SAB-1405670, Sigma), p-p65 (sc-101749), PCNA (sc-56), cyclin D1
(sc-753), p65 (sc-372) (Santa Cruz Biotechnology) and β-actin
(sc-47778, Santa Cruz Biotechnology) and then incubated with goat
anti-rabbit secondary antibodies (sc-2006, Santa Cruz
Biotechnology) for 2 h at 37°C.
Colony formation assay
To qualitatively evaluate clonoge-nicity, the
HCT-116 cells were transfected with siRNA against CST1
(CST1-siRNA; GenePharma), let-7d mimic or the control empty
vector (GenePharma) for 48 h and the HCT-116 cells were then
cultured in 6-well plates at a density of 200 cells/well. The
6-well plates were then washed with PBS, fixed in methanol for 15
min, and stained with 0.5% crystal violet for 15 min. The plates
were then photographed, and the colonies were counted with a
high-resolution microscope (DMI 3000, Leica, Wetzlar, Germany).
Statistical analysis
All statistical analyses were performed using SPSS
18.0 software. Paired t-tests were executed to compare CST1 and
let-7d expression among the sample tissues and cell lines.
Significant differences were considered if the probability of the
difference was <5 in 100 (P<0.05).
Results
Upregulation of CST1 in patients with
colon cancer
In the current study, we used GeneChip microarrays
to compare the RNA expression profiles of 6 colorectal cancer
tissues and 4 normal tissues. The microarrays revealed that
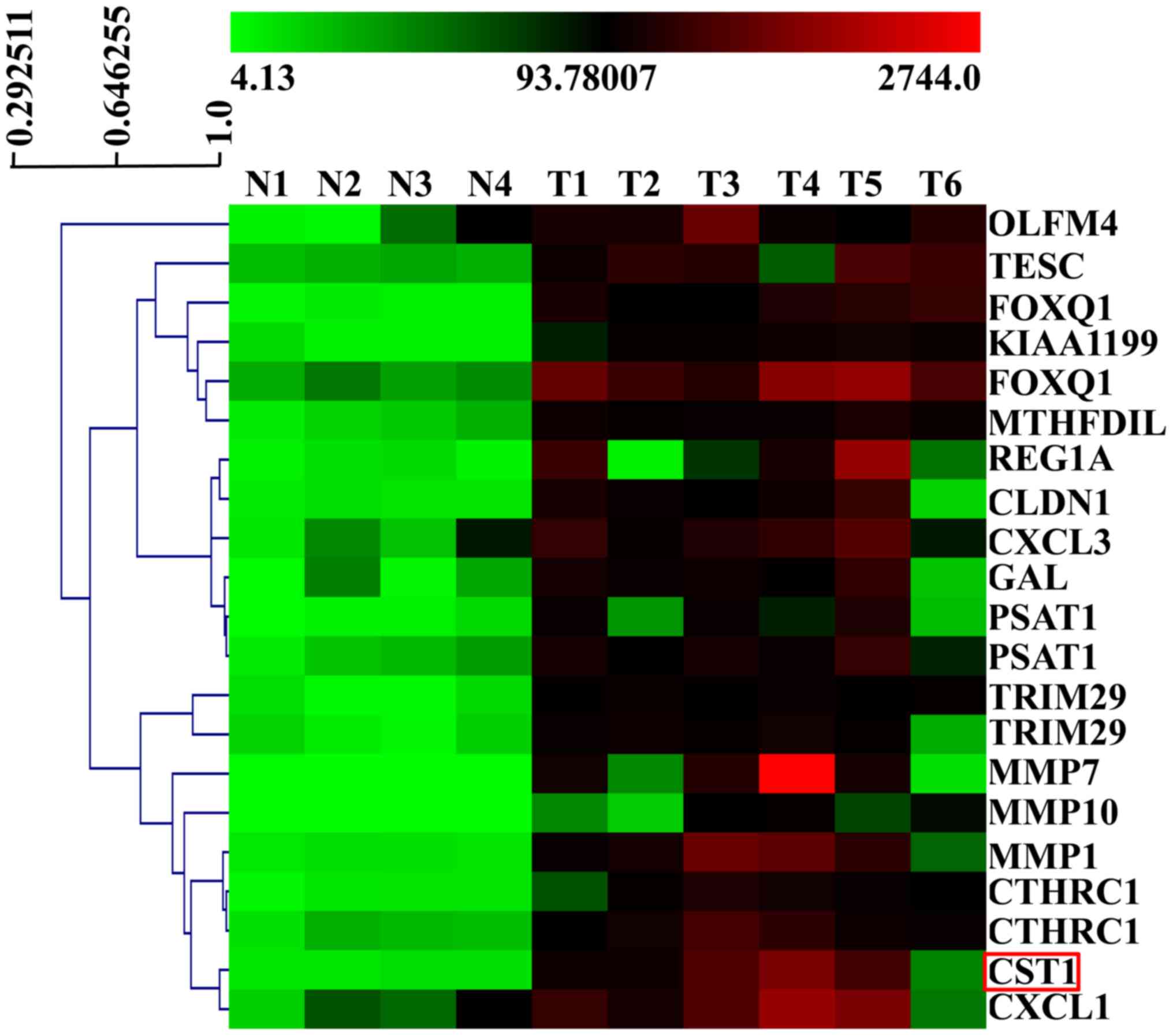
CST1 expression was significantly increased in the cancer
samples compared with the normal tissues (Fig. 1). This indicated that CST1 may be
involved in the development and/or progression of colon cancer.
