Introduction
Lung cancer is the leading cause of cancer-related
mortality and poses a highly significant risk to human health
(1). Non-small cell lung cancer
(NSCLC) accounts for almost 80% of all cases of lung cancer
(2). Despite improvements being
made in the diagnosis and therapy of NSCLC in recent years, the
overall 5-year survive rate of patients with NSCLC is 10-20%
(3). The development and the
molecular pathogenesis of NSCLC is a multi-stage process and is
associated with multiple factors, such as oncogene activation or
tumor suppression gene inactivation (4). Thus, it is crucial to explore the
molecular mechanisms of action of the genes associated with the
diagnosis and therapy of NSCLC.
Wnt inhibitory factor-1 (WIF-1) is an important
antagonist of Wnt/β-catenin signaling by binding to Wnt ligands in
verte-brate cells and is also considered a tumor suppressor
(5,6). It is known that Wnt/β-catenin
signaling plays an essential role in regulating embryonic
development and homeostasis in adult tissues (7). However, abnormal Wnt/β-catenin
signaling has been detected in the majority of NSCLC cells (69%)
and the downregulation of antagonists is a main cause of
Wnt/β-catenin signaling activation in NSCLC (8,9).
Researchers have demonstrated that the promoter hypermethylation of
the WIF-1 gene is silenced in 75% of NSCLC cases (10-12).
WIF-1 downregulation is associated with the diagnosis and a poor
prognosis of NSCLC (13,14). Moreover, the promoter demethylation
of the WIF-1 gene by epigallocatechin-3-gallate has been
shown to significantly inhibit the proliferation of NSCLC cells
(15). Transfection with
WIF-1 overexpression vector has also been shown to inhibit
colony formation and tumor growth, and increase the apoptosis of
NSCLC (16). Therefore, it can be
recognized that WIF-1, as an important antagonist of Wnt/β-catenin
signaling, is a promising therapeutic target in NSCLC. To date,
WIF-1 has been shown to inhibit Wnt down-stream members (such as
β-catenin, cyclin D1 and c-Myc) or inhibit the transcription
factors, LEF/TCF, in various types of cancer, including NSCLC
(15-19). However, the detailed mechanisms
responsible for the inhibition of Wnt/β-catenin signaling by WIF-1
have rarely been explored.
In our preliminary study (data not shown), numerous
red acid vesicles were detected by acridine orange staining in A549
cells transfected with WIF-1 overexpression vector. We
suspected these acid vesicles may be autophagic lysosomes.
Recently, Gao et al (20)
revealed that autophagy regulated Wnt/β-catenin signaling by
negatively regulating Dvl2 in vertebra embryonic cells and HeLa
cells. However, the association between autophagy and Wnt/β-catenin
signaling in NSCLC cells remains unknown. Thus, we hypothesized
that WIF-1 may induce autophagy and inhibit Wnt/β-catenin signaling
in NSCLC. In this study, we first detected the autophagy induced by
WIF-1 in two NSCLC cell lines. Subsequently, the effects of
WIF-1-mediated autophagy on the proliferation and apoptosis of
NSCLC cells were evaluated by blocking autophagy. Furthermore, the
mechanisms underlying the antitumor effects of WIF-1-mediated
autophagy were investigated. Finally, the effects of the
overexpression of the WIF-1 gene combined with treatment
with the autophagy agonist, everolimus (RAD001) against NSCLC cells
were evaluated in an A549 subcutaneous tumor xenograft model and a
pulmonary metastasis tumor model. The results first revealed a
novel mechanism through which WIF-1 inhibited Wnt/β-catenin
signaling by inducing autophagy in NSCLC. This study may also
provide a theoretical basis for the joint therapy of NSCLC with
WIF-1 and autophagic agonists in clinical practice.
Materials and methods
Mice and cell lines
A total of 66 female BALB/c nude mice (3–5 weeks
old, weighing approximately 10 g at 3 weeks and 13 g at 5 weeks)
were purchased from Beijing HFK Bioscience Co., Ltd. (Beijing,
China), and fed in a specific pathogen-free environment with a
temperature of approximately 25°C and 60% relative humidity, and
free access to food and water. All procedures were approved by the
Institute of Laboratory Animal Care and Use Committee at Sichuan
University (Chengdu, China).
The NSCLC cell lines, A549 and NCI-H460 (termed
H460), were originally obtained from the American Type Culture
Collection (ATCC, Manassas, VA, USA), and cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (both from Life
Technologies, Gaithersburg, MD, USA) at 37°C in atmosphere
containing 5% CO2.
Plasmid construction and cell
treatment
The full length cDNA of WIF-1 gene was cloned from
pBluescriptR-WIF-1 plasmid (Open Biosystems, Huntsville, AL, USA)
with a PrimeSTAR™ HS PCR kit (Takara, Dalian, China) according to
the WIF-1 cDNA coding sequence (GeneBank serial no. BC018037.1),
and subcloned into the HindIII-XbaI sites of the pVAX
vector (Invitrogen, Grand Island, NY, USA) to generate the
pVAX-WIF-1 plasmid with confirmed sequence and orientation.
The A549 and H460 cells were plated in 6-well plates and allowed to
attach by overnight incubation at 37°C. When 70-80% confluent the
cells were transfected with 2 µg of the pVAX-WIF-1 or
pVAX vector (control) using Lipofectamine 2000 (Invitrogen)
according to the manufacturer's instruction. The cells not be
treated were used as blank group. The expression of WIF-1 was
assessed by western blot analysis at 24 h following transfection.
The autophagy inhibitor, 3-methyladenine (3-MA; 1 mM; Sigma, St.
Louis, MO, USA), was used to inhibit WIF-1-mediated autophagy in
NSCLC cell lines. At 15 h following transfection, the cells were
treated with 3-MA for 1 h.
Detection of acidic vesicular organelles
(AVOs)
The cells were plated on coverslips in 6-well plates
and transfected as described above. After 24 h, the cells were
stained with 1 µg/ml acridine orange in PBS for 15 min,
washed with PBS and examined under a fluorescence microscope
(Olympus, Hamburg, Germany).
Transmission electron microscopy
(TEM)
At 24 h following transfection as described above,
the cells were harvested and fixed in 0.1% glutaraldehyde in 0.1 M
Na-cacodylate for 2 h, post-fixed with 1% OsO4 for 1.5 h and washed
with 0.1 M phosphoric acid. Finally, the samples were stained for 1
h in 3% aqueous uranyl acetate and lead citrate before they were
observed under a transmission electron microscope (Hitachi, Tokyo,
Japan) at 80 kV.
GFP-LC3 transient transfection
The cells were co-transfected with the EGFP-LC3
plasmid (referred to as GFP-LC3) and the pVAX-WIF-1 or
control vector using Lipofectamine 2000 (Invitrogen) according to
the manufacturer's instructions. At 24 h after transfection, the
fluorescence of GFP-LC3 was viewed and the rate of GFP-LC3-labeled
vacuole formation (autophagosomes) was counted under a fluorescence
micro-scope (Olympus).
Cell viability assay
The cells were seeded in 96-well culture plates. At
24 h following incubation at 37°C, the cells were transfected with
pVAX-WIF-1 or the control vector and then cultured for 48 h.
Cell viability was evaluated by 3-(4,5)-dimethylthiahi
azo(-z-y1)-3,5-di-phenytetrazoliumromide (MTT; Sigma) assay. The
absorbance was measured at 490 nm was measured using a microplate
reader (Bio-Rad, Hercules, CA, USA).
Hoechst 33258 staining assay
The cells were cultured in a 6-well plate and
transfected as described above. Before staining, the cells were
washed with PBS once and fixed by 4% fresh prepared
paraformaldehyde for 15 min. Subsequently Hoechst 33258 (Sigma)
diluted in PBS was added into each well for 10 min, followed by
washing with PBS for 5 min twice; the blue-stained nuclei were
observed under a fluorescence microscope (Olympus) immediately.
Flow cytometry assay
At 2 days following transfection as described above,
the cells were harvested following treatment with trypsin and then
stained using the Annexin V-FITC Apoptosis Detection kit (KeyGEN,
Nanjing, China) according to the manufacturer's instructions. The
stained cells were immediately analyzed using a FACScan flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
Briefly, the NSCLC cells subjected to the different
treatments were collected and lysed in RIPA buffer supplemented
with protease inhibitor cocktail Set I (Merck, Darmstadt, Germany).
The protein concentration was then determined by BCA (Pierce,
Waltham, MA, USA) ccording to the manufacturer's instructions.
