Introduction
Colorectal cancer is the second leading cause of
cancer-related mortality in the United States (1). Pre-operative chemoradiotherapy (CRT),
followed by total mesorectal excision, is the standard of care for
patients with locally advanced rectal cancer (LARC) (2-4). The
combination of radiation with capecitabine has been shown to
significantly improve local control and local recurrence-free
survival, but does not improve metastasis-free survival or overall
survival (5). To acquire better
outcomes, researchers have attempted to identify novel
radiosensitizers, such as oxaliplatin, irinotecan and some
molecular targeting agents (6).
However, the addition of oxaliplatin, in addition to capecitabine,
as a radiosensitizer has been demonstrated to have no benefit with
regard to prognoses, but does show increased toxicity (7-10).
Irinotecan (also known as CPT-11), an analog of
camptothecin (CPT), is currently used in the chemotherapy of
metastatic colorectal cancer (11). The active form of irinotecan,
SN-38, binds to and prevents topoisomerase I (TOP I) from rejoining
transient DNA single-strand breaks (SSBs) during replication
(12). Irinotecan-stabilized-TOP
I-DNA complexes interact with advancing replication forks during
the S phase, convert SSBs into irreversible DNA double-strand
breaks (DSBs) and result in cell death (13).
CPT derivatives exert radiosensitizing effects on
various cell lines (14-17). However, to date, at least to the
best of our knowledge, few studies have been published on these
effects on colorectal cancer cell lines. A previous study found
that CPT exerted radiosensitizing effects on p53-wild-type HCT116
colorectal cancer cells and proposed that p53 and p21 were the
major cellular determinants for TOP I-mediated radiation
sensitization (18). However, the
radiosensitizing effects of irinotecan on p53-mutant colon cells
remain unclear, and the potential mechanisms associated with the
DNA damage response system, cell cycle arrest and cell apoptosis
remain to be determined. Thus, in the present study, we examined
the radiosensitizing effects of irinotecan on the p53-mutant
colorectal cancer cell lines, HT29 and SW620, and explored the
potential underlying mechanisms.
Materials and methods
Chemicals
Irinotecan was purchased from Jiangsu Hengrui
Medicine Co., Ltd. (Jiangsu, China), dissolved in dimethyl
sulfoxide to obtain a 100 mg/ml stock solution and stored at
−20°C.
Cell culture
The HT29 and SW620 cell lines were purchased from
the Biochemistry and Cell Biology Institute of Shanghai, Chinese
Academy of Sciences, within 3 months of the experiments. The HT29
cells were cultured in McCoy's 5A medium, and the SW620 cells were
cultured in DMEM medium, both containing 10% heat-inactivated fetal
bovine serum, 1% penicillin (final concentration, 100 U/ml) and
streptomycin (final concentration, 0.1 mg/ml). The cells were
maintained at 37°C in a humidified atmosphere containing 5%
CO2. The experiments were conducted in the exponential
phase of growth.
Cell viability assay
The HT29 (8×103) and SW620
(8×103) cells in 100 μl of culture medium were
seeded onto 96-well plates (Corning Inc., Corning, NY, USA) and
cultured overnight. The cells were treated with irinotecan by
exchanging the medium premixed with various drug concentrations (0,
6.25, 12.5, 25, 50, 100, 200 and 400 μg/ml) for 24 h. Cell
viability was evaluated using a Cell Counting kit-8 (CCK-8) assay
(Dojindo Laboratories, Kumamoto, Japan), according to the
manufacturer's instructions. The absorbance at 450 nm was then
measured using a BioTek microplate reader and analyzed by Gen5 2.0
microplate software (BioTek Instruments, Inc., Winooski, VT, USA).
The cell growth inhibition curve was presented, and the 24-h
half-maximal inhibitory concentration (IC50) of
irinotecan was calculated. A chemotherapeutic drug exerts cytotoxic
effects on cells, which can influence the accuracy of the results
when it is combined with radiation. To examine the radiosensitizing
effects of irinotecan, the concentrations need to be low in order
to decrease the cytotoxic effects of the drug itself. According to
past practice (19), low drug
concentrations (<10% of the IC50 value or
IC10 value) were considered for use in subsequent
experiments. According to the IC50 values in this study
(shown in the Results section below), low concentrations <4
μg/ml (0, 0.5, 1, 1.5, 2, 3 and 4 μg/ml) were used to
examine the inhibitory effects of irinotecan as a single agent on
clonogenic survival. Finally, we selected the doses of 0, 1 and 2
μg/ml as the experimental concentrations for use in
combination with radiation.
Radiation exposure
The cells were irradiated with 6-MV X-rays using a
linear accelerator (Varian Medical Systems, Palo Alto, CA, USA) at
3 Gy/min. Irradiation was performed at the Experimental Irradiation
Core of the Fudan University Shanghai Cancer Center, Shanghai,
China. In clonogenic assays, the cells were irradiated with X-rays
at a dose of 0, 2, 4, 6 and 8 Gy. However, in the experiments
comparing the results of the control, irinotecan, radiation and
combined groups, there are no criteria on radiation doses.
Preliminary experiments (data not shown) based on low radiation
doses (2 and 4 Gy) revealed relatively small absolute values of the
results and the differences among groups were not so obvious. In
addition, preliminary experiments (data not shown) using high
radiation doses (8-16 Gy) revealed severe toxicity of the radiation
itself and it was hard to determine the differences between the
radiation group and the combination group. Thus, we finally
selected the dose of 6 Gy as the experimental radiation dose so
that we could obtain obvious and high-quality results.
Clonogenic assay
A clonogenic assay is the standard method to examine
the radiosensitizing effects of a drug. The cells were pre-seeded
in 6-well plates (Corning Inc., Corning, NY, USA) at several
densities (300-4,000 cells for the HT29 and 300-10,000 cells for
the SW620 cells) overnight. After exchanging with drug-containing
medium and incubation for 24 h, the cells were irradiated with
X-rays at a series of dose levels (0, 2, 4, 6 and 8 Gy). The medium
was then exchanged with culture medium without drugs, and the cells
were incubated for 10 to 14 days to allow for colony formation. The
colonies were fixed with 4% paraformaldehyde and stained with 0.1%
crystal violet (100% methanol solution) prior to counting. The
number of clones containing ≥50 cells was counted under a
stereomicroscope (Leica Microsystems, Wetzlar, Germany). The
plating efficiency (PE) was calculated in the following manner: PE
= number of colonies formed without irradiation/number of cells
inoculated ×100%. The cell survival fraction (SF) was calculated at
each irradiation dose in the following manner: SF = (number of
colonies formed at a certain irradiation dose)/(number of cells
inoculated × PE). SF curve fitting was conducted with a
linear-quadratic model (20) via
the equation y = exp[ −(a x x + b x x2)]. Currently, the
therapeutic regimen of 45-50 Gy/25-28 fractions (1.8-2.0 Gy per
fraction) is widely used in the neoadjuvant chemoradio-therapy of
locally advanced rectal cancers (21). In the present study, SFs at 2 Gy
were used to determine the sensitivity enhancement ratio (SER),
which was consistent with clinical radiotherapy. In this study, SER
referred to the fractional ratio of counts of surviving colonies
between cells treated with only 2 Gy X-rays compared to cells
treated with both 2 Gy X-rays and irinotecan.
Cell growth assay
The HT29 (4×103) and SW620
(3×103) cells with 100 μl of culture medium were
seeded onto 96-well plates (Corning Inc., Corning, New York, NY,
USA) and cultured overnight. After exchanging the medium with
drug-containing medium and incubating for 24 h, the cells were
irradiated with X-rays at 0 or 6 Gy. The medium was then exchanged
with culture medium without drugs, and the cells were incubated for
5 days. The cell growth assay was performed daily using the CCK-8
assay kit as described above.
Cell cycle analysis
The HT29 and SW620 cells were seeded onto 60-mm
plastic Petri dishes (Corning Inc.) and cultured overnight. After
exchanging the medium with drug-containing medium and incubating
the cells for 24 h, the cells were irradiated with X-rays at 0 or 6
Gy. The medium was then exchanged with culture medium without
drugs, and cells were incubated for 3 and 24 h. The cells
(discarding the culture medium) were harvested by trypsinization at
3 and 24 h following radiation. After rinsing 2 times with PBS, the
cells were fixed with 75% ethanol at −20°C overnight. The fixed
cells were incubated in PBS for 15 min and subsequently stained
with propidium iodide (PI) staining solution (50 μg/ml) for
30 min. The cell cycle was assayed with a flow cytometer
(FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA) at an
excitation wavelength of 488 nm and an emission wavelength of 585
nm and analyzed using BD CellQuest Pro software (643274 Rev. A; BD
Biosciences). A doublet discrimination module was used when using
flow cytometry to remove the doublets that can end up in the G2/M
phase.
Cell apoptosis analysis
The HT29 and SW620 cells were seeded onto 60-mm
plastic Petri dishes (Corning Inc.) and cultured overnight. After
exchanging the medium with drug-containing medium and incubating
the cells for 24 h, the cells were irradiated with X-rays at 0 or 6
Gy. The medium was then exchanged with culture medium without
drugs, and the cells were incubated for 24 h to 3 days. The cells
(including cells in the culture medium) were harvested by
trypsinization at 24, 48 and 72 h following radiation. After
rinsing twice with PBS, the cells were suspended in a binding
buffer, stained with both 4 μl of PI and 4 μl of
Annexin V-FITC per sample and loaded onto a flow cytometer
(FACSCalibur; BD Biosciences) for FL1 (Annexin V) and FL2 (PI)
bivariate analysis. Untreated samples stained with only PI or
Annexin V-FITC were also prepared. The data from 20,000
cells/sample were collected, and the quadrants were set according
to the population of viable, unstained cells in the untreated
samples. The percentage of cells in the respective quadrants was
calculated and analyzed using BD CellQuest Pro software (643274
Rev. A; BD Biosciences).
Immunofluorescence staining
A total of 5×104 HT29 and SW620 cells
were seeded separately onto 24-well plates (Corning Inc.)
containing a glass coverslip in each well and cultured overnight.
After exchanging the medium with drug-containing medium and
incubating the cells for 24 h, the cells were irradiated with
X-rays at 0 or 6 Gy. The medium was subsequently exchanged with
culture medium without the drug, and cells were incubated for 24 h.
The slides were then rinsed with PBS and fixed for 30 min in 4%
paraformal-dehyde. The cells were rinsed, treated with 1% Triton
X-100 for permeabilization of the cell membrane, and rinsed again.
