Introduction
Colorectal cancer (CRC) is the third most commonly
diagnosed type of cancer and the second most common cause of
cancer-related mortality worldwide (1). The highest incidence of CRC has been
reported in Korea and the number of patients with CRC is increasing
each year. The causes of this disease are very diverse, from
Westernized diets to genetic factors. CRC development is a
multistep process, including the accumulation of epigenetic
alterations and genetic abnormalities, such as mutations in tumor
suppressor genes or oncogenes (2).
Wnt signaling is critical for animal developmental
processes, such as tissue regeneration and morphogenesis (3). There are two distinct pathways in Wnt
signaling: Canonical and non-canonical. Signaling components of the
canonical Wnt pathway include adenomatous polyposis coli (APC),
axin, glycogen synthesis kinase 3β (GSK3β) and β-catenin, while the
non-canonical Wnt pathway is regulated by small G protein or
Ca2+ (4). The
hyperactivation of Wnt and thus the nuclear accumulation of
β-catenin are frequently observed in CRC (5). In the absence of Wnt ligands, the
destruction complex that is composed of APC, Axin, GSK3β, casein
kinase 1 (CK1) and other components captures β-catenin in the
cytoplasm. β-catenin is then phosphorylated by the destruction
complex and is ubiquitinated, followed by proteasomal degradation.
In the presence of Wnt ligands, the Frizzled receptor and
lipoprotein-related protein 5/6 (LRP5/6) receptor are activated.
The LRP5/6 receptor is phosphorylated and recruits the destruction
complex. Stabilized β-catenin translocates from the cytoplasm to
the nucleus and interacts with TCF/LEF to activate the
transcription of Wnt target genes (6). The aberrant activation of the
Wnt/β-catenin pathway has been shown to be a central oncogenic
driver in CRC by upregulating Wnt target genes, such as Myc
and cyclin D1 (7,8). These genes are associated with cell
survival and proliferation, and thus may possibly contribute to the
development of CRC (9).
The tumor suppressor protein, p53, encoded by
TP53, is a key regulator of genomic stability, apoptosis,
cell cycle and angiogenesis (10).
p53 becomes activated upon various cellular stresses, such as
oxidative stress, ribonucleotide depletion, deregulated oncogene
expression and DNA damage (11).
When DNA is damaged, p53 activates the DNA repair system to restore
damaged DNA, while terminating the cell cycle progression at
checkpoints (12). However, when
the damage is irreparable, p53 initiates apoptosis by activating
the transcription of its target genes, including BAX, PUMA
and Fas (also known as APO1) (13). Wild-type p53 is a labile protein
with a half-life of ~5–20 min in most cell types that increases by
several fold following DNA damage (14). Mouse double minute 2 homolog (MDM2)
is a major negative regulator that contributes to the instability
of p53 through ubiquitination and the subsequent proteasomal
degradation of p53 (15).
The inactivation of the p53 tumor suppressor
gene occurs in the majority of human cancers, suggesting that the
loss of p53 function is a critical step in tumorigenesis. It is
estimated that loss-of-function mutations in this gene occur in
approximately 40–50% of patients with CRC, and the loss of p53 is
considered a late event during the transition from adenoma to
carcinoma (16,17). Several observations are reminiscent
of a crosstalk between the dysregulation of Wnt signaling and p53
dysfunction: i) The strong nuclear accumulation of β-catenin is
associated with a high frequency of p53 mutations in human
CRC; ii) there is an inverse correlation between the levels of
β-catenin and p53 in a variety of cell types; and iii) activated
p53 reduces β-catenin levels via the stimulation of GSK3β (18). Intriguingly, it has recently been
shown that the C-terminus of β-catenin inhibits p53 acetylation and
its transcriptional activity in smooth muscle cells (19). However, the regulation of p53
levels by Wnt signaling is not yet fully understood in CRC.
MicroRNAs (miRNAs or miRs) are a class of non-coding
RNAs and conserved families of transcripts approximately 18–22
nucleotides (nt) in length (20).
miRNAs have been found in animals, plants and viruses (21). In animals, they are generated from
large hairpin precursors (pri-miRNA) and processed to pre-miRNAs by
nuclear RNase III Drosha in the nucleus. pre-miRNAs are exported by
exportin-5 (Exp5) from the nucleus to the cytoplasm and are cleaved
by cytoplasmic RNase III Dicer to produce mature miRNAs with the
aid of AGO (22). In plants and
viruses, the mechanism of miRNA processing is similar, although
some of the proteins involved are different.
The majority of known miRNAs have been reported to
originate from non-protein-coding regions, such as intergenic
regions and intronic regions (20,21).
Previous studies have identified transposable elements (TEs) as
important sources of miRNAs, contributing to >10% of the human
miRNA genes (23,24). TEs can be classified into two
different groups according to their mode of transposition: Class I
TEs (retrotransposons) and class II TEs (DNA transposons). The
former is transcribed into RNA that is reverse transcribed into DNA
and the copied DNA is inserted into a new site in the genome, while
the latter is cut and pasted into a different position (25). Retrotransposons consist of two
major groups, long terminal repeats (LTR) and non-LTR
retrotransposons, depending on the presence of an LTR at each end.
