Introduction
Breast cancer is one of the most prevalent female
malignancies and the second leading cause of mortality among all
cancers in women (1).
Approximately 70% of breast cancers are estrogen receptor
(ER)-positive and can be treated with endocrine therapy, such as
tamoxifen (TAM) (2). TAM is an
anti-estrogen agent, which suppresses ER activity by competitively
binding to the ER in ER-positive breast cancer (3). TAM has been used as a first-line
adjuvant treatment for patients with ER-positive breast cancer;
however, 50% of patients with ER-positive breast cancer will
eventually develop TAM resistance, presenting a huge obstacle to
breast cancer treatment (4).
Therefore, further understanding of the mechanism underlying TAM
resistance may provide novel therapeutic strategies to overcome
drug resistance.
MicroRNAs (miRNAs/miRs) function as
post-transcriptional regulators by binding to the 3′-untranslated
region (3′-UTR) of target mRNAs, thus resulting in translational
repression or degradation of the target mRNAs (5). Accumulating evidence has suggested
that dysregulation of miRNAs serves an important role in cancer
development (6,7). miR-26a is localized in the introns of
genes coding for carboxy-terminal domain RNA polymerase II
polypeptide A small phosphatase family proteins: CTD small
phosphatase like (CTDSPL; miR-26a-1 host gene) and CTD small
phosphatase 2 (CTDSP2; miR-26a-2 host gene) (8,9).
miR-26a exhibits low expression in breast cancer, and inhibits
breast carcinogenesis and metastasis by directly targeting enhancer
of zeste 2 polycomb repressive complex 2 subunit (EZH2) and
metadherin (10,11). A previous study reported that
increasing levels of miR-26a are significantly associated with
clinical outcomes of TAM treatment in breast cancer (12). Furthermore, miR-26a is
differentially expressed in TAM-resistant breast cancer cells
compared with in the parental MCF-7 cell line (13); however, the mechanism by which
miR-26a regulates TAM resistance is unclear.
The E2F transcription factor (E2F) family can be
divided into transcriptional activators (E2F1-E2F3) and repressors
(E2F4-E2F8) (14). The E2F family
members have an important role in cell cycle control (15). E2F7 overexpression in keratinocytes
and osteosarcoma cells results in an accumulation of G1 phase cells
and proliferation suppression (15–17).
Conversely, E2F7 is able to inhibit apoptosis by repressing E2F1
activity (16,18). Notably, previous studies have
revealed that E2F7 is overexpressed in numerous types of cancer,
such as squamous cell carcinoma (SCC), endometrial cancer and
ovarian cancer (16,19,20).
Furthermore, in SCC cells, inhibition of E2F7 increases sensitivity
to ultraviolet-induced DNA damage and doxorubicin-induced
cytotoxicity by antagonizing E2F1-induced apoptosis (16). In addition, E2F7 can directly
increase the transcription and activity of the sphingosine
kinase-1/sphingosine 1-phosphate axis, resulting in protein kinase
B activation and subsequent doxorubicin resistance (21). Therefore, E2F7 may serve an
important role in drug resistance during cancer therapy.
The aim of the study was to explore the role of
miR-26a and E2F7 in TAM resistance. The present study demonstrated
that miR-26a was inhibited in ER-positive breast cancer, whereas
E2F7 was significantly elevated. miR-26a directly inhibited E2F7
expression through translational inhibition and indirectly
inhibited MYC proto-oncogene, bHLH transcription factor (MYC)
expression partly through suppressing E2F7. E2F7 knockdown, in
turn, promoted miR-26a expression by decreasing MYC recruitment to
the miR-26a gene promoter. In addition, both miR-26a knockdown and
E2F7 overexpression conferred resistance to TAM in breast cancer
cells. These findings suggested that miR-26a and E2F7 may form a
double-negative feedback loop that contributes to TAM resistance in
ER-positive breast cancer.
Materials and methods
Cell culture and transfection
Breast cancer cell lines MCF-7, T47D, BT474, SKBR3,
MDA-MB-231 and Hs578T, and the normal breast cell line MCF-10A were
purchased from American Type Culture Collection (Manassas, VA,
USA). The breast cancer cells were cultured in Dulbecco's modified
Eagle's Medium (DMEM; Hyclone; GE Healthcare Life Sciences, Logan,
UT, USA) supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a
humidified atmosphere containing 5% CO2. The MCF-10A
cell line was cultured in DMEM/F12 (1:1) (Hyclone; GE Healthcare
Life Sciences) supplemented with 5% FBS, 10 µg/ml insulin,
20 ng/ml epidermal growth factor, 100 ng/ml cholera toxin and 0.5
mg/ml hydrocortisone at 37°C and 5% CO2. TAM-resistant
MCF-7 cells (MCF-7R) were established from MCF-7 cells following a
long-term culture with 1 µM 4-hydroxytamoxifen
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) as previously
described (22,23).
miR-26a mimics, miR-26a inhibitors, E2F7 small
interfering RNA (siRNA) and MYC siRNA, and the corresponding
negative controls were synthesized by Shanghai GenePharma Co., Ltd.
(Shanghai, China). The sequences of the oligonucleotides are
provided in Table I. All
transfections were conducted using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. MCF7 and T47D are widely used as
experimental models in the study of ER-positive breast cancer;
therefore, the subsequent experiments were mainly performed using
these two cell lines. Briefly, MCF-7, T47D and MCF-7R cells were
plated in 6-well plates at a density of 1×106 cells/well
overnight. Subsequently, the cells were transfected with miR-26a
mimics or inhibitors, siRNA or corresponding negative controls (50
nM). Total RNA and protein were isolated 48 or 72 h
post-transfection, respectively.
 | Table IOligonucleotides used in the present
study. |
Table I
Oligonucleotides used in the present
study.
| Gene name | Sequence
(5′-3′) |
|---|
| Primers for
RT-qPCR | |
| E2F7-F |
GTCAGCCCTCACTAAACCTAAG |
| E2F7-R |
TGCGTTGGATGCTCTTGG |
| MYC-F |
TTCGGGTAGTGGAAAACCAG |
| MYC-R |
CAGCAGCTCGAATTTCTTCC |
| CTDSPL-F |
TGCTGAGGGAGGGGAGTGAG |
| CTDSPL-R |
GCAGCATGCCACAGGTTGTC |
| CTDSP2-F |
ATGTTGGCCAGTCAAGTTCC |
| CTDSP2-R |
CTGTCACCTCTGGGAGCAG |
| ACTB-F |
CCTTCTACAATGAGCTGCGT |
| ACTB-R |
CCTGGATAGCAACGTACATG |
| miR-26a-F |
GCCCGCTTCAAGTAATCCAGG |
| miR-26a-R |
GTGCAGGGTCCGAGGT |
| U6-F |
CCTGCGCAAGGATGAC |
| U6-R |
GTGCAGGGTCCGAGGT |
|
ChIP-miR-26a1-F |
GGAGAGACTGGGAGCGAGTGT |
|
ChIP-miR-26a1-R |
CAAACTCACAACCTCCCGGT |
|
ChIP-miR-26a2-F |
CTCCATCTGTGAGCGGCC |
|
ChIP-miR-26a2-R |
AAAATAGCAAAGCTCCCGACTG |
| Primers for plasmid
construction | |
|
pcDNA3.1-E2F7-F |
CTCTAGATAGGAAAGCAGGGATGGA |
|
pcDNA3.1-E2F7-R |
GGAATTCTCACGATGTGTGCGTTGG |
| Other
oligonucleotides | |
| miR-26a mimics
(sense) |
UUCAAGUAAUCCAGGAUAGGCU |
| miR-26a mimics
(antisense) |
CCUAUCCUGGAUUACUUGAAUU |
| miR-26a
inhibitors |
AGCCUAUCCUGGAUUACUUGAA |
| E2F7 siRNA
(sense) |
GCAAAUGGCCUACCUCCAATT |
| E2F7 siRNA
(antisense) |
UUGGAGGUAGGCCAUUUGCTT |
| MYC siRNA
(sense) |
GGUGAUCCAGACUCUGACCUU |
| MYC siRNA
(antisense) |
AAGGUCAGAGUCUGGAUCACC |
Clinical samples
All clinical samples (35 pairs of ER-positive breast
cancer and normal adjacent breast tissues) were obtained from the
First Affiliated Hospital of Xi'an Jiaotong University (Xi'an,
China) between September 2015 and October 2017. The present study
was approved by the Ethics Committee of Xi'an Jiaotong University
First Affiliated Hospital and each patient provided written
informed consent. The specimens were resected and frozen in liquid
nitrogen immediately after surgery. None of the patients received
chemotherapy or radiation therapy prior to the study. Patient
characteristics are summarized in Table II. Classification of the tumors as
ER-positive breast cancer was determined according to the 2009 St.
