Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common type of cancer worldwide, with ~600,000 new cases
diagnosed each year. Treatment protocols include surgery,
chemotherapy and radiation, all of which may result in major
physical and psychological effects on the patient (1). Despite continuing research and
advances in treatment, the clinical outcomes and overall survival
rates of patients with HNSCC have not improved significantly over
the past several decades, with an overall 5-year survival rate of
60% (2,3). A high rate of metastasis is a
characteristic of HNSCC, which highlights the urgent need for the
identification of novel biomarkers to more accurately predict
recurrence and metastasis in patients with HNSCC.
MicroRNAs (miRNAs or miRs) are endogenous short
non-coding RNAs (ncRNAs) that bind to the complementary seed
sequence on the 3′-untranslated region (UTR) of specific target
protein-coding genes, known as miRNA response elements (MREs),
thereby resulting in the inhibition of target gene expression
(4). Increasing evidence suggests
that miRNA dysregulation is associated with the tumorigenesis and
progression of various cancer types (5-8). It
has been demonstrated that certain protein-coding genes and their
pseudogene sequences contain the same conserved MREs in their
3′UTRs, and their respective expression levels may be regulated
through competing for the same miRNAs through these MREs (9). A hypothesis regarding these RNA
transcripts, termed competing endogenous RNAs (ceRNAs), has been
proposed (10). According to this
hypothesis, MREs can be seen as an 'RNA language' through which
transcripts can communicate with each other to regulate their
respective expression levels. Any RNA transcript with MREs may
function as a ceRNA and compete with other RNA transcripts with
similar MREs for the same miRNAs.
Circular RNAs (circRNAs) are widespread and diverse
endogenous ncRNAs with covalently closed circular structures
(11,12). There are three main types of
circRNAs based on their biogenesis patterns, including exonic
circRNAs (13), intronic circRNAs
(14,15) and exon-intron circRNAs (15-17),
which are transcribed from pre-mRNA sequences by RNA polymerase
(Pol) II (18). circRNAs are
predominantly generated by reverse splicing between the specific
and conservative sequences in upstream and downstream regions,
respectively, a process known as back-splicing (19,20).
Of the three types, exonic circRNAs account for the largest
proportion, and are abundant in the cytoplasm (21-23).
There are multiple conserved MREs on circRNAs, known as miRNA
sponges, that sequester miRNAs from binding to targets on mRNAs,
thereby enhancing the expression of certain genes (24,25).
However, studies have demonstrated that only a few circRNAs have
multiple MREs for specific miRNAs, most of which contain only one
or two MREs (26,27). Intronic circRNAs are mainly present
in the nucleus and have almost no enrichment in MREs. They largely
accumulate around the transcription initiation sites of their
parental genes through interaction with elongation RNA Pol II
machinery, serving a cis-regulatory role on their parental coding
genes (14). Exon-intron circRNAs
are abundant in the nucleus, and interact with small nuclear
ribonucleoprotein (U1 snRNP) and Pol II at the promoter regions of
genes to promote the transcription of their parental genes
(16). At present, studies on
circRNAs are mainly focused on exonic circRNAs. Their interactions
with disease-related miRNAs indicate that circular RNAs are
important factors in disease regulation (25,28).
For instance, the circRNA ciRS-7 contains multiple and conservative
miRNA-7 binding sites, thus acting as an efficient miRNA sponge to
adsorb miRNA-7, and thus inhibiting its function (29).
Increasing evidence has demonstrated that ciRS-7
plays a crucial role in the tumorigenesis and progression of
cancers through its effect on the level of miRNA-7, thus mediating
gene expression via ceRNA mechanisms (30). Given the widespread involvement of
miR-7 in various cancer-related pathways and its potential
regulatory functions in Parkinson's and Alzheimer's disease, ciRS-7
may play a key role in other neurological disorders and
tumorigenesis (24). Exploring the
mechanisms underlying the interactions between these ceRNAs may add
a new dimension to our understanding of the mechanisms of cancer,
and may potentially provide a novel approach to cancer
treatment.
The molecular mechanisms of circRNAs in the
carcinogenesis and progression of HNSCC are largely uncharacterized
thus far. Herein, we hypothesized that differentially expressed
circRNAs could participate in the tumorigenesis and metastasis of
HNSCC. To explore the roles of circRNAs, we examined the different
expression patterns of circRNAs and mRNAs in HNSCC using microarray
analyses. Subsequently, based on our microarray data and the
circRNA-miRNA-mRNA interactions predicted with Arraystar's miRNA
target prediction software based on TargetScan (31) and miRanda (32), a network was constructed using
Cytoscape (33,34). OncomiR (35) and UALCAN (36) are online resources from The Cancer
Genome Atlas (TCGA) specifically for miRNA and protein-coding gene
analysis across many cancer types, respectively. DIANA-miRPath v3.0
(37) is an efficient tool to
analyze the combinatorial effect of miRNAs on target pathways by
P-value. According to the results obtained using these three tools,
we predicted that circRNA_036186 may promote the expression of
tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation
protein, ζ polypeptide (14-3-3ζ) by functioning as a sponge for the
inhibitory miR-193b-3p. Our data indicate that circRNA_036186 may
be suitable as a novel diagnostic marker and therapeutic target in
HNSCC, as well as providing a basis for further studies regarding
the role of circRNAs in HNSCC.
Materials and methods
Patients and samples
This study was approved and supervised by The Ethics
Committee of China Medical University (Shenyang, China). Five pairs
of HNSCC and para-carcinoma tissues were collected from patients
that had not received pre-operative chemotherapy or radiotherapy
who underwent resective surgery at the Head and Neck Tumor Center
(School of Stomatology, China Medical University). Written informed
consent was obtained from all patients. All samples were obtained
during surgery and immediately stored in liquid nitrogen. The
diagnosis of HNSCC was confirmed by a pathological examination
(Table I).
 | Table IClinicopathological characteristics
of the five patients. |
Table I
Clinicopathological characteristics
of the five patients.
| Patient no. | Sex | Age | Position | Pathological
diagnosis | Histological
grade | TNM stage |
|---|
| 1 | Male | 53 | Abdomen of the
tongue and bottom of the oral cavity | HNSCC | G2 | T2N0M0 |
| 3 | Male | 33 | Abdomen of the
tongue | HNSCC | G2 | T2N0M0 |
| 6 | Female | 63 | Lateral border of
tongue | HNSCC | G2 | T2N1M0 |
| 8 | Male | 59 | Lateral border of
tongue | HNSCC | G2 | T2N1M0 |
| 9 | Male | 43 | Lateral border of
tongue | HNSCC | G1-G2 | T2N1M0 |
RNA extraction
Total RNA was extracted from the HNSCC and
para-carcinoma tissue samples using TRIzol reagent
(Invitrogen/Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's instructions. Subsequently, the RNA
quantity and quality was measured using an ND-1000 system
(NanoDrop; Thermo Fisher Scientific, Inc., Wilmington, DE, USA).
RNA integrity was assessed via standard denaturing agarose gel
electrophoresis methods.
Microarray assay
Five pairs of carcinoma and para-carcinoma tissues
were used for a microarray assay, with the intention of identifying
differentially expressed circRNAs and mRNAs by comparing the HNSCC
samples with the adjacent control samples. Sample preparation and
microarray hybridization were performed based on Arraystar's
standard protocols. Briefly, total RNAs were digested with RNase R
(Epicentre; Illumina, Inc., San Diego, CA, USA) to remove linear
RNAs and enrich for circRNAs. Subsequently, the enriched circRNAs
were amplified and transcribed into fluorescent cRNA, utilizing a
random priming method (Arraystar Super RNA Labeling kit; Arraystar
Inc., Rockville, MD, USA). The labeled cRNAs were hybridized onto
the Arraystar Human circRNA Array V2 (8×15K; Arraystar Inc.). After
washing the slides, the arrays were scanned using an Agilent G2505C
scanner (Agilent Technologies, Inc., Santa Clara, CA, USA). Agilent
Feature Extraction software (version 11.0.1.1) was used to analyze
the acquired array images. The raw data were quantile normalized,
and subsequent data processing was performed using the limma
package in R. Significantly differentially expressed circRNAs and
mRNAs between the two groups were identified through volcano plot
filtering with the thresholds of P<0.05 and the
Benjamini-Hochberg false discovery rate (FDR) of <0.5.
Differentially expressed circRNAs and mRNAs between the HNSCC and
adjacent control groups were identified through fold change
filtering (circRNAs, |log fold change| >1.5; mRNAs, |log fold
change| >2). Hierarchical clustering was performed to show the
characteristics of the expression profiles based on the values of
all expressed transcripts, and differentially expressed
transcripts.
Gene Ontology (GO) and pathway
analyses
The GO project provides a controlled vocabulary to
describe gene and gene product attributes in any organism
(http://www.geneontology.org/). The GO
categories cover three different domains: Biological process (BP),
cellular component (CC) and molecular function (MF). The P-value
denotes the significance of GO terms enrichment in the
differentially expressed genes. The Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway analysis was used for harvesting pathway
clusters covering the molecular interaction and reaction networks
in differential gene expression profile. The P-value denotes the
significance of the pathway association with the specified
genes.
Competing endogenous RNA network
analysis
The potential miRNA response elements were searched
on the sequences of circRNAs and mRNAs, besides a measure with the
number of common miRNAs, a hypergeometric test was executed for
each ceRNA pair separately (38).
