Introduction
Adult T-cell leukemia/lymphoma (ATLL) constitutes an
aggressive T-cell malignancy characterized by the clonal expansion
of CD4+ T-cells that develops in 2–7% of individuals
infected with the retrovirus human T-cell leukemia virus type 1
(HTLV-1) (1). ATLL is associated
with a poor prognosis, and despite advances in chemotherapy, the
median survival time (8.3 and 10.6 months for acute and lymphoma
types, respectively) has not improved since HTLV-1 was first
described (2). Therefore, there is
an urgent need for more effective therapies based on a detailed
understanding of the molecular pathogenesis of this disease.
T-cells transformed by HTLV-1 are initially
dependent on interleukin (IL)-2 for survival; however, these cells
can subsequently transition into an IL-2-independent state
(3). The IL-2 receptor (IL-2R) is
composed of three subunits: IL-2Rα and -2Rβ, and the common γ
chain, with the cytoplasmic regions of IL-2Rβ and the common γ
chain being critical for IL-2 signal transduction. Although they
lack intrinsic protein tyrosine kinase (PTK) domains, these
subunits recruit various non-receptor-type PTKs, such as Janus
kinases (JAKs) and spleen tyrosine kinase (SYK) (4). In turn, JAK1 and JAK3 activate signal
transducer and activator of transcription (STAT)1, STAT3 and STAT5
(5). Notably, the transition to
IL-2 independence is associated with the activation of the JAK/STAT
signaling pathway (6,7), which is constitutively activated in
HTLV-1-transformed T-cell lines, as well as primary ATLL cells
(6–8). Accordingly, JAK inhibitors have been
shown to inhibit the proliferation of ATLL cells (9,10).
SYK is also overexpressed in HTLV-1-transformed T-cell lines
(11). Thus, therapeutic
strategies that target JAK/STAT signaling and SYK may potentially
benefit patients with ATLL.
Cerdulatinib is an orally available, ATP-competitive
small-molecule inhibitor of SYK and JAK1/3 kinases (12) that has been shown to exhibit
antitumor activity in B-cell malignancies, including diffuse large
B-cell lymphoma, Burkitt lymphoma and chronic lymphocytic leukemia
(CLL) (12–15). A phase I/II dose escalation study
in CLL, small lymphocytic lymphoma and non-Hodgkin lymphoma is
currently underway (16). In the
present study, we investigated the effects of dual SYK/JAK
inhibition on the viability of HTLV-1-transformed and ATLL-derived
T-cell lines in order to evaluate the therapeutic potential of this
approach.
Materials and methods
Reagents
Cerdulatinib (cat. no. S7634; Selleck Chemicals,
Houston, TX, USA), JAK inhibitor 1 (cat. no. 420099; Merck
Millipore, Burlington, MA, USA), PRT060318 (cat. no. SYN-1204;
SYNkinase, Parkville, Australia) and z-VAD-FMK (cat. no. G7232;
Promega Corp., Madison, WI, USA) were dissolved in dimethyl
sulfoxide (cat. no. 13407-45; Nacalai Tesque, Inc., Kyoto, Japan).
Antibodies against SYK (cat. no. 2712), phospho-SYK (Tyr525/526;
cat. no. 2710), STAT3 (cat. no. 9132), phospho-STAT3 (Tyr705; cat.
no. 9145), STAT5 (cat. no. 9363), phospho-STAT5 (Tyr694; cat. no.
4322), ERK (cat. no. 4695), phospho-ERK (Thr202/Tyr204; cat. no.
4370), phospho-p38 (Thr180/Tyr182; cat. no. 9211), Bcl-xL (cat. no.
2762), Bax (cat. no. 2772), Bak (cat. no. 3814), survivin (cat. no.
2808), AKT (cat. no. 9272), phospho-AKT (Thr308; cat. no. 13038 and
Ser473; cat. no. 4060), phospho-IκBα (Ser32/36; cat. no. 9246), IκB
kinase (IKK) α (cat. no. 2682), IKKβ (cat. no. 2684),
phospho-IKKα/β (Ser176/180 and Ser177/181; cat. no. 2694), cleaved
caspase-8 (cat. no. 9496), cleaved caspase-9 (cat. no. 9501),
cleaved caspase-3 (cat. no. 9664), cleaved poly(ADP-ribose)
polymerase (PARP; cat. no. 9541) and c-FLIP (cat. no. 3210) were
from Cell Signaling Technology, Inc. (Beverly, MA, USA). Antibodies
against cyclin B1 (cat. no. MS-868), cyclin-dependent kinase (CDK)1
(cat. no. MS-275), p53 (cat. no. MS-738) and actin (cat. no.
MS-1295) were from Neomarkers, Inc. (Fremont, CA, USA). Antibodies
against p21 (cat. no. MS-891), p27 (cat. no. MS-256) and X-linked
inhibitor of apoptosis (XIAP; cat. no. M044-3) were from Medical
& Biological Laboratories, Co. (Aichi, Japan). Antibodies
against JAK1 (cat. no. sc-277), phospho-JAK1 (Tyr1022/1023; cat.
no. sc-16773-R), IκBα (cat. no. sc-371), Mcl-1 (cat. no. sc-819)
and c-IAP2 (cat. no. sc-7944) were from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA).
Cell culture
HTLV-1-infected T-cell lines include
HTLV-1-transformed T-cell lines and ATLL-derived T-cell lines.
HTLV-1-transformed MT-2 and HUT-102, ATLL-derived TL-OmI, and
uninfected control Jurkat and CCRF-CEM T-cell lines were grown in
RPMI-1640 medium (cat. no. 30264-56, Nacalai Tesque, Inc.)
supplemented with 10% fetal bovine serum (Biological Industries,
Kibbutz Beit Haemek, Israel) and 1% penicillin/streptomycin (cat.
no. 09367-34; Nacalai Tesque, Inc.) at 37°C and 5% CO2
in a humidified incubator. The HUT-102 and Jurkat cells were
obtained from Fujisaki Cell Center, Hayashibara Laboratories, Inc.
(Okayama, Japan). The MT-2 cell line was kindly provided by Dr
Naoki Yamamoto (Tokyo Medical and Dental University, Tokyo, Japan).
The TL-OmI and CCRF-CEM cells were provided by Dr Masahiro Fujii
(Niigata University, Niigata, Japan). Frozen human peripheral blood
mononuclear cells (PBMCs) (cat. no. HC-0001) were obtained from
Lifeline Cell Technology (Frederick, MD, USA).
Cell growth and cytotoxicity assay
The cells were seeded in triplicate in 96-well flat
microtiter plates and treated with cerdulatinib (0.16–10
µM), PRT060318 (0.63–10 µM) and JAK inhibitor 1
(0.63–10 µM) for 24–72 h. Cell growth and cytotoxicity were
determined with the water-soluble tetrazolium (WST)-8 uptake assay
using a kit (Nacalai Tesque, Inc.) according to the manufacturer's
instructions. Briefly, 10 µl of WST-8 reagent (cat. no.
07553-44; Nacalai Tesque, Inc.) was added to each well. After 4 h,
WST-8 reduction was measured at 450 nm using a Wallac 1420
Multilabel Counter (PerkinElmer, Inc., Waltham, MA, USA). The
absorbance values were normalized to those of the untreated control
samples. The 50% inhibitory concentration (IC50) for
WST-8 activity was calculated by fitting data points with a
logarithmic curve using CalcuSym software (version 2.0; Biosoft,
Cambridge, UK).
Combination synergy calculation
Serial dilutions of JAK inhibitor 1 or PRT060318
alone, or in combination were tested. The combination index (CI)
was calculated using CalcuSym software, with CI<1, CI=1 and
CI>1 representing synergistic, additive or antagonistic
interactions of the two agents, respectively.
Cell cycle analysis
Cell cycle distribution was determined by staining
with propidium iodide to label nuclear DNA using the CycleTEST Plus
DNA Reagent kit (cat. no. 340242; Becton-Dickinson Immunocytometry
Systems, San Jose, CA, USA) according to the supplier's
instructions. The cells were sorted on an Epics XL flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA) and MultiCycle software
(version 3.0; Phoenix Flow Systems, San Diego, CA, USA) was used to
determine the fraction of cells at each phase of the cell
cycle.
Analysis of cell apoptosis
The cells were treated with cerdulatinib (2.5–10
µM) for up to 72 h and then permeabilized by incubation on
ice for 20 min with 100 µg/ml of digitonin, followed by
treatment with phycoerythrin-conjugated anti-APO2.7 antibody (1:10)
(cat. no. IM2088; Beckman Coulter, Inc., Marseille, France) for 15
min at room temperature. The apoptotic fraction was separated by
flow cytometry. To evaluate changes in nuclear morphology, the
cells were stained with 10 µg/ml Hoechst 33342 (cat. no.