Gallen's Consensus guidelines for ER and progesterone receptor
(PgR) markers (24). In addition,
to explore the clinical significance of miR-26a and E2F7 in
ER-positive breast cancer, RNA-Sequencing and miRNA-Sequencing data
from The Cancer Genome Atlas (TCGA) database (http://tcga-data.nci.nih.gov/) were analyzed. A total
of 431 ER-positive breast cancer cases with explicit ER, PgR, human
epidermal growth factor receptor 2 and menopausal status data were
included in the analysis.
| Table IIPatient characteristics. |
Table II
Patient characteristics.
|
Characteristics | Count |
|---|
| Age (mean ±
standard deviation) | 50.4±10.8 |
| TNM stage | |
| I–II | 25 |
| III | 10 |
| PgR status | |
| Positive | 27 |
| Negative | 8 |
| HER2 status | |
| Positive | 16 |
| Negative | 19 |
| Histological
type | |
| Ductal | 31 |
| Lobular | 4 |
Plasmid construction
To construct an E2F7-expressing plasmid
(pcDNA3.1-E2F7), a 2,795-bp DNA fragment containing the coding
sequence of E2F7 was amplified by PCR from MCF-7 cDNA and cloned
into the XbaI and EcoRI sites of a pcDNA3.1 vector
(Invitrogen; Thermo Fisher Scientific, Inc.). The cells were plated
in 6-well plates (1×106 cells/well) overnight to ensure
70% confluence. Then the cells were transfected with the vectors (4
µg/well) using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C for 6 h. The primers used
are shown in Table I. The pcDNA3.1
(+) vector was used as a negative control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. cDNA was synthesized from RNA using a
PrimeScript™ RT Reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China) as previously described (25). RT-qPCR was performed using
SYBR® Premix Ex Taq™ II (Tli RNaseH Plus 2X; Takara
Biotechnology Co., Ltd.) on a CFX96™ Real-Time PCR Detection system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The thermocycling
conditions were as follows: 30 sec at 95°C, followed by 40 cycles
of 5 sec at 95°C, 30 sec at 60°C and 72°C for 45 sec. U6 and
β-actin were used as internal controls for miR-26a and E2F7,
respectively. The relative expression levels of target genes were
calculated using the 2−ΔΔCq method (26). The PCR primer sequences are
presented in Table I.
Western blot analysis
Total protein was extracted from cells using
radioimmunoprecipitation assay buffer (Thermo Fisher Scientific,
Inc.) with proteinase inhibitor (Roche Applied Science, Mannheim,
Germany). The protein concentration was measured using a Protein
Bicinchoninic Acid Assay kit (Thermo Scientific, Inc.). An equal
amount of protein (20 µg) mixed with 2X SDS loading buffer
was loaded per lane. The proteins were separated by 10% SDS-PAGE
and transferred onto nitrocellulose membranes (Bio-Rad
Laboratories, Inc.). The membranes were incubated at room
temperature for 2 h with 5% nonfat milk to block nonspecific
binding. Subsequently, the membranes were incubated for 12 h at 4°C
with the following primary antibodies: Anti-MYC (1:1,000; cat. no.
sc-764; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and
anti-E2F7 (1:1,000; cat. no. ab56022; Abcam, Cambridge, MA, USA).
The membranes were then incubated with horseradish
peroxidase-conjugated secondary antibodies (1:5,000; cat. no.
sc-2004; Santa Cruz Biotechnology, Inc.) at room temperature for 2
h. Anti-β-actin antibody (1:5,000; A5441; Sigma-Aldrich, Merck
KGaA) was used as a loading control. The protein bands were
visualized using an enhanced chemiluminescence substrate (EMD
Millipore, Billerica, MA, USA). The results were quantified using
ImageJ software (version 1.50b; National Institutes of Health,
Bethesda, MD, USA).
MTT assay
A total of 24 h post-transfection, the cells were
seeded in 96-well plates at a density of 4×103
cells/well. Following treatment with or without 1 µM
4-hydroxytamoxifen for 24, 48, 72 and 96 h, cell viability was
detected using the MTT assay. MTT solution (20 µl; 5 mg/ml)
was added to each well and the cells were incubated at 37°C for 4
h. Subsequently, the medium containing MTT solution was discarded,
and 150 µl dimethyl sulfoxide was added to the wells. The
absorbance of each well was measured at 570 nm using a microplate
reader (Bio-Rad Laboratories, Inc.).
Cell cycle analysis
MCF-7 and MCF-7R cells were seeded in the presence
of 1 µM 4-hydroxytamoxifen or ethanol solvent. After 72 h,
the cells were trypsinized, washed with PBS and fixed with 70%
ethanol at 4°C overnight. The cells were then stained with
propidium iodide (Sigma-Aldrich; Merck KGaA) for 30 min and
analyzed by flow cytometry (FACSCalibur; BD Biosciences, Franklin
Lakes, NJ, USA) with the ModFit software (Verity Software House,
Inc., Topsham, ME, USA).
Luciferase activity assay
The Targetscan database (http://www.targetscan.org/mamm_31/) was used to
identify predicted targets of miR-26a. A fragment of the E2F7
3′-UTR containing two miR-26a-binding sites was cloned into a
pGL3-control vector (Promega Corporation, Madison, WI, USA). The
seed sequences in the E2F7 3′-UTR complementary to miR-26a were
mutated using a QuikChange II Site-Directed Mutagenesis kit
(Stratagene; Agilent Technologies GmbH, Waldbronn, Germany). MCF-7
cells were plated in 24-well plates and co-transfected with 100 ng
firefly luciferase report vector and 10 ng pRL-TK vector containing
Renilla luciferase (Promega Corporation), together with 50
nM miR-26a mimics or negative control mimics. Co-transfection was
performed with Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C for 6 h. Subsequently, 48 h
post-transfection, the cells were assayed for firefly and
Renilla luciferase activities using a Dual-Luciferase
Reporter Assay system (Promega Corporation), according to
manufacturer's protocol.
Chromatin immunoprecipitation (ChIP)
The miR-26a promoter sequence was analyzed using
UCSC Genome Browser (http://genome.ucsc.edu). ChIP was performed as
previously described (27,28). Briefly, cells were cross-linked
with 1% formaldehyde for 10 min at 37°C and resuspended in lysis
buffer on ice for 15 min. Samples were sonicated on ice at a
frequency of 20 kHz (on 5 sec and off 12 sec for 13 cycles) to
shear chromatin to fragments of 200–1,000 base pairs. The DNA
fragments were used in immunoprecipitation with the antibody
anti-MYC (cat. no. sc-764; Santa Cruz Biotechnology, Inc.). The
primers used for subsequent RT-qPCR are presented in Table I.
Statistical analysis
All statistical analyses were performed using SPSS
software (version 20.0; IBM Corp., Armonk, NY, USA). All data are
presented as the means ± standard deviation of at least three
independent experiments. The statistical significance of continuous
variables was determined using Student's t-test (two-tailed) or
Mann-Whitney U test. Multiple group comparisons were analyzed with
one-way analysis of variance followed by Dunnett's or least
significant difference post hoc tests. For categorical variables,
statistical significance was determined using the χ2
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of miR-26a and E2F7 in
ER-positive breast cancer
The present study analyzed miR-26a and E2F7
expression in 35 pairs of ER-positive breast cancer tissues and
matched adjacent normal breast tissues using RT-qPCR. The results
demonstrated that miR-26a expression was reduced in ER-positive
breast cancer compared with in the matched normal breast tissues,
whereas E2F7 expression was significantly elevated (Fig. 1A). In addition, E2F7 expression was
inversely correlated with miR-26a expression in ER-positive breast
cancer (Fig. 1B). Subsequently,
the present study determined that miR-26a levels in breast cancer
cell lines were decreased compared with in the MCF-10A cell line
(Fig. 1C). These results suggested
that miR-26a may be downregulated in ER-positive breast cancer,
whereas E2F7 may be upregulated.