Subsequently, 10 µg lysate proteins were separated by
electrophoresis on 10% SDS-polyacrylamide gels, and transferred
onto polyvi-nylidene fluoride membranes (Millipore, Bedford, MA,
USA). After blocking with 5% non-fat milk in Tris-buffered saline
containing 0.05% Tween-20, the membranes were incubated with the
primary antibodies against WIF-1 (1:1,000, #5652), Dvl2 (1:1,000,
#3224), β-catenin (1:1,000, #8480), cyclin D1 (1:1,000, #2922),
phosphoinositide 3-kinase (PI3K)(p110α) (1:200, #4255), p-AKT(S473)
(1:500, #4051, phosphorylated mammalian target of rapamycin
p-mTOR(ser2448) (1:500, #2971), β-actin(1:2000, #3700) (all from
Cell Signaling Technology, Beverly, MA, USA), AKT (1:2,000,
16-294), mTOR (1:2,000, 05-1592) (both from Millipore) at 4°C
overnight. The same isotypes of primary antibodies were then
incubated with HRP-conjugated secondary antibodies (1:2,000, #7076
and #7074; Cell Signaling Technology) for 2 h at room temperature.
The protein bands were detected with the ECA system (Millipore).
The grayscale of the protein bands was analyzed by Gel-Pro Analyzer
4.0. The level of each protein was normalized to β-actin.
Subcutaneous tumor xenografts
The A549 cells (5×106/100 µl) were
injected into the flanks of 48 female nu/nu mice, 5 weeks old, to
generate subcutaneous xenografts. At 1 week after the injection,
subcutaneous tumor volumes (V) were measured with digital calipers
(Thermo Fisher Scientific, Waltham, MA, USA) and calculated using
the following formula: V (mm3) = 0.52 × length (mm) ×
width2 (mm2). Treatment was initiated when
the mean tumor volume had reached approximately 100 mm3.
The mice were randomly divided into 6 groups (n=8 each) as follows:
5% Glc, pVAX, pVAX-WIF-1 (0.2 mg/kg), RAD001 (3 mg/kg; MCE
Technologies, San Dimas, CA, USA), pVAX + RAD001 and
pVAX-WIF-1 + RAD001, and received an intratumoral injection
of 5% Glc or pVAX or pVAX-WIF-1, or were administered RAD001
by gavage. All the treatments were administered every 2 days for a
total of 6 times. The pVAX-WIF-1 or pVAX plasmid was
transfected via a cationic liposome complex prepared by our
laboratory as previously described (21). At 2 days after the final dose, 3
mice were selected randomly from each group and anesthetized by
diethyl ether inhalation and rapidly sacrificed by cervical
dislocation. Then the tumor tissues were harvested. Parts of the
tissue were frozen in liquid nitrogen and stored at -80°C for
protein isolation and other parts were fixed in 4% paraformaldehyde
and embedded in paraffin for histological sections. In the
remaining mice, the size of the tumors was measured with calipers
every 3 days until the average tumor volume of the control group
reached approximately 1,000 mm3 or the tumors were
necrotic. The tumor tissues were removed for photographing. The
relative tumor growth ratio was calculated by the change in tumor
volumes with the designed treatment relative to that of the 5% Glc
control group.
Pulmonary metastasis tumor model
The A549 cells (2×106/200 µl) were
injected via the caudal vein into 18 female athymic nude mice, 3
weeks old. Two weeks after the cell injection, mice were randomly
divided into 6 groups (n=3 each) as follows: 5% Glc, pVAX,
pVAX-WIF-1 (0.2 mg/kg), RAD001 (3 mg/kg), pVAX + RAD001 and
pVAX-WIF-1 + RAD001, and received a caudal vein injection of
5% Glc or pVAX or pVAX-WIF-1 or RAD001 (by gavage). All
treatments were administered every 2 days for a total of 6 times.
The plasmid was transfected via a cationic liposome complex
prepared by our laboratory, as previously described (21). At 2 weeks after the final
treatment, the mice were injected intratracheally with India ink
and the lungs were fixed in AAF solution (85% ethanol, 10% acetic
acid and 5% formalin) to count the number of metastatic tumor
nodules (white dots) on the surfaces.
Immunohistochemistry
The tumor tissue samples were embedded in paraffin
and cut into 4-µm-thick sections. The slides were then
subjected to standard histological analysis. Cell proliferation in
the subcutaneous tumors was assessed by staining with primary
antibody against Ki-67 (1:500; #9027; Cell Signaling Technology).
Bright field images of all stained tissues were viewed under a
microscope (Olympus).
In situ TUNEL assay
To determine apoptosis in the subcutaneous tumor
after the different treatments, the sections of tumor tissue were
subjected to TUNEL assay using an In situ Cell Death
Detection kit (Promega, Madison, WI, USA).
Statistical analysis
To analyze the synergistic effects of WIF-1 and
RAD001 in this study, the synergistic index (SI) of
pVAX-WIF-1 and RAD001 in each experiment was calculated by
the Relative Ratio of Data (RRD) in each group of the experiment
according to a method described previously (22). Interactions between WIF-1 and
RAD001 were considered as synergism when the SI was >1.
Statistical analysis was performed by SPSS 19.0 software and
GraphPad 5.0 software. All quantitative data are presented as the
means ± standard deviation (± SD). The measurement data were tested
by one-way ANOVA with Fishers' Least Significance Difference (LSD)
as a post hoc. Statistical significance was defined as P<0.05.
Microsoft word 2010 software was used to sketch the possible
mechanism of WIF-1-mediated autophagy in Wnt/β-catenin signaling
inhibition.
Results
Overexpression of WIF-1 induces
autophagosome formation in NSCLC cells
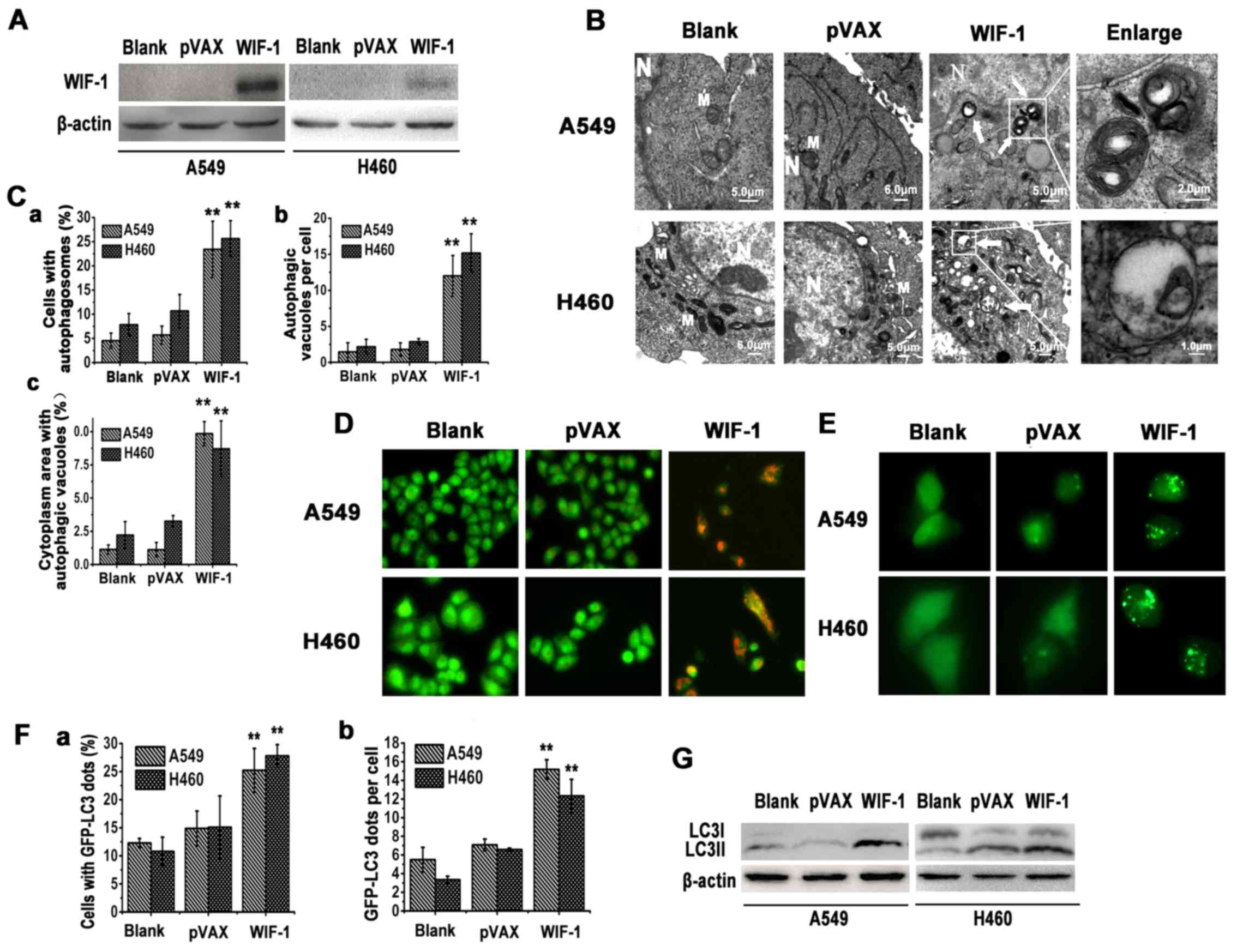
To evaluate whether WIF-1 induces autophagy in NSCLC
cells, we first analyzed the expression of WIF-1 in the A549 and
H460 cells transfected with pVAX-WIF-1 or the control vector
by western blot analysis (Fig.