The slides were blocked for 1 h in 5% bovine serum albumin (BSA)
and finally incubated with phospho-histone H2A.X (Ser139) (20E3)
rabbit monoclonal antibody (Alexa Fluor® 555 Conjugate;
#8228S, diluted 1:100 in 5% BSA; Cell Signaling Technology,
Danvers, MA, USA) at 4°C overnight. The slides were rinsed, mounted
with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher
Scientific Inc., Waltham, MA, USA) and sealed. Images were aquired
under a laser scanning confocal microscope (Leica Microsystems,
Wetzlar, Germany). The numbers of Ser139p-γH2AX foci in
the nuclei of at least 50 cells were counted in a double-blinded
manner to calculate the average number of foci per nucleus.
Western blot analysis
The HT29 and SW620 cells were seeded onto 60-mm
plastic Petri dishes (Corning Inc.) and cultured overnight. After
exchanging the medium with drug-containing medium and incubating
cells for 24 h, the cells were irradiated with X-rays at 0 or 6 Gy.
The medium was then exchanged with culture medium without drugs,
and cells were incubated for 3 and 24 h. The cells were lysed at 3
and 24 h following radiation with M-PER Mammalian Protein
Extraction Reagent (Thermo Fisher Scientific Inc.) with the
addition of protease inhibitors (cOmplete Tablets Mini; Roche,
South San Francisco, CA, USA) and phosphatase inhibitors (PhosSTOP;
Roche). Following the quantitation of the protein concentration
using a Pierce BCA Protein Assay kit (Thermo Fisher Scientific
Inc.) and protein degeneration at 100°C for 10 min, the protein
samples were resolved on SDS-polyacrylamide gels
(Ser1981p-ATM, ATM in 7.5% resolving gels,
Ser345p-Chk1, Chk1, Thr68p-Chk2, Chk2, Cyclin
B1, Ser216p-Cdc25C, Cdc25C, β-actin in 12.5% or 10%
resolving gels, Tyr15p-Cdc2, Cdc2,
Ser139p-γH2AX, Ser10p-Histone H3,
Histone H3 in 12.5% resolving gels) by electrophoresis and
subsequently transferred onto PVDF membranes (Millipore, Darmstadt,
Germany). The membranes were blocked with 5% BSA and probed with
primary antibodies at 4°C overnight. The membranes were washed with
Tris-buffered saline and Tween-20 (TBST) for 5×5 min, incubated
with goat anti-mouse and goat anti-rabbit secondary antibodies for
1 h and washed again with TBST for 5×5 min. Antibodies against
Ser1981p-ATM, ATM, Ser345p-Chk1 (#2348;
dilution, 1:1,000), Chk1 (#2360; dilution, 1:1,000),
Thr68p-Chk2 (#2197; dilution, 1:1,000), Chk2 (#6334;
dilution, 1:1,000), Ser139p-γH2AX (#9718; dilution,
1:1,000), Ser216p-Cdc25C (#4901; dilution, 1:1,000),
Cdc25C (#4688; dilution, 1:1,000), Tyr15p-Cdc2 (#4539;
dilution, 1:1,000), Cdc2 (#9116; dilution, 1:1,000), cyclin B1
(#12231; dilution, 1:1,000), Ser10p-Histone H3 (#53348;
dilution, 1:1,000), Histone H3 (#4499; dilution, 1:1,000) and
β-actin, as well as goat anti-mouse (#7056; dilution, 1:10,000) and
goat anti-rabbit (#7074; dilution, 1:10,000) secondary antibodies
were purchased from Cell Signaling Technology (Cell Signaling
Technology). The protein bands were visualized using an enhanced
chemiluminescence detection system (Thermo Fisher Scientific Inc.).
Protein bands were analyzed by ImageJ software version 1.8.0
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All experiments were repeated independently at least
3 times. SPSS version 22.0 software (SPSS Inc., Chicago, IL, USA)
was used for the statistical analysis. The data are presented as
the means ± standard deviation. Comparisons among multiple groups
were conducted using the one-way ANOVA or two-way ANOVA method for
quantitative data. Turkey's test was used for the post hoc multiple
comparisons. A P-value <0.05 was considered to indicate a
statistically significant difference. All curves and plots were
generated using GraphPad Prism 7.0 software (GraphPad Software
Inc., San Diego, CA, USA).
Results
Irinotecan inhibits the viability and
proliferation of HT29 and SW620 cells
To verify the cytotoxic effects of irinotecan on
colorectal cancer cells, we first treated both cell lines with
irinotecan as a single agent at several increasing concentrations
(0, 6.25, 12.5, 25, 50, 100, 200 and 400 μg/ml) for 24 h.
Cell viability was examined using the CCK-8 assay kit. As the
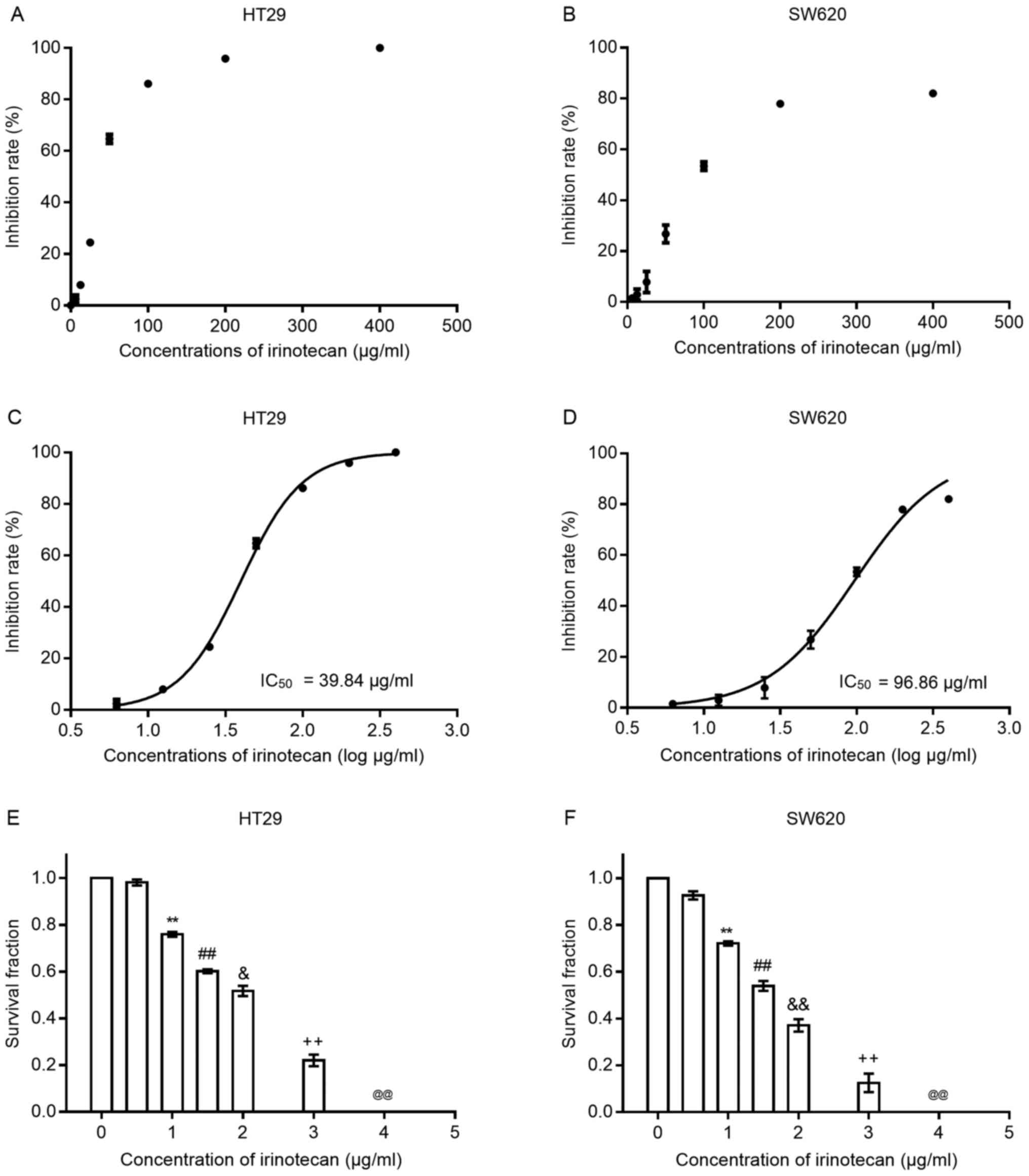
scatter grams shown in Fig. 1 A and
B, the inhibition rates increased with the increasing drug
concentration (0, 6.25, 12.5, 25, 50, 100, 200 and 400
μg/ml) in both cell lines; the HT29 cells seemed to be more
sensitive to irinotecan than the SW620 cells. We then transformed
the drug concentrations into log μg/ml style. The drug
inhibition curves (shown in Fig. 1C
and D) exhibited an S-like shape, from which IC50
values at 24 h for both cell lines were calculated. For the HT29
cells, the IC50 value was 39.84 μg/ml (95% CI,
38.27-41.48) and for the SW620 cells, it was 96.86 μg/ml
(95% CI, 89.04-105.4). Concentrations of irinotecan <10% of the
IC50 value were considered for use in the subsequent
experiments.
Subsequently, we examined the inhibitory effects of
irinotecan on clonogenic survival at low concentrations (0, 0.5, 1,
1.5, 2, 3 and 4 μg/ml) for 24 h (Fig. 1E and F). The results revealed that
the clonogenic survival rates were very low when the concentration
was >2 μg/ml; thus, we selected the dose of 0, 1 and 2
μg/ml as the experimental concentrations for use in
combination with radiation.
Low concentrations of irinotecan induce a
DNA damage response and G2/M arrest, with minimal apoptosis
Both cell lines were treated with low concentrations
of irinotecan (0, 1 and 2 μg/ml) as a single agent. The
cells were harvested after 24 h of incubation with irinotecan, and
the protein expression of Ser1981p-ATM, ATM and
Ser139p-γH2AX was measured by western blot analysis. The
cells were also submitted to flow cytometry to analyze changes in
the cell cycle and apoptosis.