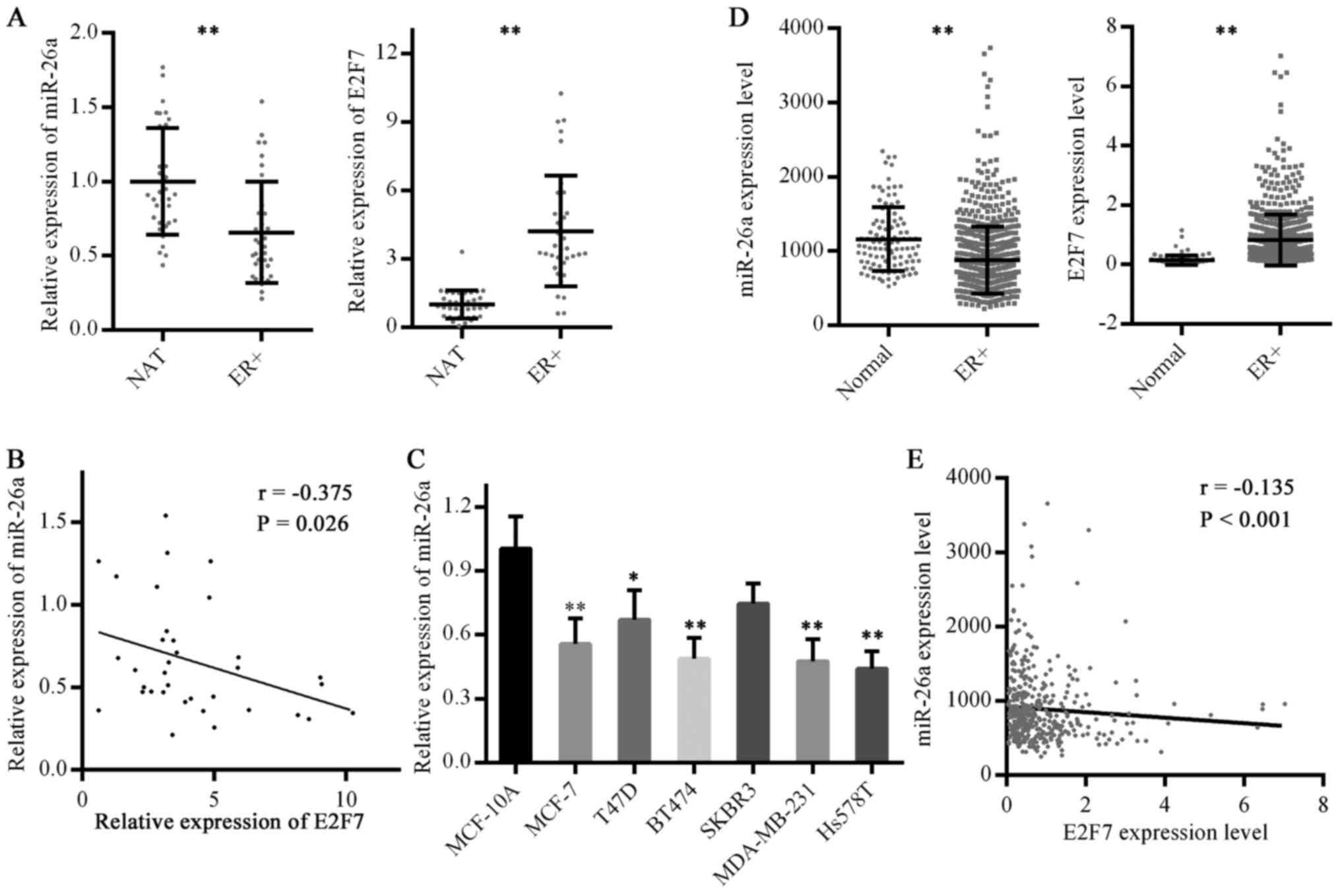 | Figure 1Expression levels of miR-26a and E2F7
in ER-positive breast cancer. (A) miR-26a and E2F7 expression in
ER-positive breast cancer was detected by RT-qPCR. NAT, n=35,
ER+, n=35. **P<0.01. (B) Correlation
between miR-26a and E2F7 expression in ER-positive breast cancer
was analyzed. (C) miR-26a levels in breast cancer cell lines and a
normal breast cell line were analyzed by RT-qPCR. Data are
presented as the means ± standard deviation of three independent
experiments. *P<0.05 and **P<0.01 vs.
MCF-10A cells (one-way analysis of variance followed by Dunnett's
test). (D) miR-26a expression data in TCGA miRNA-Seq and E2F7
expression data in TCGA RNA-Seq were analyzed. Normal, n=104 for
miRNA-Seq and n=113 for RNA-Seq; ER+, n=431.
**P<0.01. (E) Correlation between miR-26a and E2F7
expression in ER-positive breast cancer cases in TCGA database was
analyzed. E2F7, E2F transcription factor 7; ER, estrogen receptor;
miR-26a, microRNA 26a; NAT, normal adjacent breast tissues; r,
correlation coefficient; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; TCGA, The
Cancer Genome Atlas. |
To verify this conclusion, miR-26a and E2F7
expression were evaluated in ER-positive breast cancer cases
contained in TCGA) database. Consistent with the RT-qPCR results,
miR-26a exhibited reduced expression, whereas E2F7 expression was
higher, in ER-positive breast cancer compared with in normal breast
tissues (Fig. 1D). In addition, an
inverse correlation between miR-26a and E2F7 expression was
identified (Fig. 1E).
Subsequently, the association of miR-26a and E2F7 expression with
clinicopathological factors was analyzed using TCGA data. The
results demonstrated that both miR-26a and E2F7 expression levels
were associated with histological type; in addition, miR-26a levels
were correlated with tumor size and E2F7 levels with age at
diagnosis (Table III).
| Table IIIAssociation of miR-26a and E2F7
expression levels with clinicopathological factors analyzed using
The Cancer Genome Atlas datasets. |
Table III
Association of miR-26a and E2F7
expression levels with clinicopathological factors analyzed using
The Cancer Genome Atlas datasets.
| Clinicopathological
factor | miR-26a level
| E2F7 level
|
|---|
| Low (n=215) | High (n=216) | P-value | Low (n=215) | High (n=216) | P-value |
|---|
| Age (years) | | | 0.290 | | | 0.012 |
| <60 | 114 | 103 | | 95 | 122 | |
| >60 | 101 | 113 | | 120 | 94 | |
| Tumor size
(cm) | | | 0.026 | | | 0.436 |
| <5 | 189 | 172 | | 177 | 184 | |
| >5 | 26 | 44 | | 38 | 32 | |
| Lymph node
metastasis | | | 0.123 | | | 0.965 |
| Negative | 113 | 96 | | 99 | 99 | |
| Positive | 102 | 120 | | 116 | 117 | |
| Distant
metastasis | | | 0.623 | | | 0.996 |
| Negative | 214 | 213 | | 213 | 214 | |
| Positive | 1 | 3 | | 2 | 2 | |
| PgR status | | | 0.895 | | | 0.234 |
| Positive | 182 | 181 | | 186 | 177 | |
| Negative | 33 | 35 | | 29 | 39 | |
| HER2 status | | | 0.067 | | | 0.135 |
| Positive | 57 | 41 | | 42 | 56 | |
| Negative | 158 | 175 | | 173 | 160 | |
| Histological
type | | | <0.001 | | | <0.001 |
| Ductal | 183 | 138 | | 136 | 185 | |
| Lobular | 32 | 78 | | 79 | 31 | |
| Menopausal
status | | | 0.181 | | | 0.146 |
| Premenopausal | 59 | 47 | | 46 | 60 | |
|
Postmenopausal | 156 | 169 | | 169 | 156 | |
miR-26a inhibits E2F7 and MYC
expression
A previous study demonstrated that miR-26a inhibits
E2F7 expression by directly binding to its 3′-UTR in acute myeloid
leukemia (AML) cells (29). To
further investigate whether miR-26a inhibits E2F7 expression in
breast cancer cells, the present study introduced miR-26a mimics
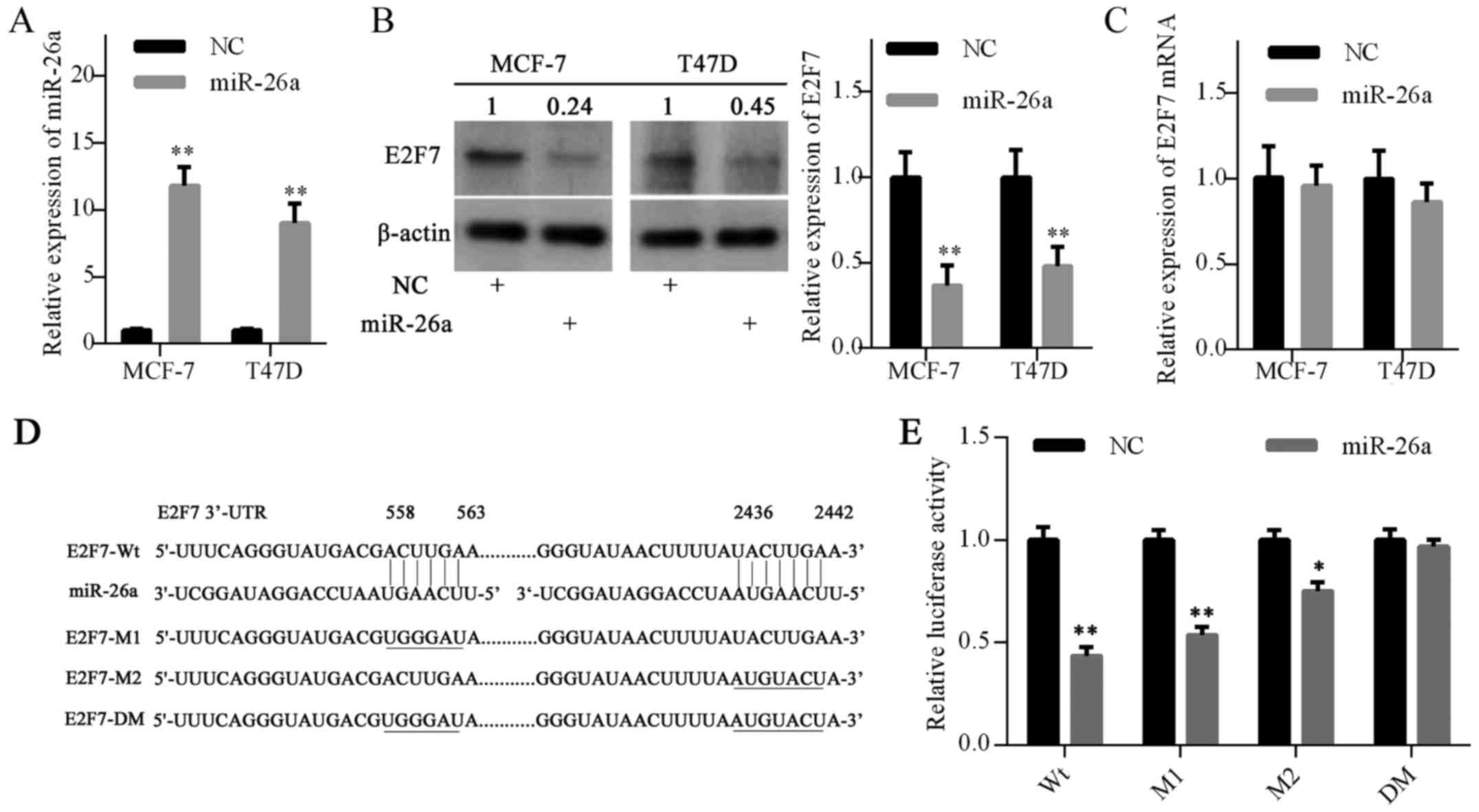
into MCF-7 and T47D cells. RT-qPCR analysis demonstrated that
miR-26a expression was elevated in cells transfected with miR-26a
mimics compared with the negative control (Fig. 2A). Furthermore, western blot
analysis confirmed that E2F7 protein expression was decreased in
the miR-26a mimics groups (Fig.