1A). The ultrastructure of the A549 and H460 cells was analyzed
by TEM at 24 h after transfection. Membrane-bound vacuoles were
observed in the cytoplasm, whereas rarely in the control vector
(pVAX) or blank group (Fig. 1B and
C). The membrane-bound vacuoles were analyzed by acridine
orange staining. As shown in Fig.
1D, the A549 and H460 cells transfected with the WIF-1
gene overexpression vector exhibited the formation of yellow-orange
AVOs. By contrast, cells transfected with the control vector (pVAX)
or blank generally exhibited green fluorescence.
LC3-II (16 kDa), localized on the membrane of
autophagosomes, is considered a marker of autophagy (23). In this study, in order to evaluate
the recruitment of LC3-II to autophagosomes following transfection
with the WIF-1 gene overexpression vector, the appearance of
a punctate GFP-LC3 signal was examined in the A549 and H460 cells.
Fluorescence microscopy revealed that punctate GFP-LC3 staining was
observed in the cytoplasm, while only diffuse LC3-associated green
fluorescence was observed in the control vector or blank groups
(Fig. 1E and F). Furthermore, an
immunoblotting-based LC3 flux assay was performed to monitor the
alteration of the WIF-1-mediated autophagic flux. LC3-II protein
was detectable in the cells transfected with the WIF-1 gene
overexpression vector, whereas this was less detectable in the
controls (Fig. 1G). Collectively,
these findings suggested that WIF-1 induced autophagy in NSCLC
cells.
Blocking of autophagy attenuates the
inhibitory effects on NSCLC cell proliferation mediated by
WIF-1
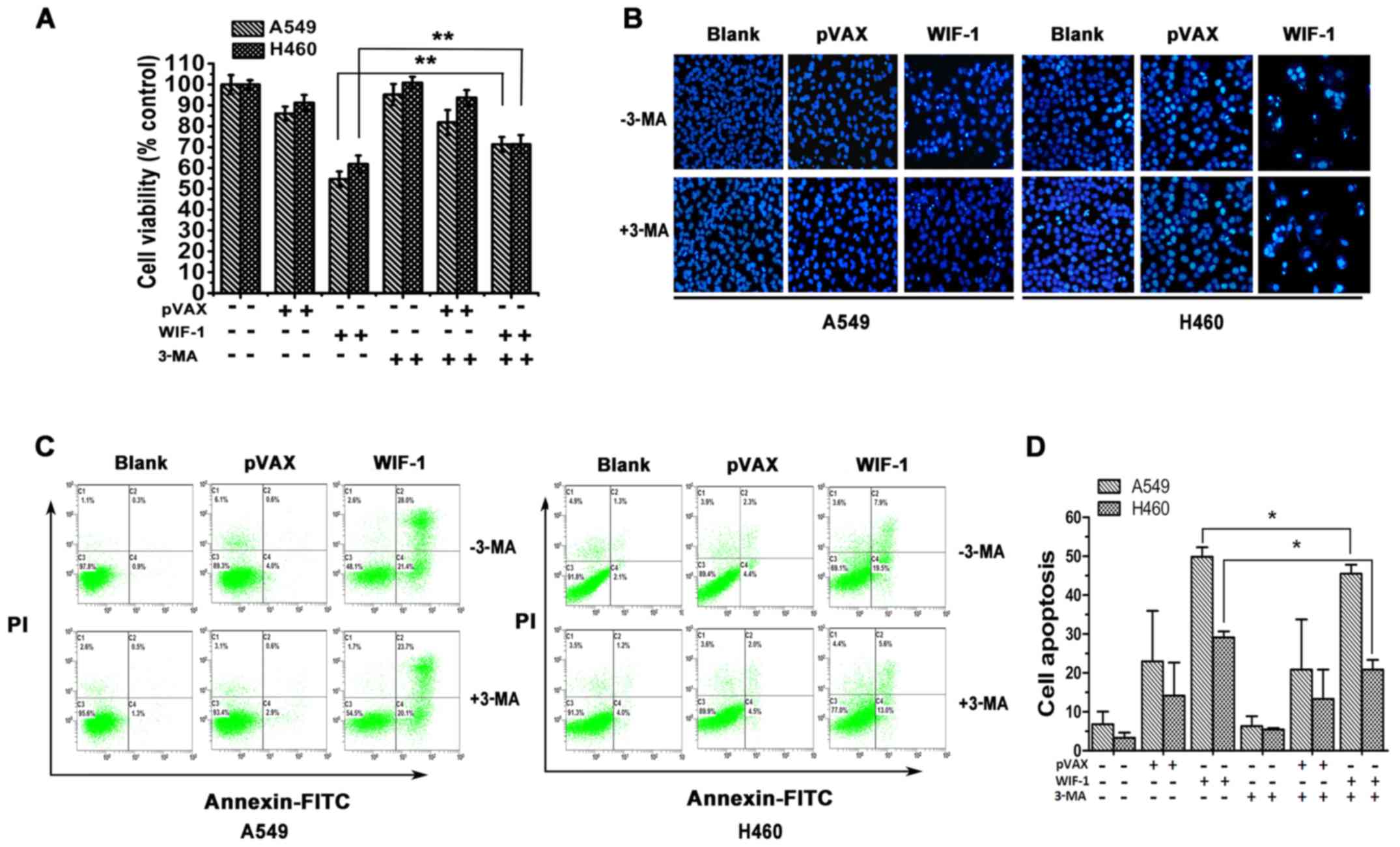
To examine the effects of WIF-1-mediated autophagy
on the proliferation of NSCLC cells, 3-MA, a common autophagy
inhibitor, was utilized to block autophagy. MTT analysis
demonstrated that WIF-1 significantly inhibited the proliferation
of the A549 and H460 cells transfected with the WIF-1 gene
over-expression vector compared with the controls. However, the
WIF-1-mediated inhibition of cell proliferation was markedly
attenuated in the presence of 3-MA, compared with the cells not
treated with 3-MA (P<0.01, Fig.
2A). This result indicated that WIF-1-mediated autophagy plays
a suppressive role against the proliferation of NSCLC cells.
Blocking autophagy attenuates the
apoptosis of NSCLC cells induced by WIF-1
To examine the effects of WIF-1-mediated autophagy
on the apoptosis of NSCLC cells, the A549 and H460 cells
transfected with pVAX-WIF-1 were stained with Hoechst 33258. The
results revealed that apoptotic bodies were evident in the cells
transfected with the WIF-1 gene overexpression vector, while
these were barely visible in the control vector or blank group.
However, the blocking of autophagy with 3-MA diminished the
formation of WIF-1-mediated apoptotic bodies (Fig. 2B). Furthermore, the ratio of
apoptosis was assessed by flow cytometry. As shown in Fig. 2C and D, the apoptotic rates of the
A549 and H460 cells transfected with the WIF-1 gene
overexpression vector were 49.87±2.47 and 29.10±1.57%,
respectively. However, following treatment with 3-MA (WIF-1 + 3-MA
group), these rates decreased to 45.50±2.33 and 20.91±2.46%,
respectively. A statistically significant difference was observed
between the WIF-1 and WIF-1 + 3-MA group (P<0.05). However, no
significant differences were observed between the blank and blank +
3-MA group, neither between the vector and vector + 3-MA group
(P>0.05). These results certified that WIF-1-mediated autophagy
contributed to the apoptosis of NSCLC cells.