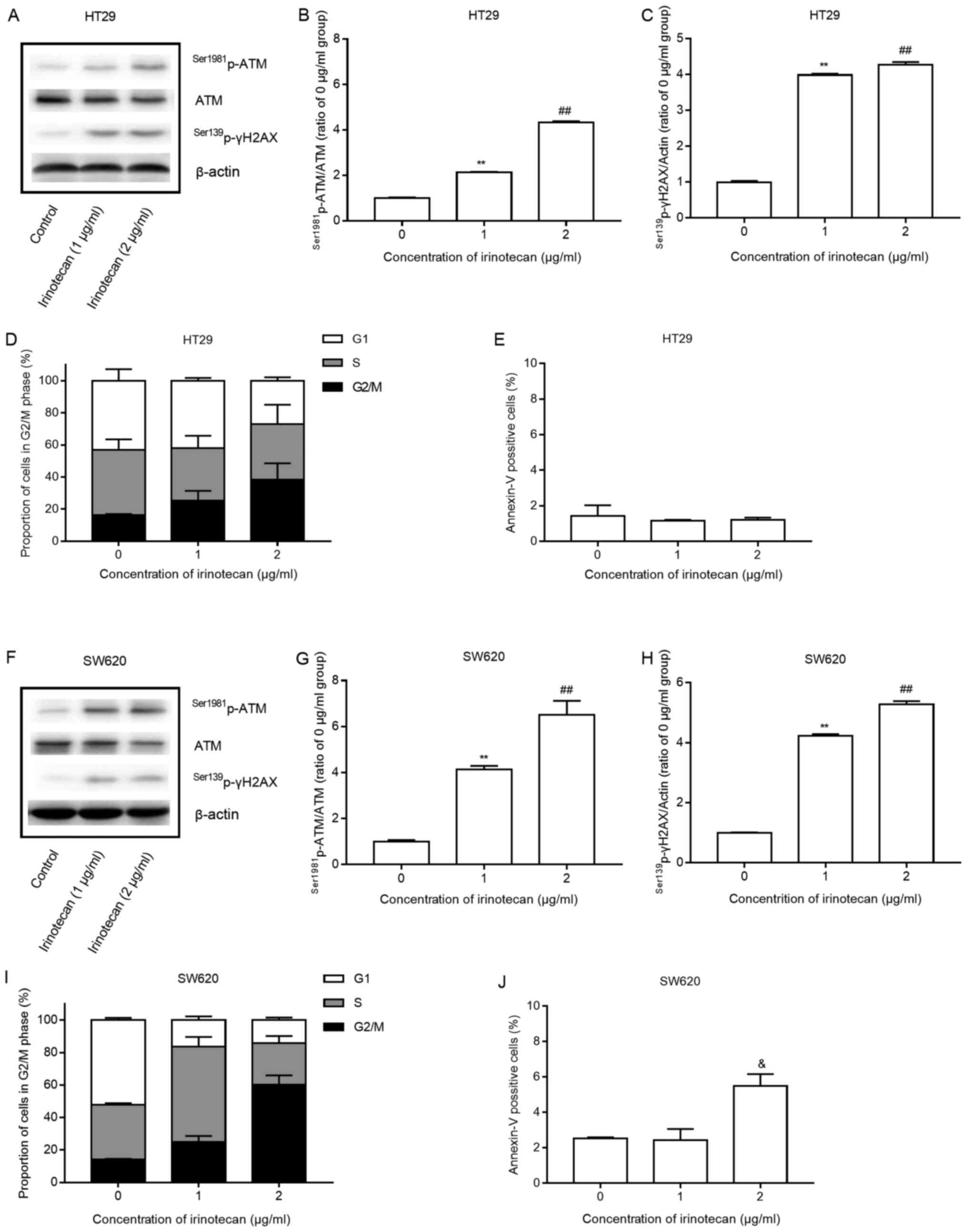
Low levels of irinotecan induced a statistically
significant increase in the expression of
Ser1981p-ATM/ATM and Ser139p-γH2AX (Fig. 2 A–C and F–H) and a marked increase
in the proportion of cells in the G2/M phase, with a corresponding
decrease in the proportion of cells in the G1 phase, all in a
concentration-dependent manner (Fig.
2D and I). The results indicated that SW620 cells were more
sensitive to delay at the G2/M phase than the HT29 cells. However,
only a significant increase (P=0.022, 2 μg/ml vs. 1
μg/ml) in apoptosis was detected at 2 μg/ml in the
SW620 cells and no increase was detected in the HT29 cells after 24
h of irinotecan treatment (Fig. 2E and
J).
These findings suggest that irinotecan activates the
DNA damage response system, induces potent G2/M phase arrest and
minimal apoptosis of the HT29 and SW620 cells, consistent with the
findings of previous studies (22,23).
These changes may potentially represent important mechanisms for
radiosensitization.
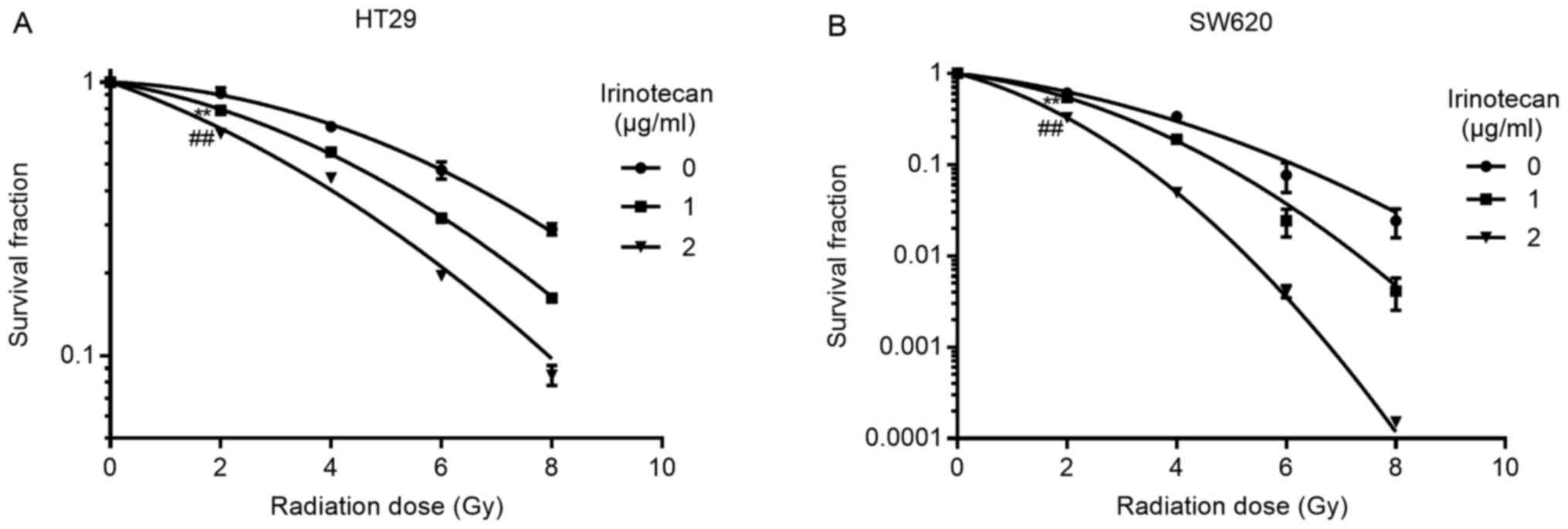
Irinotecan sensitizes the HT29 and SW620
cells to radiation
We selected 1 and 2 μg/ml of irinotecan for
use in the radiosensitization experiments. The cells were treated
with irinotecan for 24 h, followed by treatment with radiation at
different doses of up to 8 Gy. The culture medium was then
exchanged with irinotecan-free medium for 10-14 days to enable
colony formation. Following linear-quadratic analysis (LQ model),
we found that irinotecan effectively sensitized the HT29 and SW620
cells to radiation, and the SER at 2 Gy increased with the
increasing drug concentration. For the HT29 cells, the SER at 2 Gy
was 1.16 and 1.41 at 1 and 2 μg/ml, respectively, and for
the SW620 cells, the SER at 2 Gy was 1.13 and 1.87 at 1 and 2
μg/ml, respectively (Fig.
3). Thus, we concluded that irinotecan is a good
radiosensitizer against p53-mutant colorectal cancer cells.
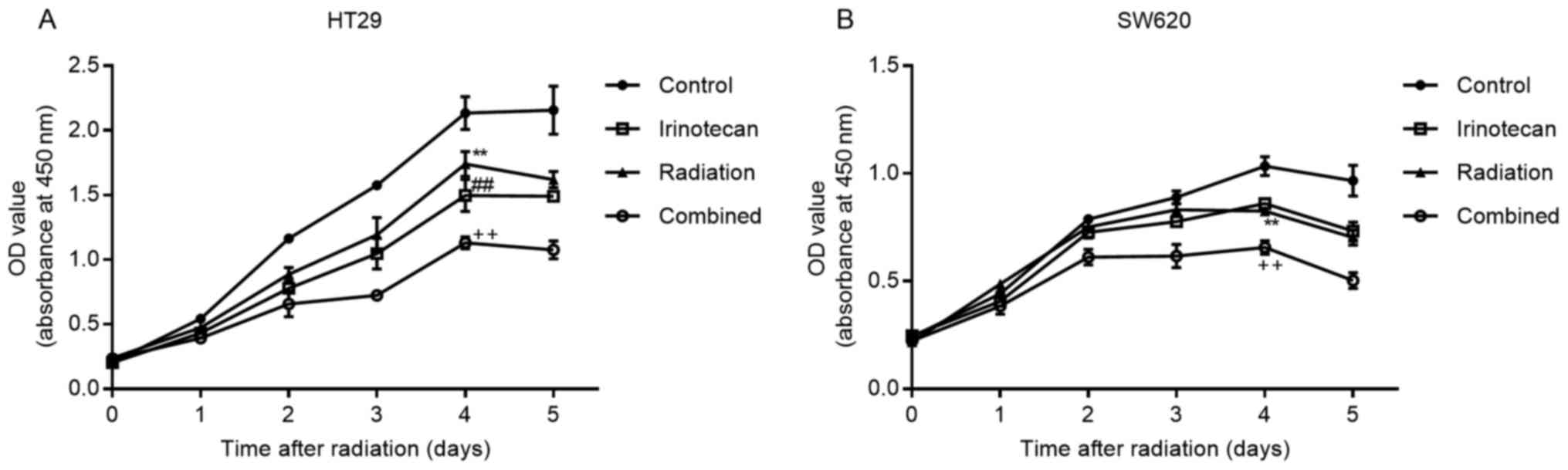
Subsequently, we verified the radiosensitizing
effects of irinotecan by drawing cell growth curves. The cells were
incubated for 5 days following 24 h of drug treatment, followed by
radiation treatment. Cell viability was measured daily using the
CCK-8 assay kit. As expected, irinotecan in combination with
radiation resulted in a reduced growth of both the HT29 and SW620
cells compared with growth in the irinotecan, radiation and control
groups, with most high significance observed on days 4 and 5
(Fig. 4).
The radiosensitizing effects of
irinotecan are associated with the activation of the DNA damage
response
The cells were treated with irinotecan (2
μg/ml), radiation (6 Gy) or irinotecan (2 μg/ml) in
combination with radiation (6 Gy). The formation of
Ser139p-γH2AX foci was illustrated by immunofluorescence
staining. Subsequently, the expression levels of proteins related
to the DNA damage response, such as Ser1981p-ATM, ATM,
Ser345p-Chk1, Chk1, Thr68p-Chk2, Chk2 and
Ser139p-γH2AX, were measured by western blot
analysis.