The overlap of the same miRNA seed sequence binding
site both on the circRNA and mRNA was predicted with the following
parameters: miRNA coverage ≥0.1; context +<
-4.999999977648258e-2; species = human; common Num ≥1; ceRNA type =
protein coding; structure >140; P<0.05; context
<-4.999999977648258e-2; energy <-10. miRNA coverage describes
the threshold value for both SeqMiRNA coverage and CeMiRNA
coverage. Context+ is the threshold value of the sum of the
context+ scores used in TargetScan from version 6.0. Common Num is
the threshold value of the number of common miRNAs between the
ceRNA gene and the gene of interest. Structure is the threshold
value of the sum of the structure scores used in miRanda. P-value
is the threshold for the P-values calculated using hypergeometric
tests. Context is the threshold value of the sum of the context
scores used in TargetScan before version 5.x. Energy is the
threshold value of the sum of the free energy predicted by
miRanda.
Exploring miRNA dysregulation in tumor
development and progression
miRNA dysregulation in tumor development and
progression was predicted using OncomiR (www.oncomir.org (35)
based on TCGA across 30 cancer types. Significance in tumor
development was determined through a paired Student's t-test
between normal and tumor tissues; and for survival, an unpaired
Student's t-test and univariate Cox proportional hazards analysis
between living and deceased patients.
Prediction of potentially functional
circRNA-miRNA-mRNA axes
The miRNA pathway investigation was carried out
based on DIANA-miRPath v3.0 (http://www.microrna.gr/miRPathv3) (37). Firstly, we searched miRNA
candidates in DIANA-miRPath v3.0-DIANA-TarBase v7.0 (39) and microT-CDS v5.0 (40,41)
to predict the potential target genes and pathways with thresholds
of P<0.05 and MicroT score >0.8. Subsequently, elaborated
signaling pathway networks based on KEGG database were constructed
gathering target genes and interacting proteins. Finally, the
expression differences for target genes in malignant tumors were
analyzed using UALCAN (http://ualcan.path.uab.edu) (36).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) validation
The microarray results were confirmed by RT-qPCR
with the same RNA samples as those used for microarray analysis.
Following RNA isolation, SuperScript™ III Reverse Transcriptase
(Invitrogen/Thermo Fisher Scientific, Inc., Waltham, MA, USA) was
used to synthesize the cDNA according to the manufacturer's
instructions. Subsequently, qPCR was performed using a ViiA 7
Real-time PCR System (Applied Biosystems/Thermo Fisher Scientific,
Inc., Foster City, CA, USA) with a total reaction volume of 10
μl, including 0.5 μl PCR forward primer (10
μM), 0.5 μl PCR reverse primer (10 μM), 2
μl cDNA, 5 μl 2X Master Mix and 2 μl double
distilled water. The thermocycling conditions were as follows: 95°C
for 10 min, then 10 sec at 95°C and 60 sec at 60°C for a total of
40 cycles. β-actin was used as an internal reference. The
experiments were run using three independent wells per condition.
For quantitative analyses, the relative expression levels of
circRNAs and mRNAs were calculated using the 2−∆∆Cq
(42) method. The primers used
were as follows: circRNA_036186 forward, 5′-CTGAAGCACCGCCCAGCT-3′
and reverse, 5′-GACGAGCCACATTCATTCCAG-3′; 14-3-3ζ forward,
5′-TGTTGTAGGAGCCCGTAG-3′ and reverse, 5′-GCAACCTCAGCCAAGTAA-3′;
β-actin (H) forward, 5′-GTGGCCGAGGACTTTGATTG-3′ and reverse,
5′-CCTGTAACAACGCATCTCATATT-3′.
Statistical analysis
All data are expressed as the means ± SD. The
statistical significance of the differentially expressed genes as
determined by RT-qPCR was estimated using the Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Identification of differentially
expressed circRNA and mRNA profiles by microarray assay
High-throughput microarray analysis was performed to
detect circRNA and mRNA expression in the HNSCC tissues. In the
present study, a total of 12,366 circRNAs and 35,252 mRNAs were
detected by microarray probes in five paired HNSCC and normal
tissues. As a result, 287 dysregulated circRNAs were detected.
Among these, 146 and 141 circRNAs were upregulated and
downregulated, respectively. Additionally, 1,053 mRNAs were found
to be differentially regulated, among which 377 mRNAs were
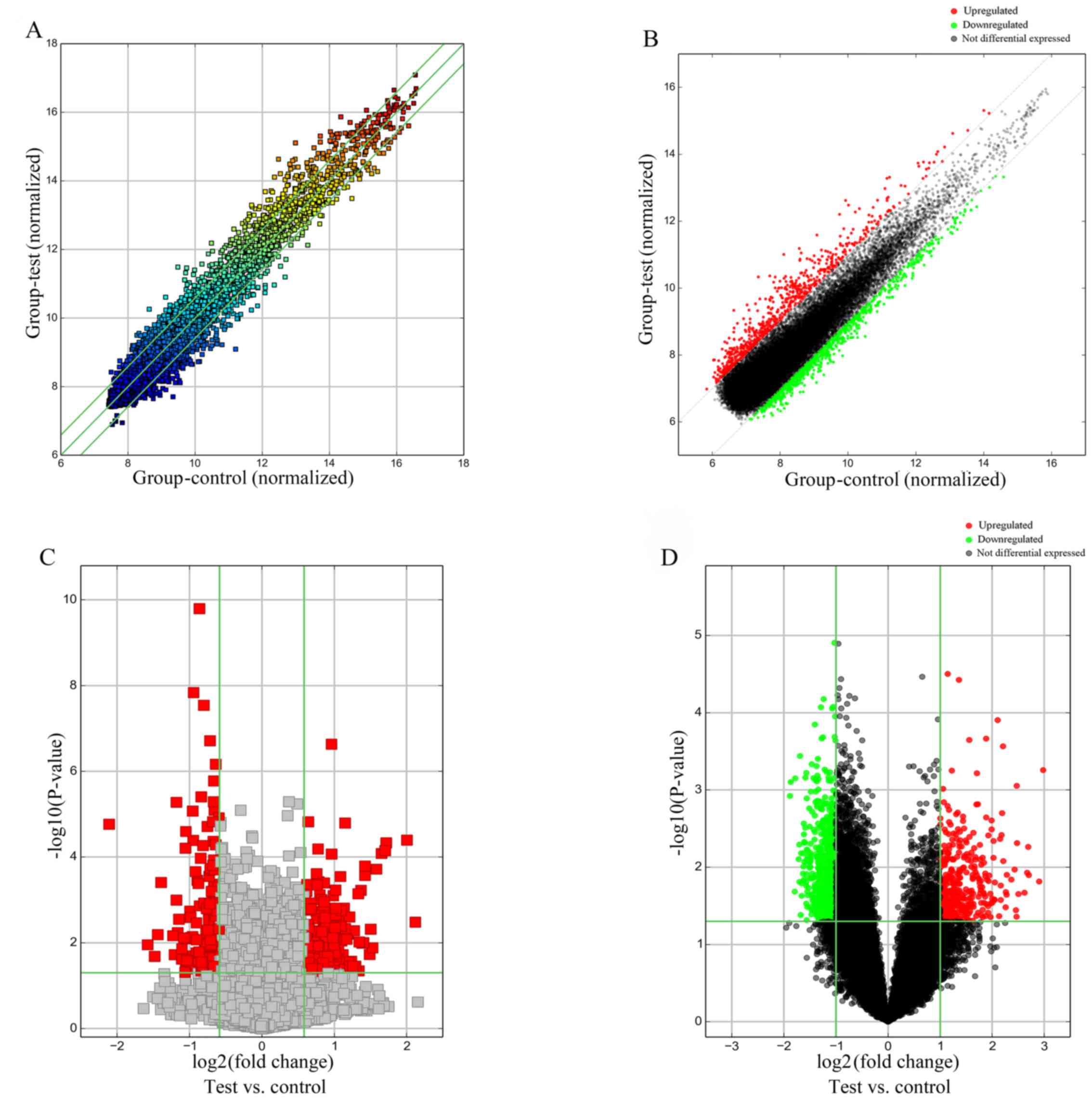
upregulated, while 676 mRNAs were downregulated. Scatter and
Volcano Plots, tools for visualizing differential expression
between two compared groups, were produced; the former was
constructed using fold change values (Fig. 1A and B), while the latter
considered both fold change and statistical significance (Fig. 1C and D).