2B). However, miR-26a overexpression did not decrease E2F7 mRNA
expression (Fig. 2C).
To further confirm the targeting of E2F7 by miR-26a,
a luciferase activity assay was performed in MCF-7 cells. Using the
TargetScan database, two putative binding sites for miR-26a were
predicted in the E2F7 mRNA 3′-UTR (Fig. 2D). Luciferase reporter vectors were
constructed with the wild-type 3′-UTR of E2F7 (E2F7-Wt), a mutant
3′-UTR of E2F7 at site 1 or site 2 (E2F7-M1 or M2) or a mutant
3′-UTR of E2F7 at both sites (E2F7-DM) (Fig. 2D). As expected, luciferase activity
was significantly reduced following trans-fection with the E2F7-Wt
vector. The E2F7-M1 vector slightly abrogated the inhibitory
effects of miR-26a; however, this effect was more significant with
the E2F7-M2 vector. Notably, E2F7-DM vector completely eradicated
the inhibitory effects of miR-26a (Fig. 2E). Collectively, these results
suggested that miR-26a may inhibit E2F7 expression by directly
targeting the 3′-UTR of E2F7 mRNA.
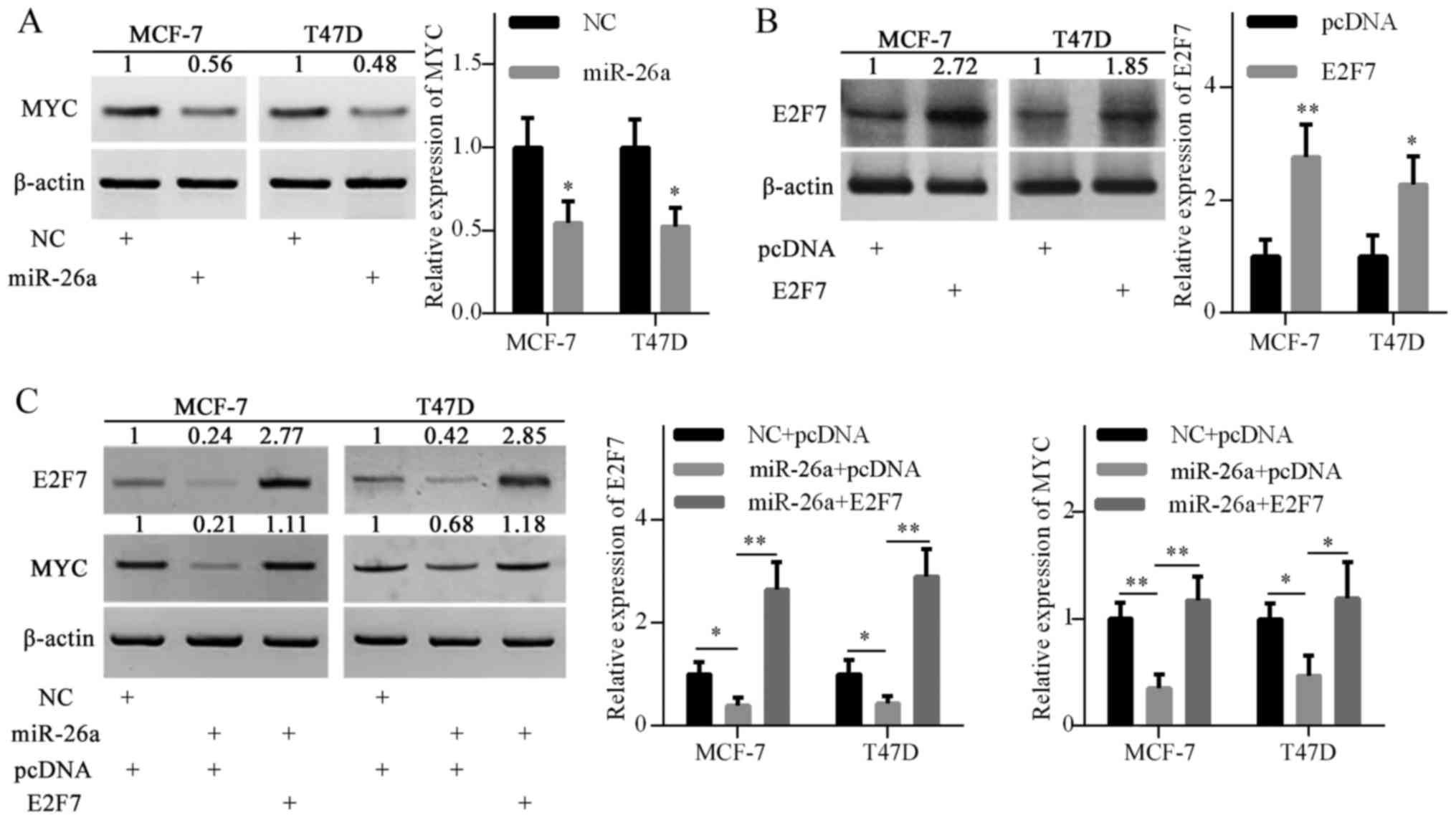
miR-26a overexpression also inhibited MYC protein
expression, as shown in Fig. 3A.
To investigate the possible mechanism, the pcDNA3.1-E2F7 plasmid
was introduced into MCF-7 and T47D cells to elevate E2F7 expression
(Fig. 3B). Ectopic expression of
E2F7 restored MYC gene expression, which was inhibited by miR-26a
overexpression (Fig. 3C). These
results suggested that miR-26a may indirectly inhibit MYC
expression, at least partly via E2F7 repression.