346-07951; Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
and observed under a DMI6000 microscope (Leica Microsystems,
Wetzlar, Germany) (17).
Measurement of caspase activity
Caspase activity was measured using Colorimetric
Caspase Assay kits (cat. nos. 4800, 4805 and 4810, Medical &
Biological Laboratories, Co.). Briefly, cell extracts were
recovered using the cell lysis buffer supplied with the kit, and
caspase-8, -9 and -3 activity was assessed using the specific
colorimetric probe. The kits are based on detection of the
chromophore ρ-nitroanilide after cleavage from caspase-specific
labeled substrates. Colorimetric measurements were made with a
Wallac 1420 Multilabel Counter.
Western blot analysis
The cells were lysed in lysis buffer containing 62.5
mM Tris-HCl (pH 6.8) (cat. no. 35434-21; Nacalai Tesque, Inc.), 2%
sodium dodecyl sulfate (SDS) (cat. no. 31607-65; Nacalai Tesque,
Inc.), 10% glycerol (cat. no. 17045-65; Nacalai Tesque, Inc.), 6%
2-mercaptoethanol (cat. no. 21438-82; Nacalai Tesque, Inc.) and
0.01% bromophenol blue (cat. no. 021-02911; Wako Pure Chemical
Industries, Osaka, Japan). The protein concentration was determined
using the DC Protein Assay kit (cat. no. 5000116JA; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Cell lysates (20 µg)
were separated by 8–15% SDS-polyacrylamide gel electrophoresis and
transferred onto a polyvinylidene difluoride membrane (cat. no.
IPVH00010EMD; Merck KGaA, Darmstadt, Germany) that was probed with
primary antibodies (1:1,000). The membranes were blocked with 4%
non-fat dry milk (cat. no. 9999; Cell Signaling Technology, Inc.)
for 1 h at room temperature. Following incubation with horseradish
peroxidase-conjugated secondary anti-mouse (1:1,000) (cat. no.
7076; Cell Signaling Technology, Inc.) or anti-rabbit (1:1,000)
(cat. no. 7074; Cell Signaling Technology, Inc.) IgG,
immunoreactivity was visualized using enhanced chemiluminescence
reagent (cat. no. RPN2232; Amersham Biosciences Corp., Piscataway,
NJ, USA).
Electrophoretic mobility shift assay
(EMSA)
To assess STAT3, STAT5, nuclear factor-κB (NF-κB)
and activator protein-1 (AP-1) activation, we prepared nuclear
extracts from the cerdulatinib-treated cells and performed EMSA, as
previously described (18).
Nuclear extracts with 5 µg of protein were incubated with
32P-labeled probes. The top strand sequences of the
oligonucleotide probes were as follows: 5′-GATCGACATTTCCCGTAAATCG-3′ (STAT3
consensus binding motif derived from the c-fos gene);
5′-GATCAGATTTCTAGGAATTCAAATC-3′ (STAT5
consensus binding motif derived from the CSN2 gene);
5′-GATCCGGCAGGGGAATCTCCCTCTC-3′ (typical
NF-κB element of the IL-2RA gene); and
5′-GATCGTGATGACTCAGGTT-3′ (consensus AP-1
element of the IL-8 gene). The oligonucleotide
5′-GATCTGTCGAATGCAAATCACTAGAA-3′ containing
the consensus sequence of the octamer (Oct) binding motif was used
to evaluate specific binding of the transcription factor Oct-1,
which regulates the transcription of a number of housekeeping
genes. STAT3, STAT5, NF-κB, AP-1 and Oct-1 binding sites are
underlined in the above sequences. It has been reported that NF-κB
(p50, RelA and RelB), AP-1 (JunB and JunD), STAT3 and STAT5
specifically bind to the above oligonucleotides in HTLV-1-infected
T-cell lines and ATLL cells (8,19).
Xenograft tumor model
C.B-17/Icr-severe combined immune deficient (SCID)
mice are a useful model for investigating the proliferative and
tumorigenic potential of HTLV-1-infected T-cell lines. HUT-102
cells (1×107/0.2 ml RPMI-1640 medium) were
subcutaneously transplanted into the postauricular region of
5-week-old female SCID mice (Kyudo, Co., Tosu, Japan) on day 0. The
mouse body weight was 16.6–18.3 g upon purchase. Animal cages were
maintained at a temperature of 24°C and a humidity of 60%. The mice
were fed a standard rodent diet (CE-2; CLEA Japan, Inc., Tokyo,
Japan) and water ad libitum. The mice were divided into 2
groups (n=5, each). Cerdulatinib was solubilized in 0.5%
methylcellulose (cat. no. 133–17815; Wako Pure Chemical Industries)
and administered at a dose of 35 mg/kg by oral gavage 5 times a
week. The treatment was continued for up to 28 days starting from
the day after cell inoculation. The control group received the
vehicle (0.3 ml of 0.5% methylcellulose) only. The tumor diameter
was measured weekly with a shifting caliper and tumor volume was
calculated using the ellipsoid volume formula (π/6 × length × width
× height) (20). Body weight was
also measured weekly. The mice were sacrificed on day 28. Tumors
were collected and their weight was measured. Experimental
procedures and animal care were in compliance with the Guidelines
for Animal Experimentation of University of The Ryukyus (Nishihara,
Japan) and were approved by the Animal Care and Use Committee of
University of The Ryukyus (reference no. A2016088).
Morphological analysis of tumor tissue
and terminal deoxynucleotidyl transferase deoxyuridine triphosphate
nick-end labeling (TUNEL) assay
Tumor specimens were collected from mice, fixed in
formalin (Wako Pure Chemical Industries) solution, dehydrated
through a graded ethanol series (Japan Alcohol Selling Co., Tokyo,
Japan) and embedded in paraffin (cat. no. 09620; Sakura Finetek
Japan Co., Tokyo, Japan). The specimens were cut into sections that
were stained with hematoxylin and eosin (H&E; cat. nos. 234-12
and 1159350025; Merck KGaA) for histological examination. DNA
fragmentation was analyzed with the TUNEL assay using a commercial
kit (cat. no. 11684817910; Roche Applied Science, Penzberg,
Germany) according to the manufacturer's instructions. Cells were
examined under a light microscope (Axioskop 2 Plus) with an
Achroplan 40×/0.65 lens (both from Zeiss, Hallbergmoos, Germany).
Images were acquired with an AxioCam 503 color camera and
AxioVision LE64 software (Zeiss GmbH, Jena, Germany).
Statistical analysis
Results are expressed as the means ± standard
deviation (SD). The Student's t-test or ANOVA with the Tukey-Kramer
test were used to compare the means of 2 groups or >2 groups,
respectively. Differences were considered statistically significant
at P<0.05.