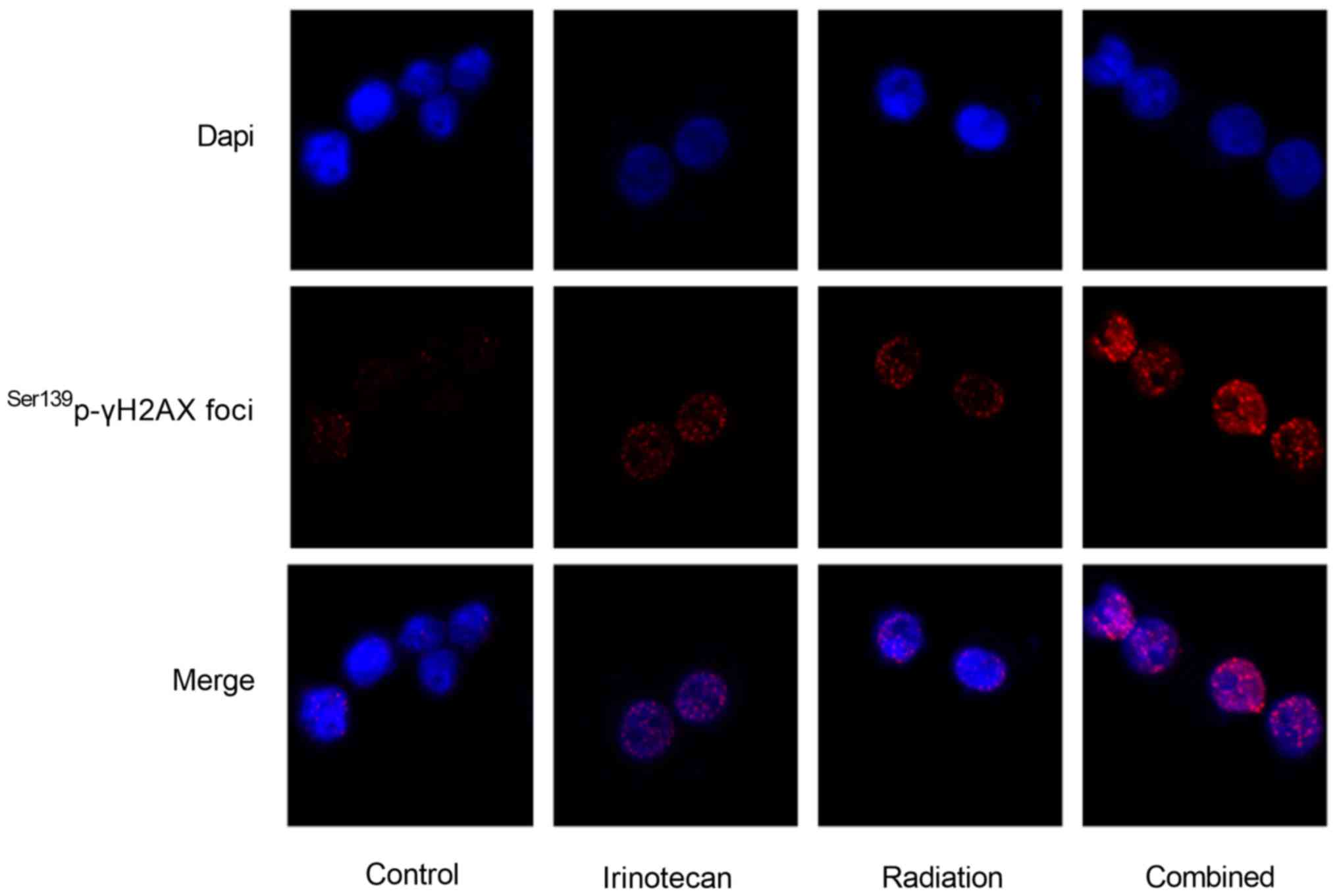
The results of immunofluorescence microscopy
revealed that the Ser139p-γH2AX foci in the nuclei of
both the HT29 and SW620 cells at 24 h following radiation were the
most obvious in the combination group. The immunofluorescence
staining images of the HT29 cells are presented in Fig. 5; however, the images of the SW620
cells are not presented (data not shown).
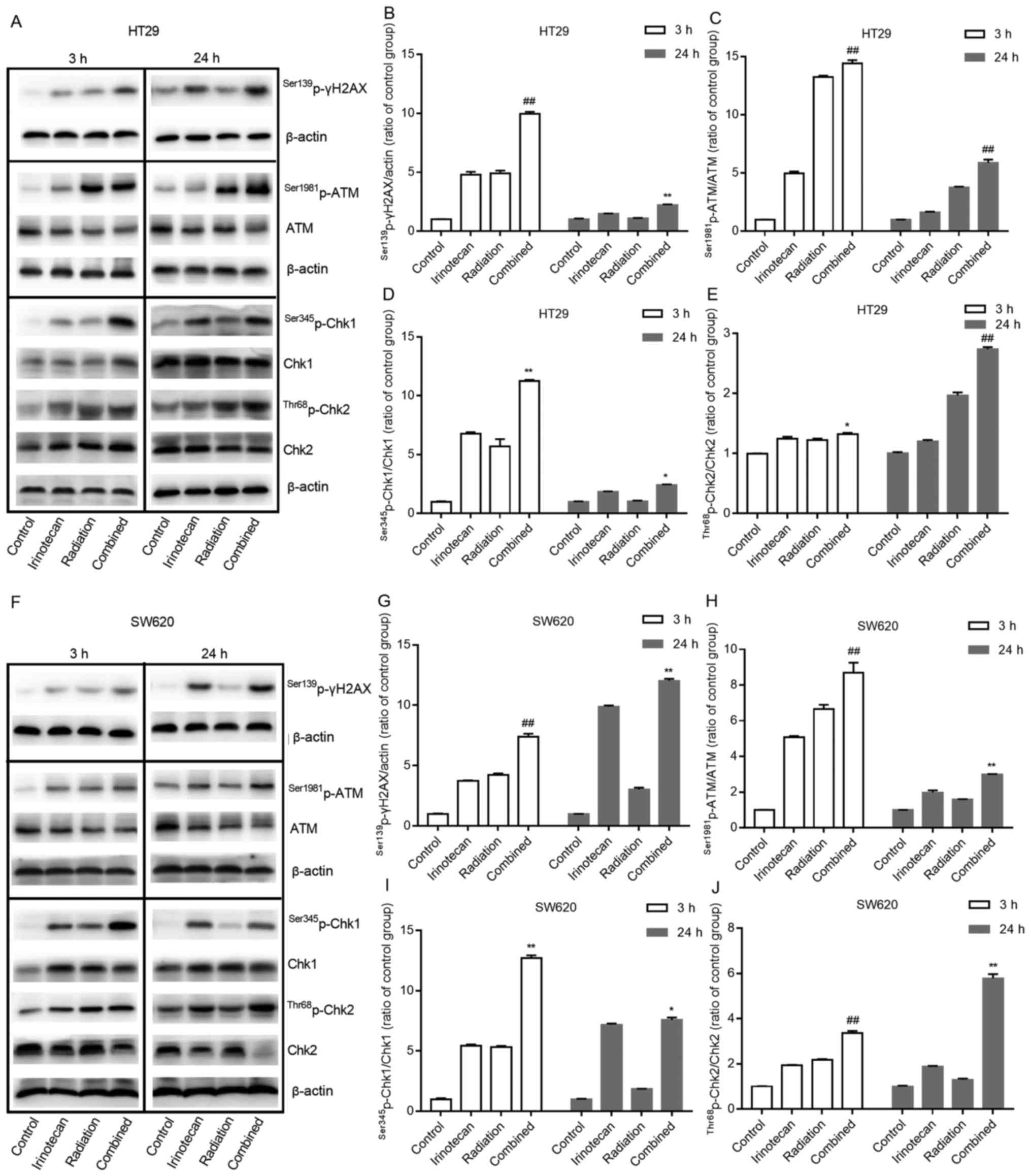
The results of western blot analysis also verified
these findings. Compared with results from the irinotecan and
radiation groups, irinotecan in combination with radiation resulted
in a marked increase in the phosphorylation levels of ATM at
Ser1981 and γH2AX at Ser139 at 3 and 24 h. Combined treatment also
increased the activation of Chk1 at Ser345 and Chk2 at Thr68 at 3
h, with significantly increased ratios of
Ser1981p-ATM/ATM, Ser345p-Chk1/Chk1 and
Thr68p-Chk2/Chk2, and the activation of these proteins
persisted for >24 h (Fig. 6A–E and
F–J). These results indicate that irinotecan could likely
enhance radiation-induced DNA double-strand damage and the DNA
damage response process through the ATM/Chk signaling pathway.
The radiosensitizing effects of
irinotecan are attributable to an enhanced G2/M phase arrest
The cells were treated with irinotecan (2
μg/ml), radiation (6 Gy) or irinotecan (2 μg/ml)
combined with radiation (6 Gy). The cell cycle distribution was
measured by flow cytometry at 3 and 24 h following radiation.
Subsequently, the expression of levels proteins related to G2/M
phase arrest, such as Tyr15p-Cdc2, Cdc2 and cyclin B1,
were measured by western blot analysis.
The proportions of cells in the G2/M phase were
elevated to a fairly high level following pre-treatment with
irinotecan. Cells in the G2/M phase are more sensitive to radiation
(24). Thus, it was suggested that
treatment with irinotecan prior to radiation could produce a
greater number of radiosensitized cells.