WIF-1-mediated autophagy inhibits
Wnt/β-catenin signaling in NSCLC cells
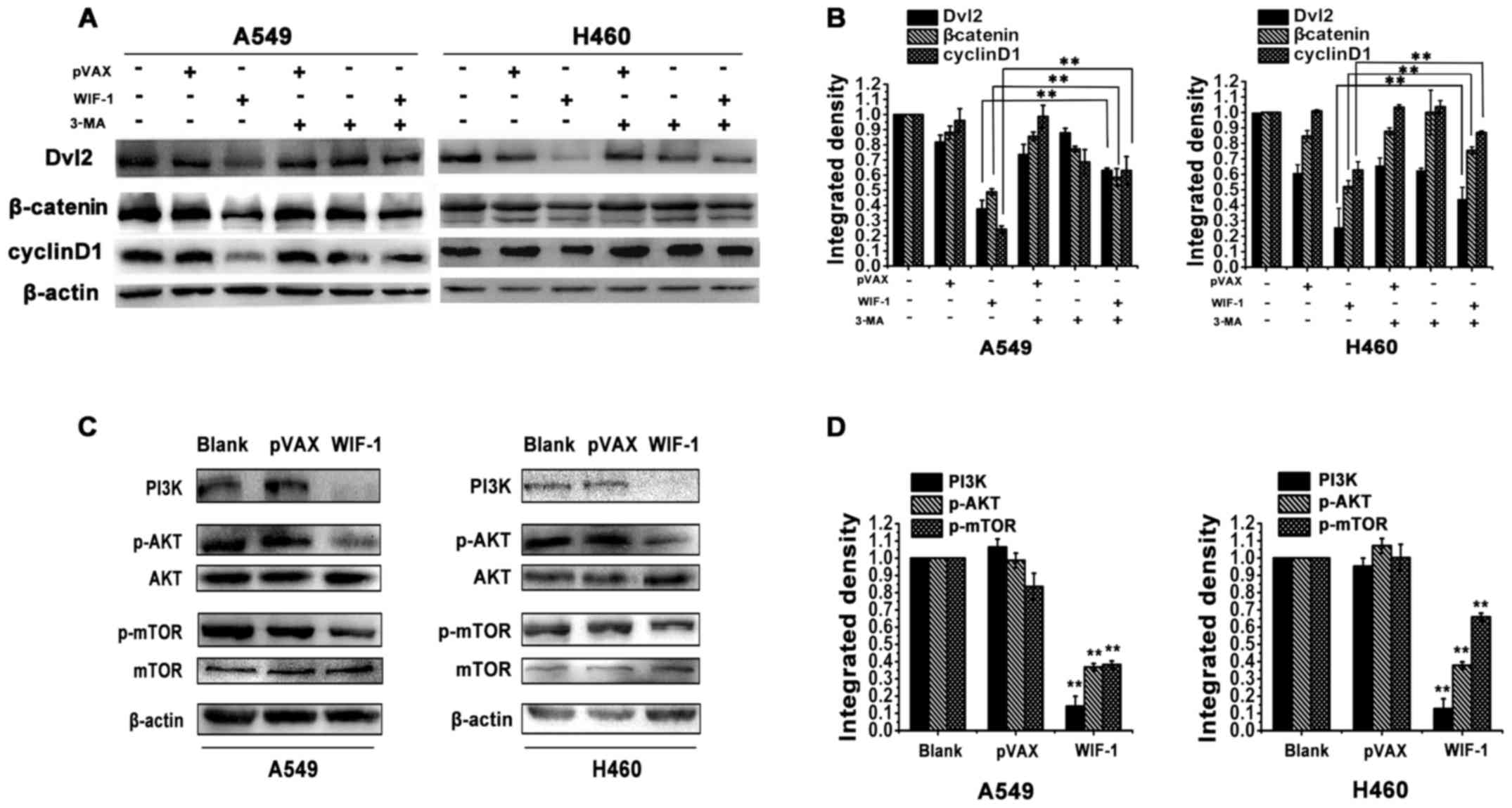
A previous study revealed that autophagy regulated
Wnt/β-catenin signaling to control cell physiological functions by
degrading Dvl2 in embryonic and HeLa cells (20). In this study, we examined whether
WIF-1-mediated autophagy inhibits Wnt/β-catenin signaling through
Dvl2 in NSCLC cells. Western blot analysis revealed a notable
decrease in Dvl2 expression in the A549 and H460 cells transfected
with the WIF-1 gene overexpression vector compared with the
control cells. However, the blocking of autophagy with 3-MA
markedly increased the protein level of Dvl2 in the cells
transfected with the WIF-1 gene overexpression vector
(Fig. 3A and B). β-catenin and
cyclin D1 are the downstream members of Wnt/β-catenin signaling
(6). The results of western blot
analysis also revealed that the β-catenin and cyclin D1 expression
levels were decreased in the cells transfected with the
WIF-1 gene overexpression vector. However, the decrease in
the levels of β-catenin and cyclin D1 was significantly reversed in
the presence of 3-MA (Fig. 3A and
B). These results indicated that WIF-1 induced autophagy to
inhibit Wnt/β-catenin signaling through Dvl2 in NSCLC cells.
The PI3K/Akt/mTOR pathway is involved in
WIF-1-mediated autophagy in NSCLC cells
mTOR is the gating mechanism of autophagy and the
PI3K/Akt pathway is an important upstream regulator of mTOR
(24). As shown in Fig. 3C and D, transfection with the
WIF-1 gene overexpression vector significantly inhibited the
phosphorylation of both mTOR and Akt in the A549 and H460 cells,
compared with the control cells. Moreover, PI3K expression was
markedly decreased following WIF-1 overexpression (Fig. 3C and D). This result inferred that
the mechanism of autophagy induction by WIF-1 may be related to
PI3K/Akt/mTOR pathway inhibition in NSCLC cells.
Combination treatment with WIF-1 and the
autophagy agonist enhances the tumor growth inhibitory effects of
WIF-1 in vivo
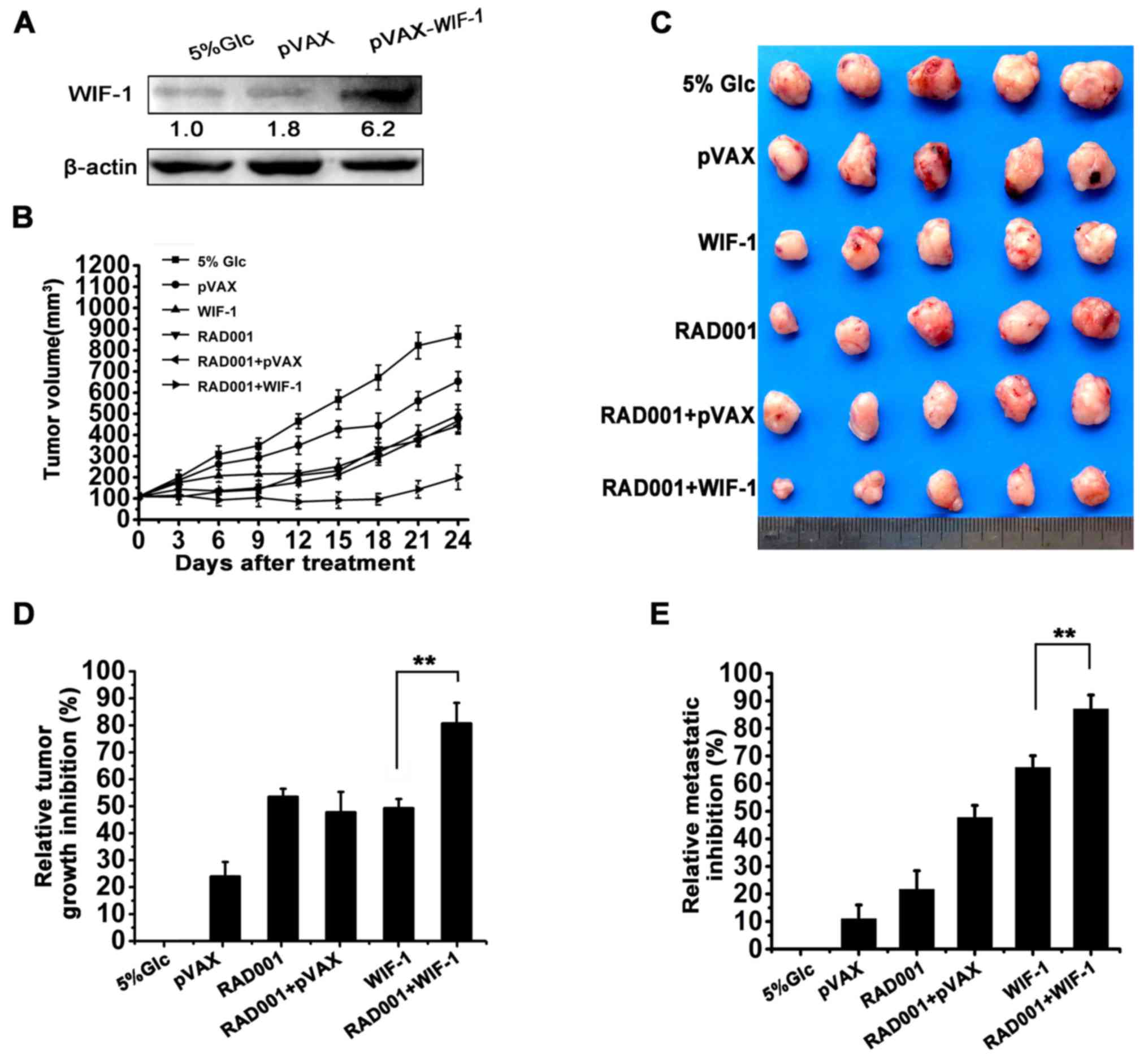
Since WIF-1-mediated autophagy was demonstrated to
contribute to the inhibition of cell proliferation, the effects of
combination treatment with pVAX-WIF-1 and RAD001 were
evaluated in a subcutaneous tumor model and pulmonary metastasis
tumor model, respectively. The protein expression level of WIF-1 in
the subcutaneous tumors was upregulated with pVAX-WIF-1
treatment (Fig. 4A). As shown in
Fig. 4B–D, WIF-1 or RAD001
individual treatment inhibited subcutaneous tumor growth with a
50.41 and 54.72% reduction, respectively, compared to treatment