Results
HTLV-1-infected T-cell lines are
sensitive to dual SYK/JAK inhibition with cerdulatinib
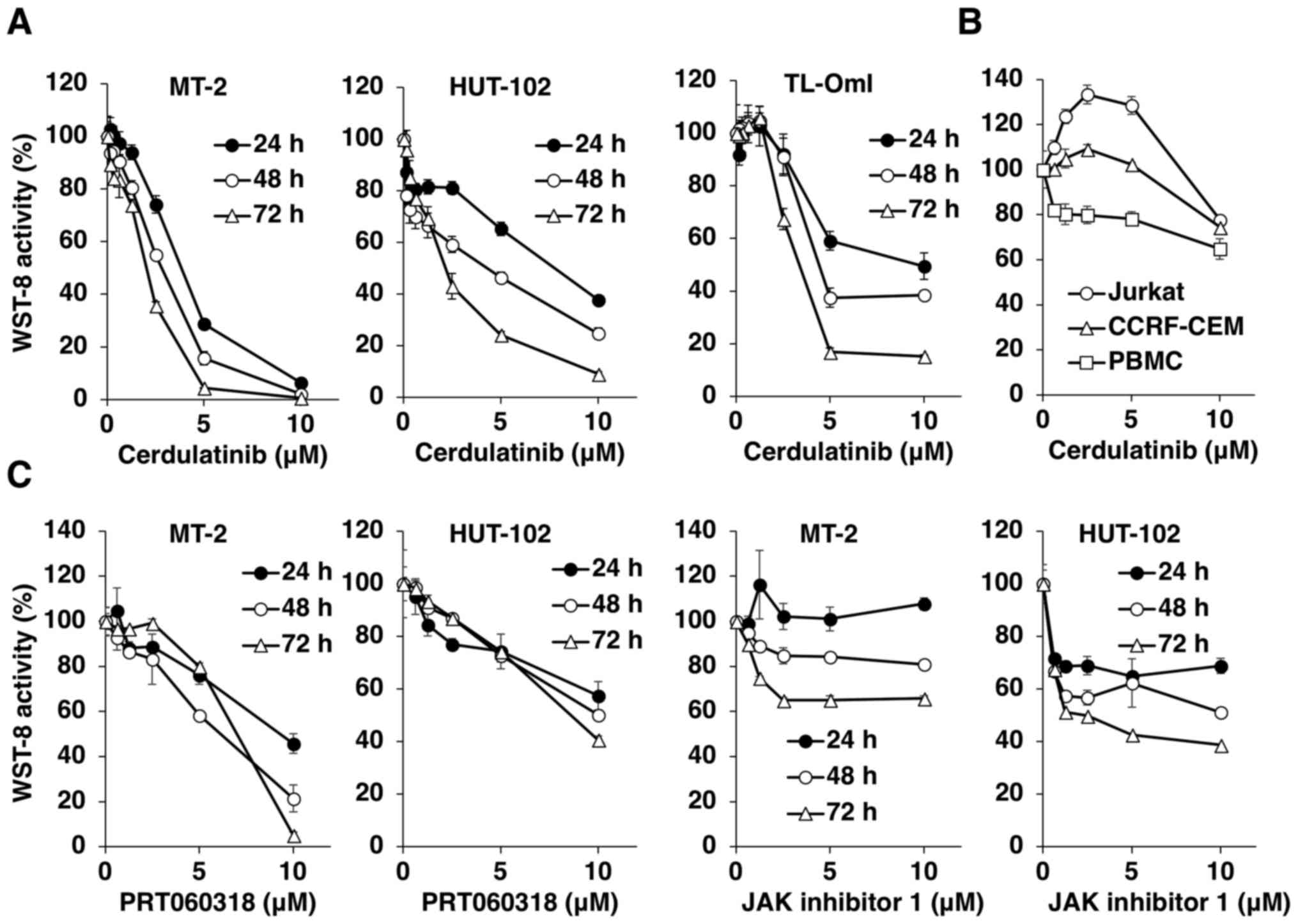
Using the WST-8 assay, we first examined the
cytotoxicity of various concentrations of cerdulatinib in
HTLV-1-infected T-cell lines up to 72 h after treatment (Fig. 1A). Cerdulatinib decreased WST-8
activity (and thus, cell viability) in a dose- and time-dependent
manner in all HTLV-1-infected T-cell lines. Notably, the uninfected
Jurkat and CCRF-CEM T-cell lines were resistant to the cytotoxic
effects of cerdulatinib. The effects of cerdulatinib on PBMCs from
a healthy donor were less pronounced (Fig. 1B). The IC50 values at 24
h were 3.5 µM for the MT-2, 7.3 µM for the HUT-102
and 8.4 µM for the TL-OmI cells, as compared to 62.9
µM for the normal PBMCs. In the Jurkat and CCRF-CEM cells,
the IC50 value was not attained with the administered
doses. We compared the effects of cerdulatinib with those of two
reference compounds, the SYK-selective inhibitor, PRT060318, and
the pan-JAK inhibitor, JAK inhibitor 1, on the MT-2 and HUT-102
cells, and found that cerdulatinib decreased cell viability more
potently than either single agent (Fig. 1, compare parts A and C). As
PRT060318 or JAK inhibitor 1 decreased the viability of
HTLV-1-infected T-cell lines, we further tested the combination of
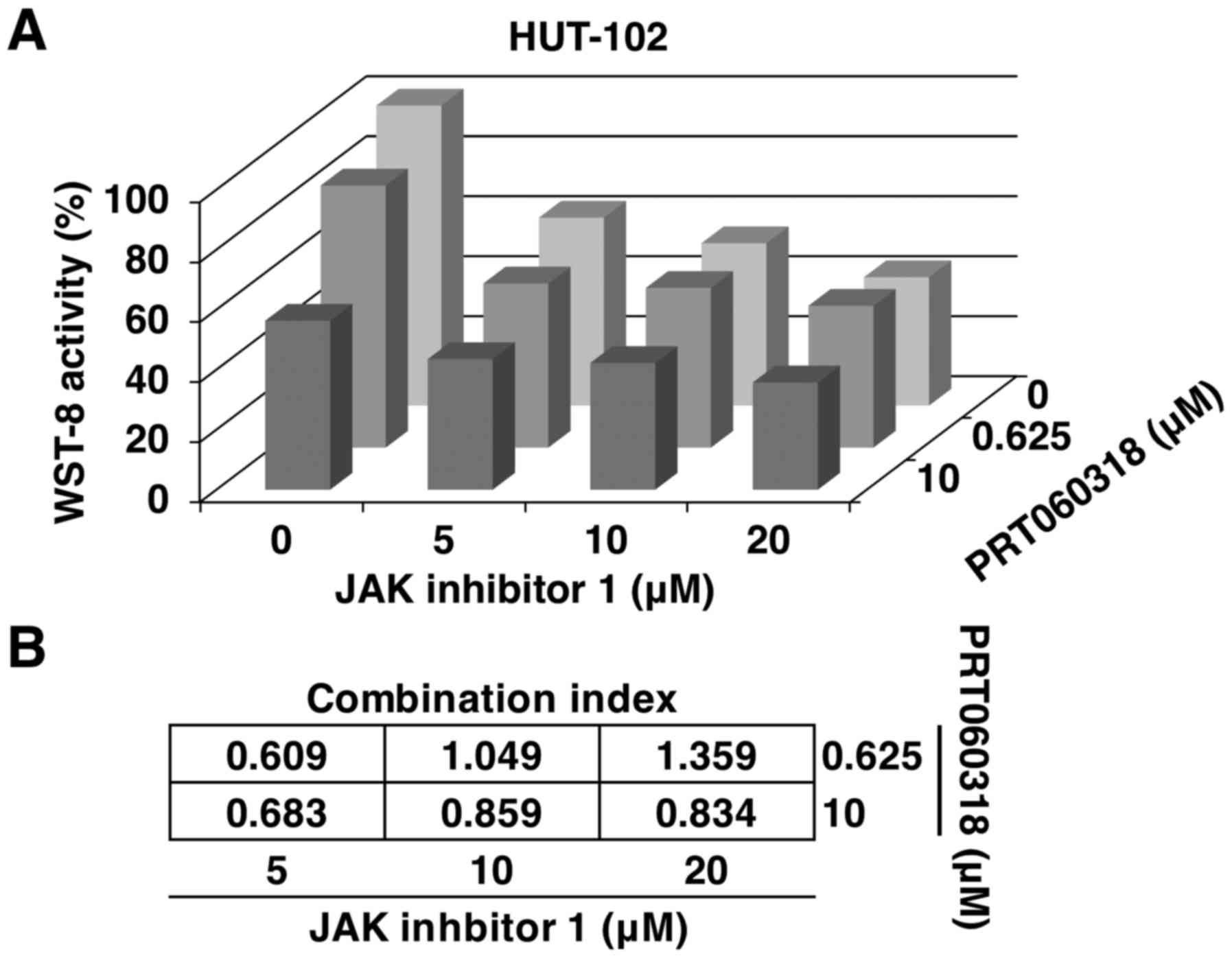
the two agents to determine whether they acted synergistically to
suppress the viability of HTLV-1-infected T-cells. The CI was used
to determine whether the effect of the combined treatment was
synergistic, additive or antagonistic. Instead of a simple additive
effect, the combination of PRT060318 and JAK inhibitor 1 exerted a
synergistic cytotoxic effect on HUT-102 cells, with the optimal CI
of 0.609 (Fig. 2).