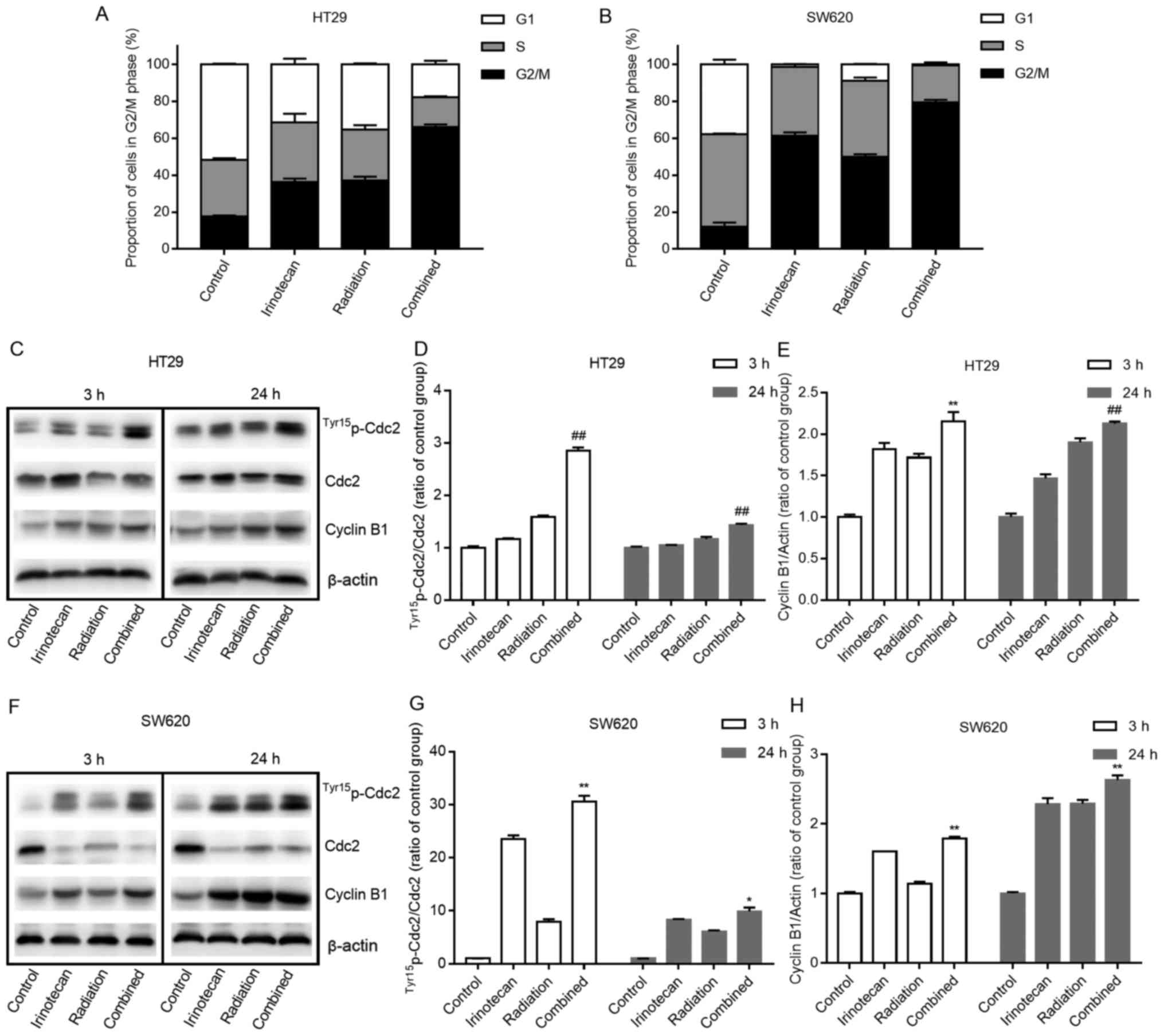
At 3 h following radiation, the combination group
exhibited a slight increase in the number of cells undergoing G2/M
phase arrest compared with the irinotecan group (data not shown).
However, 24 h later, a marked increase in the number of cells
undergoing G2/M phase arrest was observed in the radiation group
and the combination group, indicating that it took some time for
radiation to induce cell cycle arrest. As expected, the combination
group exhibited the highest rates of G2/M phase arrest at 24 h when
compared with those in the other 3 groups. The rates of G2/M phase
arrest were 66.08±1.42% for the HT29 cells and 79.35±1.49% for the
SW620 cells, indicating that the addition of irinotecan enhanced
the activation of the cell cycle checkpoint and delayed the growth
of the cells. The cell cycle distributions at 24 h are presented in
Fig. 7A and B.
Cdc2 and cyclin B1 are two key regulators of the
G2-to-M phase transition. We found a significant increase in the
expression levels of Tyr15p-Cdc2 (and in the ratio of
Tyr15p-Cdc2/Cdc2) and cyclin B1 at 3 h in the
combination group compared with values in the other 3 groups, and
this increase was enhanced over 24 h (Fig. 7C–E and F–H).
The radiosensitizing effects of
irinotecan are associated with the ATM/Chk/Cdc25C/Cdc2 pathway in
p53-mutant colorectal cancer cells
The tumor suppressor p53 is a key protein in
checkpoint pathways in normal cells, and the mutation of the p53
gene is considered an important step in colorectal cancer
formation. There are two pathways related to G2/M phase arrest, the
p21-dependent pathway in p53-wild-type cancer cells and the
Chk-dependent pathway in p53-deficient cancer cells (24-27).
For p53-mutant HT29 and SW620 cells, we hypothesized that the
Chk-dependent pathway plays an important role in irinotecan-induced
or/and radiation-induced G2/M phase arrest. The key intermediate
factor between Chk proteins and the Cdc2-cyclin B1 complex is
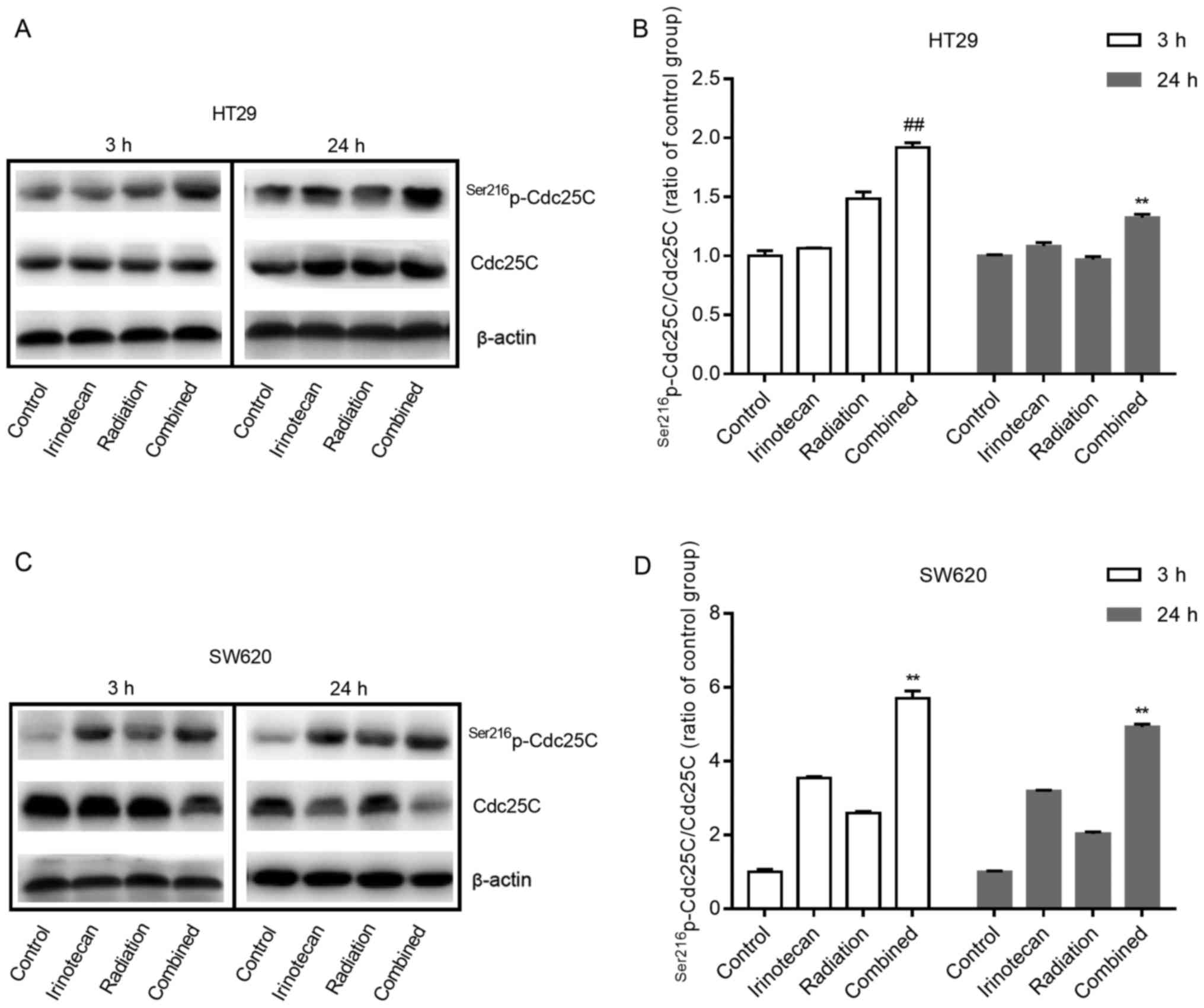
Cdc25. The negative regulation of Cdc25C by phosphorylation at
Ser216 can subsequently inhibit the activity of the Cdc2-cyclin B1
complex and lead to G2/M phase arrest. Therefore, we examined the
effects of irinotecan and radiation on the expression of
Ser216p-Cdc25C and Cdc25C in p53-mutant HT29 and SW620
cells.
As shown in Fig. 8A and
B, and C and D, for both cell lines, irinotecan and radiation
as single treatments slightly increased the expression of
Ser216p-Cdc25C; however, combined treatment
significantly enhanced phosphorylation, with an increasing ratio of
p-Cdc25C/Cdc25C. These results suggest that the addition of
irinotecan to radiation treatment promotes cycle arrest through the
negative regulation of Cdc25C by phosphorylation at Ser216, and the
ATM/Chk/Cdc25C/Cdc2/cyclin B1 pathway likely plays an important
role in the mechanism of G2/M phase arrest.