with 5% GLC treatment (P<0.01). Notably however, combination
treatment with WIF-1 and RAD001 exerted synergistic effects, and an
82.58% decrease was observed (Table
I, SI >1). However, systemic administration delivery is a
more effective and practical method for lung cancer treatment
(25). Considering the clinical
perspective, a model of pulmonary metastasis was established in
mice which were treated with pVAX-WIF-1 by tail vein
injection. As shown in Fig. 4E,
combination treatment with WIF-1 by systemic administration or
RAD001 by gavage independently resulted in a lung metastatic tumor
nodule reduction rate of 66 and 22%, respectively, compared with
the 5% GLC control group (P<0.001). However, a synergistic lung
metastatic tumor nodules reduction (87.06%) was observed following
combined treatment with WIF-1 and RAD001 (P<0.001, Fig. 4E; Table I, SI >1). These results
indicated that combination treatment with WIF-1 and RAD001 in NSCLC
enhanced the antitumor effects in vivo.
 | Table ISynergistic indices of combination
treatment with WIF-1 and RAD001 calculated by the relative ratio of
data (RRD)a in each experiment. |
Table I
Synergistic indices of combination
treatment with WIF-1 and RAD001 calculated by the relative ratio of
data (RRD)a in each experiment.
| Experiment | WIF-1 groupa | RAD001
groupa | Combination
treatment with WIF-1 and RAD001 group
|
|---|
| Expectb | Observed | Ratio (SI)c |
|---|
| Lung cancer
metastasis model | | | | | |
| Number of tumor
nodules | 0.35 | 0.79 | 0.27 | 0.13 | 2.07 |
| Lung cancer
subcutaneous model | | | | | |
| Tumor volume | 0.50 | 0.45 | 0.23 | 0.17 | 1.35 |
| Cell
proliferation | 0.50 | 0.57 | 0.29 | 0.18 | 1.61 |
| TUNEL assay | 10.11 | 2.26 | 22.85 | 13.36 | 1.71 |
Combination treatment with WIF-1 and
RAD001 results the inhibition of proliferation and the promotion of
apoptosis in vivo
To investigate the mechanisms involved in the
effects of combination treatment with WIF-1 and RAD001 in lung
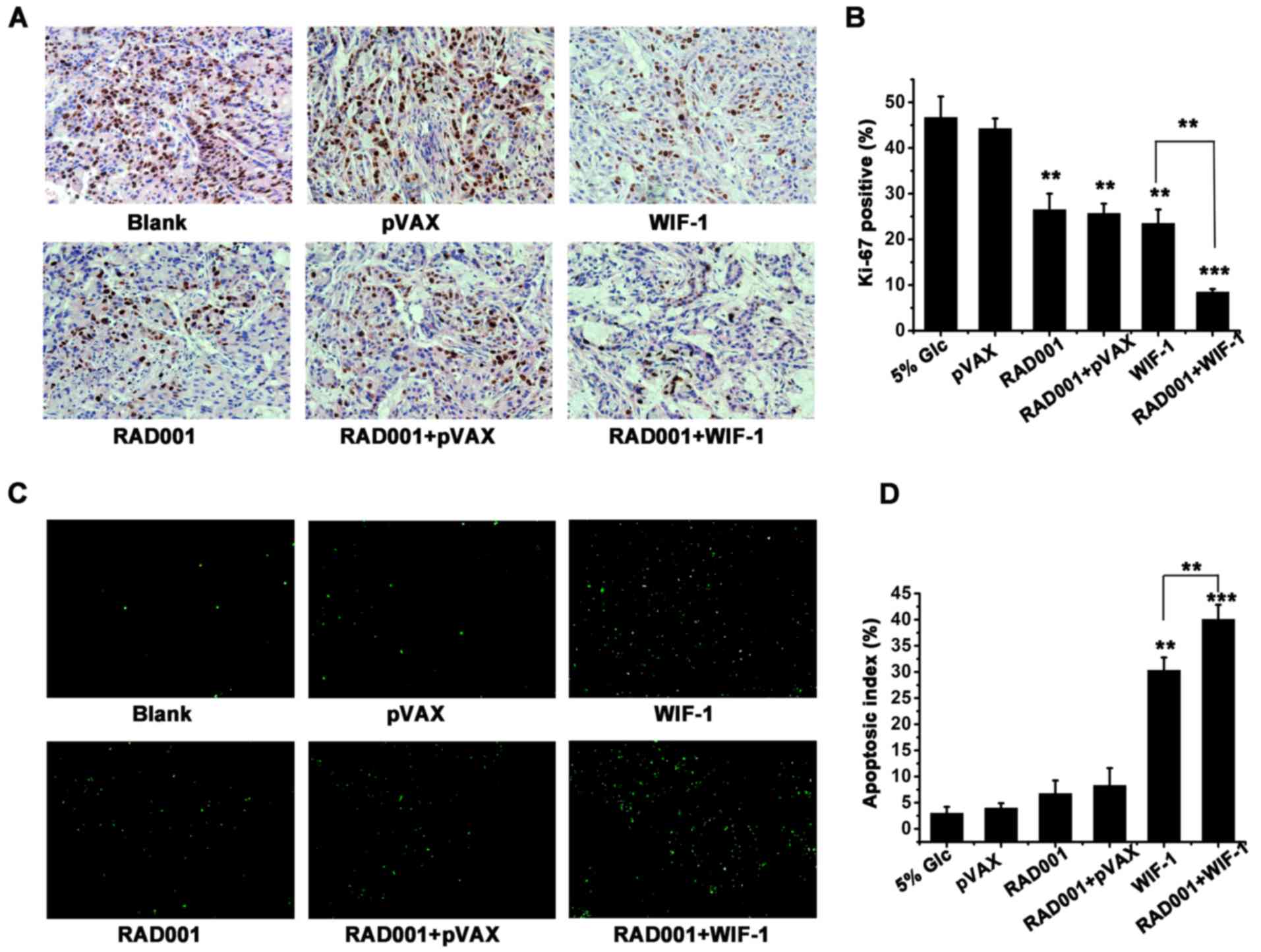
cancer, Ki-67 staining and TUNEL assay were carried out to examine
the tumor tissues excised from mice subjected to the different
treatments. The analysis of the Ki-67 index revealed that the
combination treatment was clearly more potent in the inhibition of
tumor cell proliferation compared to either individual treatment
(WIF-1 or RAD001) (P<0.01, Fig. 5A
and B). Furthermore, a high apoptotic rate was detected in the
WIF-1 treatment group (30.34%), but was seldom detected in the
RAD001 group. Moreover, the induction of apoptosis (40%) was
apparently increased by combination treatment with WIF-1 and RAD001
compared with the other groups (Fig.