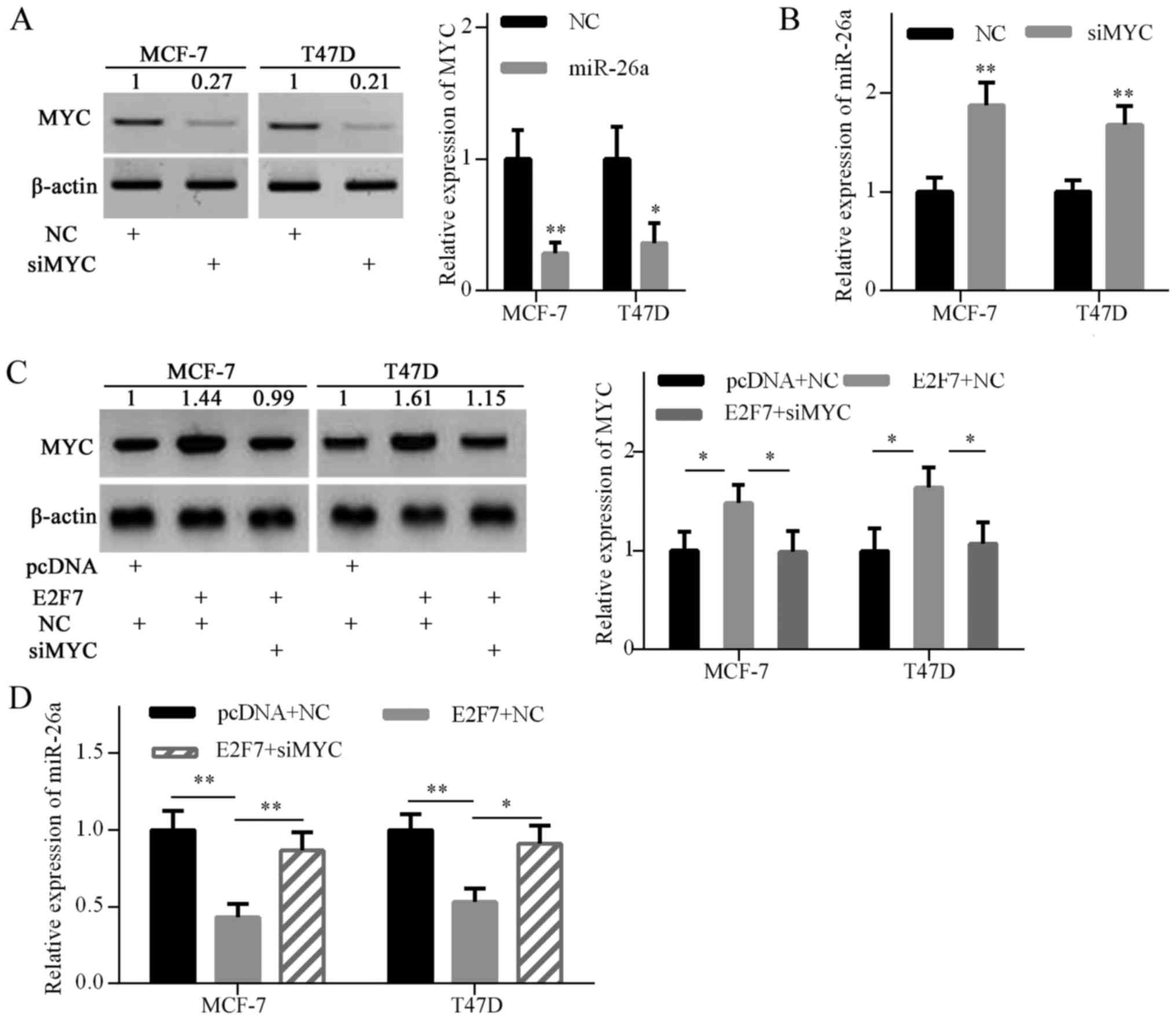
E2F7 overexpression inhibits miR-26a via
MYC upregulation
To confirm whether MYC is involved in E2F7-induced
miR-26a repression, MYC gene expression was knocked down with siRNA
in breast cancer cells (Fig. 4A).
As expected, MYC knockdown significantly increased miR-26a
expression in MCF-7 and T47D cells (Fig. 4B). To further explore whether E2F7
inhibited miR-26a expression via upregulating MYC expression,
E2F7-expressing plasmids and MYC siRNA were cotransfected into
MCF-7 and T47D cells. Western blot analysis confirmed that MYC
expression was elevated by E2F7 overexpression in both breast
cancer cell lines, and decreased by siRNA in E2F7-overexpressing
cells (Fig. 4C). RT-qPCR analysis
indicated that E2F7 overexpression significantly decreased miR-26a
expression, and MYC knockdown abrogated E2F7-induced miR-26a
repression (Fig. 4D). These
findings suggested that E2F7 overexpression inhibited miR-26a
expression via MYC upregulation.
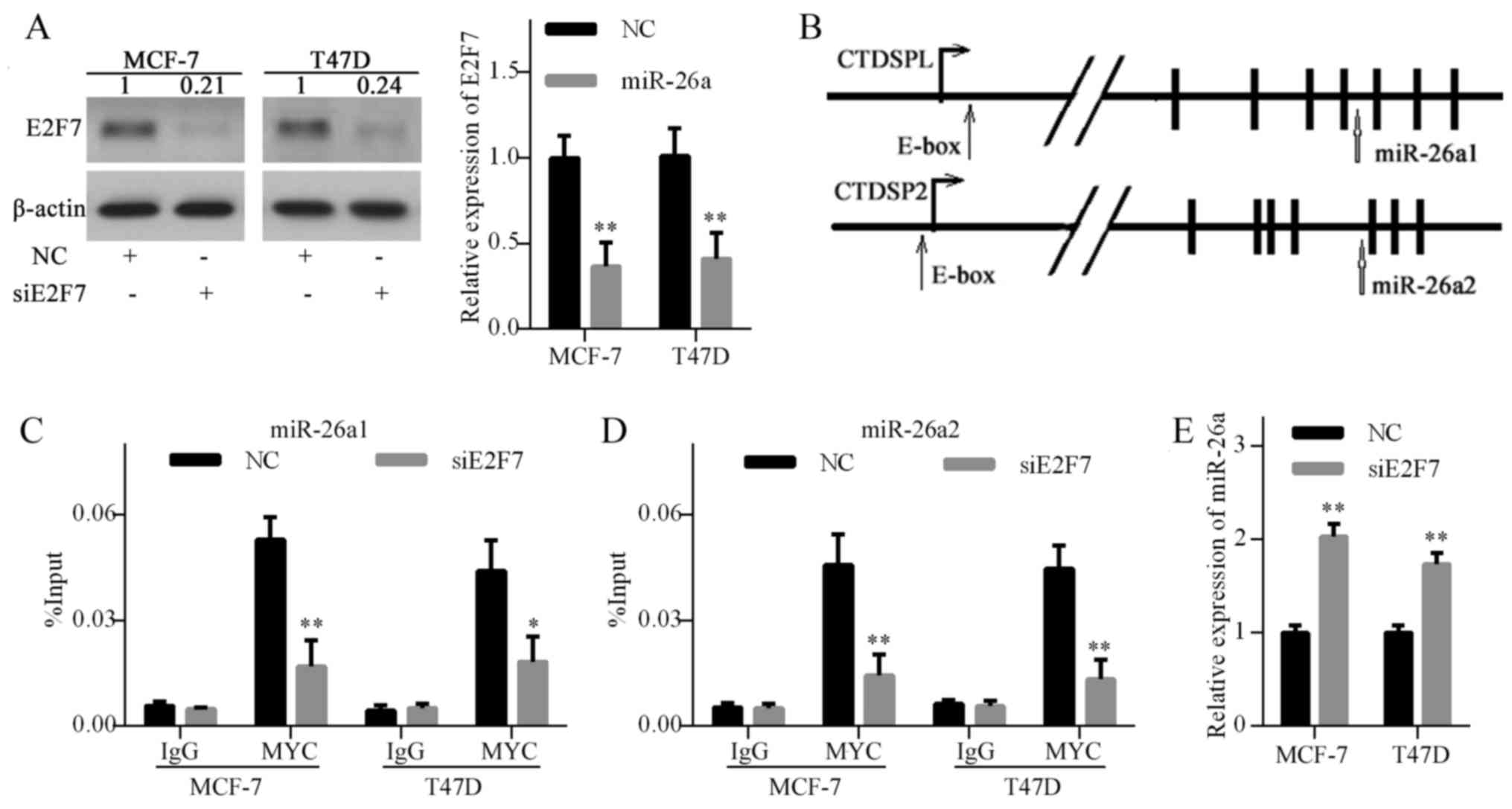
E2F7 knockdown decreases MYC recruitment
to the miR-26a promoter
Previously, MYC has been implicated in the
regulation of a series of miRNAs, including miR-26a (30). A previous study reported that MYC
suppressed miR-26a expression by recruiting EZH2 to the miR-26a
promoter (31). To explore the
mechanism underlying E2F7-induced miR-26a repression, E2F7
expression was knocked down using siRNA in MCF-7 and T47D cells
(Fig. 5A). Subsequently, the
promoter regions of CTDSPL (miR-26a1) and CTDSP2 (miR-26a2) were
analyzed using the UCSC Genome Browser and two MYC-binding E-box
sites were identified (Fig. 5B).
Subsequently, ChIP was performed to determine the effects of E2F7
knockdown on MYC enrichment on the miR-26a promoter. The results
demonstrated that E2F7 knockdown decreased MYC binding to the
miR-26a promoter (Fig. 5C and D).
Furthermore, E2F7 knockdown significantly increased miR-26a levels
in breast cancer cells (Fig. 5E).