Non-LTR retrotransposons can be subdivided into long and short
interspersed elements (LINEs and SINEs). It has been reported that
LINEs comprise approximately 17% of the human genome; however, only
a small percentage is active and capable of retrotransposition.
Although LINEs are autonomous and can transpose autonomously, SINEs
are non-autonomous and depend on the machinery of other
retrotransposons for transposition (25).
miRNAs play important roles in diverse biological
processes, such as apoptosis, organ development and cell
proliferation (26). They mostly
downregulate gene expression by base pairing to
3′-or-5′-untranslated regions (UTRs) in target mRNAs, which can
result in the inhibition of target gene translation or promotion of
mRNA degradation (27). It has
been shown that the aberrant expression of miRNAs is associated
with the development of various types of cancer, including CRC
(28), and these miRNAs affect
different aspects of colon carcinogenesis (29). The targeting of E2F1 by miR34a,
that of Rb by miR-675, and that of Ras by miR-143 and let-7 inhibit
the proliferation of CRC cells; miR-26, miR-145 and miR-196a
regulate invasiveness and metastasis; miR-107 induced by p53
inhibits tumor angiogenesis, the process of new blood vessel
development, by blocking the hypoxia-inducible factor (HIF)
1-mediated expression of vascular endothelial growth factor (VEGF);
and the levels of miR-17-3p and miR-92a in plasma samples from
patients with CRC have been found to be markedly elevated and these
miRNAs may potentially be developed into novel diagnostic
markers.
Previous studies have indicated that the
dysregulation of miR-552 is linked to increased Wnt signaling in
CRC cells (30,31). Given the frequent inactivating
mutations in APC and abnormal Wnt signals, in this study, we
further characterized the association between miR-552 and the Wnt
pathway. We hypothesized that miR-552 acts as an oncogene and
mediator that transduces the activation of Wnt signaling to the
downregulation of the p53 tumor suppressor. Revealing the
underlying mechanism of the aberrations in miR-552 expression and
functions may provide novel therapeutic targets for CRC.
Materials and methods
Cell culture, antibodies and
reagents
The human CRC cell lines HCT116 (10247; Korean Cell
Line Bank, Seoul, Korea) and DLD-1 (10221; Korean Cell Line Bank)
were cultured in DMEM (Welgene, Gyeongsan, Korea) and RPMI-1640
medium (Gibco/Thermo Fisher Scientific, Waltham, MA, USA),
respectively, supplemented with 10% fetal bovine serum (FBS,
Capricorn Scientific, Ebsdorfergrund, Germany), 1%
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 1%
L-glutamine and 1% penicillin/streptomycin (PEN/STR) at 37°C in a
humidified 5% CO2 incubator.
The following primary antibodies were used in this
study: Anti-β-actin [1:5,000 dilution; sc-47778; Santa Cruz
Biotechnology (SCB), Dallas, TX, USA], anti-phospho-GSK3β ser9
(1:2,000 dilution; 5558; Cell Signaling Technology, Danvers, MA,
USA), anti-TCF4 (1:1,000 dilution; 05-511; MilliporeSigma,
Burlington, MA, USA), anti-β-catenin (1:2,000 dilution; sc-7199;
SCB), anti-p53 (1:1,000 dilution; sc-126; SCB), anti-Myc (1:2,000
dilution; ab32072; Abcam, Cambridge, UK) antibodies. HRP-conjugated
anti-rabbit (A120-101p) and -mouse (A90-116p-33) secondary
antibodies were from Bethyl (1:5,000 dilution; Montgomery, TX,
USA).
The chemicals used in this study were doxorubicin
(sc-200923; SCB), LiCl (L4408; Sigma, St. Louis, MO, USA), a BET
bromodomain inhibitor JQ1 (A1910; APExBIO; Boston, MA, USA), a
Myc-Max dimerization inhibitor 10058-F4 (475956; MilliporeSigma,
Burlington, MA, USA), crystal violet (C0775; Sigma).
Reverse transcription-quantitative PCR
(RT-qPCR)
The transcription level of miR-552 was measured by
RT-qPCR as previously described (32). Total RNA was extracted using TRIzol
reagent (Favorgen, Ping-Tung, Taiwan) and reverse transcription was
performed to synthesize cDNA using the High Capacity cDNA Reverse
Transcription kit, TaqMan MicroRNA Assay Probes and Universal
Master Mix II (Applied Biosystems, Foster City, CA, USA). The
thermocy-cling condition for RT-qPCR were 10 m at 95°C for the
initial denaturation followed by 39 cycles of 15 sec at 95°C and 60
sec at 60°C. Relative gene expression was analyzed using the
2−ΔΔCq method (33).