5C and D). Both the inhibition of proliferation and the
promotion of apoptosis following combination treatment produced a
synergistic effect (Table I, SI
>1). It was thus suggested that combination treatment with WIF-1
and RAD001 enhanced the antitumor effects of WIF-1 by inhibiting
proliferation and promoting apoptosis in vivo.
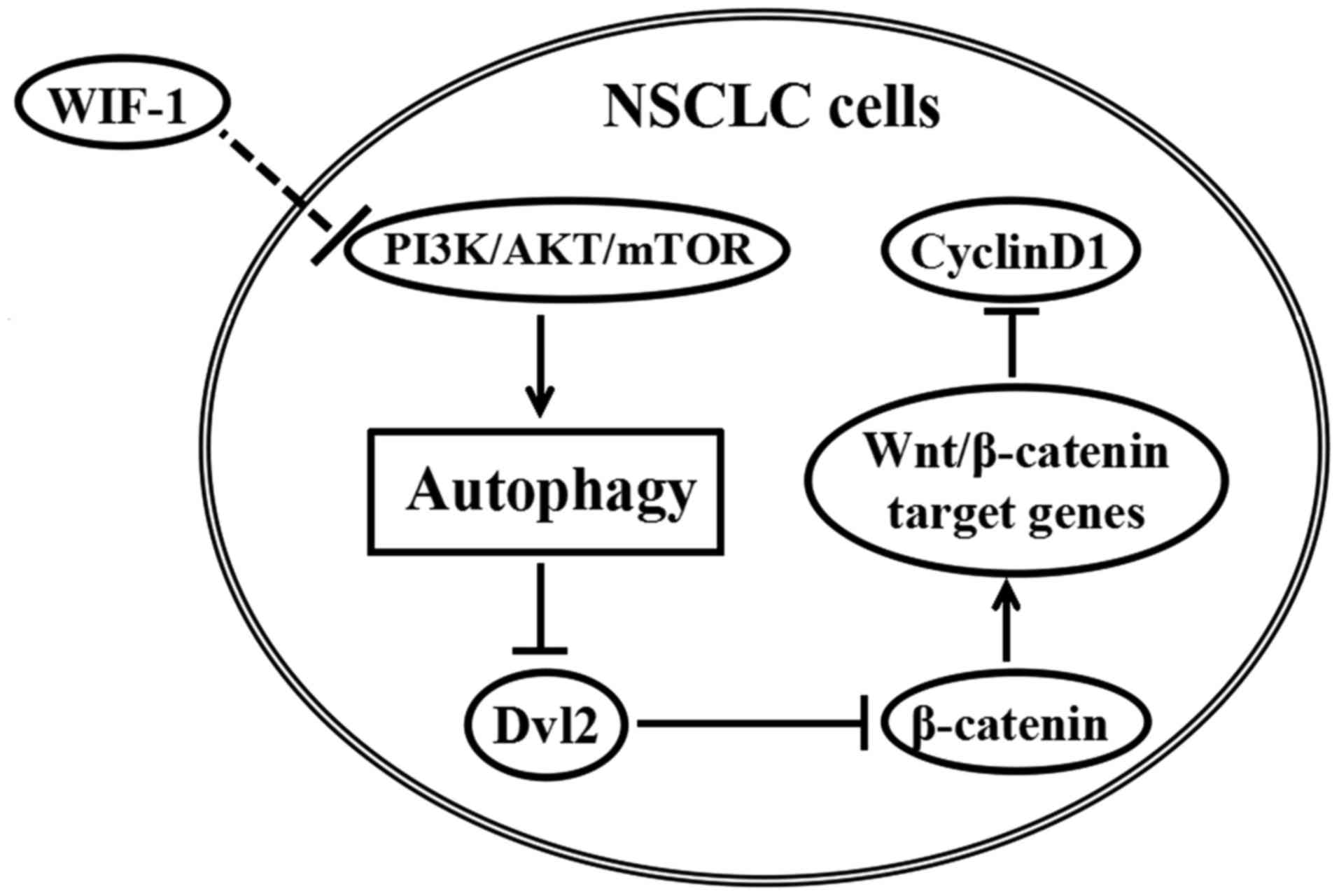
The probable mechanism of WIF-1 in
Wnt/β-catenin signaling inhibiton
To elucidate the role of WIF-1-mediated autophagy in
Wnt/β-catenin signaling inhibition, we sketched a map (Fig. 6) according to the results in the
western blot analysis mentioned above (Fig. 3A and C). The map interpreted that
WIF-1 induced autophagy probably via PI3K/Akt/mTOR. WIF-1-induced
autophagy led to Dvl2 downregulation, which reduced the level of
downstream protein (β-catenin). The downregulation of β-catenin
inhibited some Wnt/β-catenin target gene expression, one of which
was the downregulation of cyclin D1, as analyzed in Fig. 3A.
Discussion
NSCLC development is a stepwise progression and
tumor suppression gene inactivation is a main cause of NSCLC
development (4). Hence, a thorough
investigation of the molecular mechanisms of tumor suppressor will
supply the foundation for clinical application in NSCLC.
Previous studies have demonstrated that the
restoration of WIF-1 expression significantly inhibits
proliferation and promotes cell apoptosis in a number of
malignancies, including NSCLC (16,19,26-29).
However, studies on the antitumor mechanisms of WIF-1 have been
limited to the evaluation of Wnt pathway downstream members (such
as β-catenin, cyclin D1 and c-Myc) or the inhibition of
transcription factors (LEF/TCF) (16-19).
Only Tang et al (28)
reported that WIF-1 inhibited cell growth by binding to Wnt1 and
subsequently inhibited Wnt/β-catenin signaling in bladder cancer
cells. However, few of the details of Wnt/β-catenin signaling
inhibition by WIF-1 (for example, the research about which Wnt
proteins can bind with WIF-1 in cancer cells) have been extensively
investigated to date. In this study, a series of autophagy-related
incidents occurred in the A549 and H460 transfected with the
WIF-1 gene overexpression vector, such as the formation of
yellow-orange AVOs detected by acridine orange and membrane-bound
vacuoles detected by TEM. In addition, LC3II is also a reliable
marker of autophagosomes and is localized on the membrane of
autophagosomes (23). In our
results, an increase in the number of GFP-LC3 punctate dots and
LC3II expression were observed in the cells transfected with the
WIF-1 gene overexpression vector. These results indicated
that WIF-1 induced autophagy in NSCLC cells.
In order to determine whether WIF-1-mediated
autophagy involves the inhibition of Wnt/β-catenin signaling, we
first focused on the effects of WIF-1-mediated autophagy on the
proliferation and apoptosis of NSCLC cells. Autophagy is used by
eukaryotic cells to self-digest their long-lived proteins and
dysfunctional organelles and to provide nutrients in response to
cellular metabolic stress (30).
The aberration of autophagy has been shown to be associated with
oncogenesis (31). Currently, a
number of anticancer agents have been documented to induce
autophagy (32-36) and blocking autophagy weakened the
therapeutic efficacy of anticancer drugs (37-39).
In this study, we demonstrated that WIF-1 inhibited the
proliferation and promoted the apoptosis of NSCLC cells, while
these anti-proliferative and pro-apoptotic effects were attenuated
by the blocking of WIF-1-mediated autophagy. These results
suggested that WIF-1-mediated autophagy played a positive role in
the inhibition of proliferation and the promotion of apoptosis in
NSCLC.
Furthermore, we wished to determine whether the
anti-tumor mechanisms of WIF-1-mediated autophagy are related to
the inhibition of Wnt/β-catenin signaling. The mechanisms through
which autophagy contributes to antitumor effects are complex
(34). Recently, Gao et al
(20) revealed that autophagy
negatively regulated Wnt/β-catenin signaling by promoting Dvl
degradation in embryonic cells and HeLa cells. In this study, Dvl2
was downregulated in the A549 and H460 cells transfected with the
WIF-1 gene overexpression vector, while the blocking of
autophagy reversed the reduction of Dvl2 expression in these cells
transfected with the WIF-1 gene over-expression vector.
Moreover, β-catenin and cyclin D1 are the downstream members of
Wnt/β-catenin signaling (8). Thus,
we further detected the protein expression levels of β-catenin and
cyclin D1 when autophagy was blocked in the A549 and H460
transfected with the WIF-1 gene overexpression vector. The
results revealed that the levels of both these Wnt pathway
downstream members were downregulated in the cells trans-fected
with the WIF-1 gene overexpression vector. However, the
decrease in β-catenin and cyclin D1 expression was reversed by the
blocking of autophagy in the cells transfected with the WIF-1 gene
overexpression vector. Notably, the changes in the expression of
β-catenin and cyclin D1 were positively associated with those of
Dvl2. Hence, these results indicated that WIF-1-mediated autophagy
inhibited Wnt/β-catenin signaling by downregulating Dvl2 in NSCLC
cells.