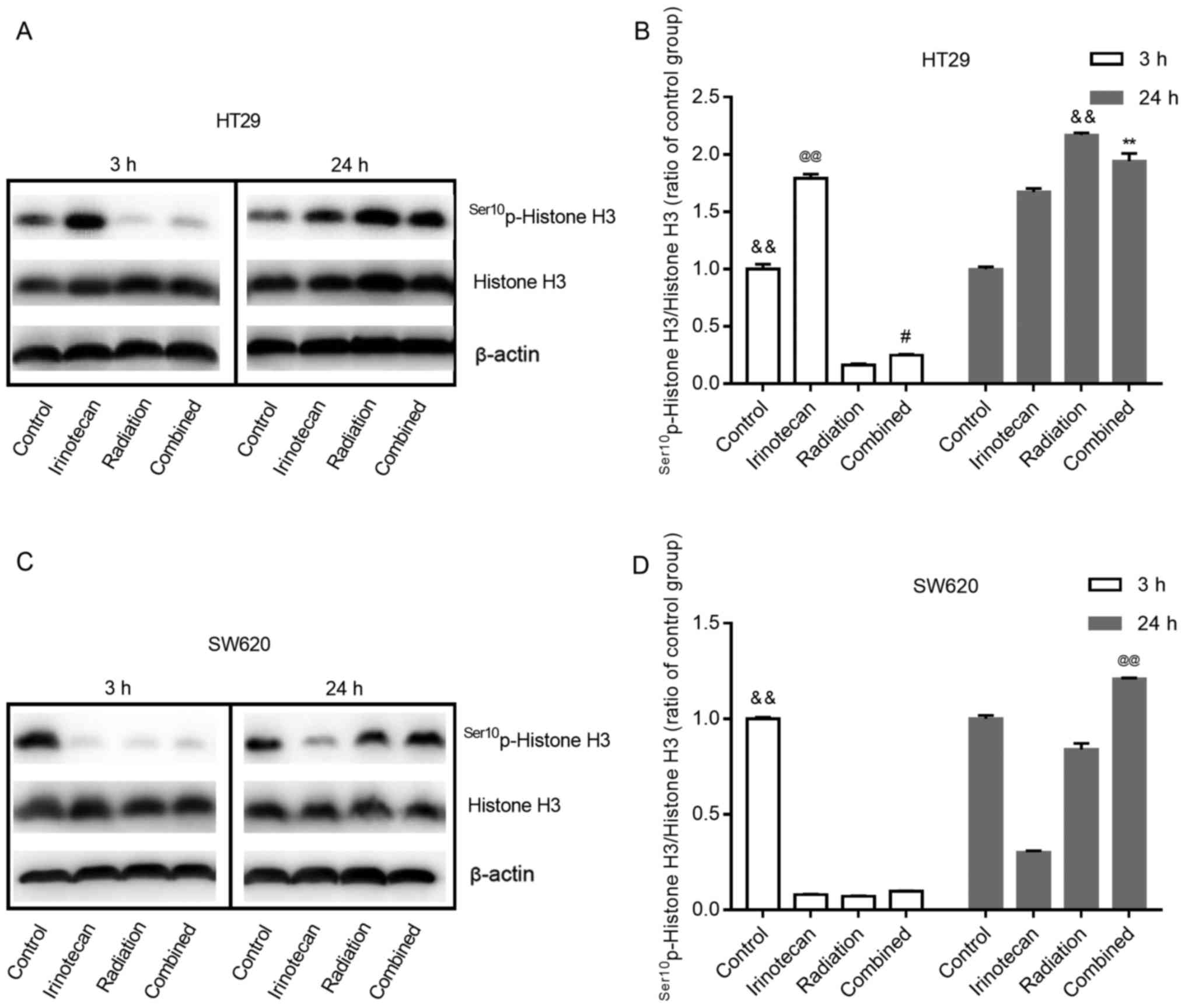
Radiosensitization by irinotecan results
in a decreased and then increased M phase arrest
Histone H3 phosphorylation is a well-established
marker of mitosis (28). In this
study, to determine whether radiation combined with irinotecan
causes the arrest of cells in the G2 phase or M phase, we examined
the expression of Ser10p-Histone H3 at 3 h and 24 h
following treatment.
As shown in Fig. 9A and
B, and C and D, for both cell lines, the cells in the control
group exhibited some degree of expression of
Ser10p-Histone H3. Of note, the effects of irinotecan
alone on the HT29 cells and SW620 cells were not consistent. For
the HT29 cells, irinotecan alone led to an enhanced expression of
Ser10p-Histone H3 at 3 and 24 h. However, for the SW620
cells, irinotecan alone reduced the expression of
Ser10p-Histone H3 at 3 h and lasted for >24 h.
For both cell lines, treatment with radiation alone
inhibited the expression of Ser10p-Histone H3 for a
short period of time, showing a low-level expression of
Ser10p-Histone H3 at 3 h. However, its expression
increased in the following 24 h, showing an increased level at 24 h
after treatment. When radiation was combined with irinotecan, we
found the same trend at 3 and 24 h following combined treatment.
For the SW620 cells, the expression of Ser10p-Histone H3
in the combination group at 24 h was higher than that in the
irinotecan, radiation and control groups. However, for the HT29
cells, the expression of Ser10p-Histone H3 in the
combination group at 24 h was lower than that in the radiation
group, but higher than that in the irinotecan and control group.
However, the expression of Ser10p-Histone H3 in the
combination group exhibited an increasing trend, probably exceeding
the radiation group in the following hours. Thus, we found that
irinotecan in combination with radiation resulted in a decreased
and then increased M phase arrest.
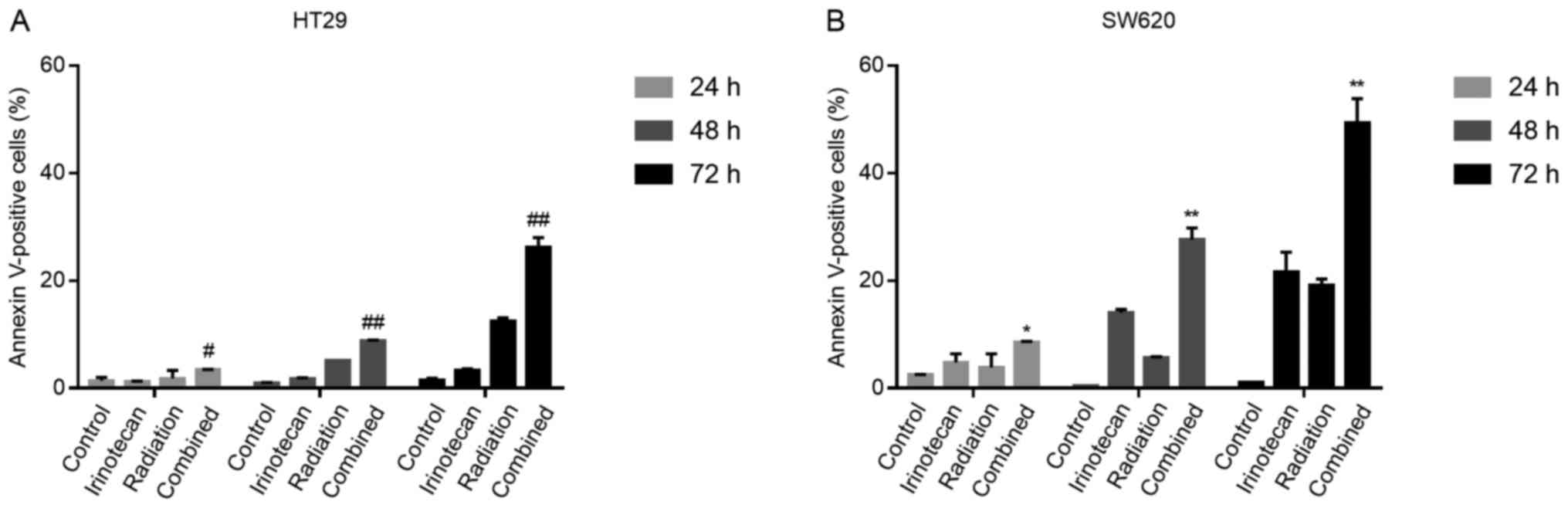
The radiosensitizing effects of
irinotecan are attributable to enhanced apoptosis following the
cycle arrest of p53-mutant colorectal cancer cells
It has been previously demonstrated that the DNA
damage induced by the TOP I inhibitor, irinotecan, triggers the
long-term cell cycle arrest of p53-wild-type colorectal carcinoma
cells and transient arrest followed by apoptosis in p53-mutant
cells (29). Thus, for the two
p53-mutant cell lines used in the present study, we examined
whether the G2/M phase arrest caused by irinotecan and radiation
was followed by apoptosis.
The cells were treated with irinotecan (2
μg/ml), radiation (6 Gy) or irinotecan (2 μg/ml) in
combination with radiation (6 Gy). Cell apoptosis was measured by
flow cytometry at a series of time points (24, 48 and 72 h)
following radiation.
As expected, the rates of cell apoptosis were
highest in the combination group, compared with the control group,
irinotecan group and radiation group. However, at 24 h, the rates
of apoptosis were relatively low and increased significantly when
the cells were incubated for a further 24 or 48 h. The rates of
apoptosis at 7 h were 26.25±1.77% for the HT29 and 49.40±4.45% for
the SW620 cells (Fig. 10). Thus,
the radiosensitizing effects of irinotecan are attributable to
short-term DNA damage-induced G2/M cycle arrest, followed by
enhanced apoptosis, which significantly inhibits cell viability and
proliferation.
Discussion
Colorectal cancer is the second leading cause of
cancer-related mortality in the United States (1). For patients with LARC, pre-operative
CRT followed by total mesorectal excision is the standard treatment
according to the NCCN Clinical Practice Guidelines in Oncology
Rectal Cancer Version 2.2018 (21). The combined use of radiation with
capecitabine has been shown to achieve a pathologically complete
response in 15 to 25% of cases and to result in downstaging in 50
to 60% of cases (30). Researchers
have attempted to identify novel radiosensitizers to achieve better
outcomes, and the addition of oxaliplatin, in addition to
capecitabine, as a radiosensitizer has been demonstrated to have no
benefit with regard to prognosis and resulted in increased toxicity
(7-10). Thus, other radiosensitizing agents
are required.
Irinotecan (also known as CPT-11), an analog of CPT,
which is an inhibitor of TOP I, is currently used in the
chemotherapy of metastatic colorectal cancer. The active form of
irinotecan, SN-38, can bind to and prevent TOP I from rejoining
transient DNA breaks during DNA replication, RNA transcription and
DNA damage repair, transforming the concealed 'potentially
sublethal' DNA damage into 'sublethal' DNA damage (13). TOP I inhibitors exhibit
S-phase-specific cytotoxicity, which has been explained by the
'Folk Collision Model'; according to Chen et al (31). It is hypothesized that the
collision between the replication machinery and the drug-trapped
TOP I cleavable complex leads to eventual G2-phase cell cycle
arrest and cell death (32), and
the addition of radiation can convert this 'sublethal' DNA damage
into 'lethal' DNA damage.
In previous studies, CPT derivatives were
demonstrated to exert radiosensitizing effects on many cell types
(14-17). Chen et al demonstrated that
CPT derivatives radiosensitized human breast cancer MCF-7 cells
when drug treatments were administered prior to, but not following,
radiation (33). However, few
studies have been published on the effects on colorectal cancer
cell lines. Chen et al (18) found that CPT exerted
radiosensitizing effects on parental HCT116 colorectal cancer
cells, with an SER of 2.0. Omura et al demonstrated
radiosensitization with SN-38 in HT29 spheroids (34).
The molecular mechanisms of the radiosensitization
of TOP I inhibitors are largely unknown. Chen et al proposed
that the drug-trapped TOP I cleavable complex may initiate TOP
I-mediated radiosensitization by 'interacting' with the replication
fork during active DNA synthesis, leading to at least three major
biochemical events, including DNA DSBs, replication fork arrest and
an aborted 'cleaved' TOP I-DNA complex (31). The induction of TOP I-mediated
radiosensitization likely requires one or more of these three
events.
When DNA damage occurs, a variety of kinases are
activated and connect the checkpoint with the cell cycle machinery.
The activation of the DNA damage checkpoint, involving ATM kinase
activation and ATM autophosphorylation at Ser1981, can lead to cell
cycle arrest to prevent mitosis in the presence of damaged DNA.
G2/M phase arrest delays the proliferation of cancer cells, and
cells in the G2/M phase are more sensitive to radiation (24). Thus, the G2/M transition is
considered a specific target, and this arrest may be a useful
strategy in CRT.
In this study, we found that low levels of
irinotecan led to a certain degree of activation of the DNA damage
response, followed by a marked increase in the number of cells
undergoing G2/M phase arrest, inducing the transformation of many
cells into a more radiosensitive status. Radiation also results in
DNA DSBs in cells, followed by G2/M phase arrest and apoptosis.