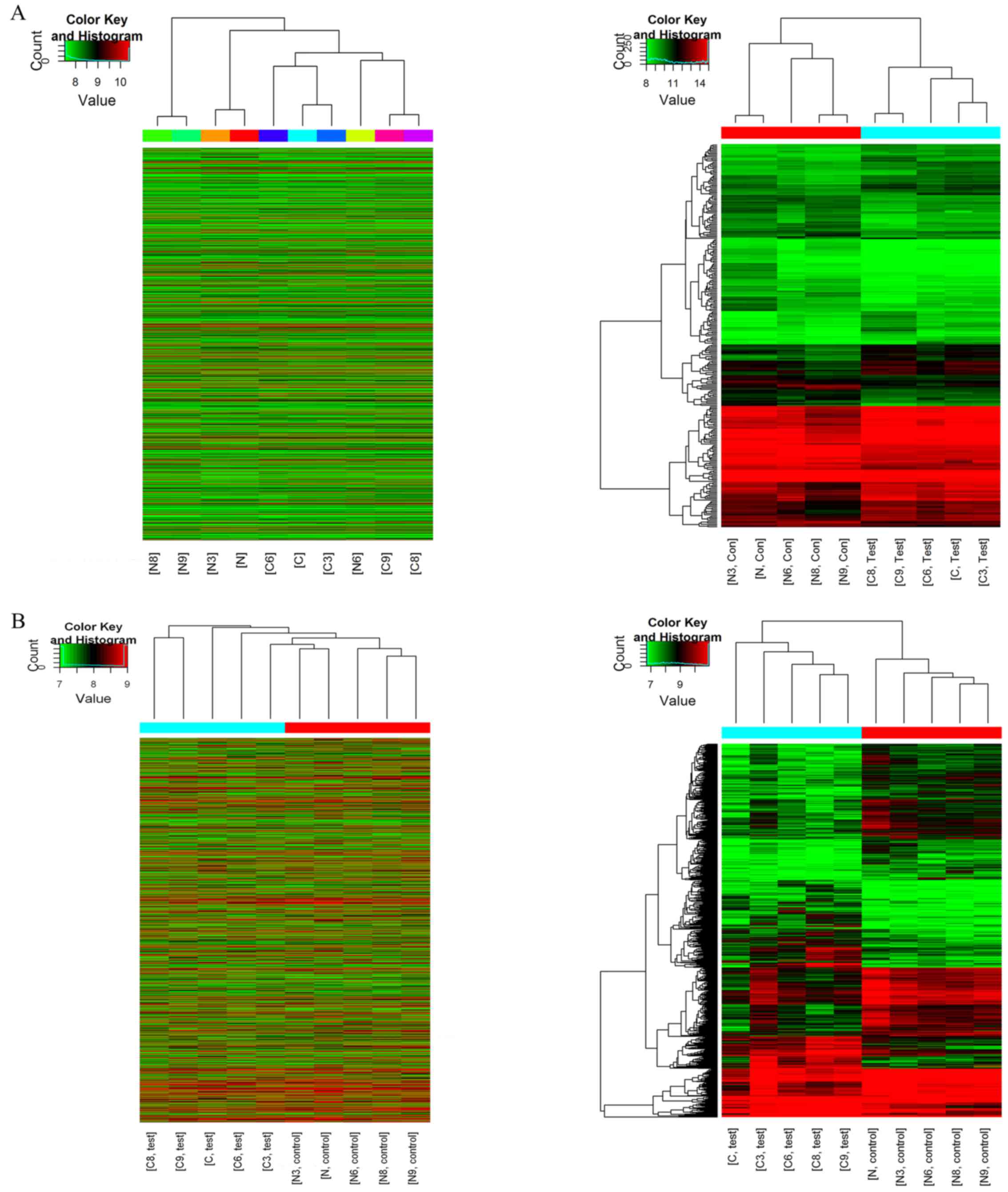
Hierarchical clustering analysis indicated that the
circRNA and mRNA expression patterns were distinguishable between
the groups (Fig. 2). The
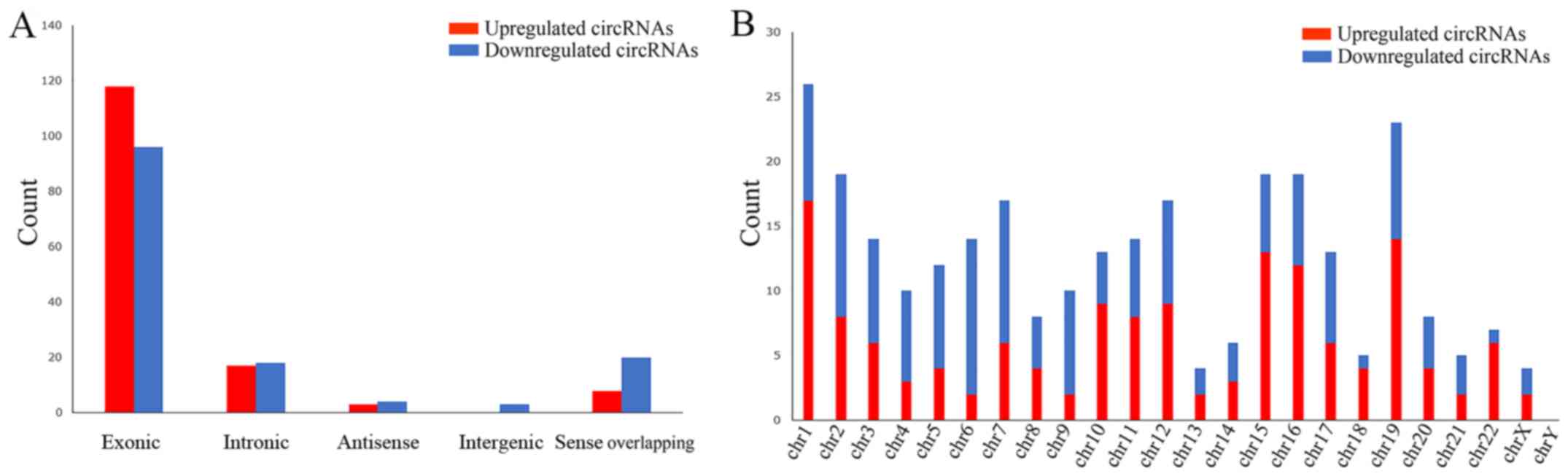
distribution of the dysregulated circRNAs on human chromosomes was
summarized, and the results revealed that these circRNAs were
distributed among all chromosomes, apart from the Y sex chromosome
(Fig. 3B). According to the
associations with the protein-coding genes, the dysregulated
circRNAs were classified into five categories: 214 were exonic, 35
were intronic, 7 were antisense, 3 were intergenic and 28 were
sense overlapping (Fig. 3A). Thus,
the expression profiles of the circRNAs and mRNAs in the HNSCC
tissues were determined to differ from those in the matched
tumor-adjacent tissues.
GO term and KEGG and pathway enrichment
analyses
To date, the functions of the majority circRNAs have
not been annotated. The functional prediction of circRNAs is
largely based on the annotations of their interacting
protein-coding genes. Therefore, the GO term and KEGG pathway
enrichment analyses of dysregulated mRNAs can reveal the roles of
significantly differentially expressed circRNAs, to a certain
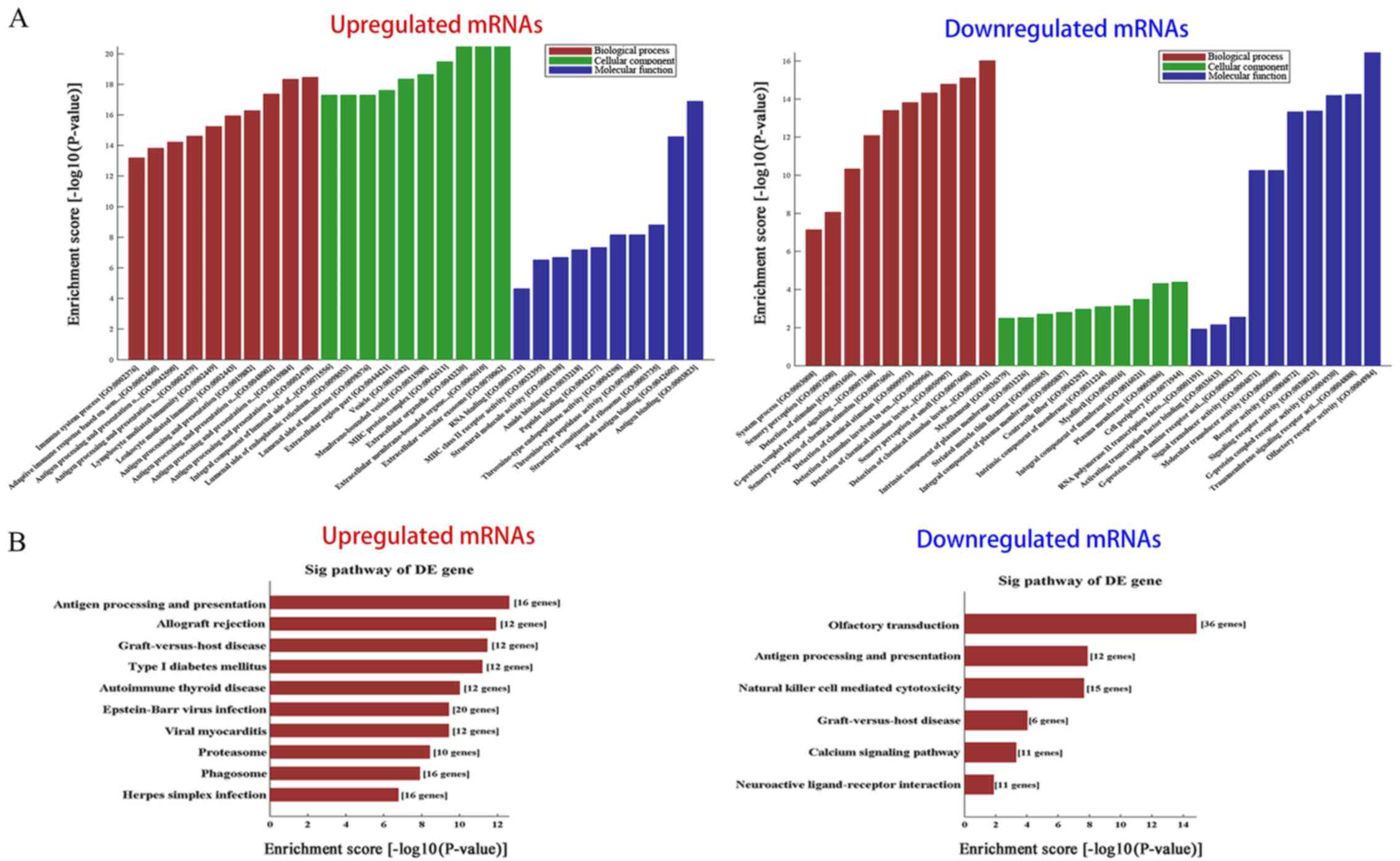
extent. According to the results of a GO term enrichment analysis,
the principally enriched significant BP terms included immune
system process, immune response and signaling transduction, such as
'antigen processing and presentation of exogenous peptide antigen'
(GO:0002478), 'lymphocyte mediated immunity' (GO:0002449) and
G-protein coupled receptor signaling pathway (GO:0007186). The most
enriched CC terms were mostly regarding membranes, such as
'integral component of membrane' (GO:0016021), 'plasma membrane'
(GO:0005886) and 'extracellular membrane-bounded organelle'
(GO:0065010). Furthermore, the most enriched MF terms were
associated with receptor activity and receptor binding, including
'signaling receptor activity' (GO:0038023), 'signal transducer
activity' (GO:0004871), 'activating transcription factor binding'
(GO:0033613) and 'calcium-dependent protein binding' (GO:0048306).