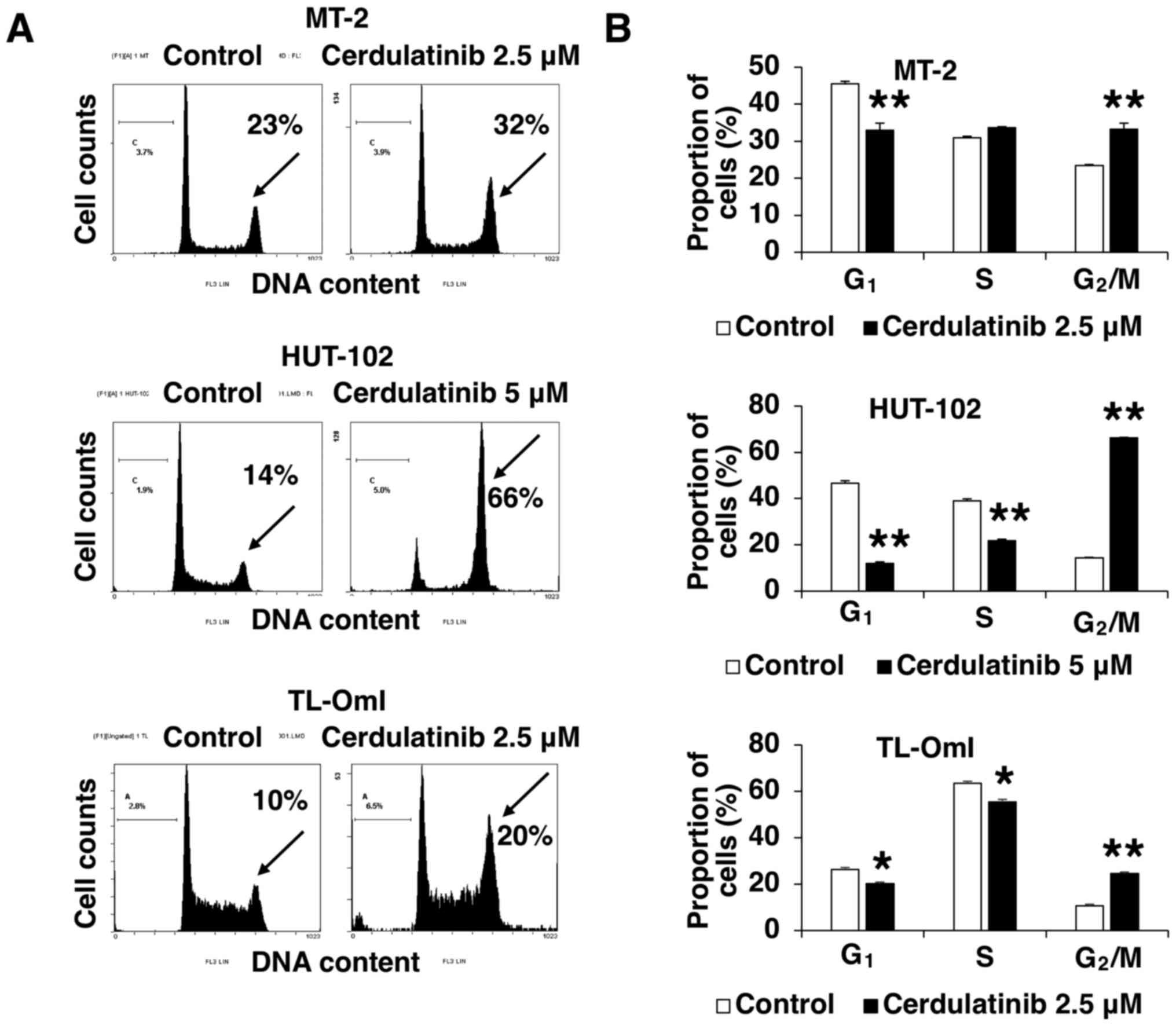
Cerdulatinib inhibits cell cycle
progression
The negative effect of cerdulatinib on cell
viability (as determined using the WST-8 assay) may result from
cell growth inhibition, death or both. To clarify the mechanisms
underlying the inhibition of cell growth by cerdulatinib, we
evaluated cell cycle distribution in the MT-2, HUT-102 and TL-OmI
cells treated with 2.5 and 5 µM cerdulatinib for 24 h.
Treatment with cerdulatinib led to a significant accumulation of
cells in the G2/M-phase, with concurrent decreases in
the G1- and/or S-phase fractions (Fig. 3). These results indicated that
cerdulatinib inhibits cell proliferation by targeting the
G2/M checkpoint and blocking cell cycle progression.
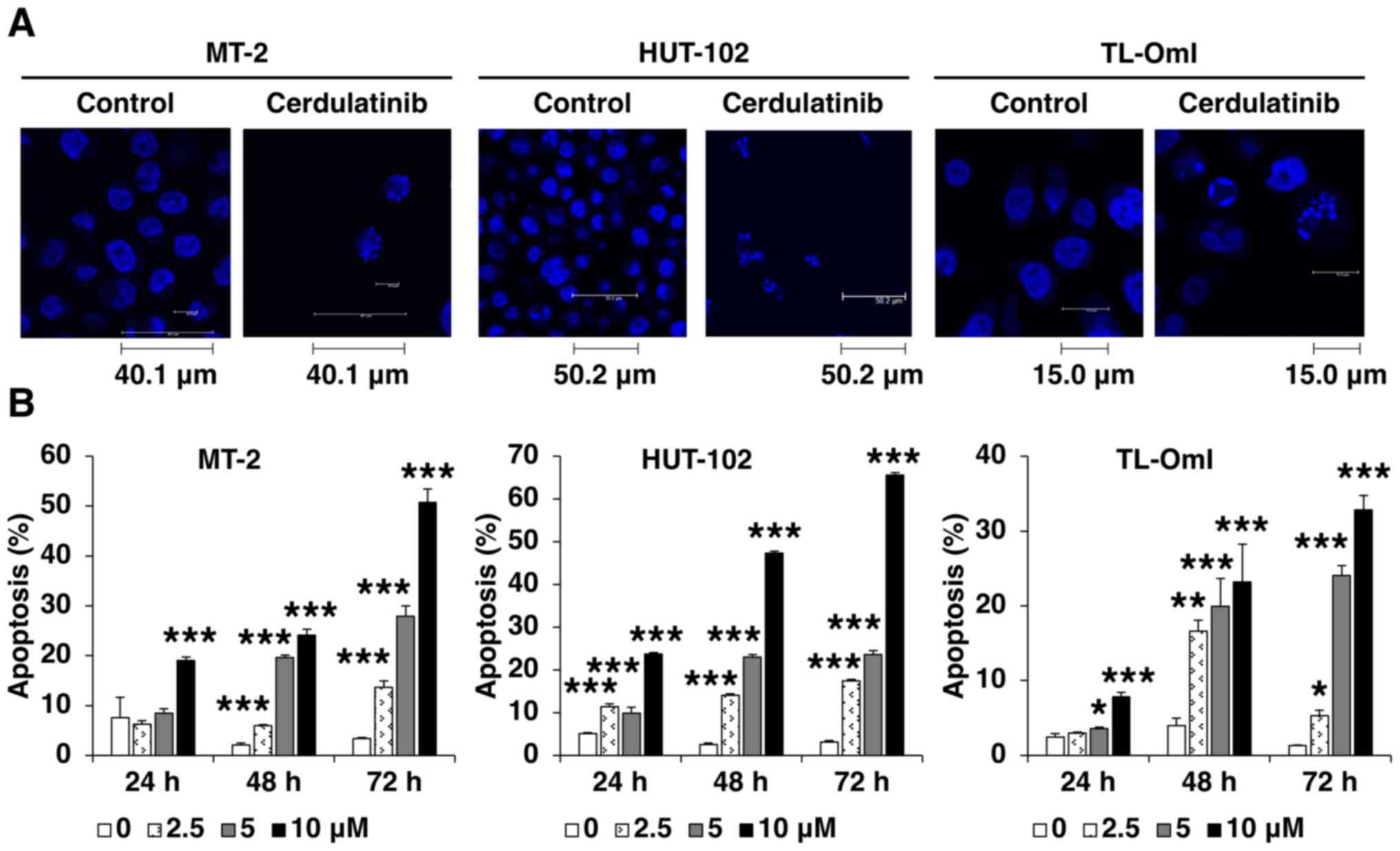
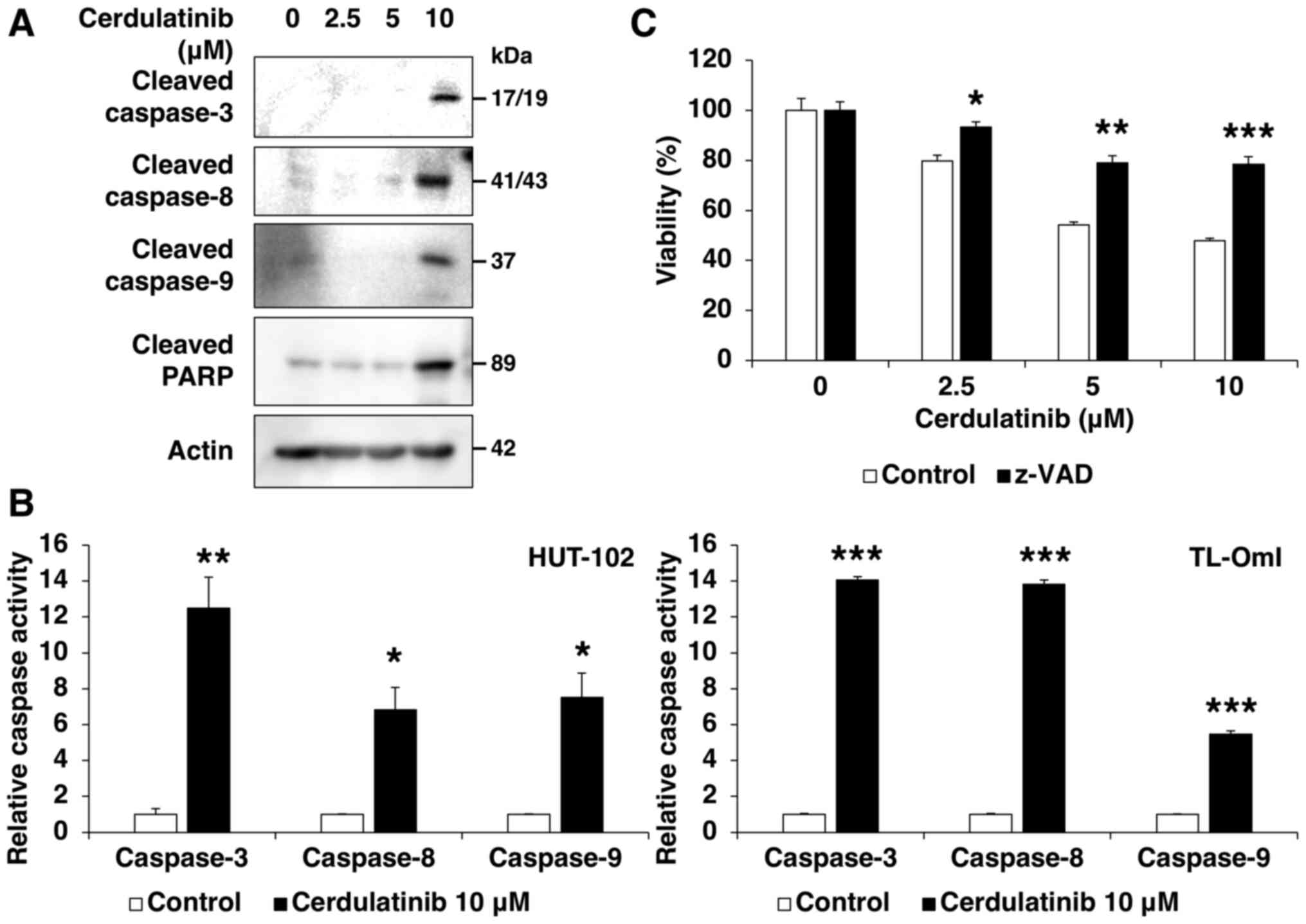
Cerdulatinib induces apoptosis
We then examined the effect of cerdulatinib on cell
death. A morphological analysis using Hoechst staining revealed
chromatin condensation and nuclear fragmentation in the cells
cultured with cerdulatinib (Fig.
4A). APO2.7 immunolabeling indicated that apoptosis was induced
by cerdulatinib in a concentration- and time-dependent manner
(Fig. 4B). To investigate the role
of caspases in the response to cerdulatinib, cell lysates were
examined by western blot analysis using specific antibodies. The