To measure the transcriptional level of LINEs, the
PrimeScript™ RT reagent kit with gDNA Eraser (Perfect Real Time)
(Takara, Shiga, Japan) was used to synthesize the cDNA. qPCR was
conducted using TOPreal™ qPCR 2X PreMIX (SYBR-Green with low ROX)
(Enzynomics, Daejeon, Korea) and target primers. The thermocycling
condition for qPCR were 15 m at 95°C for the initial denaturation
followed by 40 cycles of 15 sec at 95°C, 15 sec at 60°C and 30 sec
at 72°C and a final extension for 10 sec at 95°C. The sequences for
the TBP-, LINE- and Myc-specific primers were as follows: TBP
forward, 5′-TATAATCCCAAGCGGTTTGCTGCG-3′ and reverse,
5′-AATTGTTGGTGGGTGAGCACAAGG-3′; LINE forward,
5′-CAAATCACACACCTGAAAAGGA-3′ and reverse,
5′-CATGGTTTTAATTTGCATTACCC-3′; Myc forward,
5′-CTCCTGGCAAAAGGTCAGAG-3′ and reverse,
5′-TCGGTTGTTGCTGATCTGTC-3′.
Western blot analysis
To perform western blot analysis, relevant cells at
a density of two million cells per well in 6-well plates were
washed with PBS and lysed in lysis buffer [Protein Extraction
Solution (Elpis Biotech, Daejeon, Korea), Na-vanadate (1 mM),
β-glycerol phosphate (50 mM), protease inhibitor (G-Biosciences),
EDTA (5 mM) and β-mercaptoethanol (142.7 mM; Bioworld, Irving, TX,
USA)]. The samples were boiled at 100°C for 10 min and loaded on
10% polyacrylamide gels. Twenty micrograms of proteins were
transferred onto membranes using the Mini Trans-Blot®
Cell and Criterion™ Blotter (Bio-Rad, Hercules, CA, USA), and
membranes were blocked in 1% BSA dissolved in Tris-buffered saline
containing 0.1% Tween-20 (TBST). The membranes were probed with the
indicated primary antibodies overnight at 4°C, washed for 5 min in
TBST, and incubated with anti-mouse or anti-rabbit secondary
antibodies for 1 h at room temperature. After washing 3 times for
10 min each with TBST at room temperature, the bands were detected
with a chemiluminescent substrate (Ez West Lumi Plus, ATTO
Technology, Amherst, NY, USA) and visualized on the
Chemiluminescence Imaging system (Luminograph II, ATTO
Technology).
Transfection of short interfering RNA
(siRNA), short hairpin RNA (shRNA) and expression vectors
A total of 100 pM of Myc siRNA (sense,
5′-GACAGUGUCAGAGUCCUGA-3′ and antisense,
5′-UCAGGACUCUGACACUGUC-3′), 100 pM of a non-targeting negative
control siRNA (Bioneer, Daejeon, Korea), 1 µg of a
non-targeting shRNA control vector, or 1 µg of shRNAs
targeting β-catenin or TCF4, 1 µg of empty pcDNA control
vector, or 1 µg of pcDNA-Myc vector (please see the
Acknowledgements section below) were transfected into two million
HCT116 cells in 6-well plates using Lipofectamine 2000 (Invitrogen,
Carlsbad, CA, USA) (34), followed
by incubation for 24 h. Similar transfection efficiencies were
confirmed by FACS analysis of the expression of green fluorescent
protein (GFP).
Stable and transient expression of
miR-552
To generate HCT116 and DLD-1 cells stably expressing
miR-552, a fragment (~300 bp) containing the miR-552 sequence was
PCR-amplified using primers as follows: Forward,
5′-TTTTTAGATCTAAACCCAGCATGCCTATGAC-3′ and reverse,
5′-TTTGAATTCCCTCCACCTCACCACATTCT-3′. The PCR product was
directionally cloned into the BglII and EcoRI sites
of the murine stem cell virus (MSCV)-puro retroviral vector.
Retrovirus was generated by co-transfection of the MSCV constructs
with pKAT and VSVG (vesicular stomatitis virus G glycoprotein),
which were a kind gift from Dr R. Aguiar (University of Texas
Health Science Center at San Antonio, TX, USA), into 293 cells
(21573; Korean Cell Line Bank) using Lipofectamine 2000
(Invitrogen). At 48 h following transfection, cells were infected
with the viral supernatant and selected using puromycin (1 mg/ml).
For the transient expression of miR-552, the HCT116 cells were
transfected with 100 pM of miR-552 mimic or miRNA negative control
(Bioneer, Daejeon, Korea) using Lipofectamine 2000
(Invitrogen).
Cell counting and colony forming
assays
For cell counting, two million cells were seeded in
a 6-well cell culture plate (SPL Life Sciences, Pocheon, Korea) at
day 0 and counted every 24 h. For colony forming assays,
miR-552-expressing cells or control cells were seeded in a 6-well
plate (1,000 cells/well), and 1 week later colonies were fixed and
stained with 0.5% crystal violet at room temperature for 10
min.