The Dvl family often is regarded as a cytoplasmic
mediator of Wnt/β-catenin signaling to destruct the APC/Axin/GSK3β
complex and break down β-catenin degradation, which finally leads
to Wnt/β-catenin signaling activation (40). The overexpression of Dvl2 has been
detected in NSCLC (41). It has
also been demonstrated that the knockdown of Dvl2 expression
inhibits Wnt/β-catenin signaling and the growth of NSCLC cells
(42). Moreover, it has been
reported that blocking Wnt-1 activity induces the tumor-specific
apoptosis of NSCLC cells by regulating the Wnt-Dvl-β-catenin
signaling pathway (43). In this
study, we revealed that the overexpression of WIF-1
significantly inhibited the proliferation and promoted the
apoptosis of A549 and H460, which was markedly attenuated by the
blocking of autophagy. Moreover, it was observed that Dvl2
expression was downregulated in the cells transfected with the
WIF-1 gene overexpression vector, but this effect was
reversed by the blocking of autophagy. Therefore, these findings
suggest that negatively regulating Dvl2 by WIF-1-mediated autophagy
leads to the inhibition of the proliferation and the promotion of
the apoptosis of NSCLC cells.
The upstream signaling of autophagy regulation
contained mTOR signaling and non-mTOR signaling (24). Among these, PI3K-Akt-mTOR pathway
was a general regulator of autophagy (30). In our study, we found the level of
p-mTOR and p-Akt were attenuated, and the expression of PI3K was
significantly reduced by WIF-1. Moreover, the decrease of β-catenin
and cyclin D1 could be reversed by treated with 3-MA, which was
reported as a PI3K inhibitor to regulate autophagy. These results
suggested that the mechanism of autophagy induction and the
following Wnt/β-catenin pathway regulation by WIF-1 might be
related to PI3K-Akt-mTOR signaling in NSCLC cells.
Increasing evidence suggests that combining
antitumor drugs with autophagy agonists in clinical experiments
exerts more potent effects (44-46).
Moreover, autophagy agonists have been manifested to induce
autophagy in leukemia, papillary thyroid cancer and lung cancer
(47-49). Among these, RAD001 has been
reported to be an autophagy agonist and to induce autophagy in
vivo (48), and it has been
approved by the US Food and Drug Administration (FDA) and the
European Medicines Agency (EMA) for the treatment of advanced renal
cell carcinoma (RCC) in 2009 and pancreatic neuroendocrine tumors
(PNET) in 2011 (50,51). The results of this study indicated
that WIF-1-mediated autophagy contributed to the antitumor effects
of WIF-1 in vitro. Therefore, the effects of both
transfection with WIF-1 gene overexpression vector and
treatment with RAD001 against NSCLC were further evaluated in A549
subcutaneous tumor xenografts and in a pulmonary metastasis tumor
model. The results demonstrated that treatment with RAD001 alone
inhibited tumor growth; however, combination treatment with WIF-1
and RAD001 exerted significant synergistic effects against
subcutaneous tumor growth compared with WIF-1 or RAD001 individual
treatment. More importantly, systemic administration delivery is a
more effective and practical method for lung cancer treatment
(25). Therefore, combination
treatment with WIF-1 by systemic administration and RAD001 by
gavage exerted more potent antitumor effects, suppressing lung
metastasis in our experimental model. Our results suggested that
combination treatment with WIF-1 by systemic administration and
RAD001 by gavage may be a novel and potential strategy for lung
cancer therapy.
In conclusion, to the best of our knowledge, this is
the first study demonstrating that WIF-1-mediated autophagy
inhibits Wnt/β-catenin signaling through the downregulation of
Dvl2, which may be independent of Wnt ligands, then further
inhibits the proliferation and promotes the apoptosis of NSLCL
cells. Moreover, the mechanisms of the induction of autophagy by
WIF-1 are related to PI3K/AKT/mTOR signaling (Fig. 6). Furthermore, combination
treatment involving transfection with the WIF-1 gene
overexpression vector and treatment with the autophagy agonist,
RAD001, exerted a synergistic antitumor effect in vivo.
Collectively, the data from this study reveal a novel molecular
mechanism involving the WIF-1-mediated inhibition of Wnt/β-catenin
signaling by the induction of autophagy; this may provide the
theoretical basis for the joint therapy of NSCLC with WIF-1 and
autophagy agonists in clinical practice in the future.
Abbreviations:
|
WIF-1
|
Wnt inhibitory factor-1
|
|
NSCLC
|
non-small cell lung cancer
|
|
Dvl
|
dishevelled
|
|
3-MA
|
3-methyladenine
|
|
MTT
|
3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide
|
|
RAD001
|
everolimus
|
Acknowledgments
The authors would like to thank Ms. Qiaorong Huang
(State Key Laboratory of Biotherapy and Cancer Center, West China
Hospital, Sichuan University) for providing technical
assis-tance.
Funding
This study was partly supported by grants from the
National Science and Technology Major Projects of New Drugs (no.
2012ZX09103301-009) and the National 863 Plan Project (no.
2012AA020802)
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XL was involved in the conception and design,
collection and/or assembly of data, data analysis and
interpretation, manuscript writing, and the final approval of the
manuscript; SY was involved in the conception and design,
collection and/or assembly of data; WZ was involved in the
conception and design, financial support, administrative support,
provision of study material, data analysis and interpretation,
manuscript writing, and the final approval of the manuscript; QJ,
YG, YY, XH and XS were involved in the collection and/or assembly
of data, data analysis and interpretation, technical support. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experimental procedures were approved by
the Institute of Laboratory Animal Care and Use Committee at
Sichuan University (Chengdu, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pao W and Hutchinson KE: Chipping away at
the lung cancer genome. Nat Med. 18:349–351. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Morgensztern D, Pennell NA, Subramanian J
and Govindan R: Summary of presentations from the 46th annual
meeting of the American Society Of Clinical Oncology (2010): Focus
on developmental therapeutics related to lung cancer. Clin Lung
Cancer. 12:94–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heist RS and Engelman JA: SnapShot:
Non-small cell lung cancer. Cancer Cell. 21:448–448e2. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsieh JC, Kodjabachian L, Rebbert ML,
Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB and Nathans J:
A new secreted protein that binds to Wnt proteins and inhibits
their activities. Nature. 398:431–436. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Malinauskas T, Aricescu AR, Lu W, Siebold
C and Jones EY: Modular mechanism of Wnt signaling inhibition by
Wnt inhibitory factor 1. Nat Struct Mol Biol. 18:886–893. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Amerongen R and Nusse R: Towards an
integrated view of Wnt signaling in development. Development.
136:3205–3214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mazieres J, He B, You L, Xu Z and Jablons
DM: Wnt signaling in lung cancer. Cancer Lett. 222:1–10. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Licchesi JD, Westra WH, Hooker CM, Machida
EO, Baylin SB and Herman JG: Epigenetic alteration of Wnt pathway
antagonists in progressive glandular neoplasia of the lung.
Carcinogenesis. 29:895–904. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wissmann C, Wild PJ, Kaiser S, Roepcke S,
Stoehr R, Woenckhaus M, Kristiansen G, Hsieh JC, Hofstaedter F,
Hartmann A, et al: WIF1, a component of the Wnt pathway, is
down-regulated in prostate, breast, lung, and bladder cancer. J
Pathol. 201:204–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mazieres J, He B, You L, Xu Z, Lee AY,
Mikami I, Reguart N, Rosell R, McCormick F and Jablons DM: Wnt
inhibitory factor-1 is silenced by promoter hypermethylation in
human lung cancer. Cancer Res. 64:4717–4720. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Korobko EV, Kalinichenko SV, Shepelev MV,
Zborovskaia IB, Allakhverdiev AK, Zinov'eva MV, Vinogradova TV,
Sverdlov ED and Korobko IV: Suppression of the WIF1 transcript and
protein in non-small cell lung carcinomas. Mol Gen Mikrobiol
Virusol. 22:53–58. 2007.In Russian.
|
|
13
|
Yang TM, Leu SW, Li JM, Hung MS, Lin CH,
Lin YC, Huang TJ, Tsai YH and Yang CT: WIF-1 promoter region
hypermethylation as an adjuvant diagnostic marker for non-small
cell lung cancer-related malignant pleural effusions. J Cancer Res
Clin Oncol. 135:919–924. 2009. View Article : Google Scholar
|
|
14
|
Yoshino M, Suzuki M, Tian L, Moriya Y,
Hoshino H, Okamoto T, Yoshida S, Shibuya K and Yoshino I: Promoter
hypermethylation of the p16 and Wif-1 genes as an independent
prognostic marker in stage IA non-small cell lung cancers. Int J
Oncol. 35:1201–1209. 2009.PubMed/NCBI
|
|
15
|
Gao Z, Xu Z, Hung MS, Lin YC, Wang T, Gong
M, Zhi X, Jablon DM and You L: Promoter demethylation of WIF-1 by
epigallocatechin-3-gallate in lung cancer cells. Anticancer Res.