induction of apoptosis of the HUT-102 cells at increasing
concentrations of cerdulatinib was accompanied by the cleavage of
caspase-8, -9 and -3, and its substrate PARP (Fig. 5A). We also examined the enzymatic
activity of caspase-8, -9 and -3, as this is not always associated
with pro-caspase processing, and found the significant activation
of all three proteins in the HUT-102 and TL-OmI cells following 72
h of treatment with 10 µM cerdulatinib (Fig. 5B). These results suggested that
cerdulatinib induces apoptosis via the activation of extrinsic and
intrinsic pathways. Pre-treatment with the pan-caspase inhibitor,
z-VAD-FMK, partly abrogated cerdulatinib-induced cytotoxicity
(Fig. 5C), suggesting that the
latter occurs through caspase-dependent apoptosis.
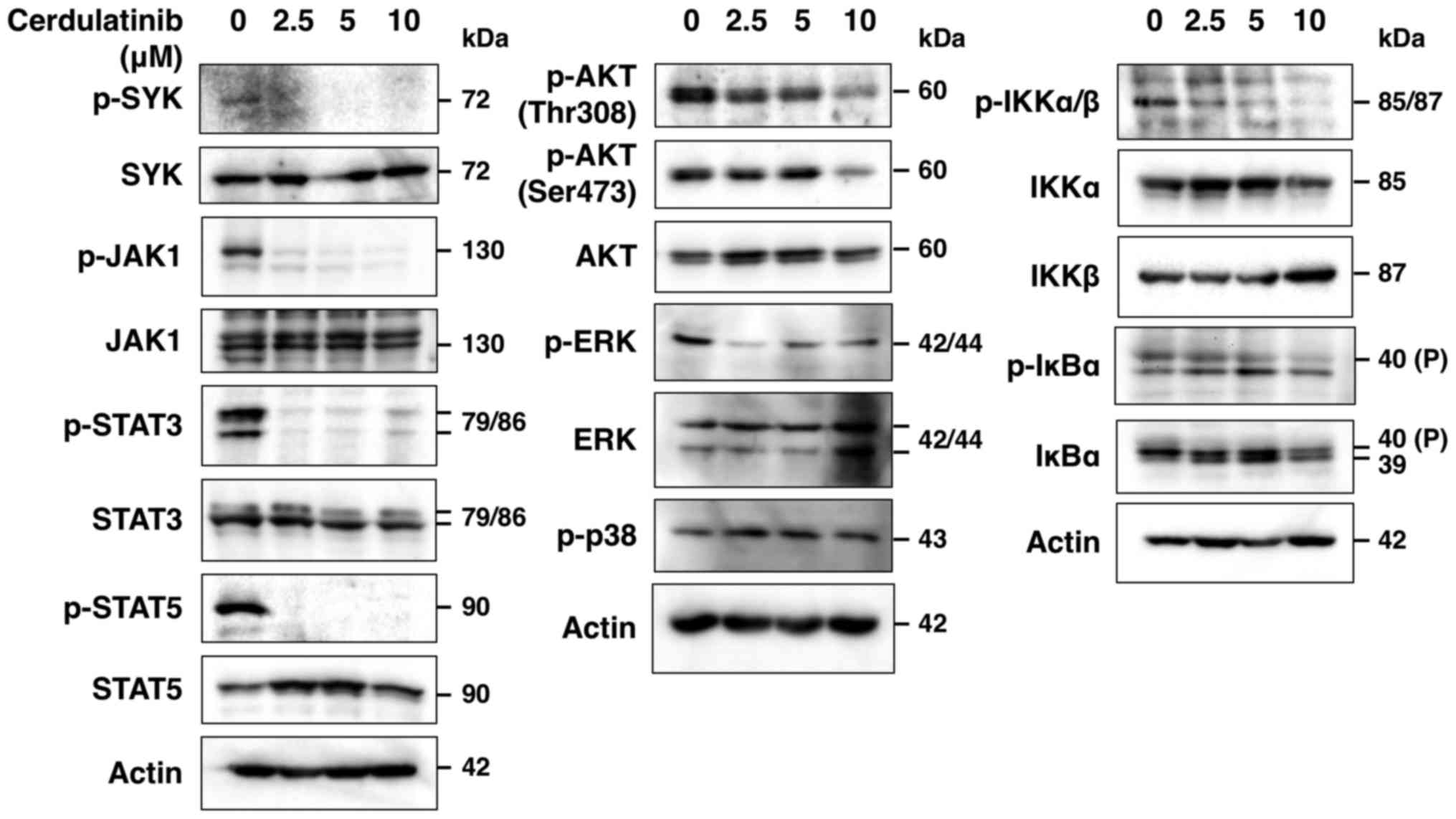
Cerdulatinib blocks SYK, JAK/STAT, AKT,
ERK, NF-κB and AP-1 signaling
To determine whether cerdulatinib inhibits the SYK
and JAK/STAT signaling pathways, we evaluated the phosphorylation
status of SYK and JAK-STAT in the HUT-102 cells treated with
cerdulatinib by western blot analysis, and found that SYK, JAK1,
STAT3 and STAT5 phosphorylation was decreased (Fig. 6). SYK is associated with various
immunoreceptors (21) and is
thought to act upstream of phosphoinositide 3-kinase
(PI3K)-AKT-mediated IKK-IκBα activation (21–23).