Chromatin immunoprecipitation (ChIP)
assays
ChIP assays were performed as previously described
(35). Briefly, approximately
1.0×107 to 2.0×107 HCT116 cells were
harvested and proteins were cross-linked to DNA with 1%
formaldehyde, followed by fixation and sonication to shear the DNA
to an average size of ~300 nucleotides. The fragmented chromatin
was reacted with antibodies in a volume of 1,000 µl and
immu-noprecipitated with protein A/G agarose beads (sc2003; SCB).
The immune complexes were washed 5 times before eluting the DNA.
The primary antibodies used were anti-Myc antibody (sc764: SCB),
normal rabbit IgG (sc2027; SCB), anti-TCF4 antibody (05-511;
MilliporeSigma), and normal mouse IgG (12-371; MilliporeSigma). The
obtained DNA was analyzed by RT-qPCR using the primers described
below. Primers for amplifying the 3 potential TCF4 binding sites in
the miR-552 promoter were designated as 1, 2, and 3: Myc forward,
5′-TTTTGCTAGCTCAAAACTCAATGGTAAA-3′ and reverse,
5′-TTTTCTCGAGACAATTTTACATCCCATC-3′; TCF4 forward 1,
5′-TTCTTTTTTCCAAATTGATTCAAACAT-3′ and reverse 1,
5′-TCCACCCTCTTCTTCTTTAGC-3′; TCF4 forward 2,
5′-GGGCTTTTACGGATGTCAGA-3′ and reverse 2,
5′-GACAGGCTCCTGGATTGAAG-3′; TCF4 forward 3,
5′-CTCCCACTTGTCACCAGCTC-3′ and reverse 3,
5′-GGCAGTAGGAGAGGGAGAGAG-3′.
p53 3′-UTR luciferase reporter assays and
site-directed mutagenesis
The 3′UTR of p53 with a probable miR-552 binding
site was cloned into the psiCHECK-2 vector (Promega, Madison, WI,
USA) using the following primers: Forward,
5′-TTTCTCGAGGAGACTGGGTCTCGCTTTGT-3′ and reverse,
5′-TTTGCGGCCGCAAATGCAGATGTGCTTGCAG-3′. The putative miR-552 binding
site was mutated using a QuikChange Site-Directed Mutagenesis kit
(Stratagene, Santa Clara, CA, USA). Primers for mutagenesis were as
follows: Forward, 5′-GGCTCAGGCGATCGAGGTCTCTCAGCCTCCCAG-3′ and
reverse, 5′-CTGGGAGGCTGAGAGACCTCGATCGCCTGAGCC-3′. Sanger sequencing
was performed to verify the sequences of the mutated construct. For
luciferase assays, 500 ng of p53 3′-UTR construct with the
wild-type or mutant miR-552 binding site were co-transfected with
20 pM of miR-552 mimic or control oligonucleotides (Bioneer, Seoul,
Korea). The cells were harvested at 72 h post-transfection and dual
luciferase reporter assays were performed (Promega).
Statistical analysis
The data are presented as the means ± standard
deviation (SD). Statistical analyses were performed using a
non-parametric Mann-Whitney U test to identify statistically
significant differences (P<0.05). Prism software was used to
perform the statistical analysis. All experiments were performed at
least 3 times independently.
Software programs
The software programs used in this study are
TRANSFAC version 8.0 (genexplain/transfac) to identify potential
transcription factor-binding sites, TargetScan Version 7.1
(www.targetscan.org) to analyze the
targets of miRNAs, and STarMir (http://sfold.wadsworth.org/cgi-bin/starmirtest2.pl) to
predict miRNA binding sites.