Fig. 4A shows the top 10 most
significantly enriched GO terms in the BP, CC and MF categories for
the up- and downregulated mRNAs.
Pathway analysis was performed with the KEGG
database. A total of 31 pathways associated with the upregulated
mRNAs, and 6 related to the downregulated mRNAs were identified,
including 'antigen processing and presentation' (hsa04612),
'natural killer cell mediated cytotoxicity' (hsa04650) and 'calcium
signaling pathway' (hsa04020), among others. The most enriched
pathways for the up- and downregulated protein-coding genes are
listed in Fig. 4B. These
biological processes, molecular functions and pathways likely
contribute to the occurrence and development of HNSCC.
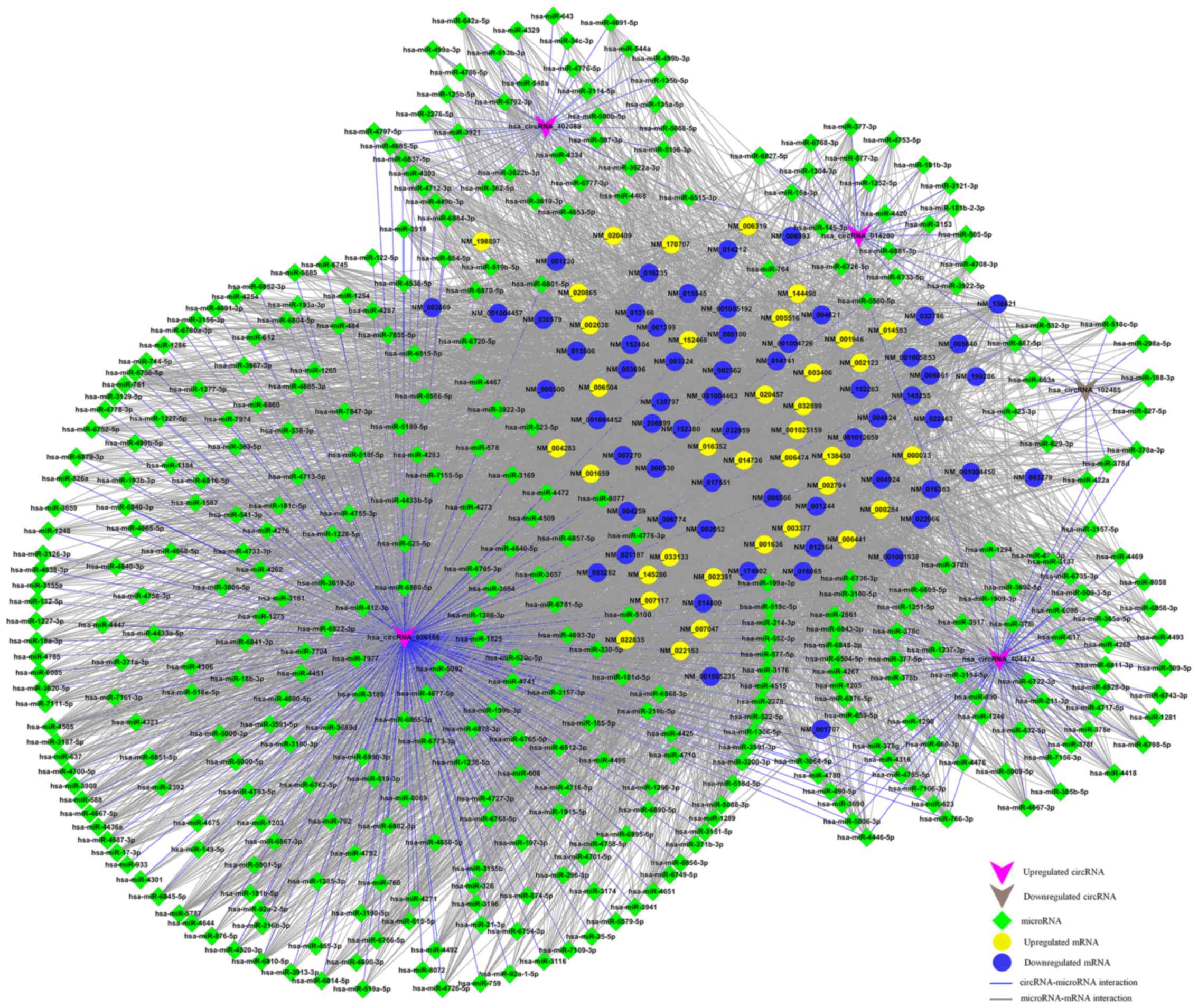
Construction of circRNA-miRNA-mRNA
network
According to the ceRNA hypothesis, the members of
each ceRNA group compete for the same MREs to regulate each other.
We selected the top five upregulated and downregulated circRNAs as
ranked by fold change from the microarray data (Table II), and then identified the
potential ceRNA interactions via various bioinformatics methods. A
summary of ceRNAs, including 5 circRNAs, 385 miRNAs and 5,148
mRNAs, was produced by merging the common targeting miRNAs. To
further investigate the interactions between these ceRNAs in HNSCC
tissues, we used only the differentially expressed mRNAs from the
mRNA microarray data, and a specific ceRNA network, consisting of
five circRNAs, 385 miRNAs and 96 mRNAs, for HNSCC was established
(Fig. 5).
| Table IITop 5 up- and downregulated circRNAs
ranked by fold change >3.0, P<0.01 and FDR <0.05 in the
microarray data. |
Table II
Top 5 up- and downregulated circRNAs
ranked by fold change >3.0, P<0.01 and FDR <0.05 in the
microarray data.
| circRNA | Regulation | Absolute fold
change | P-value | FDR | Source | Alias | Chrom | circRNA type |
|---|
|
hsa_circRNA_014280 | Up | 4.005569 | 4.01E-05 | 0.017333277 | circBase |
hsa_circ_0014280 | chr1 | Exonic |
|
hsa_circRNA_402089 | Up | 3.2867219 | 4.64E-05 | 0.018507674 | 25242744 | _ | chr19 | Exonic |
|
hsa_circRNA_036186 | Up | 3.2503065 | 7.05E-05 | 0.022943867 | circBase |
hsa_circ_0036186 | chr15 | Exonic |
|
hsa_circRNA_404474 | Up | 3.1449976 | 8.22E-05 | 0.024789243 | 25070500 | _ | chr1 | Exonic |
|
hsa_circRNA_102485 | Down | 4.3172347 | 1.71E-05 | 0.009591013 | circBase |
hsa_circ_0050102 | chr19 | Exonic |
Exploring miRNAs associated with tumor
development and patient survival
miRNAs have been shown to play a crucial role in the
pathogenesis and development of cancer (5-8).
OncomiR online software (http://www.oncomir.org/) was used to systematically
identify the miRNAs associated with development, staging and
overall survival in 30 cancer types, and these data were used to
identify the miRNAs associated with tumor formation and patient
survival in HNSCC. The results revealed that there were 376 miRNAs
associated with tumor development and 172 miRNAs related to tumor
prognosis in HNSCC. Among these, 80 overlapping miRNAs were found
in both the tumor development and prognosis groups, suggesting that
these miRNAs were likely to be involved in the regulation of both
HNSCC occurrence and metastasis. Finally, we retained the miRNAs
associated with both tumor development and patient survival from
the ceRNA network (Fig. 6A and
Table III), and thus constructed
a network consisting of 4 circRNAs, 8 miRNAs and 51 mRNAs (Fig. 6B).