These results suggested that E2F7 repression elevated miR-26a
expression by inhibiting MYC binding to the miR-26a promoter.
miR-26a and E2F7 form a feedback
loop
Concomitant expression of miR-26a with host genes
has been identified in physiological and pathological conditions
(9). To avoid interference of
exogenous miR-26a mimics, CTDSPL and CTDSP2 levels were detected,
instead of miR-26a, when conducting a miR-26a self-induction
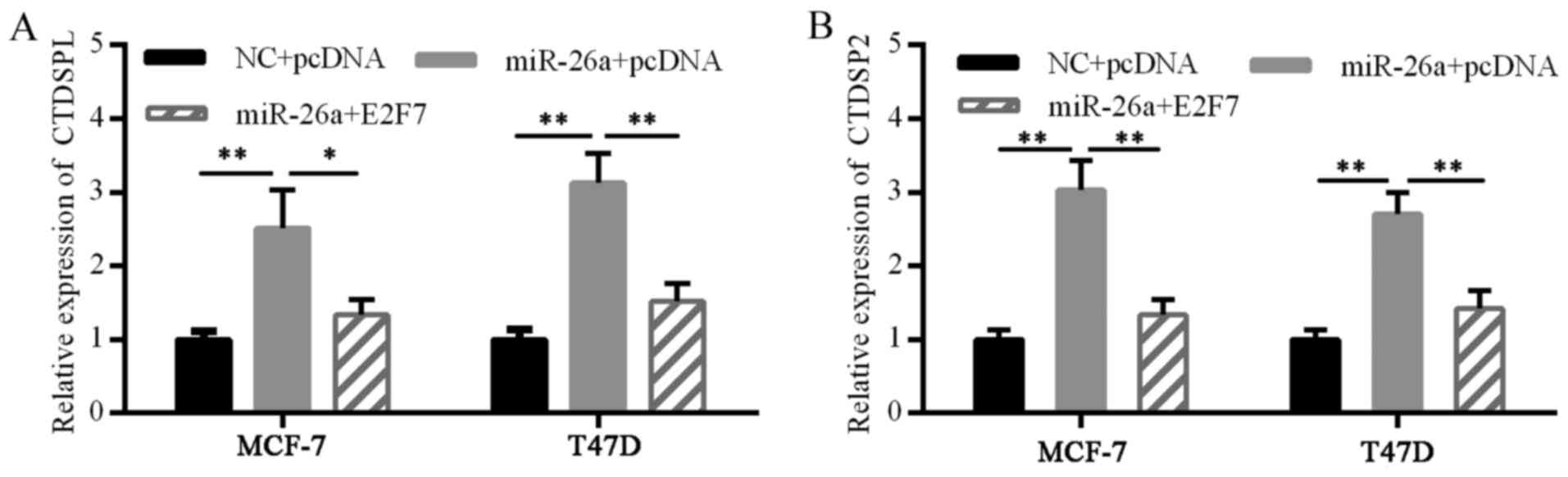
experiment. The miR-26a mimics markedly increased CTDSPL and CTDSP2
mRNA expression, suggesting a self-perpetuating loop of miR-26a.
Furthermore, E2F7 overexpression disturbed this loop (Fig. 6). These results suggested the
existence of a double-negative feedback loop of E2F7 and miR-26a in
ER-positive breast cancer cells.
miR-26a/E2F7 feedback loop induces TAM
resistance in ER-positive breast cancer cells
The present study investigated whether
overexpression of miR-26a or knockdown of E2F7 affected
proliferation of MCF-7 and T47D cells. A cell viability assay
indicated that miR-26a overexpression or E2F7 knockdown inhibited
the viability of MCF-7 and T47D cells (Fig. 7A and B).
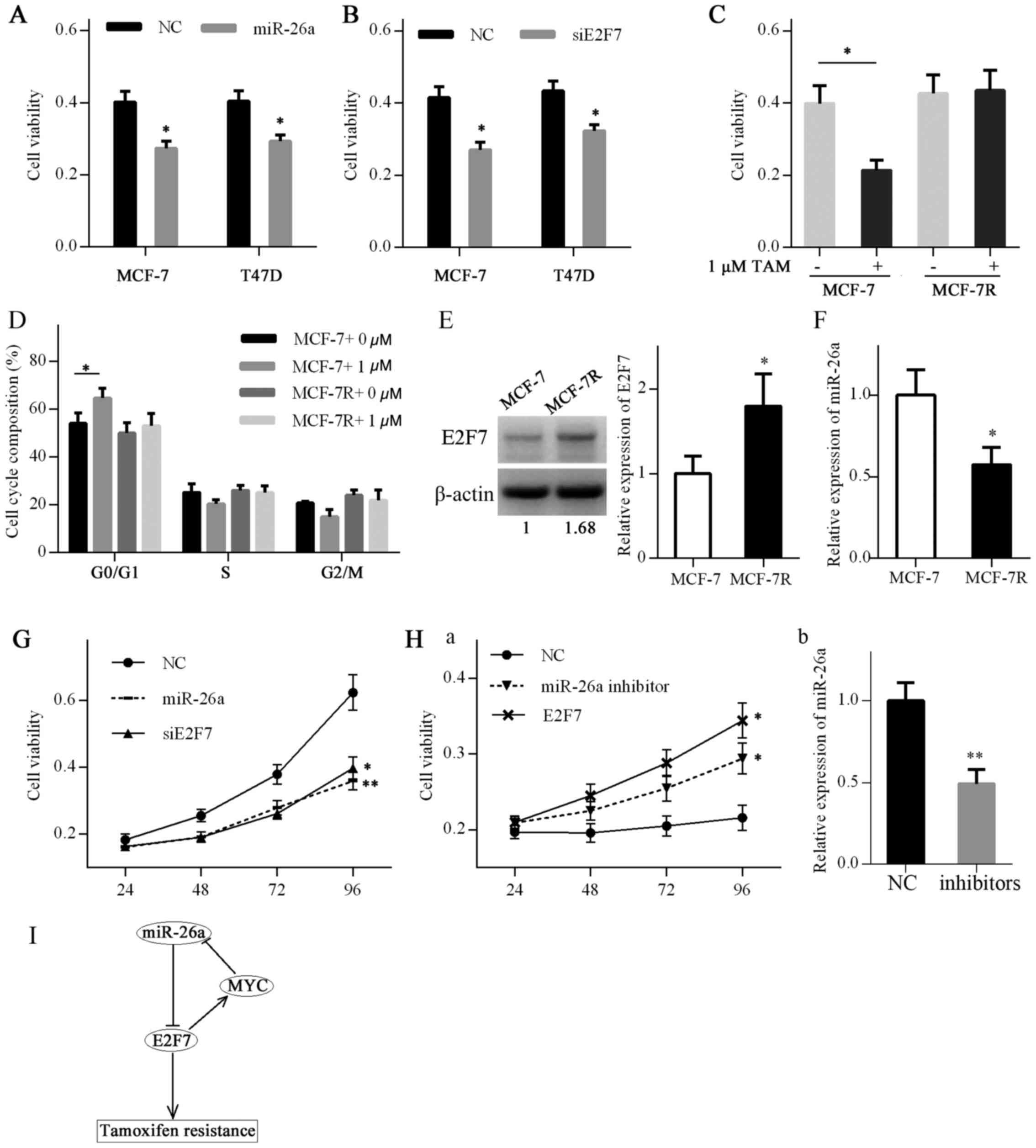 | Figure 7miR-26a/E2F7 feedback loop induces
TAM resistance in ER-positive breast cancer. (A) Viability of MCF-7
and T47D cells transfected with miR-26a mimics or NC for 72 h was
detected by MTT assay. (B) Viability of MCF-7 and T47D cells
transfected with siE2F7 or NC for 72 h was detected by MTT assay.
(C) Viability of MCF-7 and MCF-7R cells treated with 1 μM TAM or
ethanol for 72 h was detected by MTT assay. (D) Cell cycle
distribution of MCF-7 and MCF-7R cells treated with 1 µM TAM
or ethanol for 48 h was analyzed by flow cytometry.
*P<0.05 (one-way analysis of variance followed by
least significant difference test). (E) E2F7 expression in MCF-7
and MCF-7R cells was detected by western blotting. (F) miR-26a
expression in MCF-7 and MCF-7R cells was detected by RT-qPCR. (G)
MCF-7R cells were transfected with NC mimics plus siRNA NC, miR-26a
mimics plus siRNA NC and siE2F7 plus NC mimics. The viability of
MCF-7R cells was detected by MTT assay. (H) MCF-7 cells were
transfected with inhibitor NC plus pcDNA3.1 vector, miR-26a
inhibitors plus pcDNA3.1 vector and E2F7 vector plus inhibitor NC.
(a) Viability of MCF-7 cells was detected by MTT assay. (b) miR-26a
knockdown by transfection of MCF-7 cells with inhibitors was
confirmed by RT-qPCR. (I) A schematic diagram illustrating the
feedback loop of miR-26a and E2F7 in regulating TAM resistance.