Expression of miRNAs in human colorectal
cancer
We analyzed the expression of miR15b, miR-145,
miR-126, miR-21 and let-7d in 5 cancer tissues and 5 normal tissues
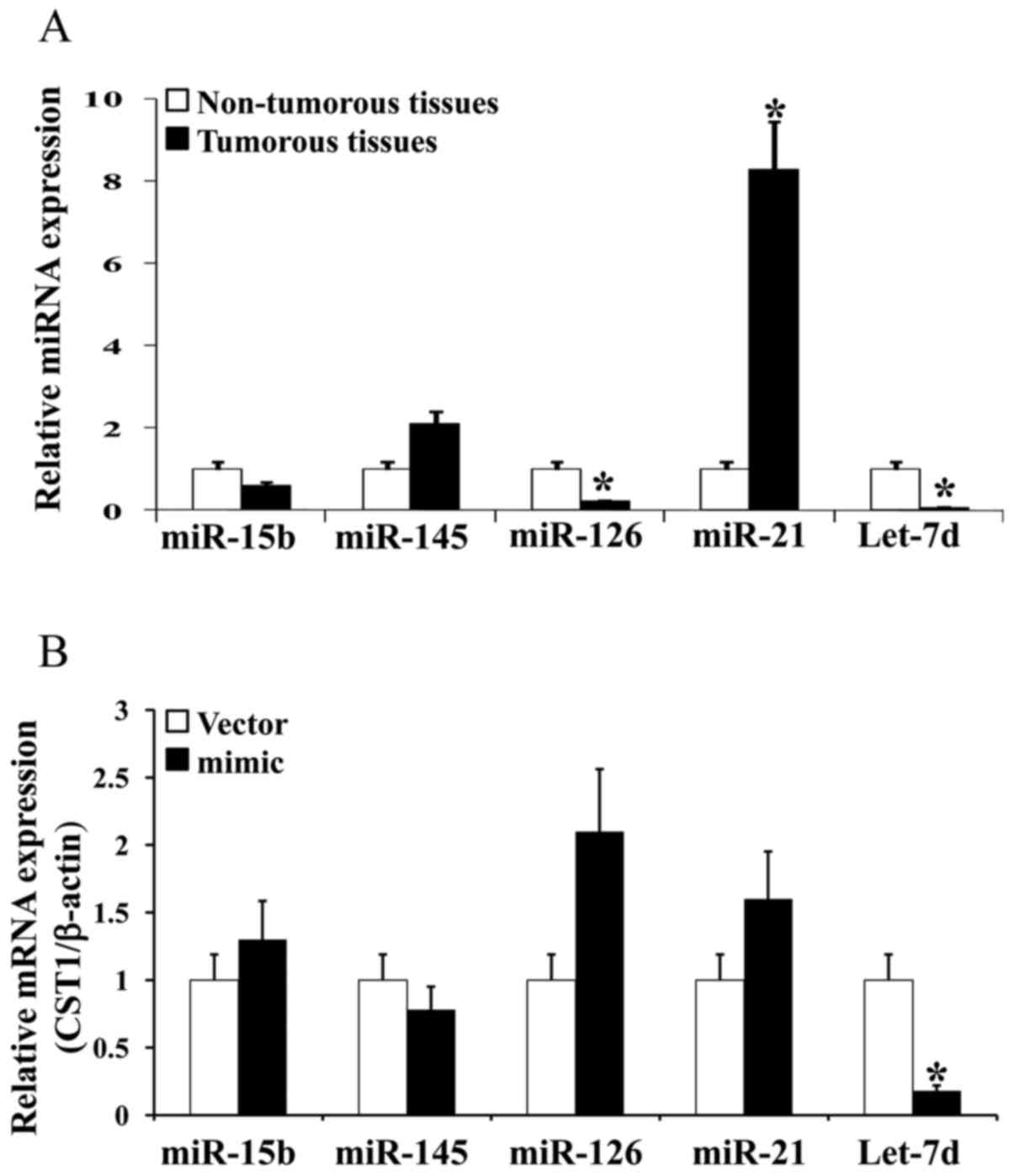
by RT-qPCR. The results revealed that let-7d expression was
significantly decreased by approximately 20-fold in the cancer
tissues (Fig. 2A). We then used
different miRNA mimics to increase the expression of 5 miRNAs in
the HCT116 cells and examine the effects on miRNA expression. The
results revealed that CST1 expression was markedly decreased when
using let-7d-mimic (Fig.
2B). Thus, our findings indicate that let-7d expression was
decreased in the colon cancer tissues and hypothesized that it was
related to CST1.