Thus, the addition of low concentrations of irinotecan as a
sensitizer to radiation treatment may be feasible.
The tumor suppressor p53 is one of the key proteins
in checkpoint pathways (26). The
activation of p53 may lead to growth arrest at both the G1 and G2/M
phases. In the canonical p53-dependent G2/M phase arrest pathway,
overexpressed p53 binds to the promoter of the p21 Cdk inhibitor,
causing p21 to accumulate. The resulting inhibition of Cdc2-cyclin
B1 activity leads to cell cycle arrest. However, several studies
have indicated that the absence of p53 (via mutation or deficiency)
is sufficient to induce G2/M arrest, and this pathway is switched
from p21-dependent to Chk-dependent in p53-deficient cells
(24,25,35).
At the G2/M transition, the removal of inhibitory
phosphorylation at Thr14 and Tyr15 results in Cdc2 activation, and
this reaction is mediated by Cdc25 phosphatases (36). In the Chk-dependent pathway, in
response to DNA damage, activated ATM phosphorylates/activates Chk1
at Ser345 and Chk2 at Thr68 (37).
The two phosphorylated Chk proteins subsequently phosphorylate
Cdc25C at Ser216, leading to the binding of 14-3-3 proteins and the
sequestration of Cdc25C in the cytoplasm, inactivating the
phosphatase activity of Cdc25C. The negative regulation of Cdc25C
by phosphorylation at Ser216 can subsequently inhibit the activity
of the Cdc2-cyclin B1 complex and lead to G2/M phase arrest, which
is an important regulatory mechanism used by cells to block mitotic
entry in response to DNA damage.
As for the mechanisms responsible for the
radiosensitizing effects induced by CPT derivatives, few studies
have examined the signaling pathways associated with the DNA damage
response system, the cell cycle and apoptosis. Chen et al
(18) demonstrated that the SERs
decreased from 2.0 in p53-wild-type HCT-116 cells to 1.6 in HCT-116
(p53−/−) cells and to 1.0 in HCT-116 (p21−/−)
cells. These findings indicated that TOP I-mediated sublethal
damage may activate DNA damage responses, including p53 and p21
regulatory pathways, and eventually lead to radiosensitization.
In the present study, both the HT29 and SW620 were
p53-mutant cell lines. Irinotecan exerted a radiosensitizing effect
on the HT29 and SW620 cells, and the SERs increased with increasing
drug concentration (SER at 2 Gy, 1.41 for the HT29 cells; SER at 2
Gy, 1.87 for the SW620 cells, with irinotecan at 2 μg/ml).
Compared with the control group, the irinotecan group and radiation
group, the combination group exhibited the slowest cell growth rate
and the most obvious foci of Ser139p-γH2AX. Combined
treatment resulted in the most significant G2/M phase arrest, and
the effects lasted for >24 h, followed by the most significant
increase in apoptosis. The results of western blot analysis
indicated that the expression of proteins related to the DNA damage
response system (Ser1981p-ATM, Ser345p-Chk1,
Thr68p-Chk2, Ser139p-γH2AX), as well as of
those related to the cell cycle (Tyr15p-Cdc2, cyclin
B1), was increased in the combination group. We propose that in
p53-mutant colorectal cancer cells, the mechanism for G2/M phase
arrest of radiosensitization is Chk-dependent and that Cdc25C is
involved. As expected, the expression of Ser216p-Cdc25C
was also increased in the combination group, indicating that
irinotecan likely radiosensitized the p53-mutant HT29 and SW620
cells through the ATM/Chk/Cdc25C/Cdc2 pathway.
In addition to G2/M phase arrest, the levels of
Histone H3 phosphorylation (Ser10), a well-established marker of
mitosis, were determined to quantify the cells in the mitotic phase
of the cell cycle. Phosphorylation at Ser10 of Histone H3 is
tightly associated with chromosome condensation during mitosis
(28), and it can act as part of a
molecular mechanism driving mitotic chromosomal condensation during
M phase entry (38). In this
study, we found that irinotecan in combination with radiation
resulted in a decreased and then increased M phase arrest. This
indicated that the arrested cells were mostly present in the G2
phase at tge early time points after treatment, and arrested cells
in M phase increased in the following 24 h. The cells probably
underwent G2 phase arrest for a short period time and both G2 and M
phase arrest occurred later. The cells arrested in the G2 or M
phase probably died through mitotic catastrophe and apoptosis. The
gradually enhanced apoptosis after cycle arrest in both cell lines
was also confirmed in our experiments.
The present study has several limitations. First, we
did not perform xenograft experiments to examine whether irinotecan
exerts radiosensitizing effects on animals, which represents a
condition more similar to the clinical setting. Second, experiments
related to gene regulation were not performed in this study,
including the upregulation and knockout of specific genes (such as
Cdc25C and Wee1), which can aid in the verification of relevant
molecular pathways. Third, experiments related to the mechanisms of
apoptosis were not performed. The associations between the p53
status, cell cycle arrest and apoptosis in terms of
irinotecan-induced radiosensitization remain unknown. Previous
studies using irinotecan as a single agent have demonstrated that
the DNA damage induced by irinotecan triggers long-term cell cycle
arrest in p53-wild-type colorectal cancer cells and transient
arrest, which is followed by the apoptosis of p53-mutant cells
(29,39). Additionally, in p53-wild-type
cells, the DNA damage-induced expression of p53 suppresses the
mitotic checkpoint kinase hMps1 (human ortholog of the yeast
monopolar spindle 1 kinase), and the lack of this suppression in
p53-mutant cells contributes to apoptosis (40). In this study, we examined
irinotecan as a radiosensitizer to and found that it induced marked
G2/M phase arrest, followed by the enhanced apoptosis of p53-mutant
colorectal cancer cells. However, whether this drug leads to the
long-term cell cycle arrest of p53-wild-type colorectal cancer
cells remains unclear. Thus, further studies are warranted to
explore the above-mentioned mechanisms and to verify the results in
animals.
The present study indicated that irinotecan could be
used as an effective radiosensitizer in the pre-operative CRT of
LARCs and may increase tumor regression rates, local control and,
potentially, overall survival. To date, a number of phase I-II
clinical trials have achieved pathologically complete response
rates ranging from 13.7 to 37% (41-44),
with a weekly irinotecan dose of 50-60 mg/m2 in
combination with fluoropyrimidine. Irinotecan should be used with
caution in clinical treatments as of its adverse effects, such as
diarrhea and neutropenia occur, particularly in UGT1A1⁄28
*1*28 and *28 *28
genotypes (45-47). Recently, Zhu et al (48) reported that the maximum tolerated
doses (MTDs) of weekly irinotecan in neoadjuvant CRT for LARC were
80 mg/m2 in patients with the *1*1
genotype and 65 mg/m2 in those with the
*1*28 genotype. A randomized, controlled
phase III trial is currnelty ongoing to examine whether high-dose
irinotecan can improve the clinical benefit under the guidance of
the UGT1A1*28 genotype in neoadjuvant therapy compared
with standard capecitabine-based CRT (CinClare, NCT02605265).
In conclusion, this study demonstrates that
irinotecan exerts radiosensitizing effects on HT29 and SW620 cells,
and the SER increases with the increasing drug concentration. This
effect is attributed to the activation of the DNA damage response
system, leading to significant G2/M phase arrest, followed by
enhanced apoptosis, and this process likely occurs through the
ATM/Chk/Cdc25C/Cdc2 pathway in p53-mutant colorectal cancer cells.
Large-scale clinical trials are required to investigate not only
the efficacy of irinotecan, but also its effects as a
radiosensitizer.
Acknowledgments
The authors would like to thank American Journal
Experts (https://www.aje.com) for assisting
with language editing.
Abbreviations:
|
IC50
|
half-maximal inhibitory
concentration
|
|
SER
|
sensitivity enhancement ratio
|
|
DSB
|
double-strand breaks
|
Funding
This study was supported by grants from the
National Natural Science Foundation of China (no. 81572955).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
YW, LY and JZ have full access to all of the
findings in the study and take responsibility for the integrity of
the data and the accuracy of the data analysis. ZZ, LL, MZ and YY
provided the study concept and design. YW, LY, JZ and MZ were major
contributors in performing the experiments. YW, LS, HZ, WY, RH and
WD analyzed and interpreted the data. YW wrote the manuscript. MZ,
RH, WY and YY reviewed and edited the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sauer R, Becker H, Hohenberger W, Rödel C,
Wittekind C, Fietkau R, Martus P, Tschmelitsch J, Hager E, Hess CF,
et al German Rectal Cancer Study Group: Preoperative versus
postoperative chemoradiotherapy for rectal cancer. N Engl J Med.
351:1731–1740. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bosset JF, Calais G, Mineur L, Maingon P,
Radosevic-Jelic L, Daban A, Bardet E, Beny A, Briffaux A and
Collette L: Enhanced tumorocidal effect of chemotherapy with
preoperative radiotherapy for rectal cancer: Preliminary results -
EORTC 22921. J Clin Oncol. 23:5620–5627. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bosset JF, Collette L, Calais G, Mineur L,
Maingon P, Radosevic-Jelic L, Daban A, Bardet E, Beny A and Ollier
JC; EORTC Radiotherapy Group Trial 22921: Chemotherapy with
preoperative radiotherapy in rectal cancer. N Engl J Med.
355:1114–1123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma B, Gao P, Wang H, Xu Q, Song Y, Huang
X, Sun J, Zhao J, Luo J, Sun Y, et al: What has preoperative
radio(chemo) therapy brought to localized rectal cancer patients in
terms of perioperative and long-term outcomes over the past
decades? A systematic review and meta-analysis based on 41,121
patients. Int J Cancer. 141:1052–1065. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Greenhalgh TA, Dearman C and Sharma RA:
Combination of novel agents with radiotherapy to treat rectal
cancer. Clin Oncol (R Coll Radiol). 28:116–139. 2016. View Article : Google Scholar
|
|
7
|
Aschele C, Cionini L, Lonardi S, Pinto C,
Cordio S, Rosati G, Artale S, Tagliagambe A, Ambrosini G, Rosetti
P, et al: Primary tumor response to preoperative chemoradiation
with or without oxaliplatin in locally advanced rectal cancer:
Pathologic results of the STAR-01 randomized phase III trial. J
Clin Oncol. 29:2773–2780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gérard JP, Azria D, Gourgou-Bourgade S,
Martel-Laffay I, Hennequin C, Etienne PL, Vendrely V, François E,
de La Roche G, Bouché O, et al: Comparison of two neoadjuvant
chemoradiotherapy regimens for locally advanced rectal cancer:
Results of the phase III trial ACCORD 12/0405-Prodige 2. J Clin
Oncol. 28:1638–1644. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rödel C, Liersch T, Becker H, Fietkau R,
Hohenberger W, Hothorn T, Graeven U, Arnold D, Lang-Welzenbach M,
Raab HR, et al German Rectal Cancer Study Group: Preoperative
chemo-radiotherapy and postoperative chemotherapy with fluorouracil
and oxaliplatin versus fluorouracil alone in locally advanced
rectal cancer: Initial results of the German CAO/ARO/AIO-04
randomised phase 3 trial. Lancet Oncol. 13:679–687. 2012.