Data are presented as the means ± standard deviation of three
independent experiments. *P<0.05 and
**P<0.01. E2F7, E2F transcription factor 7; MCF-7R,
TAM-resistant MCF-7 cells; miR-26a, microRNA 26a; MYC, MYC
proto-oncogene, bHLH transcription factor; NC, negative control;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; siE2F7, E2F7 small interfering RNA; TAM, tamoxifen. |
To further determine the association between
miR-26a, E2F7 and TAM resistance, a TAM-resistant breast cancer
cell line, MCF-7R, was developed. TAM resistance of MCF-7R cells
was verified by cell viability and cell cycle assays. TAM inhibited
cell viability and induced a G1 cell cycle arrest in MCF-7 cells,
whereas MCF-7R cells were resistant to the drug (Fig. 7C and D). Subsequently, E2F7 and
miR-26a expression were evaluated in MCF-7 cells and MCF-7R cells
by western blot analysis and RT-qPCR, respectively. E2F7 expression
in MCF-7R cells was elevated compared with that in MCF-7 cells,
whereas miR-26a expression was decreased (Fig. 7E and F).
MCF-7R cells were subsequently transfected with
miR-26a mimics, E2F7 siRNA or corresponding controls, and cell
viability was monitored by the MTT assay. The results demonstrated
that re-expression of miR-26a or knockdown of E2F7 was capable of
resensitizing MCF-7R cells to TAM (Fig. 7G). In addition, MCF-7 cells were
transfected with miR-26a inhibitors, E2F7-expressing plasmids or
corresponding controls. A cell viability assay indicated that
miR-26a knockdown or E2F7 overexpression conferred resistance to
TAM in MCF-7 cells (Fig. 7H). The
schematic diagram presented in Fig.
7I explains the proposed feedback loop of miR-26a and E2F7
based on the present results.
Discussion
TAM is highly effective in the treatment of
ER-positive breast tumors, which works by inhibiting the ER pathway
(3); however, TAM resistance
remains a major challenge in the treatment of ER-positive breast
cancer. Previous studies have revealed the important roles of
miRNAs in the TAM resistance of breast cancer. Re-expression of
miR-375, miR-873 and miR-27b has been reported to reverse TAM
resistance, whereas miR-221/222 upregulation confers TAM resistance
(23,32–34).
The present study reported that miR-26a expression was inhibited in
ER-positive breast cancer, whereas E2F7 expression was elevated.
Subsequently, the results demonstrated that a miR-26a/E2F7 feedback
loop resulted in miR-26a downregulation and E2F7 overexpression,
and thereby contributed to TAM resistance in ER-positive breast
cancer cells.
miR-26a is differently expressed in cancer tissues
and exhibits diverse functions dependent on cancer type. miR-26a
expression is reduced in gastric cancer, hepatocellular carcinoma
and gallbladder cancer compared with in normal adjacent tissue
(35–37). Conversely, miR-26a expression is
elevated in lung cancer, glioma and ovarian cancer (38–40).
The present study revealed that miR-26a expression was reduced in
ER-positive breast cancer, whereas E2F7 expression was elevated, as
previously described (10,41). miR-26a and E2F7 expression levels
were detected by RT-qPCR analysis of clinical specimens, and
findings were confirmed using TCGA datasets. Furthermore, an
inverse correlation between miR-26a and E2F7 expression was
identified in ER-positive breast cancer tissues, suggesting a
possible crosstalk between the two molecules.
A previous study demonstrated that E2F7 is a direct
target of miR-26a in AML cells (29). miRNAs repress target gene
expression through modulation of translation efficiency or
degradation of mRNAs (5). The
present study revealed that ectopic expression of miR-26a markedly
decreased E2F7 protein, but not mRNA, expression. These results
indicated that miR-26a may directly inhibit E2F7 via translational
inhibition. Notably, miR-26a overexpression also led to MYC
repression in breast cancer cells. A previous study reported that
ectopic miR-26a expression inhibits MYC expression in aggressive
B-cell lymphomas (42); however,
the underlying mechanism remains unknown. Furthermore, E2F7
silencing increases expression of the miR-17–92 cluster by
inhibiting MYC transcriptional activity in AML cells (29). In addition, E2F7 has been revealed
to increase MYC expression by decreasing E2F1/2/3 recruitment to
the MYC gene promoter region (43). In the present study, it was
demonstrated that ectopic expression of E2F7 abrogated
miR-26a-induced MYC repression. These results suggested that
miR-26a overexpression may inhibit MYC expression by repressing
E2F7 in breast cancer cells.
It has been reported that E2F7 inhibits the activity
of other E2F family members, including E2F1, E2F2 and E2F3
(15,18,43).
E2F7 knockdown elevates miR-26a expression by increasing the
recruitment of E2F3 to the miR-26a promoter (43). A recent study demonstrated that MYC
represses miR-26a expression by recruiting EZH2 to the gene
promoter (31). The present study
aimed to determine whether E2F7 regulated miR-26a expression by
modulating recruitment of MYC to the miR-26a promoter. The results
demonstrated that ectopic expression of E2F7 inhibited miR-26a
expression, at least partly by increasing MYC expression.
Furthermore, E2F7 knockdown significantly reduced the recruitment
of MYC to the miR-26a gene promoter and led to miR-26a
re-expression.
The present study revealed that miR-26a mimics
increased the expression of the miR-26a host genes, CTDSPL and
CTDSP2, whereas E2F7 overexpression abrogated miR-26a-induced
CTDSPL and CTDSP2 expression. These findings indicated that miR-26a
and E2F7 formed a feedback loop in ER-positive breast cancer.
During breast carcinogenesis, the balance of the feedback loop may
be disrupted and result in miR-26a suppression and E2F7
overexpression. In the present study, downregulation of miR-26a and
upregulation of E2F7 were also detected in TAM-resistant cells. A
previous study reported that E2F7 overexpression led to TAM
resistance by suppressing the miR-15a/16 cluster in breast cancer
cells (38). Furthermore, not only
E2F7 overexpression, but also miR-26a knockdown, conferred
resistance to TAM in MCF-7 cells. Conversely, miR-26a
overexpression and E2F7 silencing resensitized MCF-7R cells to
TAM.
In conclusion, the present study demonstrated that
crucial functional crosstalk may exist between miR-26a and E2F7 via
a double-negative feedback loop in ER-positive breast cancer, which
may have an important role in TAM resistance. Therefore, it may be
hypothesized that re-expression of miR-26a or inhibition of E2F7
are potential therapeutic approaches for the treatment of
TAM-resistant breast cancer in the future.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81272418; Hong Ren),
the National Science Foundation for Young Scientists of China
(grant no. 81602597; Xin Sun) and the National Science Foundation
for Young Scientists of China (grant no. 81402506; Sida Qin).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YP, HR and JL designed the study. JL, XL, MW, GX and
GY performed the experiments. XL, MW, GX and YL contributed to data
acquisition and analysis. JL, XL and YP wrote the manuscript. HW,
YL and XS collected the clinical samples and contributed to
clinical data analysis. GX, YL and XS contributed to bioinformatics
analysis. SQ and ND contributed to statistical analysis and revised
the manuscript critically for important intellectual content. XS,
SQ and ND were involved in drafting the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Xi'an Jiaotong University First Affiliated Hospital
and each patient provided written informed consent.
Patient consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vargo-Gogola T and Rosen JM: Modelling
breast cancer: One size does not fit all. Nat Rev Cancer.
7:659–672. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mandlekar S and Kong AN: Mechanisms of
tamoxifen-induced apoptosis. Apoptosis. 6:469–477. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qadir MA, Kwok B, Dragowska WH, To KH, Le
D, Bally MB and Gorski SM: Macroautophagy inhibition sensitizes
tamoxifen-resistant breast cancer cells and enhances mitochondrial
depolarization. Breast Cancer Res Treat. 112:389–403. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun X, Jiao X, Pestell TG, Fan C, Qin S,
Mirabelli E, Ren H and Pestell RG: MicroRNAs and cancer stem cells:
The sword and the shield. Oncogene. 33:4967–4977. 2014. View Article : Google Scholar
|
|
7
|
Di Leva G, Garofalo M and Croce CM:
MicroRNAs in cancer. Annu Rev Pathol. 9:287–314. 2014. View Article : Google Scholar :
|
|
8
|
Fatica A and Fazi F: MicroRNA-regulated
pathways in hemato-logical malignancies: How to avoid cells playing
out of tune. Int J Mol Sci. 14:20930–20953. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu Y, Lu Y, Zhang Q, Liu JJ, Li TJ, Yang
JR, Zeng C and Zhuang SM: MicroRNA-26a/b and their host genes
cooperate to inhibit the G1/S transition by activating the pRb
protein. Nucleic Acids Res. 40:4615–4625. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang B, Liu XX, He JR, Zhou CX, Guo M, He
M, Li MF, Chen GQ and Zhao Q: Pathologically decreased miR-26a
antagonizes apoptosis and facilitates carcinogenesis by targeting
MTDH and EZH2 in breast cancer. Carcinogenesis. 32:2–9. 2011.