Upregulation of CST1 in human colon
cancer
To confirm that CST1 was upregulated in the colon
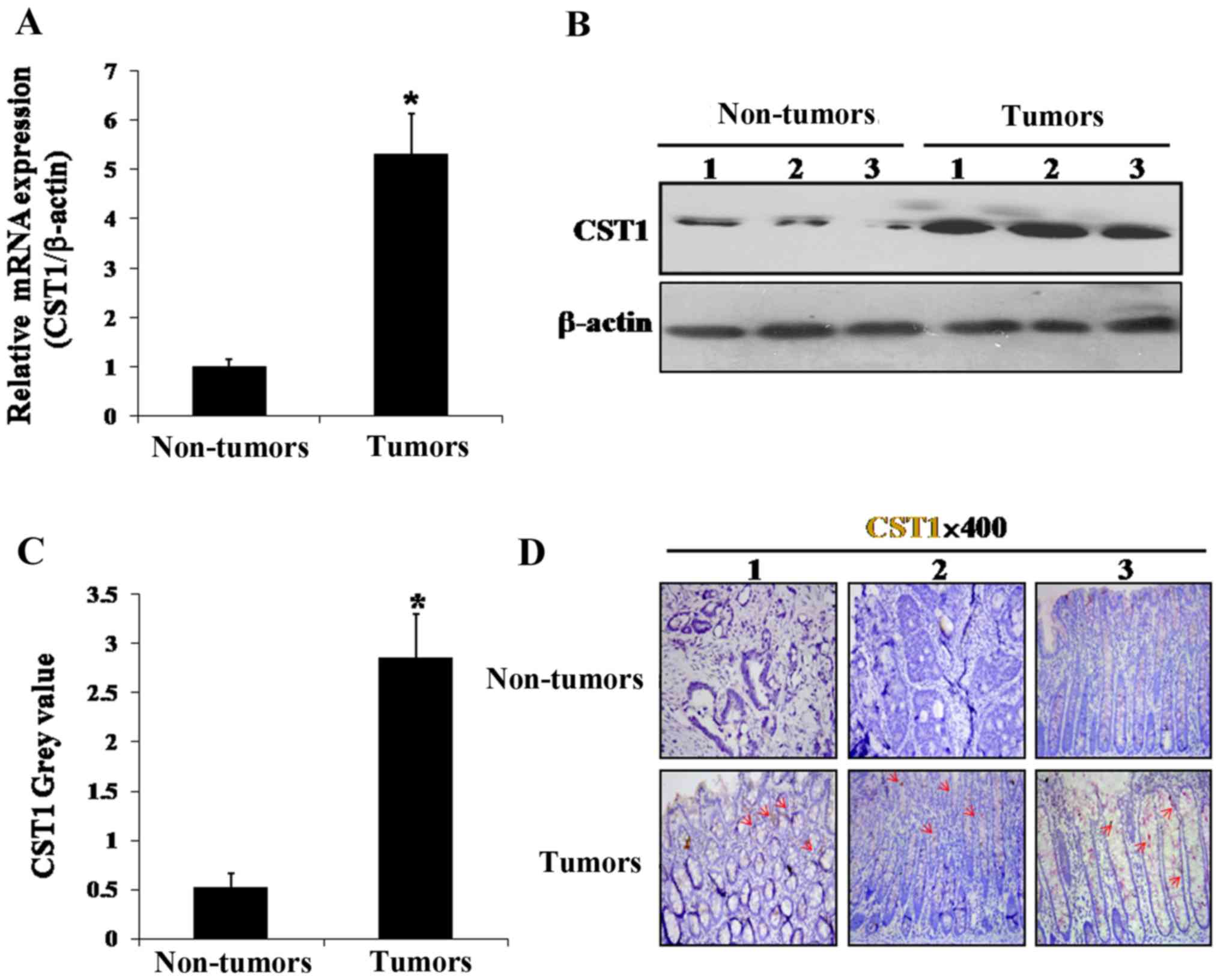
cancer tissues, we verified its mRNA expression in the colon cancer
tissues and normal tissues by RT-qPCR. CST1 mRNA expression
was found to be markedly increased by ~5-fold in the cancer
compared with the normal tissues, consistent with the results of
the microarray analysis (Fig. 3A).
Moreover, western blot analysis revealed that CST1 protein
expression was also highly upregulated in the cancer tissues, but
downregulated in the normal tissues (Fig. 3B and C). Furthermore, by using
immunohistochemistry, CST1 protein expression was found to be
increased conspicuously in the tumor tissues, but not in the normal
tissues (Fig. 3D). These data thus
suggest that CST1 plays a role in the initiation and development of
color cancer.
Overexpression of let-7d suppresses
colorectal cancer cell proliferation
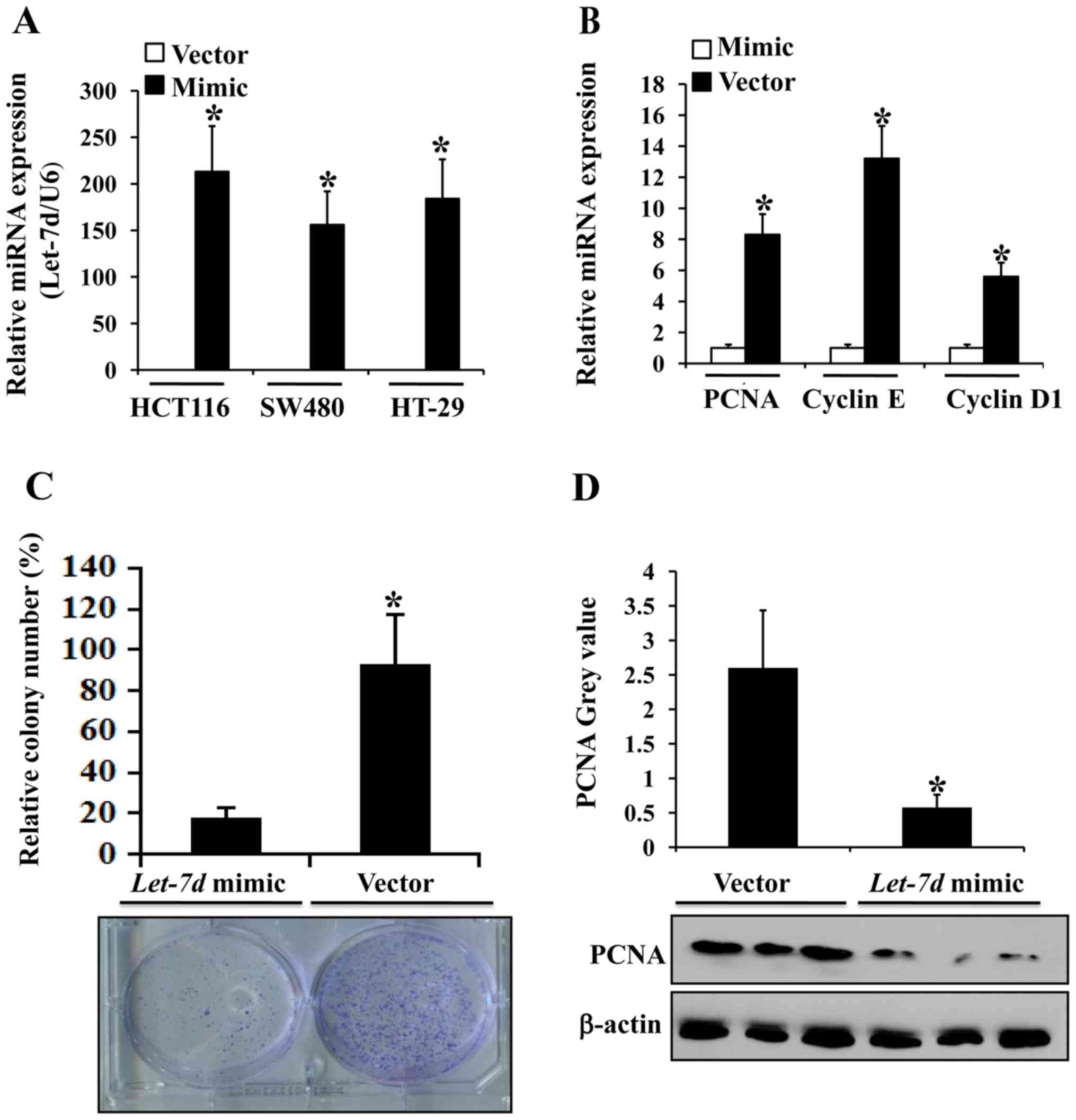
We screened the most efficient and effective let-7d
mimics for upregulating let-7d expression for use in the subsequent
experiments (Fig. 4A). The mRNA