29:2025–2030. 2009.PubMed/NCBI
|
|
16
|
Kim J, You L, Xu Z, Kuchenbecker K, Raz D,
He B and Jablons D: Wnt inhibitory factor inhibits lung cancer cell
growth. J Thorac Cardiovasc Surg. 133:733–737. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deng Y, Yu B, Cheng Q, Jin J, You H, Ke R,
Tang N, Shen Q, Shu H, Yao G, et al: Epigenetic silencing of WIF-1
in hepatocellular carcinomas. J Cancer Res Clin Oncol.
136:1161–1167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kawakami K, Hirata H, Yamamura S, Kikuno
N, Saini S, Majid S, Tanaka Y, Kawamoto K, Enokida H, Nakagawa M,
et al: Functional significance of Wnt inhibitory factor-1 gene in
kidney cancer. Cancer Res. 69:8603–8610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang J, Zhou B, Liu Y, Chen K, Bao P,
Wang Y, Wang J, Zhou Z, Sun X and Li Y: Wnt inhibitory factor-1
functions as a tumor suppressor through modulating Wnt/β-catenin
signaling in neuroblastoma. Cancer Lett. 348:12–19. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T,
Fu W, Zhang J, Wu W, Zhang X, et al: Autophagy negatively regulates
Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol.
12:781–790. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ren J, Yu C, Wu S, Peng F, Jiang Q, Zhang
X, Zhong G, Shi H, Chen X, Su X, et al: Cationic liposome mediated
delivery of FUS1 and hIL-12 coexpression plasmid demonstrates
enhanced activity against human lung cancer. Curr Cancer Drug
Targets. 14:167–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dings RP, Yokoyama Y, Ramakrishnan S,
Griffioen AW and Mayo KH: The designed angiostatic peptide anginex
synergis-tically improves chemotherapy and antiangiogenesis therapy
with angiostatin. Cancer Res. 63:382–385. 2003.PubMed/NCBI
|
|
23
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang YP, Liang ZQ, Gu ZL and Qin ZH:
Molecular mechanism and regulation of autophagy. Acta Pharmacol
Sin. 26:1421–1434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Templeton NS, Lasic DD, Frederik PM, Strey
HH, Roberts DD and Pavlakis GN: Improved DNA: Liposome complexes
for increased systemic delivery and gene expression. Nat
Biotechnol. 15:647–652. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin YC, You L, Xu Z, He B, Yang CT, Chen
JK, Mikami I, Clément G, Shi Y, Kuchenbecker K, et al: Wnt
inhibitory factor-1 gene transfer inhibits melanoma cell growth.
Hum Gene Ther. 18:379–386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu J, Dong A, Fernandez-Ruiz V, Shan J,
Kawa M, Martínez-Ansó E, Prieto J and Qian C: Blockade of Wnt
signaling inhibits angiogenesis and tumor growth in hepatocellular
carcinoma. Cancer Res. 69:6951–6959. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang Y, Simoneau AR, Liao WX, Yi G, Hope
C, Liu F, Li S, Xie J, Holcombe RF, Jurnak FA, et al: WIF1, a Wnt
pathway inhibitor, regulates SKP2 and c-myc expression leading to
G1 arrest and growth inhibition of human invasive urinary bladder
cancer cells. Mol Cancer Ther. 8:458–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu J, Fang J, Yang Z, Chen F, Liu J and
Wang Y: Wnt inhibitory factor-1 regulates glioblastoma cell cycle
and proliferation. J Clin Neurosci. 19:1428–1432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Klionsky DJ: Autophagy revisited: A
conversation with Christian de Duve. Autophagy. 4:740–743. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang ZJ, Chee CE, Huang S and Sinicrope F:
Autophagy modulation for cancer therapy. Cancer Biol Ther.
11:169–176. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shen S, Kepp O, Michaud M, Martins I,
Minoux H, Métivier D, Maiuri MC, Kroemer RT and Kroemer G:
Association and dissociation of autophagy, apoptosis and necrosis
by systematic chemical study. Oncogene. 30:4544–4556. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maycotte P and Thorburn A: Autophagy and
cancer therapy. Cancer Biol Ther. 11:127–137. 2011. View Article : Google Scholar :
|
|
37
|
Fulda S: Autophagy and cell death.
Autophagy. 8:1250–1251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Park EJ, Choi KS and Kwon TK:
β-Lapachone-induced reactive oxygen species (ROS) generation
mediates autophagic cell death in glioma U87 MG cells. Chem Biol
Interact. 189:37–44. 2011. View Article : Google Scholar
|
|
39
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bilic J, Huang YL, Davidson G, Zimmermann
T, Cruciat CM, Bienz M and Niehrs C: Wnt induces LRP6 signalosomes
and promotes dishevelled-dependent LRP6 phosphorylation. Science.
316:1619–1622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wei Q, Zhao Y, Yang ZQ, Dong QZ, Dong XJ,
Han Y, Zhao C and Wang EH: Dishevelled family proteins are
expressed in non-small cell lung cancer and function differentially
on tumor progression. Lung Cancer. 62:181–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Uematsu K, He B, You L, Xu Z, McCormick F
and Jablons DM: Activation of the Wnt pathway in non small cell
lung cancer: Evidence of dishevelled overexpression. Oncogene.
22:7218–7221. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He B, You L, Uematsu K, Xu Z, Lee AY,
Matsangou M, McCormick F and Jablons DM: A monoclonal antibody
against Wnt-1 induces apoptosis in human cancer cells. Neoplasia.
6:7–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nahta R and O'Regan RM: Evolving
strategies for overcoming resistance to HER2-directed therapy:
Targeting the PI3K/Akt/mTOR pathway. Clin Breast Cancer. 10(Suppl
3): S72–S78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Racanelli AC, Rothbart SB, Heyer CL and
Moran RG: Therapeutics by cytotoxic metabolite accumulation:
Pemetrexed causes ZMP accumulation, AMPK activation, and mammalian
target of rapamycin inhibition. Cancer Res. 69:5467–5474. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Margolin K, Longmate J, Baratta T, Synold
T, Christensen S, Weber J, Gajewski T, Quirt I and Doroshow JH:
CCI-779 in metastatic melanoma: A phase II trial of the California
Cancer Consortium. Cancer. 104:1045–1048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Crazzolara R, Cisterne A, Thien M, Hewson
J, Baraz R, Bradstock KF and Bendall LJ: Potentiating effects of
RAD001 (Everolimus) on vincristine therapy in childhood acute
lymphoblastic leukemia. Blood. 113:3297–3306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lin CI, Whang EE, Donner DB, Du J, Lorch
J, He F, Jiang X, Price BD, Moore FD Jr and Ruan DT: Autophagy
induction with RAD001 enhances chemosensitivity and
radiosensitivity through Met inhibition in papillary thyroid
cancer. Mol Cancer Res. 8:1217–1226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marinov M, Ziogas A, Pardo OE, Tan LT,
Dhillon T, Mauri FA, Lane HA, Lemoine NR, Zangemeister-Wittke U,
Seckl MJ, et al: AKT/mTOR pathway activation and BCL-2 family
proteins modulate the sensitivity of human small cell lung cancer
cells to RAD001. Clin Cancer Res. 15:1277–1287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Thompson LA, Kim M, Wenger SD and O'Bryant
CL: Everolimus: A new treatment option for advanced pancreatic
neuroendocrine tumors. Ann Pharmacother. 46:1212–1219. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tan X, Liu Y, Hou J and Cao G: Targeted
therapies for renal cell carcinoma in Chinese patients: Focus on
everolimus. Onco Targets Ther. 8:313–321. 2015.PubMed/NCBI
|