Results
miR-552 is an oncogene in CRC
A previous study analyzed the expression of miR-552
in 183 pairs of non-cancerous colon samples and CRC tissues and
found a significantly higher expression of this miRNA (36), which suggests that miR-552 acts as
an oncogene in CRC. To directly test this, in this study, we
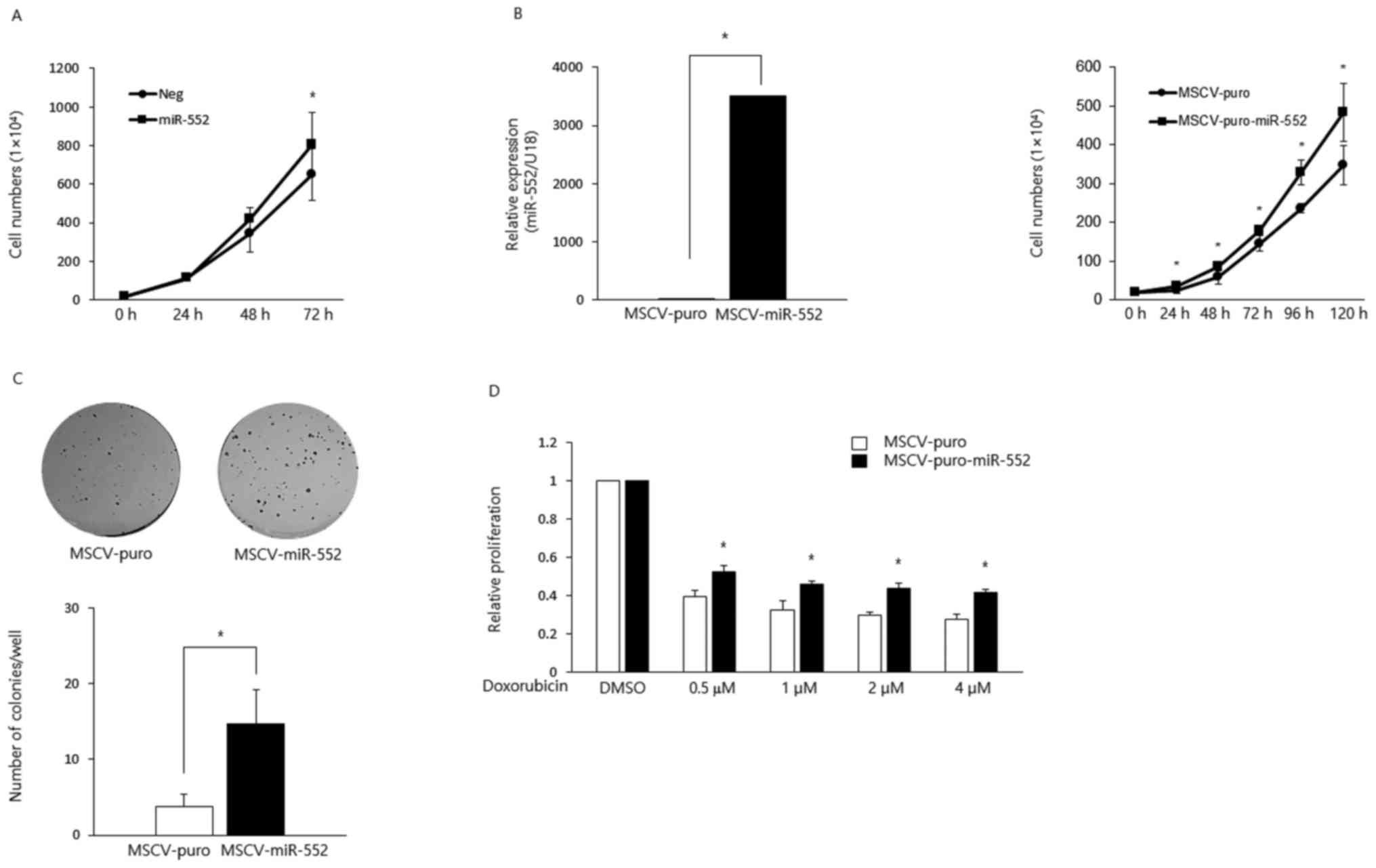
transiently overexpressed this miRNA in HCT116 CRC cells and found
that the ectopic expression of miR-552 increased cell proliferation
(Fig. 1A). To further characterize
its oncogenic potential, we generated HCT116 stable cell lines to
ectopically express MSCV-puro or MSCV-puro-miR-552 using retroviral
constructs (Fig. 1B, left panel).
The overexpression of miR-552 promoted cell proliferation (Fig. 1B, right panel) and colony formation
(Fig. 1C). Moreover,
miR-552-expressing cells exhibited resistance to the
doxorubicin-induced suppression of proliferation (Fig. 1D).