expression levels of cancer proliferation-associated proteins, such
as PCNA, cyclin D1 and cyclin E, were markedly diminished in the
HCT116 cells transfected with let-7d-mimics (Fig. 4B). Colony formation assays were
executed using the HCT116 cells transfected with
let-7d-mimics or the empty vector and found that compared
with the controls, the mean colony number was significantly
decreased in the group transfected with let-7d-mimics
(Fig. 4C). Combined with the
decrease in the expression of the proliferation-associated protein,
PCNA, in the group transfected with let-7d-mimics (Fig. 4D), it was suggested that let-7d
regulates colorectal cancer cell proliferation.
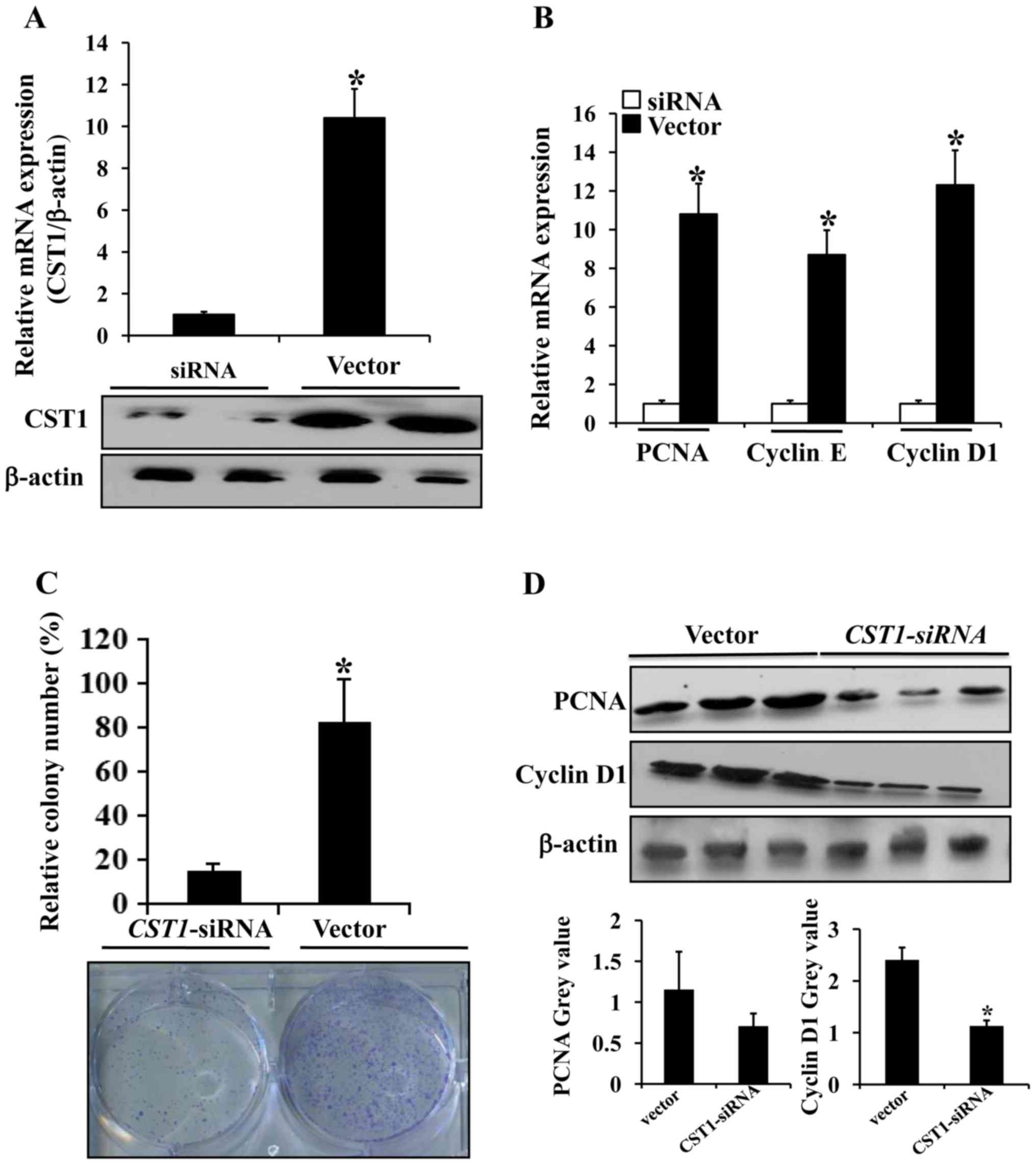
CST1 contributes to human colorectal
cancer cell proliferation
We screened the most efficient and effective
CST1-siRNA from 3 siRNAs for inhibiting CST1 expression for
use in the subsequent experiments (Fig. 5A). The mRNA expression levels of
the cancer proliferation-associated proteins, PCNA, cyclin D1 and
cyclin E, were markedly decreased in the HCT116 cells transfected
with CST1-siRNA compared with the cells transfected with the
control vector (Fig. 5B). The