View Article : Google Scholar
|
|
11
|
Liu P, Tang H, Chen B, He Z, Deng M, Wu M,
Liu X, Yang L, Ye F and Xie X: miR-26a suppresses tumour
proliferation and metastasis by targeting metadherin in triple
negative breast cancer. Cancer Lett. 357:384–392. 2015. View Article : Google Scholar
|
|
12
|
Jansen MPHM, Reijm EA, Sieuwerts AM,
Ruigrok-Ritstier K, Look MP, Rodríguez-González FG, Heine AA,
Martens JW, Sleijfer S, Foekens JA, et al: High miR-26a and low
CDC2 levels associate with decreased EZH2 expression and with
favorable outcome on tamoxifen in metastatic breast cancer. Breast
Cancer Res Treat. 133:937–947. 2012. View Article : Google Scholar :
|
|
13
|
Joshi T, Elias D, Stenvang J, Alves CL,
Teng F, Lyng MB, Lykkesfeldt AE, Brünner N, Wang J, Gupta R, et al:
Integrative analysis of miRNA and gene expression reveals
regulatory networks in tamoxifen-resistant breast cancer.
Oncotarget. 7:57239–57253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DeGregori J and Johnson DG: Distinct and
overlapping roles for E2F family members in transcription,
proliferation and apoptosis. Curr Mol Med. 6:739–748.
2006.PubMed/NCBI
|
|
15
|
Logan N, Delavaine L, Graham A, Reilly C,
Wilson J, Brummelkamp TR, Hijmans EM, Bernards R and La Thangue NB:
E2F-7: A distinctive E2F family member with an unusual organization
of DNA-binding domains. Oncogene. 23:5138–5150. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Endo-Munoz L, Dahler A, Teakle N, Rickwood
D, Hazar-Rethinam M, Abdul-Jabbar I, Sommerville S, Dickinson I,
Kaur P, Paquet-Fifield S, et al: E2F7 can regulate proliferation,
differentiation, and apoptotic responses in human keratinocytes:
Implications for cutaneous squamous cell carcinoma formation.
Cancer Res. 69:1800–1808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Di Stefano L, Jensen MR and Helin K: E2F7,
a novel E2F featuring DP-independent repression of a subset of
E2F-regulated genes. EMBO J. 22:6289–6298. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zalmas LP, Zhao X, Graham AL, Fisher R,
Reilly C, Coutts AS and La Thangue NB: DNA-damage response control
of E2F7 and E2F8. EMBO Rep. 9:252–259. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Q, Qiu XM, Li QH, Wang XY, Li L, Xu M,
Dong M and Xiao YB: MicroRNA-424 may function as a tumor suppressor
in endometrial carcinoma cells by targeting E2F7. Oncol Rep.
33:2354–2360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reimer D, Sadr S, Wiedemair A, Stadlmann
S, Concin N, Hofstetter G, Müller-Holzner E, Marth C and Zeimet AG:
Clinical relevance of E2F family members in ovarian cancer - an
evaluation in a training set of 77 patients. Clin Cancer Res.
13:144–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hazar-Rethinam M, de Long LM, Gannon OM,
Topkas E, Boros S, Vargas AC, Dzienis M, Mukhopadhyay P, Simpson F,
Endo-Munoz L, et al: A novel E2F/sphingosine kinase 1 axis
regulates anthracycline response in squamous cell carcinoma. Clin
Cancer Res. 21:417–427. 2015. View Article : Google Scholar
|
|
22
|
Thrane S, Lykkesfeldt AE, Larsen MS,
Sorensen BS and Yde CW: Estrogen receptor α is the major driving
factor for growth in tamoxifen-resistant breast cancer and
supported by HER/ERK signaling. Breast Cancer Res Treat. 139:71–80.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cui J, Yang Y, Li H, Leng Y, Qian K, Huang
Q, Zhang C, Lu Z, Chen J, Sun T, et al: MiR-873 regulates ERα
transcriptional activity and tamoxifen resistance via targeting
CDK3 in breast cancer cells. Oncogene. 34:3895–3907. 2015.
View Article : Google Scholar
|
|
24
|
Goldhirsch A, Ingle JN, Gelber RD, Coates
AS, Thürlimann B and Senn HJ; Panel members: Thresholds for
therapies: Highlights of the St Gallen International Expert
Consensus on the primary therapy of early breast cancer 2009. Ann
Oncol. 20:1319–1329. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pang Y, Liu J, Li X, Zhang Y, Zhang B,
Zhang J, Du N, Xu C, Liang R, Ren H, et al: Nano Let 7b
sensitization of eliminating esophageal cancer stem like cells is
dependent on blockade of Wnt activation of symmetric division. Int
J Oncol. 51:1077–1088. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-DeltaDeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Lim YY, Wright JA, Attema JL, Gregory PA,
Bert AG, Smith E, Thomas D, Lopez AF, Drew PA, Khew-Goodall Y, et
al: Epigenetic modulation of the miR-200 family is associated with
transition to a breast cancer stem-cell-like state. J Cell Sci.
126:2256–2266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Licchesi JD, Van Neste L, Tiwari VK, Cope
L, Lin X, Baylin SB and Herman JG: Transcriptional regulation of
Wnt inhibitory factor-1 by Miz-1/c-Myc. Oncogene. 29:5923–5934.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Salvatori B, Iosue I, Mangiavacchi A,
Loddo G, Padula F, Chiaretti S, Peragine N, Bozzoni I, Fazi F and
Fatica A: The microRNA-26a target E2F7 sustains cell proliferation
and inhibits monocytic differentiation of acute myeloid leukemia
cells. Cell Death Dis. 3:e4132012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tao J, Zhao X and Tao J: c-MYC-miRNA
circuitry: A central regulator of aggressive B-cell malignancies.
Cell Cycle. 13:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao X, Lwin T, Zhang X, Huang A, Wang J,
Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E and Tao J:
Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell
lymphoma survival and clonogenicity. Leukemia. 27:2341–2350. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ward A, Balwierz A, Zhang JD, Küblbeck M,
Pawitan Y, Hielscher T, Wiemann S and Sahin Ö: Re-expression of
microRNA-375 reverses both tamoxifen resistance and accompanying
EMT-like properties in breast cancer. Oncogene. 32:1173–1182. 2013.
View Article : Google Scholar
|
|
33
|
Zhu J, Zou Z, Nie P, Kou X, Wu B, Wang S,
Song Z and He J: Downregulation of microRNA-27b-3p enhances
tamoxifen resistance in breast cancer by increasing NR5A2 and CREB1
expression. Cell Death Dis. 7:e24542016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miller TE, Ghoshal K, Ramaswamy B, Roy S,
Datta J, Shapiro CL, Jacob S and Majumder S: MicroRNA-221/222
confers tamoxifen resistance in breast cancer by targeting p27Kip1.
J Biol Chem. 283:29897–29903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deng M, Tang HL, Lu XH, Liu MY, Lu XM, Gu
YX, Liu JF and He ZM: miR-26a suppresses tumor growth and
metastasis by targeting FGF9 in gastric cancer. PLoS One.
8:e726622013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Liang L, Zhang XF, Jia HL, Qin Y,
Zhu XC, Gao XM, Qiao P, Zheng Y, Sheng YY, et al: MicroRNA-26a
suppresses tumor growth and metastasis of human hepatocellular
carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology.
58:158–170. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou H, Guo W, Zhao Y, Wang Y, Zha R, Ding
J, Liang L, Hu J, Shen H, Chen Z, et al: MicroRNA-26a acts as a
tumor suppressor inhibiting gallbladder cancer cell proliferation
by directly targeting HMGA2. Int J Oncol. 44:2050–2058. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin G, Liu B, Meng Z, Liu Y, Li X, Wu X,
Zhou Q and Xu K: MiR-26a enhances invasive capacity by suppressing
GSK3β in human lung cancer cells. Exp Cell Res. 352:364–374. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qian X, Zhao P, Li W, Shi ZM, Wang L, Xu
Q, Wang M, Liu N, Liu LZ and Jiang BH: MicroRNA-26a promotes tumor
growth and angiogenesis in glioma by directly targeting prohibitin.
CNS Neurosci Ther. 19:804–812. 2013.PubMed/NCBI
|
|
40
|
Shen W, Song M, Liu J, Qiu G, Li T, Hu Y
and Liu H: MiR-26a promotes ovarian cancer proliferation and
tumorigenesis. PLoS One. 9:e868712014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chu J, Zhu Y, Liu Y, Sun L, Lv X, Wu Y, Hu
P, Su F, Gong C, Song E, et al: E2F7 overexpression leads to
tamoxifen resistance in breast cancer cells by competing with E2F1
at miR-15a/16 promoter. Oncotarget. 6:31944–31957. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T,
Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al: Coordinated
silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic
target of histone modification in aggressive B-Cell lymphomas.
Cancer Cell. 22:506–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mitxelena J, Apraiz A, Vallejo-Rodríguez
J, Malumbres M and Zubiaga AM: E2F7 regulates transcription and
maturation of multiple microRNAs to restrain cell proliferation.
Nucleic Acids Res. 44:5557–5570. 2016. View Article : Google Scholar :
|