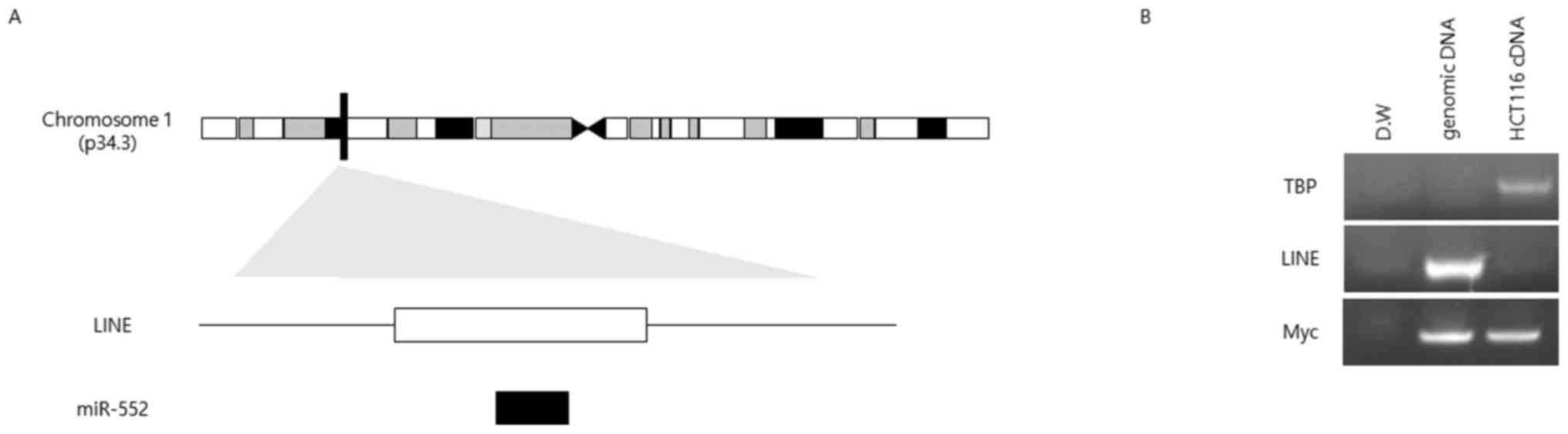
miR-552 is located in a LINE
Our sequence analysis suggested that the sequences
of miR-552 and LINE L1 overlapped (Fig. 2A); therefore, we evaluated whether
the expression of miR-552 is affected by that of L1. To that end,
we designed primers for specifically amplifying the LINE L1
sequence to test whether this specific LINE was active or produced
RNA transcripts. No PCR product was detected when cDNA was used as
a template, whereas genomic DNA as a positive control produced a
single band of the expected size (Fig.
2B).
miR-552 is directly regulated by the
Wnt-Myc axis
To investigate the transcriptional control of
miR-552, we identified transcription factors that potentially bind
to the promoter using TRANSFAC software (Fig. 3A). Among the many TFs that may
influence miR-552 transcription, we were interested in
characterizing the TCF4- and Myc-binding sites as the inactivation
of APC, thereby the hyperactivation of Wnt signaling, and the
dysregulation of Myc (8), a
critical downstream target gene of the Wnt pathway, are frequently
observed in CRC. We wished to determine whether the miR-552 levels
were affected by Wnt signaling. GSK3β, a component of the
destruction complex, has been known to catalyze the phosphorylation
and ubiquitination of β-catenin in the absence of Wnt protein,
marking it for degradation by the proteasome (6). LiCl is a well-characterized inhibitor
of GSK3β, and its addition in HCT116 cells led to an increase in
the phosphorylation of GSK3β (ser9) and concomitant increase of
miR-552 (Fig. 3B). To further
elucidate the role for the Wnt signaling in the regulation of
miR-552 expression, we genetically silenced β-catenin and its
binding partner, TCF4, using shRNA constructs, which markedly
inhibited the expression of this miRNA, suggesting that the Wnt
pathway regulates the expression of miR-552 (Fig. 3C).
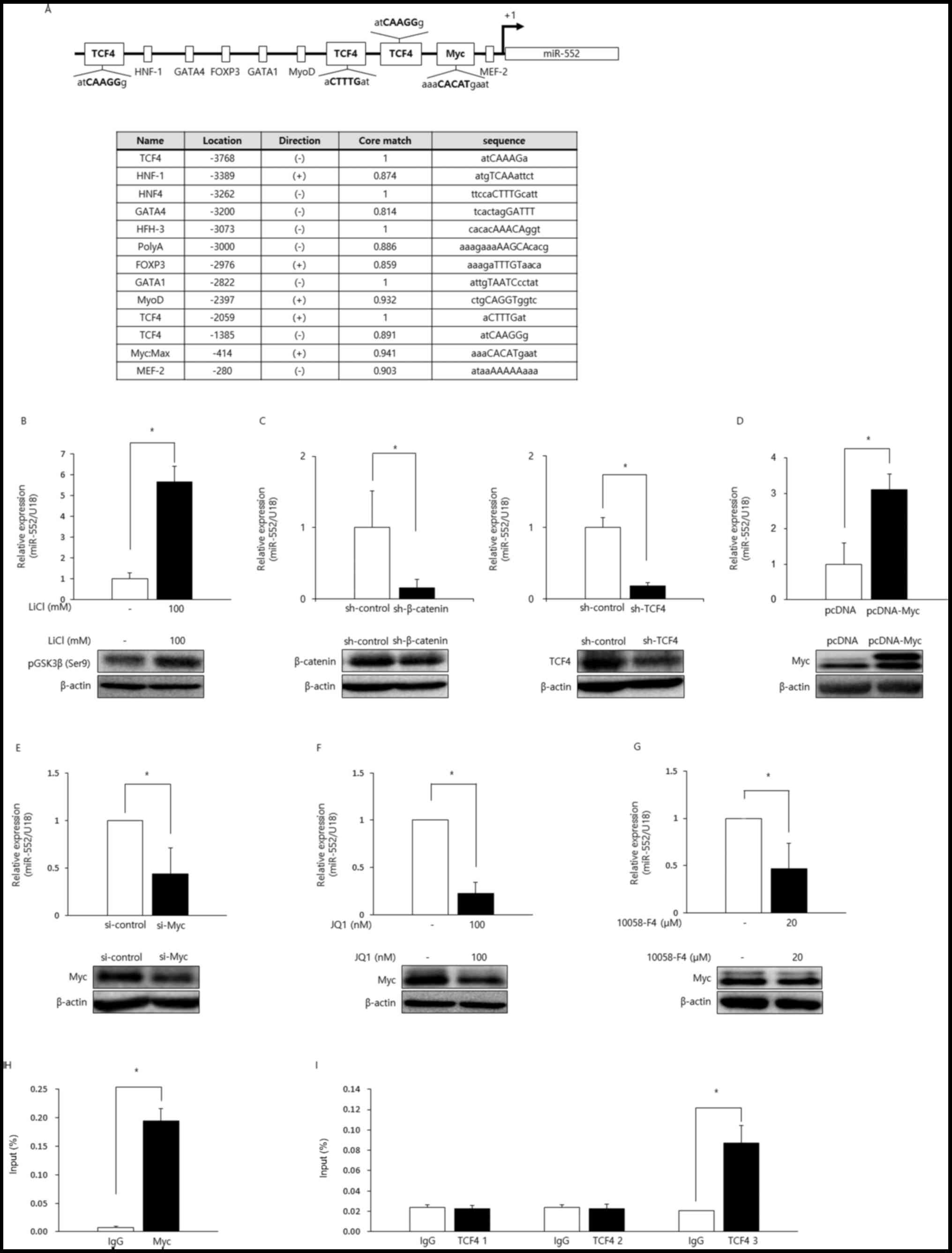 | Figure 3miR-552 is a target of the Wnt/Myc
axis. (A) Transcription factor-binding sites in the miR-552
promoter region were identified using TRANSFAC software. Relative
positions of TCF4- and Myc-binding sites are shown in the
schematic, and the +1 represents the transcriptional start site of
miR-552. Direction indicates whether the transcription factor binds
to the (+) or (−) strand of the DNA. The core match is defined as
the binding probability of transcription factors to their potential
DNA binding sites. When the core match for the indicated