results of colony formation assay revealed that compared with the
control, the mean colony number was significantly decreased in the
HCT116 cells transfected with CST1-siRNA (Fig. 5C). We then evaluated the expression
of proliferation-associated proteins and found that the expression
of PCNA and cyclin D1 was markedly decreased in the HCT116 cells
transfected with CST1-siRNA (Fig. 5D). Thus, these findings suggest
that CST1 promotes colorectal cancer cell proliferation by
increasing PCNA and cyclin D1 expression.
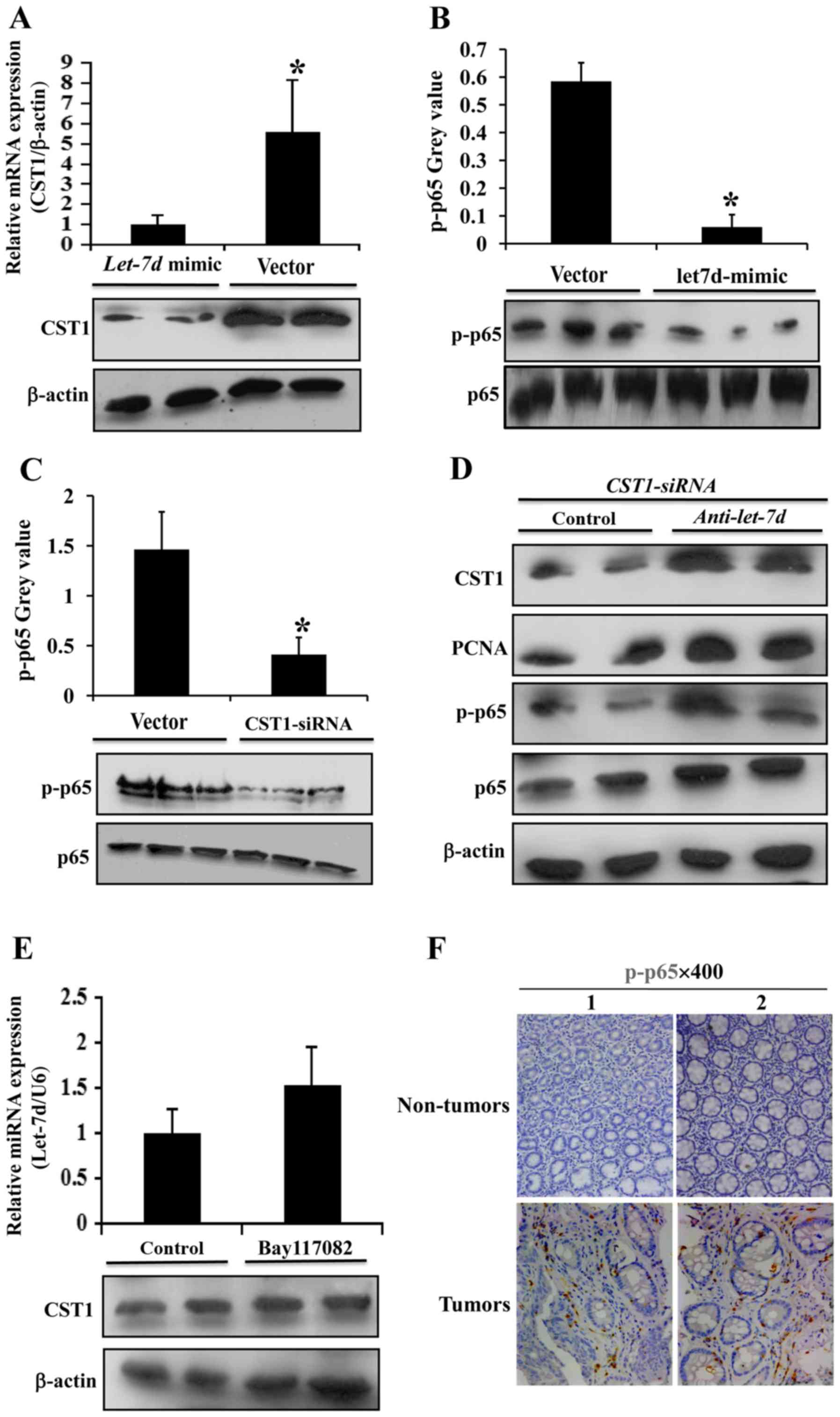
Let-7d inhibits colorectal cancer cell
proliferation through CST1/p65
We used let-7d-mimics to increase let-7d expression
in the HCT116 cells and we found that in this process, CST1
expression was markedly decreased (Fig. 6A). We also used CST1-siRNA
to silence CST1 expression in the HCT116 cells; however, the
silencing of CST1 had no marked effect on let-7d expression
(data not shown); this indicated that let-7d may regulate CST1
expression in colorectal cancer. NNF-κB p65 phosphorylation was
then analyzed, and it was found to decrease following transfection
with let-7d-mimics (Fig.