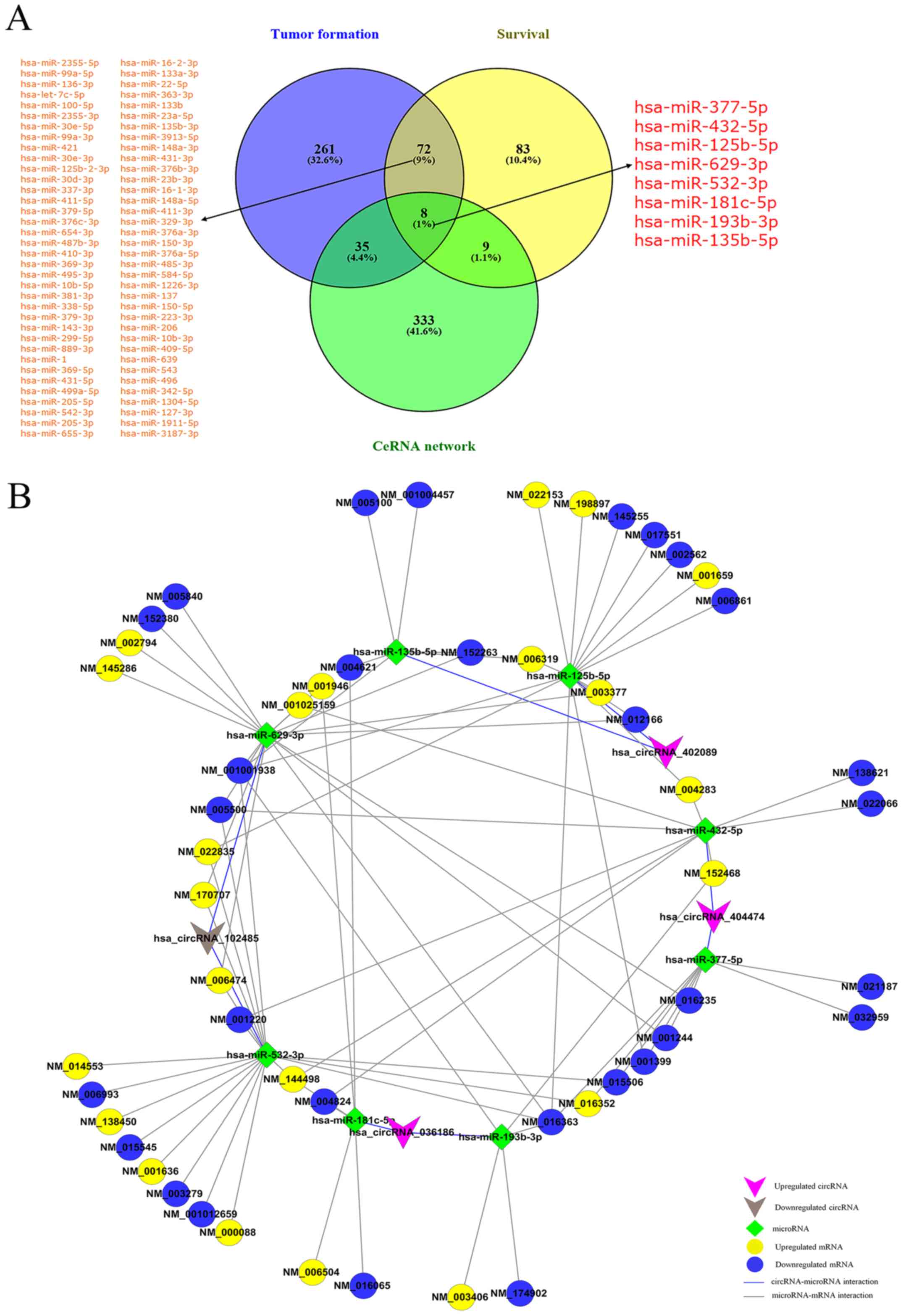 | Figure 6miRNAs associated with tumor
development and patient survival. (A) Venn diagram indicating that
the predicted miRNAs derived from the results of OncomiR and ceRNA
analyses. miR-377-5p, miR-432-5p, miR-125b-5p, miR-629-3p,
miR-532-3p, miR-181c-5p, miR-193b-3p and miR-135b-5p were
intersecting. (B) The circRNA-miRNA-mRNA network consists of 4
circRNAs, 8 microRNAs and 51 mRNAs. circRNA, circular RNA; miRNA,
microRNA; ceRNA, competing endogenous RNA. |
| Table IIImiRNAs associated with both tumor
development and overall survival in the ceRNA network. |
Table III
miRNAs associated with both tumor
development and overall survival in the ceRNA network.
| miRNA | Cancer | Tumor development
| Tumor progression
|
|---|
| t-test P-value | t-test FDR | Upregulated
in: | Log-rank
P-value | Log-rank FDR | Z-score |
|---|
| hsa-miR-432-5p | HNSCC | 0.00177a | 0.00458 | Normal | 0.000457a | 0.143 | 3.496 |
|
hsa-miR-181c-5p | HNSCC | 0.00723a | 0.0166 | Normal | 0.00159a | 0.106 | 3.18 |
| hsa-miR-377-5p | HNSCC | 0.000000119a | 0.000000861 | Normal | 0.00334a | 0.154 | 3.017 |
|
hsa-miR-125b-5p | HNSCC | 7.03E-11a | 1.23E-09 | Normal | 0.00775a | 0.416 | 2.672 |
|
hsa-miR-193b-3p | HNSCC | 3.41E-13a | 1.47E-11 | Tumor | 0.0285a | 0.686 | 2.163 |
| hsa-miR-532-3p | HNSCC | 0.000438a | 0.00127 | Tumor | 0.0314a | 0.174 | 2.156 |
| hsa-miR-629-3p | HNSCC | 2.38E-10a | 3.32E-09 | Tumor | 0.0389a | 0.183 | 2.059 |
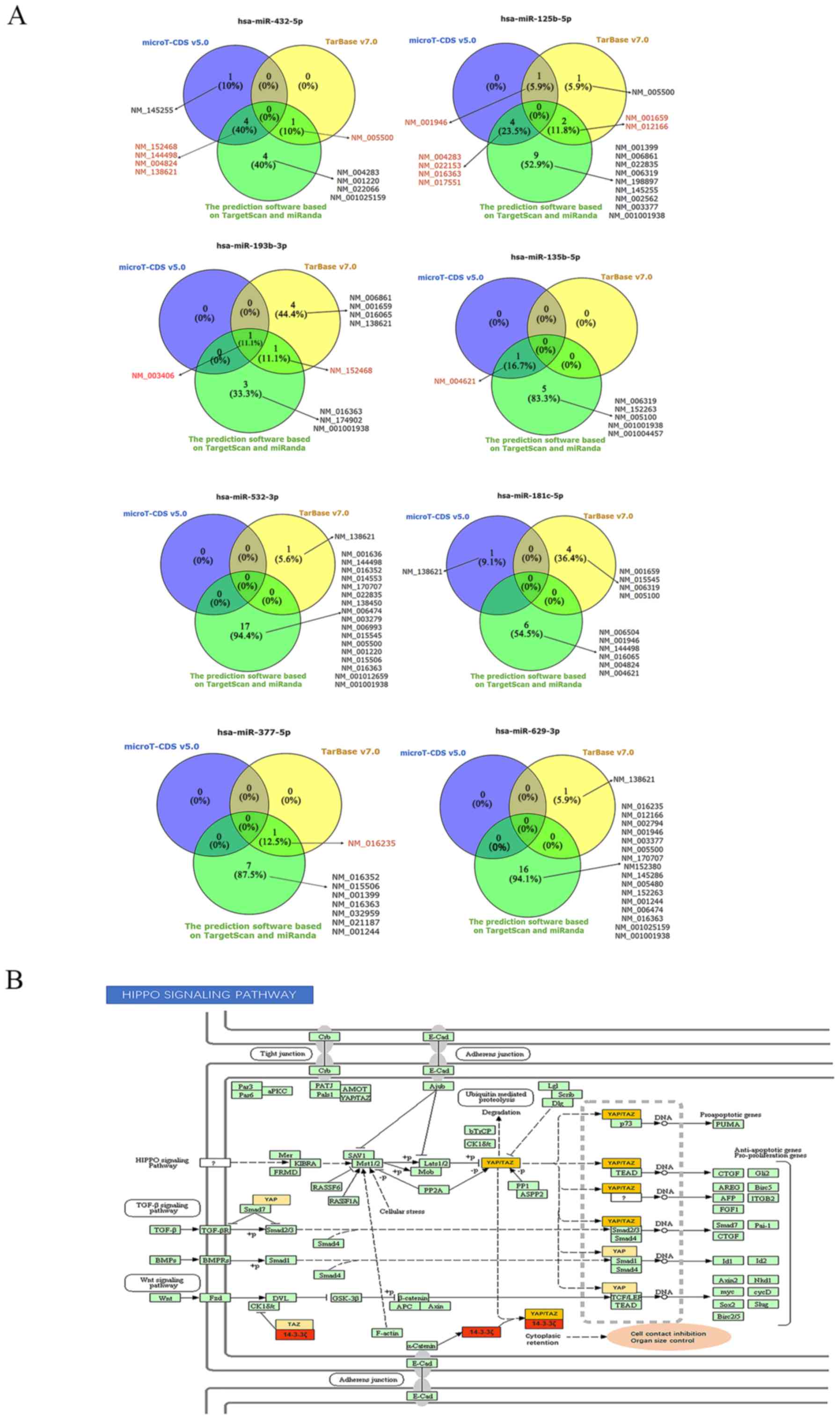
Prediction of responsible and functional
miRNA-mRNA axes
To explore the molecular mechanisms of action of
circRNAs in HNSCC, we predicted the circRNA-miRNA axes associated
with cancer-related pathways. DIANA-miRPath v3.0 allows the
analysis of miRNA-associated pathways as well as the prediction of
miRNA targets using DIANA-microT-CDS v5.0 and/or DIANA-TarBase
v7.0. We queried with the 8 miRNAs from our network, including
miR-377-5p, -432-5p, -125b-5p, -629-3p, -532-3p, -181c-5p, -193b-3p
and -135b-5p, in DIANA-miRPath v3.0-DIANA-TarBase v7.0 and
microT-CDS v5.0 to predict the potential target genes and pathways,
with thresholds of P<0.05 and MicroT score >0.8. The
overlapping results from DIANA-TarBase v7.0 and DIANA-microT-CDS
v5.0, and the aforementioned analyses on ceRNAs revealed that
miR-193b-3p was matched with 14-3-3ζ according to the results of
all three methods, indicating that miR-193b-3p had a significant
potential to interact with 14-3-3ζ (Fig. 7A). Therefore, 14-3-3ζ and
miR-193b-3p were analyzed, and the results from pathway
intersection analysis indicated that miR-193b-3p was likely to
participate in the Hippo signaling pathway (hsa04390)
(P=5.442667e-06) by binding to 14-3-3ζ in HNSCC (Fig. 7B).