transcription factor is closer to 1, it has a better chance to bind
to the respective binding site. (B) Transcriptional levels of
miR-552 were analyzed by RT-qPCR following treatment with LiCl (100
mM, 24 h; upper panel). Phosphorylation levels of the inactive form
GSK3β at Ser9 were measured by western blot analysis (lower panel).
β-actin was used as a loading control. The inhibition of GSK3β led
to increments in miR-552 expression (*P<0.05,
non-parametric Mann-Whitney U test). (C) Relative miR-552
transcriptional levels in HCT116 cells were examined by RT-qPCR 48
h post-transfection with an shRNA control vector, or shRNAs
targeting β-catenin or TCF4. The downregulation of β-catenin and
TCF4 was confirmed by western blot analysis (lower panel) and
β-actin was used as a loading control. The inhibition of the Wnt
signaling pathway significantly suppressed miR-552 levels (upper
panel; *P<0.05). (D and E) A Myc-expressing vector
construct or siRNA targeting Myc was transfected into HCT116 cells.
Twenty-four hours later, relative miR-552 transcriptional levels
were assessed by RT-qPCR. The ectopic expression or knockdown of
Myc was confirmed by western blot analysis (lower panel), which led
to the up- and downregulation of miR-552 (upper panel;
*P<0.05), respectively. β-actin was used as a loading
control. (F and G) HCT116 cells were treated with JQ1 (100 nM, 48
h) or 10058F4 (20 µM, 24 h), followed by the analysis of the
transcriptional levels of miR-552. Myc levels following treatment
with JQ1 or 10058-F4 were analyzed by western blot analysis (lower
panel). β-actin was used as a loading control. The inhibition of
Myc decreased the expression of miR-552 (upper panel;
*P<0.05). (H and I) ChIP assays were performed using
anti-Myc and anti-TCF4 antibodies. DNA obtained from the ChIP assay
was analyzed by RT-qPCR (upper panel) using primers listed in the
Materials and methods. The IgG antibody was used as a negative
control. All experiments were performed 3 times independently
(*P<0.05, non-parametric Mann-Whitney U test). |
Subsequently, we investigated whether the miR-552
levels would be modulated by Myc in CRC. The ectopic expression of
Myc in HCT116 cells resulted in the upregulation of miR-552
(Fig. 3D). Consistently, genetic
knockdown using Myc-targeting siRNAs or the pharmacological
inhibition of Myc using JQ1, a BET bromodomain inhibitor, or
10058-F4, a Myc-Max dimerization inhibitor, downregulated the
expression of miR-552 (Fig.
3E–G).