6B), indicating that let-7d may be connected with the NNF-κB
p65 pathway. Subsequently, following the silencing of CST1, NNF-κB
p65 phosphorylation was analyzed, and it was found to markedly
decrease (Fig. 6C). Moreover,
following the use of let-7d inhibitor (anti-let-7d) in the HCT116
cells, the downregulation of CST1, PCNA and p-p65 observed with the
CST1-siRNA was abrogated (Fig.
6D). In addition, we used Bay117082 (a NNF-κB p65 inhibitor) in
the HCT116 cells; it was not found to have any marked effect on
let-7d and CST1 expression (Fig.
6E). The results of immunohistochemistry confirmed that p-p65
expression was upregulated in the human colon cancer tissues
(Fig. 6F), compared with the
normal tissues. Thus, the above-mentioned data demonstrate that the
inhibitory effects of let-7d on colorectal cancer progression are
partly mediated by the targeting of CST1 via NNF-κB p65.
Discussion
Colon cancer is one of the most malignant types of
cancer; however, the molecular mechanisms that underlie the
development of colon cancer remain unclear. Let-7 miRNAs comprise
one of the largest and most highly expressed families of miRNAs,
possessing potent anti-carcinogenic properties in a variety of
tissues (12). Although a number
of studies have demonstrated that let-7d plays a pivotal role in
the initiation and development of several malignant neoplasms, the
association between let-7d and colon cancer has rarely been
reported. Madison et al reported that let-7d was negatively
associated with intestinal cancer and that let-7 suppressed
carcinogenesis and the stem cell phenotypic regulation of Hmga2
(26). The transcription factor,
NNF-κB, a master regulator of cell survival, inflammation and
immunity, has been shown to comprise a key link between
inflammation and cancer (27). The
association between let-7d and NNF-κB p65 has not yet been
reported, at least to the best of our knowledge; our study thus
focused on the mechanisms of action of let-7d in colon
carcinogenesis via NNF-κB p65.
Cysteine proteases, including cathepsins and papain
are proteolytic enzymes that are highly expressed in different
tissues and have been found to participate in several
pathophysiological procedures, such as tissue reconstitution,
inflammatory tissue damage, regulation of the immune response and
the migration of monocytes and cancer cells (28–30).
The cystatin (CST) superfamily function as inhibitors in the
proteolytic activity of cysteine proteases by constituting tight,
but reversible complexes (31,32).
Previous studies have revealed that cystatins exert a significant
effect on malignant tumor invasion and metastasis. The type 2
cystatin superfamily contains CST1, CST2, CST3, CST4, CST5, CSTP1,
CSTP2 and CST1 (CST SN).
Moreover, CST2 (CST SA) and CST4 (CST S) are known
as S-type cystatins. CST1 encoding CST SN is a member of type 2
cystatins, which are highly expressed in the uterus, submandibular
gland and gallbladder, but not in the colon, according to previous
findings (33). Our previous study
indicated that CST1 expression was highly elevated in pancreatic
cancer, contributing to pancreatic cancer cell proliferation, and
was a potential biomarker for the early detection of pancreatic
cancer (6). Previous studies have
found that CST1 plays a critical role in gastric cancer, colorectal
cancer and esophageal squamous cell carcinoma (6–9). An
elevated CST1 expression in colorectal cancer was found to
contribute to colorectal carcinogenesis by counteracting the
inhibitory effects of CST3 on the proteolytic activity of cathepsin
B (9,34). The association between let-7d and
the CST1/p65 pathway has not yet been reported, at least to the
best of our knowledge. Thus, our study focused on the mutual
regulation of let-7d and CST1 in colon cancer, with an aim to
explore a novel mechanism of tumor cell proliferation in colon
carcinogenesis.
In the current study, our microarrays comparing gene
expression in colon cancer and control tissues indicated that
CST1 was one of the most highly and conspicuously expressed
of the 22,000 genes on the array (Fig.