In addition, JAK inhibition has been reported to suppress
IL-2-mediated T-cell growth and STAT, AP-1 and ERK activation by
IL-2 (24). In this study, we
found that cerdulatinib blocked the phosphorylation of AKT, IKKα/β,
IκBα and ERK, but not that of p38 (Fig. 6), and nuclear extracts obtained
from the cerdulatinib-treated cells exhibited a reduced DNA binding
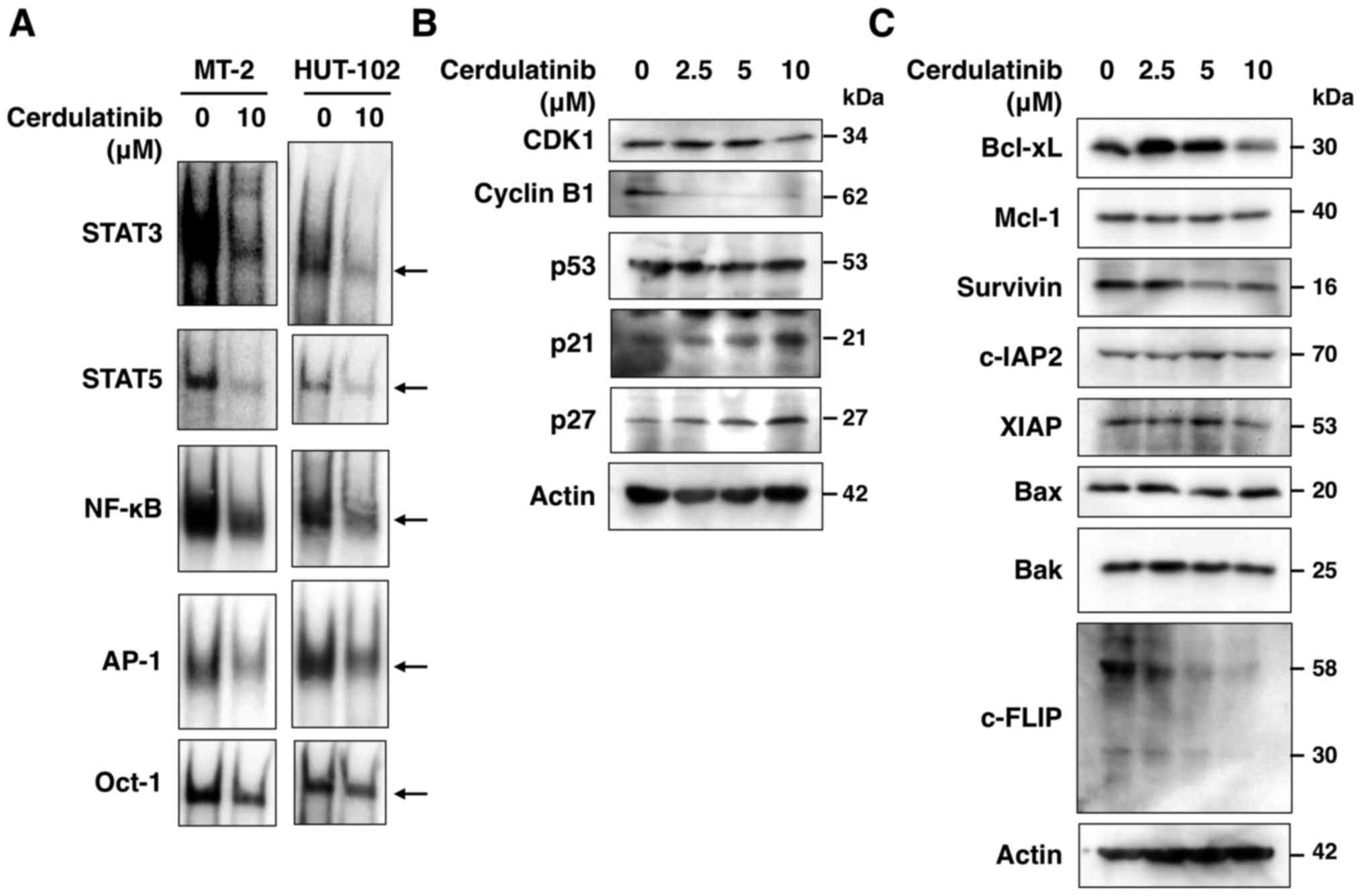
of STAT3, STAT5, NF-κB and AP-1, but not Oct-1 (Fig. 7A). Thus, SYK and JAK/STAT, as well
as downstream signaling pathways were effectively inhibited by
cerdulatinib.
Cerdulatinib modulates key proteins
related to cell cycle progression and apoptosis
To further clarify the molecular mechanisms
underlying the growth inhibitory and cytotoxic effects of
cerdulatinib on HTLV-1-infected T-cell lines, we evaluated the
expression of proteins associated with cell cycle progression and
apoptosis. The results of western blot analysis revealed that
cerdulatinib treatment increased the expression of p21 and p27, and
decreased that of CDK1, cyclin B1, Bcl-xL, survivin, XIAP and
c-FLIP (Fig. 7B and C). However,
it had no effect on the levels of Mcl-1, c-IAP2, and the
pro-apoptotic proteins, Bak and Bax. As p21 and p27 expression is
modulated by p53, we also examined p53 expression, but found no
obvious changes in the level of this protein in the
cerdulatinib-treated cells.
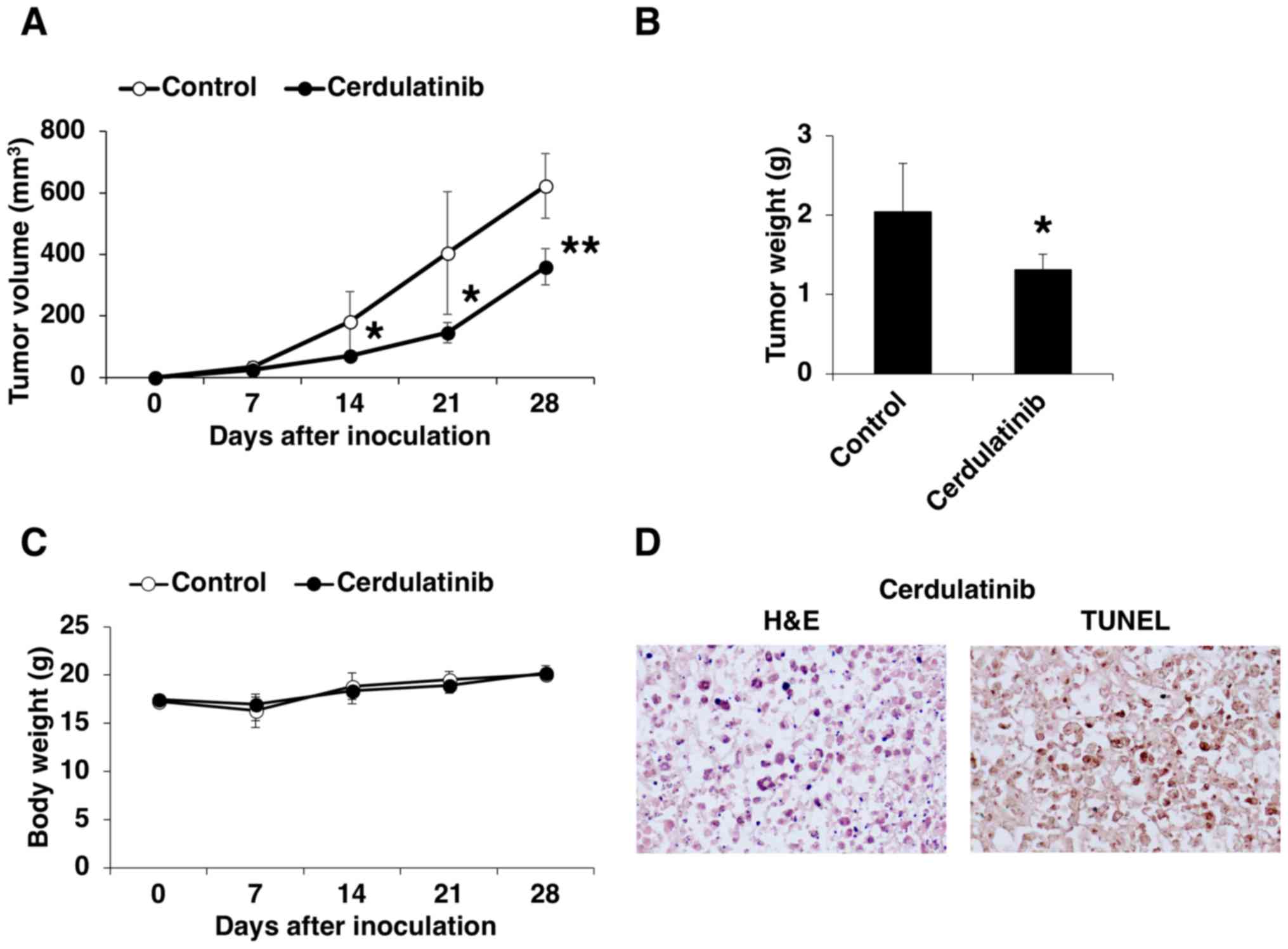
Pre-clinical evaluation of cerdulatinib
in a mouse xenograft model of ATLL
We examined the in vivo efficacy of
cerdulatinib in a mouse xenograft model of ATLL. HUT-102 cells were
subcutaneously transplanted into SCID mice, which were then orally
administered the vehicle or 35 mg/kg cerdulatinib 5 times weekly.