Prediction of the potential function of
circRNA_036186 regarding 14-3-3ζ expression
According to GO, 14-3-3ζ may act through 'protein
targeting' (GO:0006605), 'anti-apoptosis' (GO:0006916) and 'signal
transduction' (GO:0007165). All these GO biological processes are
associated with the occurrence and development of cancer. We
analyzed the expression differences for miR-193b-3p and 14-3-3ζ in
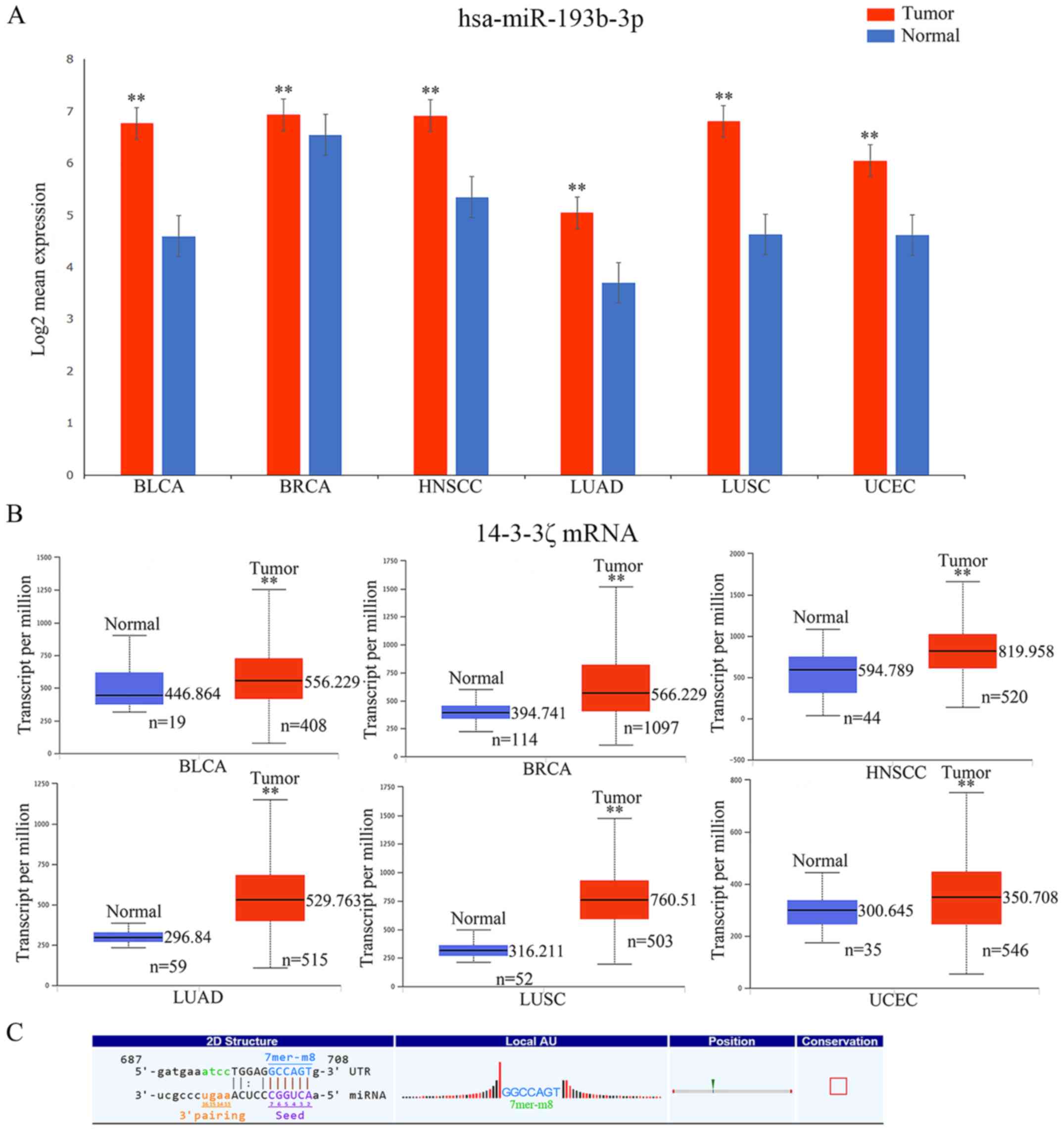
malignant tumors using OncomiR and UALCAN, respectively. The
results revealed that the expression levels of miR-193b-3p and
14-3-3ζ were both upregulated (P<0.01) in the six malignant
tumor samples compared with in the adjacent tissues (Fig. 8A and B). However, evidence has
indicated that the mRNA expression level is negatively associated
with its corresponding miRNA (4),
and we hypothesized that this effect may be attributed to the
participation of circRNA expression. As a miRNA sponge, circRNAs
can counteract, and even reverse, the functional differences caused
by an alteration in miRNA expression. In this study, according to
the results of ceRNA analysis, there was an evolutionarily
conserved regulatory region (7mer-m8) in circRNA_036186 for binding
to miR-193b-3p (Fig. 8C), and the
expression of circRNA_036186 was significantly higher in the HNSCC
tumor tissues compared with the para-carcinoma tissues in the
microarray data. Therefore, we proposed that the
circRNA_036186-miR-193b-3p-14-3-3ζ axis was likely crucial in the
occurrence and prognosis of a variety of cancer types, including
HNSCC. Furthermore, our results from microarray analysis revealed
that the level of 14-3-3ζ was upregulated in HNSCC, compared with
the control, which was consistent with the data from TCGA (36). This indicated that circRNA_036186
probably serves as a crucial and positive regulator of 14-3-3ζ and
is significantly engaged in the Hippo signaling pathway in
HNSCC.
Validation of the differential expression
levels of circRNA_036186 and 14-3-3ζ
The results of microarray analysis of circRNA_036186
and 14-3-3ζ were verified by RT-qPCR with the same RNA samples used
for the micro-array analysis. The results of RT-qPCR assay revealed
that the expression levels of circRNA_036186 and 14-3-3ζ were each
upregulated in HNSCC compared with the controls. The results are
consistent with those of the microarray assay (Table IV); therefore, the RT-qPCR data
verified the accuracy of the microarray results. These results
provide evidence for our hypothesis that circRNA_036186
participates in the pathogenesis and development of HNSCC.
| Table IVMicroarray and RT-qPCR analyses of
selected circRNAs and mRNAs. |
Table IV
Microarray and RT-qPCR analyses of
selected circRNAs and mRNAs.
| Detection
method | hsa_circRNA_036186
| 14-3-3ζ
|
|---|
| Absolute fold
change | Regulation | P-value | Absolute fold
change | Regulation | P-value |
|---|
| Microarray | 3.250307 | Up | 0.000071a | 2.318862 | Up | 0.021603a |
| RT-qPCR | 2.836309 | Up | 0.001582a | 1.7610711 | Up | 0.011234a |
Discussion
As a group of functional molecules with abundant
biological information, RNAs have particular value in the
prevention and treatment of human diseases. In eukaryotic cells,
protein coding RNA accounts for only 2% of the genome, and the vast
majority of transcripts are ncRNAs (43). In recent years, the biogenesis and
functions of ncRNAs have attracted increasing attention (44). The malignant behaviors of cancer
cells are usually regulated by multiple gene products, and,
theoretically, these genes can be grouped into 'networks' based on
their interactions (10,33,45,46).
These interactions may provide new insight into the potential
mechanisms of neoplasia and lay the groundwork for a new approach
to cancer treatment.
miRNAs are the most well-studied ncRNAs, and are a
class of small ncRNAs ~22 nucleotides (nt) in length that, by
binding to MREs, can block protein translation or modulate mRNA
stability on a post-transcriptional level (4,47,48).
Unlike miRNAs, the functions and regulatory mechanisms of circRNAs
remain largely unknown. Fortunately, the application of circRNA
microarray techniques has allowed for the identification of
differentially expressed circRNAs and the exploration of their
functions (49). An increasing
number of circRNAs have been found to be cell type- or
tissue-specific, rendering them more predictive of the potential
biological functions (50,51). Evidence has indicated that the
functions of circRNAs stem from three main aspects: Absorbing miRNA
sponge (24,29,30,52),
binding RNA-binding proteins (RBPs) (16) and translating peptides (53,54).
To date, the miRNA sponge theory remains the most popular
hypothesis.
HNSCC is an aggressive tumor that often occurs with
recurrence and metastasis, and the survival rate has changed little
in recent decades. Over the past few years, an increasing number of
experimental and clinical studies have suggested that ncRNAs play
an important role in HNSCC pathophysiology (55 and refs. therein).
For instance, miR-150-5p and miR-150-3p, antitumor miRNAs, have
been shown to significantly inhibit cell aggressiveness by directly
targeting SPARC (osteonectin), cwcv and kazal like domains
proteoglycan 1 (SPOCK1) in HNSCC (56). miR-206 inhibited cell proliferation
by arresting the cell cycle in S-phase and targeting histone
deacetylase 6 (HDAC6) via the phosphatase and tensin homolog
(PTEN)/AKT/mammalian target of rapamycin (mTOR) pathway (57). Additionally, by evaluating the
associations between miRNA polymorphisms and HNSCC risk according
to the cancer site, miR-605 rs2043556 and miR-196a2 rs11614913 have
been shown as likely to affect genetic susceptibility to oral
squamous cell carcinoma (58).