1) and we then verified CST1 mRNA and protein expression in
colon cancer tissues compared with non-cancer control tissues and
the results were consistent with those of the microarray. CST1 was
upregulated at both the mRNA and protein level (Fig. 3). We found that let-7d expression
was downregulated in the colon cancer tissues compared with the
non-cancer controls (Fig. 2A) and
it was the only miRNA that had an effect on CST1 expression when
using miRNA-mimics among the 5 miRNAs (miR15b, miR-145, miR-126,
miR-21 and let-7d) (Fig. 2B). We
further used let-7d-mimic to increase its expression in the
colorectal cancer cell lines, HCT116, SW480 and HT-29 (Fig. 4A). In addition, let-7d-mimic
was used to increase let-7d expression in the HCT-116 cells
(Fig. 4C), and we found that
let-7d overexpression suppressed the expression of
proliferation-associated proteins, such as PCNA, cyclin D1 and
cyclin E (Fig. 4B) suggesting that
let-7d may be involved in colorectal cancer cell proliferation. The
suppression of CST1 by CST1-siRNA in the HCT116 cells
in colony formation assays revealed that CST1 participates in
colorectal cancer cell proliferation (Fig. 5A and C). Consistent with this
finding, CST1 silencing decreased the expression of
malignancy-associated proteins, such as PCNA, cyclin D1 and cyclin
E (Fig. 5B and D). On the basis of
the results mentioned above, we hypothesized that maybe let-7d was
associated with CST1 and played a role in colon carcinogenesis.
However, we then wished to determine the pathway involved. We
further used let-7d mimic to increase its expression in the HCT116
cells, and found that CST1 expression decreased significantly
(Fig. 6A). A high expression of
let-7d inhibited NNF-κB p65 phosphorylation (Fig. 6B) and the silencing of CST1 also
inhibited NNF-κB p65 phosphorylation (Fig. 6C). We then silenced CST1, and also
used let-7d inhibitor in the HCT116 cells; the use of let-7d
inhibitor abrogated the suppressive effects of the silencing of
CST1 on CST1, PCNA and p-p65 expression (Fig. 6D). We also used Bay117082 (an
NNF-κB p65 inhibitor) in the HCT116 cells, and it found to have no
marked effect on let-7d and CST1 expression (Fig. 6E). Finally, our immunohistochemical
analysis confirmed that p-p65 expression was upregulated in the
human colon cancer tissues (Fig.
6F).
In conclusion, this study demonstrates that the
upregulation of CST1 in colorectal cancer contributes to cell
proliferation. Let-7d inhibits colorectal carcinogenesis through
the CST1/p65 pathway, and may thus be prove to be a potential
target for clinical therapy. The results of this study were
obtained from in vitro experiments. In the future, we aim to
examine the effects of let-7d on colorectal cancer proliferation
and growth in vivo in order to confirm the findings of this
study.
Abbreviations:
|
CST1
|
cystatin 1
|
|
miRNA or miR
|
microRNA
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
NNF-κB
|
nuclear factor-κB
|
Acknowledgments
The authors would like to express their gratitude
for the helpful comments on this manuscript received by the
reviewers.
Funding
This study was partly supported by grants from the
National Natural Science Foundation of China (nos. U1501224 and
81370511), and the Natural Science Foundation of Guangdong Province
(no. S2013010016541).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JJ and HLL were the major contributors in performing
the experiments and the writing of the manuscript. LT assisted in
performing the experiments. XYL provided technical assistance. YDY
and SWT provided assistance with the collection of the clinical
samples, as well as technical assistance. BW was involved in the
design of the study, and was also in charge of our laboratory. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board at The Third Affiliated Hospital of Sun Yat-sen University.
Written informed consent was obtained from each patient.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schwitalla S, Fingerle AA, Cammareri P,
Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ,
Moreaux G, et al: Intestinal tumorigenesis initiated by
dedifferentiation and acquisition of stem-cell-like properties.
Cell. 152:25–38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li G, Ji XD, Gao H, Zhao JS, Xu JF, Sun
ZJ, Deng YZ, Shi S, Feng YX, Zhu YQ, et al: EphB3 suppresses
non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt
signalling complex. Nat Commun. 3:6672012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hessmann E, Zhang JS, Chen NM, Hasselluhn
M, Liou GY, Storz P, Ellenrieder V, Billadeau DD and Koenig A:
NFATc4 regulates Sox9 gene expression in acinar cell plasticity and
pancreatic cancer initiation. Stem Cells Int. 2016:52724982016.
View Article : Google Scholar
|
|
5
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jiang J, Liu HL, Liu ZH, Tan SW and Wu B:
Identification of cystatin SN as a novel biomarker for pancreatic
cancer. Tumour Biol. 36:3903–3910. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi EH, Kim JT, Kim JH, Kim SY, Song EY,
Kim JW, Kim SY, Yeom YI, Kim IH and Lee HG: Upregulation of the
cysteine protease inhibitor, cystatin SN, contributes to cell
proliferation and cathepsin inhibition in gastric cancer. Clin Chim
Acta. 406:45–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen YF, Ma G, Cao X, Luo RZ, He LR, He
JH, Huang ZL, Zeng MS and Wen ZS: Overexpression of cystatin SN
positively affects survival of patients with surgically resected
esophageal squamous cell carcinoma. BMC Surg. 13:152013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yoneda K, Iida H, Endo H, Hosono K,
Akiyama T, Takahashi H, Inamori M, Abe Y, Yoneda M, Fujita K, et
al: Identification of Cystatin SN as a novel tumor marker for
colorectal cancer. Int J Oncol. 35:33–40. 2009.PubMed/NCBI
|
|
10
|
Kumar MS, Pester RE, Chen CY, Lane K, Chin
C, Lu J, Kirsch DG, Golub TR and Jacks T: Dicer1 functions as a
haploinsufficient tumor suppressor. Genes Dev. 23:2700–2704. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lambertz I, Nittner D, Mestdagh P,
Denecker G, Vandesompele J, Dyer MA and Marine JC: Monoallelic but
not biallelic loss of Dicer1 promotes tumorigenesis in vivo. Cell
Death Differ. 17:633–641. 2010. View Article : Google Scholar
|
|
12
|
Büssing I, Slack FJ and Grosshans H: let-7
microRNAs in development, stem cells and cancer. Trends Mol Med.