The volume and weight of the transplanted tumors were lower in the
mice treated with cerdulatinib than in the control group (Fig. 8A and B). Cerdulatinib was well
tolerated, as it did not cause significant weight loss or other
clinical manifestations (Fig. 8C).
Tumor tissue from the mice treated with cerdulatinib examined by
H&E staining (Fig. 8D, left
panel) exhibited evidence of apoptosis, including cytoplasmic
condensation, chromatin hyperchromatism and condensation, and
nuclear fragmentation. Apoptosis in the tumor tissue from the
cerdulatinib-treated mice was confirmed by TUNEL assay (Fig. 8D, right panel), which was
consistent with our in vitro findings. These results
demonstrated the effectiveness of cerdulatinib in controlling ATLL
progression in vivo.
Discussion
In this study, we evaluated the therapeutic
potential of cerdulatinib, a potent inhibitor of SYK and JAK1/3, in
the treatment of ATLL. We demonstrated that this compound was
effective in suppressing the growth of HTLV-1-infected T-cell lines
in vitro and ATLL xenograft tumors in vivo. The
HTLV-1-infected T-cell lines were more sensitive to cerdulatinib
than uninfected T-cell lines and normal PBMCs. Moreover,
cerdulatinib exhibited greater cytotoxicity than either a SYK or
JAK single inhibitor alone.
Cerdulatinib induced apoptosis in addition to
causing cell cycle arrest in HTLV-1-infected T-cell lines. The
inhibition of JAK can lead to the suppression of STAT, ERK and
AP-1, thereby blocking the proliferation of IL-2-stimulated T-cells
(24), whereas SYK is known to
induce PI3K-AKT activation, which is critical for NF-κB signaling
(21–23). In this study, we observed that
cerdulatinib treatment suppressed both SYK and JAK-STAT signaling
along with the activity of the downstream factors, AKT, ERK, AP-1
and NF-κB.
To investigate the mechanistic basis for the cell
cycle arrest at the G2/M phase induced by cerdulatinib,
we examined the CDK1 and cyclin B1 levels in cerdulatinib-treated
cells, as the CDK1/cyclin B1 complex constitutes a major regulator
of the G2/M transition. Cerdulatinib induced a marked
decrease in the CDK1 and cyclin B1 levels. p21 and p27 are CDK
inhibitors that negatively regulate cell cycle progression, with
their suppression resulting in increased cell proliferation
(25). p21 plays a well-documented
role as a G1/S checkpoint protein, but is also known to
modulate the G2/M checkpoint (26,27)
through the inhibition of the CDK1/cyclin B1 complex (28). p27 is an inhibitor of CDK1
(27). AKT, ERK, AP-1 and NF-κB
regulate cyclin B1 expression in cell proliferation (29,30),
and AKT and ERK have also been shown to suppress p21 and p27
(31–33). In this study, following treatment
with cerdulatinib, the p21 and p27 protein levels were upregulated
relative to those of the control group. Our results indicated that
cerdulatinib suppressed HTLV-1-infected T-cell proliferation by the
downregulation of CDK1 and cyclin B1, and the upregulation of the
CDK inhibitors, p21 and p27, causing p53-independent cell cycle
arrest at the G2/M phase by suppressing the AKT, ERK,
AP-1 and NF-κB pathways.
Cerdulatinib blocked the expression of the
anti-apoptotic factors, Bcl-xL, survivin, XIAP and c-FLIP, which
prevent caspase activation; their inhibition therefore increases
caspase-3, -8 and -9 levels (34,35).
STAT and NF-κB are translocated to the nucleus where they bind to
specific DNA sequences in the promoters of target genes, including
BCLXL, BIRC5 and XIAP, thereby inducing their
transcription (36,37). c-FLIP expression is regulated by
AKT and NF-κB (35). AKT activates
NF-κB (22,31,37)
but also inhibits caspase-9 activation along with that of ERK
(31,34,38).
AKT activity is regulated by XIAP (39), whereas survivin is a downstream
target of AKT signaling (40,41).
Thus, the increase in caspase activity in cerdulatinib-treated
cells suggested that this compound promotes apoptosis by
suppressing AKT, ERK, STAT and NF-κB pathways. Notably, the safety
and clinical efficacy of cerdulatinib was recently demonstrated in
patients with CLL, as well as in those with another type of B-cell
non-Hodgkin lymphoma (42),
further supporting its potential for application as a treatment for
ATLL.
In conclusion, the findings of this study
demonstrate that cerdulatinib exerts anti-ATLL effects in
vitro and in vivo. Although the detailed mechanisms
underlying the modulation of CDK1 by cerdulatinib require further
investigation, our results indicate that cerdulatinib induces
G2/M arrest and subsequent apoptosis by inhibiting the
SYK and JAK/STAT signaling pathways and their downstream effectors,
AKT, ERK, AP-1 and NF-κB. The preclinical findings presented herein
provide a rationale for evaluating the efficacy of cerdulatinib in
patients with ATLL.
Acknowledgments
The authors would like to thank the Fujisaki Cell
Center, Hayashibara Biochemical Laboratories, Inc. (Okayama, Japan)
for providing the HUT-102 and Jurkat cells, Dr Naoki Yamamoto
(Tokyo Medical and Dental University, Tokyo, Japan) for providing
MT-2 cells, Dr Masahiro Fujii (Niigata University, Niigata, Japan)
for providing TL-OmI and CCRF-CEM cells, and Editage (www.editage.jp) for English language editing.
Funding
This study was supported in part by a JSPS KAKENHI
grants (nos. 15K18414 and 17K07175).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CI and NM were responsible for the study design, the
drafting and editing of the manuscript, and data acquisition and
analysis. MS was responsible for data acquisition and analysis. All
authors have read and approved this manuscript.
Ethics approval and consent to
participate
Experimental procedures and animal care were in
compliance with the Guidelines for Animal Experimentation of
University of The Ryukyus (Nishihara, Japan) and were approved by
the Animal Care and Use Committee of University of The Ryukyus
(reference no. A2016088).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Iwanaga M, Watanabe T and Yamaguchi K:
Adult T-cell leukemia: A review of epidemiological evidence. Front
Microbiol. 3:3222012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Katsuya H and Ishitsuka K: Treatment
advances and prognosis for patients with adult T-cell
leukemia-lymphoma. J Clin Exp Hematop. 57:87–97. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yssel H, de Waal Malefyt R, Duc Dodon MD,
Blanchard D, Gazzolo L, de Vries JE and Spits H: Human T cell
leukemia/lymphoma virus type I infection of a CD4+
proliferative/cytotoxic T cell clone progresses in at least two
distinct phases based on changes in function and phenotype of the
infected cells. J Immunol. 142:2279–2289. 1989.PubMed/NCBI
|
|
4
|
Miyazaki T and Taniguchi T: Coupling of
the IL2 receptor complex with non-receptor protein tyrosine
kinases. Cancer Surv. 27:25–40. 1996.PubMed/NCBI
|
|
5
|
Rochman Y, Spolski R and Leonard WJ: New
insights into the regulation of T cells by gamma(c) family
cytokines. Nat Rev Immunol. 9:480–490. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Migone TS, Lin JX, Cereseto A, Mulloy JC,
O'Shea JJ, Franchini G and Leonard WJ: Constitutively activated
Jak-STAT pathway in T cells transformed with HTLV-I. Science.