View Article : Google Scholar
|
|
10
|
Allegra CJ, Yothers G, O'Connell MJ, Beart
RW, Wozniak TF, Pitot HC, Shields AF, Landry JC, Ryan DP, Arora A,
et al: Neoadjuvant 5-FU or capecitabine plus radiation with or
without oxaliplatin in rectal cancer patients: A phase III
randomized clinical trial. J Natl Cancer Inst. 107:1072015.
View Article : Google Scholar
|
|
11
|
Fujita K, Kubota Y, Ishida H and Sasaki Y:
Irinotecan, a key chemotherapeutic drug for metastatic colorectal
cancer. World J Gastroenterol. 21:12234–12248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chabot GG: Clinical pharmacokinetics of
irinotecan. Clin Pharmacokinet. 33:245–259. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen AY, Choy H and Rothenberg ML: DNA
topoisomerase I-targeting drugs as radiation sensitizers. Oncology
(Williston Park). 13(Suppl 5): 39–46. 1999.
|
|
14
|
Mattern MR, Hofmann GA, McCabe FL and
Johnson RK: Synergistic cell killing by ionizing radiation and
topoisomerase I inhibitor topotecan (SK&F 104864). Cancer Res.
51:5813–5816. 1991.PubMed/NCBI
|
|
15
|
Kim JH, Kim SH, Kolozsvary A and Khil MS:
Potentiation of radiation response in human carcinoma cells in
vitro and murine fibrosarcoma in vivo by topotecan, an inhibitor of
DNA topoisomerase I. Int J Radiat Oncol Biol Phys. 22:515–518.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boothman DA, Wang M, Schea RA, Burrows HL,
Strickfaden S and Owens JK: Posttreatment exposure to camptothecin
enhances the lethal effects of x-rays on radioresistant human
malignant melanoma cells. Int J Radiat Oncol Biol Phys. 24:939–948.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hennequin C, Giocanti N, Balosso J and
Favaudon V: Interaction of ionizing radiation with the
topoisomerase I poison camptothecin in growing V-79 and HeLa cells.
Cancer Res. 54:1720–1728. 1994.PubMed/NCBI
|
|
18
|
Chen AY, Scruggs PB, Geng L, Rothenberg ML
and Hallahan DE: p53 and p21 are major cellular determinants for
DNA topoisomerase I-mediated radiation sensitization in mammalian
cells. Ann N Y Acad Sci. 922:298–300. 2000. View Article : Google Scholar
|
|
19
|
Yin L, Wu J, Wu J, Ye J, Jiang X, Chen M,
Wang D, Wang X, Zong D, Gu J, et al: Radiosensitization effect of
nedaplatin on nasopharyngeal carcinoma cells in different status of
Epstein-Barr virus infection. BioMed Res Int. 2014:7136742014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jones L, Hoban P and Metcalfe P: The use
of the linear quadratic model in radiotherapy: A review. Australas
Phys Eng Sci Med. 24:132–146. 2001. View Article : Google Scholar
|
|
21
|
NCCN Clinical Practice Guidelines in
Oncology Rectal Cancer Version 2.2018. J Natl Compr Canc Netw.
16:874–901. 2018. View Article : Google Scholar
|
|
22
|
Haug K, Kravik KL and De Angelis PM:
Cellular response to irinotecan in colon cancer cell lines showing
differential response to 5-fluorouracil. Anticancer Res.
28A:583–592. 2008.
|
|
23
|
Kaku Y, Tsuchiya A, Kanno T and Nishizaki
T: Irinotecan induces cell cycle arrest, but not apoptosis or
necrosis, in Caco-2 and CW2 colorectal cancer cell lines.
Pharmacology. 95:154–159. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pawlik TM and Keyomarsi K: Role of cell
cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol
Biol Phys. 59:928–942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stark GR and Taylor WR: Control of the
G2/M transition. Mol Biotechnol. 32:227–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Taylor WR and Stark GR: Regulation of the
G2/M transition by p53. Oncogene. 20:1803–1815. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2, and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Preuss U, Landsberg G and Scheidtmann KH:
Novel mitosis-specific phosphorylation of histone H3 at Thr11
mediated by Dlk/ZIP kinase. Nucleic Acids Res. 31:878–885. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhonde MR, Hanski ML, Notter M, Gillissen
BF, Daniel PT, Zeitz M and Hanski C: Equivalent effect of DNA
damage-induced apoptotic cell death or long-term cell cycle arrest
on colon carcinoma cell proliferation and tumour growth. Oncogene.
25:165–175. 2006. View Article : Google Scholar
|
|
30
|
Das P, Skibber JM, Rodriguez-Bigas MA,
Feig BW, Chang GJ, Wolff RA, Eng C, Krishnan S, Janjan NA and Crane
CH: Predictors of tumor response and downstaging in patients who
receive preoperative chemoradiation for rectal cancer. Cancer.
109:1750–1755. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen AY, Chou R, Shih SJ, Lau D and
Gandara D: Enhancement of radiotherapy with DNA topoisomerase
I-targeted drugs. Crit Rev Oncol Hematol. 50:111–119. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li TK and Liu LF: Tumor cell death induced
by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol.
41:53–77. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen AY, Okunieff P, Pommier Y and
Mitchell JB: Mammalian DNA topoisomerase I mediates the enhancement
of radiation cytotoxicity by camptothecin derivatives. Cancer Res.
57:1529–1536. 1997.PubMed/NCBI
|
|
34
|
Omura M, Torigoe S and Kubota N: SN-38, a
metabolite of the camptothecin derivative CPT-11, potentiates the
cytotoxic effect of radiation in human colon adenocarcinoma cells
grown as spheroids. Radiother Oncol. 43:197–201. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jo HJ, Song JD, Kim KM, Cho YH, Kim KH and
Park YC: Diallyl disulfide induces reversible G2/M phase arrest on
a p53-independent mechanism in human colon cancer HCT-116 cells.
Oncol Rep. 19:275–280. 2008.
|
|
36
|
Sur S and Agrawal DK: Phosphatases and
kinases regulating CDC25 activity in the cell cycle: Clinical
implications of CDC25 overexpression and potential treatment
strategies. Mol Cell Biochem. 416:33–46. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bartek J and Lukas J: Chk1 and Chk2
kinases in checkpoint control and cancer. Cancer Cell. 3:421–429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hendzel MJ, Wei Y, Mancini MA, Van Hooser
A, Ranalli T, Brinkley BR, Bazett-Jones DP and Allis CD:
Mitosis-specific phosphorylation of histone H3 initiates primarily
within pericentromeric heterochromatin during G2 and spreads in an
ordered fashion coincident with mitotic chromosome condensation.
Chromosoma. 106:348–360. 1997. View Article : Google Scholar
|
|
39
|
Magrini R, Bhonde MR, Hanski ML, Notter M,
Scherübl H, Boland CR, Zeitz M and Hanski C: Cellular effects of
CPT-11 on colon carcinoma cells: Dependence on p53 and hMLH1
status. Int J Cancer. 101:23–31. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bhonde MR, Hanski ML, Budczies J, Cao M,
Gillissen B, Moorthy D, Simonetta F, Scherübl H, Truss M, Hagemeier
C, et al: DNA damage-induced expression of p53 suppresses mitotic
checkpoint kinase hMps1: The lack of this suppression in p53MUT
cells contributes to apoptosis. J Biol Chem. 281:8675–8685. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mohiuddin M, Winter K, Mitchell E, Hanna
N, Yuen A, Nichols C, Shane R, Hayostek C and Willett C; Radiation
Therapy Oncology Group Trial 0012: Randomized phase II study of
neoadjuvant combined-modality chemoradiation for distal rectal
cancer: Radiation Therapy Oncology Group Trial 0012. J Clin Oncol.
24:650–655. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wong SJ, Moughan J, Meropol NJ, Anne PR,
Kachnic LA, Rashid A, Watson JC, Mitchell EP, Pollock J, Lee RJ, et
al: Efficacy endpoints of radiation therapy group protocol 0247: A
randomized, phase 2 study of neoadjuvant radiation therapy plus
concurrent capecitabine and irinotecan or capecitabine and
oxaliplatin for patients with locally advanced rectal cancer. Int J
Radiat Oncol Biol Phys. 91:116–123. 2015. View Article : Google Scholar
|
|
43
|
Mehta VK, Cho C, Ford JM, Jambalos C, Poen
J, Koong A, Lin A, Bastidas JA, Young H, Dunphy EP, et al: Phase II
trial of preoperative 3D conformal radiotherapy, protracted venous
infusion 5-fluorouracil, and weekly CPT-11, followed by surgery for
ultrasound-staged T3 rectal cancer. Int J Radiat Oncol Biol Phys.
55:132–137. 2003. View Article : Google Scholar
|
|
44
|
Navarro M, Dotor E, Rivera F,
Sánchez-Rovira P, Vega-Villegas ME, Cervantes A, García JL, Gallén
M and Aranda E: A Phase II study of preoperative radiotherapy and
concomitant weekly irinotecan in combination with protracted venous
infusion 5-fluorouracil, for resectable locally advanced rectal
cancer. Int J Radiat Oncol Biol Phys. 66:201–205. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu X, Cheng D, Kuang Q, Liu G and Xu W:
Association of UGT1A1*28 polymorphisms with
irinotecan-induced toxicities in colorectal cancer: A meta-analysis
in Caucasians. Pharmacogenomics J. 14:120–129. 2014. View Article : Google Scholar
|
|
46
|
Toffoli G, Cecchin E, Corona G, Russo A,
Buonadonna A, D'Andrea M, Pasetto LM, Pessa S, Errante D, De
Pangher V, et al: The role of UGT1A1*28 polymorphism in
the pharmacodynamics and pharmacokinetics of irinotecan in patients
with metastatic colorectal cancer. J Clin Oncol. 24:3061–3068.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Innocenti F, Undevia SD, Iyer L, Chen PX,
Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM,
et al: Genetic variants in the UDP-glucuronosyltransferase 1A1 gene
predict the risk of severe neutropenia of irinotecan. J Clin Oncol.
22:1382–1388. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhu J, Li X, Shen Y, Guan Y, Gu W, Lian P,
Sheng W, Cai S and Zhang Z: Genotype-driven phase I study of weekly
irinotecan in combination with capecitabine-based neoadjuvant
chemo-radiation for locally advanced rectal cancer. Radiother Oncol
Radiother Oncol. S0167-8140:32749–32744. 2017.
|