Evidence has suggested that circRNA_100290 may serve as a sponge of
the miR-29 family to regulate cyclin-dependent kinase 6 (CDK6)
expression in oral squamous cell carcinoma (59).
In the present study, microarray screening and
RT-qPCR verification were used to identify circRNA and mRNA
expression profiles, and ceRNA analysis was performed to evaluate
the circRNA-miRNA-mRNA interactions. The results of microarray
analysis and RT-qPCR revealed that circRNA_036186 and 14-3-3ζ were
highly expressed in HNSCC compared with the controls. Using
bioinformatics analysis, we found that circRNA_036186 and 14-3-3ζ
had the same seed sequence binding site to for interaction with
miR-193b-3p. It has been previously reported that miR-193b-3p and
14-3-3ζ are closely related to the occurrence and prognosis of
cancer (60-64). Existing evidence for miR-193b-3p
has been largely focused on the negative regulation of the
expression of oncogenes in cancer development (65-67),
and the effect on carcinogenesis and prognosis is the most
well-characterized aspect of 14-3-3ζ (68-70).
Furthermore, 14-3-3ζ mRNA and protein expression have been reported
to be upregulated in HNSCC tissue samples, and the overexpression
of 14-3-3ζ promotes cell growth, as well as morphological changes.
By contrast, reduced 14-3-3ζ levels increase the relative
proportion of cells in the G1/G0-phase (71).
The results from DIANA-miRPath v3.0 revealed that
miR-193b-3p significantly engaged in the Hippo signaling pathway
via 14-3-3ζ with the function of cell contact inhibition and organ
size control by combining with yes-associated protein 1 (YAP)/WW
domain-containing transcription regulator protein 1 (TAZ). A
growing body of evidence has suggested that the Hippo pathway may
act as a critical role in organ size control by promoting cell
growth, cell proliferation and preventing apop-tosis (72-75).
A recent study reported that the interaction between 14-3-3ζ and
YAP, as a negative regulatory feedback loop, may play an important
role in cell proliferation and survival in gastric cancer (76). Moreover, using TCGA data, we
identified a group of miRNAs that were relevant to both cancer
development and patient survival in HNSCC. The result revealed that
the expression of miR-193b-3p in HNSCC tissues was significantly
higher than that in the adjacent tissues. However, to date, at
least to the best of our knowledge, there have been no reports on
the functions and mechanisms of the
circRNA_036186-miR-193b-3p-14-3-3ζ pathway in patients with HNSCC.
Considering the data from previous studies in addition to the
results of the current study, the emerging endogenous circRNA
molecule circRNA_036186 probably serves as a crucial regulator of
14-3-3ζ by acting as a miRNA sponge to inhibit miR-193b-3p
function, and hence participates in the Hippo signaling pathway to
exert an effect on the course of HNSCC.
In this study, due to the limited number of
available tissues samples, we only analyzed circRNA and mRNA
expression profiles in five paired HNSCC tissues and their normal
controls. More samples should be obtained and analyzed in the
future to verify our results. Additionally, our results were mainly
based on bioinformatics predictions; whether they can be extended
to wider applications remains undefined. The main limitation of the
present study was that at present, there is no convincing
functional study to verify that circRNA_036186 inhibited
miR-193b-3p to result in regulation of the Hippo signaling pathway;
further functional studies to verify the roles of circRNA_036186
are required.
In conclusion, the findings of this study revealed
the circRNA and mRNA expression characteristics in HNSCC and
highlighted the possibility that circRNA-036186 may serve as a
diagnostic and therapeutic biomarker in HNSCC.
Acknowledgments
Not applicable.
Abbreviations:
|
circRNA
|
circular RNA
|
|
miRNA or miR
|
microRNA
|
|
ncRNA
|
non-coding RNA
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MRE
|
miRNA response element
|
|
ceRNA
|
competing endogenous RNA
|
|
UTR
|
untranslated region
|
|
RBP
|
RNA-binding protein
|
|
TCGA
|
The Cancer Genome Atlas
|
|
BLCA
|
bladder urothelial carcinoma
|
|
BRCA
|
breast invasive carcinoma
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
LUAD
|
lung adenocarcinoma
|
|
LUSC
|
lung squamous cell carcinoma
|
|
UCEC
|
uterine corpus endometrial
carcinoma
|
|
YAP
|
yes-associated protein 1
|
|
TAZ
|
WW domain-containing transcription
regulator protein 1
|
|
14-3-3ζ
|
tyrosine 3-monooxygenase/tryptophan
5-monooxygenase activation protein, ζ polypeptide
|
|
HDAC6
|
histone deacetylase 6
|
|
SPOCK1
|
sparc/osteonectin, cwcv and kazal-like
domains proteoglycan 1
|
|
CDK6
|
cyclin-dependent kinase 6
|
Funding
This study was supported by grants from the National
Natural Science Foundation of China (no. 81372877), the National
Young Scholars Science Foundation of China (no. 81102058), the
Foundation of Education Bureau of Liaoning Province (no. L2014317),
the Natural Science Foundation of Liaoning Province (no.
2014021096), the Excellent Talent Fund Project of Higher Education
of Liaoning Province (no. LJQ2014087), the Doctoral Scientific
Research Foundation of Liaoning Province (no. 201501002), and the
Young and Middle-aged Science and Technology Innovation Talent
Support Project of Shenyang (no. RC170489).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WLW, CFS and FYL conceived and designed the
experiments. ZY and YJZ carried out the experiments. WLW analyzed
the data, prepared the figures and wrote the manuscript. PL and YKN
were mainly involved in the collection and pathological
examinations of patient specimens. CFS supervised the whole
project. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved and supervised by The Ethics
Committee of China Medical University (Shenyang, China). Written
informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Marur S and Forastiere AA: Head and neck
squamous cell carcinoma: Update on epidemiology, diagnosis, and
treatment. Mayo Clin Proc. 91:386–396. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fabian MR, Sonenberg N and Filipowicz W:
Regulation of mRNA translation and stability by microRNAs. Annu Rev
Biochem. 79:351–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li H, Xie S, Liu M, Chen Z, Liu X, Wang L,
Li D and Zhou Y: The clinical significance of downregulation of
mir-124-3p, mir-146a-5p, mir-155-5p and mir-335-5p in gastric
cancer tumorigenesis. Int J Oncol. 45:197–208. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao J, Cai J, Huang D, Han Q, Yang Q, Li
T, Ding H and Wang Z: miR-335 represents an invasion suppressor
gene in ovarian cancer by targeting Bcl-w. Oncol Rep. 30:701–706.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Poliseno L, Salmena L, Zhang J, Carver B,
Haveman WJ and Pandolfi PP: A coding-independent function of gene
and pseudogene mRNAs regulates tumour biology. Nature.
465:1033–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rybak-Wolf A, Stottmeister C, Glažar P,
Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss
R, et al: Circular RNAs in the mammalian brain are highly abundant,
conserved, and dynamically expressed. Mol Cell. 58:870–885. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qu S, Yang X, Li X, Wang J, Gao Y, Shang
R, Sun W, Dou K and Li H: Circular RNA: A new star of noncoding
RNAs. Cancer Lett. 365:141–148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jeck WR, Sorrentino JA, Wang K, Slevin MK,
Burd CE, Liu J, Marzluff WF and Sharpless NE: Circular RNAs are
abundant, conserved, and associated with ALU repeats. RNA.
19:141–157. 2013. View Article : Google Scholar :
|
|
14
|
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin
QF, Xing YH, Zhu S, Yang L and Chen LL: Circular intronic long
noncoding RNAs. Mol Cell. 51:792–806. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen T, Han M, Wei G and Ni T: An
intriguing RNA species - perspectives of circularized RNA. Protein
Cell. 6:871–880. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Z, Huang C, Bao C, Chen L, Lin M, Wang
X, Zhong G, Yu B, Hu W, Dai L, et al: Exon-intron circular RNAs
regulate transcription in the nucleus. Nat Struct Mol Biol.
22:256–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen LL: The biogenesis and emerging roles
of circular RNAs. Nat Rev Mol Cell Biol. 17:205–211. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: circRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilusz JE and Sharp PA: Molecular biology.
A circuitous route to noncoding RNA. Science. 340:440–441. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vicens Q and Westhof E: Biogenesis of
circular RNAs. Cell. 159:13–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL
and Yang L: Complementary sequence-mediated exon circularization.
Cell. 159:134–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Westholm JO, Miura P, Olson S, Shenker S,
Joseph B, Sanfilippo P, Celniker SE, Graveley BR and Lai EC:
Genome-wide analysis of drosophila circular RNAs reveals their
structural and sequence properties and age-dependent neural
accumulation. Cell Reports. 9:1966–1980. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Suzuki H, Aoki Y, Kameyama T, Saito T,
Masuda S, Tanihata J, Nagata T, Mayeda A, Takeda S and Tsukahara T:
Endogenous multiple exon skipping and back-splicing at the DMD
mutation hotspot. Int J Mol Sci. 17:172016. View Article : Google Scholar
|
|
24
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen
B, Luo Y, Lyu D, Li Y, Shi G, et al: Circular RNA profiling reveals
an abundant circHIPK3 that regulates cell growth by sponging
multiple miRNAs. Nat Commun. 7:112152016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo JU, Agarwal V, Guo H and Bartel DP:
Expanded identification and characterization of mammalian circular
RNAs. Genome Biol. 15:4092014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ghosal S, Das S, Sen R, Basak P and
Chakrabarti J: Circ2Traits: A comprehensive database for circular
RNA potentially associated with disease and traits. Front Genet.