269:79–81. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu X, Kang SH, Heidenreich O, Okerholm M,
O'Shea JJ and Nerenberg MI: Constitutive activation of different
Jak tyrosine kinases in human T cell leukemia virus type 1 (HTLV-1)
tax protein or virus-transformed cells. J Clin Invest.
96:1548–1555. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takemoto S, Mulloy JC, Cereseto A, Migone
T-S, Patel BKR, Matsuoka M, Yamaguchi K, Takatsuki K, Kamihira S,
White JD, et al: Proliferation of adult T cell leukemia/lymphoma
cells is associated with the constitutive activation of JAK/STAT
proteins. Proc Natl Acad Sci USA. 94:13897–13902. 1997. View Article : Google Scholar
|
|
9
|
Ju W, Zhang M, Jiang J-K, Thomas CJ, Oh U,
Bryant BR, Chen J, Sato N, Tagaya Y, Morris JC, et al: CP-690,550,
a therapeutic agent, inhibits cytokine-mediated Jak3 activation and
proliferation of T cells from patients with ATL and HAM/TSP. Blood.
117:1938–1946. 2011. View Article : Google Scholar :
|
|
10
|
Zhang M, Mathews Griner LA, Ju W, Duveau
DY, Guha R, Petrus MN, Wen B, Maeda M, Shinn P, Ferrer M, et al:
Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL
blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad
Sci USA. 112:12480–12485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weil R, Levraud J-P, Dodon MD, Bessia C,
Hazan U, Kourilsky P and Israël A: Altered expression of tyrosine
kinases of the Src and Syk families in human T-cell leukemia virus
type 1-infected T-cell lines. J Virol. 73:3709–3717.
1999.PubMed/NCBI
|
|
12
|
Coffey G, Betz A, DeGuzman F, Pak Y,
Inagaki M, Baker DC, Hollenbach SJ, Pandey A and Sinha U: The novel
kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in
models of autoimmunity and B-cell cancer. J Pharmacol Exp Ther.
351:538–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma J, Xing W, Coffey G, Dresser K, Lu K,
Guo A, Raca G, Pandey A, Conley P, Yu H, et al: Cerdulatinib, a
novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity
in both ABC and GCB types of diffuse large B cell lymphoma.
Oncotarget. 6:43881–43896. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Blunt MD, Koehrer S, Dobson RC, Larrayoz
M, Wilmore S, Hayman A, Parnell J, Smith LD, Davies A, Johnson PWM,
et al: The dual Syk/JAK inhibitor cerdulatinib antagonizes B-cell
receptor and microenvironmental signaling in chronic lymphocytic
leukemia. Clin Cancer Res. 23:2313–2324. 2017. View Article : Google Scholar
|
|
15
|
Guo A, Lu P, Coffey G, Conley P, Pandey A
and Wang YL: Dual SYK/JAK inhibition overcomes ibrutinib resistance
in chronic lymphocytic leukemia: Cerdulatinib, but not ibrutinib,
induces apoptosis of tumor cells protected by the microenvironment.
Oncotarget. 8:12953–12967. 2017.PubMed/NCBI
|
|
16
|
Liu D and Mamorska-Dyga A: Syk inhibitors
in clinical development for hematological malignancies. J Hematol
Oncol. 10:1452017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stapper NJ, Stuschke M, Sak A and Stüben
G: Radiation-induced apoptosis in human sarcoma and glioma cell
lines. Int J Cancer. 62:58–62. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mori N and Prager D: Transactivation of
the interleukin-1alpha promoter by human T-cell leukemia virus type
I and type II Tax proteins. Blood. 87:3410–3417. 1996.PubMed/NCBI
|
|
19
|
Ishikawa C, Senba M and Mori N: Butein
inhibits NF-κB, AP-1 and Akt activation in adult T-cell
leukemia/lymphoma. Int J Oncol. 51:633–643. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tomayko MM and Reynolds CP: Determination
of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother
Pharmacol. 24:148–154. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mócsai A, Ruland J and Tybulewicz VL: The
SYK tyrosine kinase: A crucial player in diverse biological
functions. Nat Rev Immunol. 10:387–402. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Z, Li Z, Chen S, Pan J and Ma X:
Tetramethylpyrazine attenuates TNF-α-induced iNOS expression in
human endothelial cells: Involvement of Syk-mediated activation of
PI3K-IKK-IκB signaling pathways. Exp Cell Res. 319:2145–2151. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hatton O, Lambert SL, Krams SM and
Martinez OM: Src kinase and Syk activation initiate PI3K signaling
by a chimeric latent membrane protein 1 in Epstein-Barr virus
(EBV)+ B cell lymphomas. PLoS One. 7:e426102012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang LH, Kirken RA, Erwin RA, Yu C-R and
Farrar WL: JAK3, STAT, and MAPK signaling pathways as novel
molecular targets for the tyrphostin AG-490 regulation of
IL-2-mediated T cell response. J Immunol. 162:3897–3904.
1999.PubMed/NCBI
|
|
25
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karimian A, Ahmadi Y and Yousefi B:
Multiple functions of p21 in cell cycle, apoptosis and
transcriptional regulation after DNA damage. DNA Repair (Amst).
42:63–71. 2016. View Article : Google Scholar
|
|
27
|
Hu X and Moscinski LC: Cdc2: A monopotent
or pluripotent CDK? Cell Prolif. 44:205–211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guadagno TM and Newport JW: Cdk2 kinase is
required for entry into mitosis as a positive regulator of
Cdc2-cyclin B kinase activity. Cell. 84:73–82. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yadav S, Kalra N, Ganju L and Singh M:
Activator protein-1 (AP-1): A bridge between life and death in lung
epithelial (A549) cells under hypoxia. Mol Cell Biochem.
436:99–110. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y-X, Li X-F, Yuan G-Q, Hu H, Song
X-Y, Li J-Y, Miao X-K, Zhou T-X, Yang W-L, Zhang X-W, et al:
β-Arrestin 1 has an essential role in neurokinin-1
receptor-mediated glioblastoma cell proliferation and
G2/M phase transition. J Biol Chem. 292:8933–8947. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roy SK, Srivastava RK and Shankar S:
Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of
FOXO transcription factor, leading to cell cycle arrest and
apoptosis in pancreatic cancer. J Mol Signal. 5:102010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Das D, Pintucci G and Stern A:
MAPK-dependent expression of p21(WAF) and p27(kip1) in PMA-induced
differentiation of HL60 cells. FEBS Lett. 472:50–52. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim R, Tanabe K, Uchida Y, Emi M, Inoue H
and Toge T: Current status of the molecular mechanisms of
anticancer drug-induced apoptosis. The contribution of
molecular-level analysis to cancer chemotherapy. Cancer Chemother
Pharmacol. 50:343–352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krueger A, Baumann S, Krammer PH and
Kirchhoff S: FLICE-inhibitory proteins: Regulators of death
receptor-mediated apoptosis. Mol Cell Biol. 21:8247–8254. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Catlett-Falcone R, Dalton WS and Jove R:
STAT proteins as novel targets for cancer therapy. Signal
transducer an activator of transcription. Curr Opin Oncol.
11:490–496. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng HC: The molecular mechanisms of
chemoresistance in cancers. Oncotarget. 8:59950–59964.
2017.PubMed/NCBI
|
|
38
|
Allan LA, Morrice N, Brady S, Magee G,
Pathak S and Clarke PR: Inhibition of caspase-9 through
phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 5:647–654.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Asselin E, Mills GB and Tsang BK: XIAP
regulates Akt activity and caspase-3-dependent cleavage during
cisplatin-induced apoptosis in human ovarian epithelial cancer
cells. Cancer Res. 61:1862–1868. 2001.PubMed/NCBI
|
|
40
|
Liang YL, Wang LY, Wu H, Ma DZ, Xu Z and
Zha XL: PKB phosphorylation and survivin expression are
cooperatively regulated by disruption of microfilament
cytoskeleton. Mol Cell Biochem. 254:257–263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vaira V, Lee CW, Goel HL, Bosari S,
Languino LR and Altieri DC: Regulation of survivin expression by
IGF-1/mTOR signaling. Oncogene. 26:2678–2684. 2007. View Article : Google Scholar
|
|
42
|
Hamlin PA, Farber CM, Fenske TS,
Khatcheressian JL, Miller CB, Munoz J, Patel MR, Schreeder MT,
Smith SM, Stevens DA, et al: The dual SYK/JAK inhibitor
cerdulatinib demonstrates rapid tumor responses in a phase 2 study
in patients with relapsed/refractory B-cell malignancies. Hematol
Oncol. 35:742017. View Article : Google Scholar
|