14:400–409. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HMGA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Boyerinas B, Park SM, Shomron N, Hedegaard
MM, Vinther J, Andersen JS, Feig C, Xu J, Burge CB and Peter ME:
Identification of let-7-regulated oncofetal genes. Cancer Res.
68:2587–2591. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gurtan AM, Ravi A, Rahl PB, Bosson AD,
JnBaptiste CK, Bhutkar A, Whittaker CA, Young RA and Sharp PA:
Let-7 represses Nr6a1 and a mid-gestation developmental program in
adult fibroblasts. Genes Dev. 27:941–954. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiao LR, Frampton AE, Jacob J, Pellegrino
L, Krell J, Giamas G, Tsim N, Vlavianos P, Cohen P, Ahmad R, et al:
MicroRNAs targeting oncogenes are down-regulated in pancreatic
malignant transformation from benign tumors. PLoS One.
7:e320682012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Volinia S, Galasso M, Sana ME, Wise TF,
Palatini J, Huebner K and Croce CM: Breast cancer signatures for
invasiveness and prognosis defined by deep sequencing of microRNA.
Proc Natl Acad Sci USA. 109:3024–3029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang CJ, Hsu CC, Chang CH, Tsai LL, Chang
YC, Lu SW, Yu CH, Huang HS, Wang JJ, Tsai CH, et al: Let-7d
functions as novel regulator of epithelial-mesenchymal transition
and chemoresistant property in oral cancer. Oncol Rep.
26:1003–1010. 2011.PubMed/NCBI
|
|
19
|
Ramberg H, Alshbib A, Berge V, Svindland A
and Taskén KA: Regulation of PBX3 expression by androgen and Let-7d
in prostate cancer. Mol Cancer. 10:502011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Childs G, Fazzari M, Kung G, Kawachi N,
Brandwein-Gensler M, McLemore M, Chen Q, Burk RD, Smith RV,
Prystowsky MB, et al: Low-level expression of microRNAs let-7d and
miR-205 are prognostic markers of head and neck squamous cell
carcinoma. Am J Pathol. 174:736–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tezcan G, Tunca B, Bekar A, Yalcin M,
Sahin S, Budak F, Cecener G, Egeli U, Demir C, Guvenc G, et al:
Ficus carica latex prevents invasion through induction of let-7d
expression in GBM cell lines. Cell Mol Neurobiol. 35:175–187. 2015.
View Article : Google Scholar
|
|
22
|
Nuovo GJ, Garofalo M, Valeri N, Roulstone
V, Volinia S, Cohn DE, Phelps M, Harrington KJ, Vile R, Melcher A,
et al: Reovirus-associated reduction of microRNA-let-7d is related
to the increased apoptotic death of cancer cells in clinical
samples. Mod Pathol. 25:1333–1344. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ivo D'Urso P, Fernando D'Urso O, Damiano
Gianfreda C, Mezzolla V, Storelli C and Marsigliante S: miR-15b and
miR-21 as circulating biomarkers for diagnosis of glioma. Curr
Genomics. 16:304–311. 2015. View Article : Google Scholar
|
|
24
|
Yu Y, Nangia-Makker P, Farhana L, G
Rajendra S, Levi E and Majumdar AP: miR-21 and miR-145 cooperation
in regulation of colon cancer stem cells. Mol Cancer. 14:982015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang W, Lin J and Zhang H: miR-126: A
novel regulator in colon cancer. Biomed Rep. 4:131–134. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Madison BB, Jeganathan AN, Mizuno R,
Winslow MM, Castells A, Cuatrecasas M and Rustgi AK: Let-7
represses carcinogenesis and a stem cell phenotype in the intestine
via regulation of Hmga2. PLoS Genet. 11:e10054082015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin M and Greten FR: NNF-kappaB: Linking
inflammation and immunity to cancer development and progression.
Nat Rev Immunol. 5:749–759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lah TT, Babnik J, Schiffmann E, Turk V and
Skaleric U: Cysteine proteinases and inhibitors in inflammation:
Their role in periodontal disease. J Periodontol. 64(Suppl):
485–491. 1993.PubMed/NCBI
|
|
29
|
Travis J and Potempa J: Bacterial
proteinases as targets for the development of second-generation
antibiotics. Biochim Biophys Acta. 1477:35–50. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koblinski JE, Ahram M and Sloane BF:
Unraveling the role of proteases in cancer. Clin Chim Acta.
291:113–135. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barrett AJ: The cystatins: A diverse
superfamily of cysteine peptidase inhibitors. Biomed Biochim Acta.
45:1363–1374. 1986.PubMed/NCBI
|
|
32
|
Lindahl P, Abrahamson M and Björk I:
Interaction of recombinant human cystatin C with the cysteine
proteinases papain and actinidin. Biochem J. 281:49–55. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dickinson DP, Thiesse M, Dempsey LD and
Millar SJ: Genomic cloning, physical mapping, and expression of
human type 2 cystatin genes. Crit Rev Oral Biol Med. 4:573–580.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim JT, Lee SJ, Kang MA, Park JE, Kim BY,
Yoon DY, Yang Y, Lee CH, Yeom YI, Choe YK, et al: Cystatin SN
neutralizes the inhibitory effect of cystatin C on cathepsin B
activity. Cell Death Dis. 4:e9742013. View Article : Google Scholar : PubMed/NCBI
|