4:2832013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lukiw WJ: Circular RNA (circRNA) in
Alzheimer's disease (AD). Front Genet. 4:3072013. View Article : Google Scholar
|
|
30
|
Hansen TB, Kjems J and Damgaard CK:
Circular RNA and miR-7 in cancer. Cancer Res. 73:5609–5612. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in Drosophila. Genome Biol.
5:R12003. View Article : Google Scholar
|
|
32
|
Pasquinelli AE: MicroRNAs and their
targets: Recognition, regulation and an emerging reciprocal
relationship. Nat Rev Genet. 13:271–282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Assenov Y, Ramírez F, Schelhorn SE,
Lengauer T and Albrecht M: Computing topological parameters of
biological networks. Bioinformatics. 24:282–284. 2008. View Article : Google Scholar
|
|
34
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wong NW, Chen Y, Chen S and Wang X:
OncomiR: An online resource for exploring pan-cancer microRNA
dysregulation. Bioinformatics. 34:713–715. 2018. View Article : Google Scholar
|
|
36
|
Chandrashekar DS, Bashel B, Balasubramanya
SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi BVSK and
Varambally S: UALCAN: A portal for facilitating tumor subgroup gene
expression and survival analyses. Neoplasia. 19:649–658. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Vlachos IS, Zagganas K, Paraskevopoulou
MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T and
Hatzigeorgiou AG: DIANA-miRPath v3.0: Deciphering microRNA function
with experimental support. Nucleic Acids Res. 43W1:W460–W466. 2015.
View Article : Google Scholar
|
|
38
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42D1:D92–D97. 2014. View Article : Google Scholar
|
|
39
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
indexing more than half a million experimentally supported
miRNA:mRNA interactions. Nucleic Acids Res. 43D:D153–D159. 2015.
View Article : Google Scholar
|
|
40
|
Paraskevopoulou MD, Georgakilas G,
Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C,
Dalamagas T and Hatzigeorgiou AG: DIANA-microT web server v5.0:
Service integration into miRNA functional analysis workflows.
Nucleic Acids Res. 41W:W169–W173. 2013. View Article : Google Scholar
|
|
41
|
Reczko M, Maragkakis M, Alexiou P, Grosse
I and Hatzigeorgiou AG: Functional microRNA targets in protein
coding sequences. Bioinformatics. 28:771–776. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
43
|
Alexander RP, Fang G, Rozowsky J, Snyder M
and Gerstein MB: Annotating non-coding regions of the genome. Nat
Rev Genet. 11:559–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mei Y, Yang JP and Qian CN: For robust big
data analyses: A collection of 150 important pro-metastatic genes.
Chin J Cancer. 36:162017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qian CN, Mei Y and Zhang J: Cancer
metastasis: Issues and challenges. Chin J Cancer. 36:382017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Carthew RW and Sontheimer EJ: Origins and
mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Caiment F, Gaj S, Claessen S and Kleinjans
J: High-throughput data integration of RNA-miRNA-circRNA reveals
novel insights into mechanisms of benzo[a]pyrene-induced
carcinogenicity. Nucleic Acids Res. 43:2525–2534. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia S, Feng J, Lei L, Hu J, Xia L, Wang J,
Xiang Y, Liu L, Zhong S, Han L, et al: Comprehensive
characterization of tissue-specific circular RNAs in the human and
mouse genomes. Brief Bioinform. 18:984–992. 2017.
|
|
51
|
Salzman J, Chen RE, Olsen MN, Wang PL and
Brown PO: Cell-type specific features of circular RNA expression.
PLoS Genet. 9:e10037772013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Granados-Riveron JT and Aquino-Jarquin G:
The complexity of the translation ability of circRNAs. Biochim
Biophys Acta. 1859:1245–1251. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang Y and Wang Z: Efficient backsplicing
produces translatable circular mRNAs. RNA. 21:172–179. 2015.
View Article : Google Scholar :
|
|
55
|
Sannigrahi MK, Sharma R, Panda NK and
Khullar M: Role of non-coding RNAs in head and neck squamous cell
carcinoma: A narrative review. Oral Dis. Sep 21–2017.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Koshizuka K, Hanazawa T, Kikkawa N, Katada
K, Okato A, Arai T, Idichi T, Osako Y, Okamoto Y and Seki N:
Antitumor miR-150-5p and miR-150-3p inhibit cancer cell
aggressiveness by targeting SPOCK1 in head and neck squamous cell
carcinoma. Auris Nasus Larynx. 45:854–865. 2018. View Article : Google Scholar
|
|
57
|
Liu F, Zhao X, Qian Y, Zhang J, Zhang Y
and Yin R: miR-206 inhibits head and neck squamous cell carcinoma
cell progression by targeting HDAC6 via PTEN/AKT/mTOR pathway.
Biomed Pharmacother. 96:229–237. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Miao L, Wang L, Zhu L, Du J, Zhu X, Niu Y,
Wang R, Hu Z, Chen N, Shen H, et al: Association of microRNA
polymorphisms with the risk of head and neck squamous cell
carcinoma in a Chinese population: A case-control study. Chin J
Cancer. 35:772016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen L, Zhang S, Wu J, Cui J, Zhong L,
Zeng L and Ge S: circRNA_100290 plays a role in oral cancer by
functioning as a sponge of the miR-29 family. Oncogene.
36:4551–4561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rauhala HE, Jalava SE, Isotalo J, Bracken
H, Lehmusvaara S, Tammela TL, Oja H and Visakorpi T: miR-193b is an
epigenetically regulated putative tumor suppressor in prostate
cancer. Int J Cancer. 127:1363–1372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Leivonen SK, Rokka A, Ostling P, Kohonen
P, Corthals GL, Kallioniemi O and Perälä M: Identification of
miR-193b targets in breast cancer cells and systems biological
analysis of their functional impact. Mol Cell Proteomics.
10:M110.0053222011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mitra AK, Chiang CY, Tiwari P, Tomar S,
Watters KM, Peter ME and Lengyel E: Microenvironment-induced
downregulation of miR-193b drives ovarian cancer metastasis.
Oncogene. 34:5923–5932. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhao GY, Ding JY, Lu CL, Lin ZW and Guo J:
The overexpression of 143-3ζ and Hsp27 promotes non-small cell lung
cancer progression. Cancer. 120:652–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rehman SK, Li SH, Wyszomierski SL, Wang Q,
Li P, Sahin O, Xiao Y, Zhang S, Xiong Y, Yang J, et al: 14-3-3ζ
orchestrates mammary tumor onset and progression via
miR-221-mediated cell proliferation. Cancer Res. 74:363–373. 2014.
View Article : Google Scholar
|
|
65
|
Li J, Kong F, Wu K, Song K, He J and Sun
W: miR-193b directly targets STMN1 and uPA genes and suppresses
tumor growth and metastasis in pancreatic cancer. Mol Med Rep.
10:2613–2620. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Mets E, Van der Meulen J, Van Peer G,
Boice M, Mestdagh P, Van de Walle I, Lammens T, Goossens S, De
Moerloose B, Benoit Y, et al: MicroRNA-193b-3p acts as a tumor
suppressor by targeting the MYB oncogene in T-cell acute
lymphoblastic leukemia. Leukemia. 29:798–806. 2015. View Article : Google Scholar
|
|
67
|
Zhang J, Qin J and Su Y: miR-193b-3p
possesses anti-tumor activity in ovarian carcinoma cells by
targeting p21-activated kinase 3. Biomed Pharmacother.
96:1275–1282. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Matta A, Bahadur S, Duggal R, Gupta SD and
Ralhan R: Over-expression of 14-3-3zeta is an early event in oral
cancer. BMC Cancer. 7:1692007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Nishimura Y, Komatsu S, Ichikawa D, Nagata
H, Hirajima S, Takeshita H, Kawaguchi T, Arita T, Konishi H,
Kashimoto K, et al: Overexpression of YWHAZ relates to tumor cell
proliferation and malignant outcome of gastric carcinoma. Br J
Cancer. 108:1324–1331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Murata T, Takayama K, Urano T, Fujimura T,
Ashikari D, Obinata D, Horie-Inoue K, Takahashi S, Ouchi Y, Homma
Y, et al: 14-3-3zeta, a novel androgen-responsive gene, is
upregulated in prostate cancer and promotes prostate cancer cell
proliferation and survival. Clin Cancer Res. 18:5617–5627. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lin M, Morrison CD, Jones S, Mohamed N,
Bacher J and Plass C: Copy number gain and oncogenic activity of
YWHAZ/14-3-3zeta in head and neck squamous cell carcinoma. Int J
Cancer. 125:603–611. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yao CB, Zhou X, Chen CS and Lei QY: The
regulatory mechanisms and functional roles of the Hippo signaling
pathway in breast cancer. Yi Chuan. 39:617–629. 2017.PubMed/NCBI
|
|
73
|
Patel SH, Camargo FD and Yimlamai D: Hippo
signaling in the liver regulates organ size, cell fate, and
carcinogenesis. Gastroenterology. 152:533–545. 2017. View Article : Google Scholar :
|
|
74
|
Ji XY, Zhong G and Zhao B: Molecular
mechanisms of the mammalian Hippo signaling pathway. Yi Chuan.
39:546–567. 2017.PubMed/NCBI
|
|
75
|
Pan D: The hippo signaling pathway in
development and cancer. Dev Cell. 19:491–505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang B, Gong A, Shi H, Bie Q, Liang Z, Wu
P, Mao F, Qian H and Xu W: Identification of a novel YAP-14-3-3ζ
negative feedback loop in gastric cancer. Oncotarget.
8:71894–71910. 2017.PubMed/NCBI
|