Given that the miR-552 levels were regulated by the
Wnt/Myc axis, we wished to delineate the underlying mechanisms. We
suspected that the regulation of miR-552 by Myc and the Wnt signals
was accomplished via the Myc E-box and TCF4-binding sites in the
promoter region of this miRNA. To directly investigate whether the
Myc E-box and TCF4-binding sites were functional, we performed ChIP
assays using normal IgG, anti-Myc, or anti-TCF4 antibodies. The
promoter region of miR-552 containing the Myc E-box was enriched by
antibodies against Myc, suggesting that Myc directly binds to the
E-box in the miR-552 promoter (Fig.
3H). Anti-TCF4 antibodies efficiently immunoprecipitated one of
the three probable TCF4-binding sites, indicating that only one,
but not the other two, TCF4-binding site was functional (Fig. 3I). Taken together, these data
clearly demonstrated that miR-552 was directly upregulated by Myc
and TCF4 transcription factors via their corresponding binding
sites in the miR-552 promoter region.
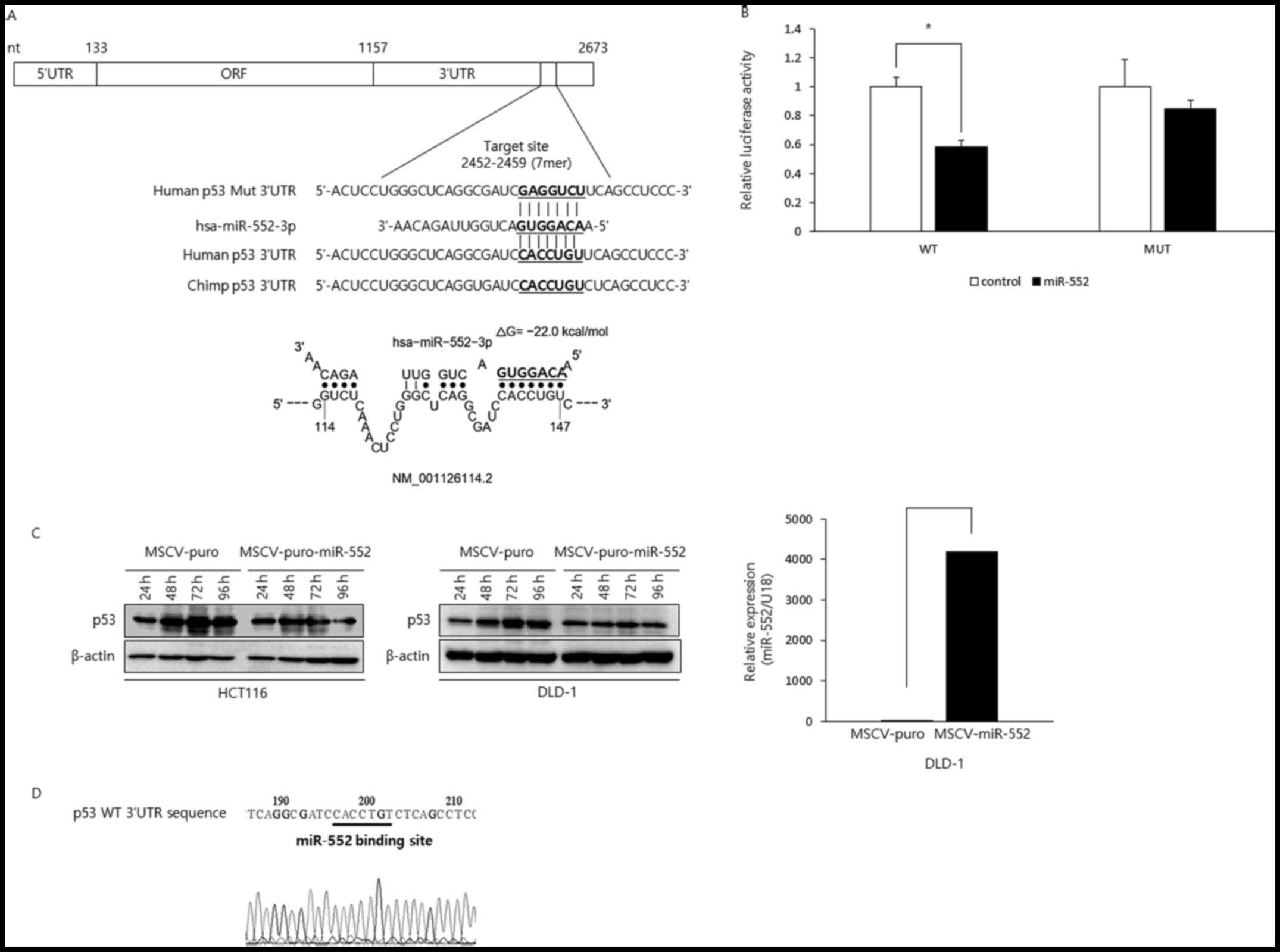
The p53 tumor suppressor is a target of
miR-552
The results described above clearly demonstrated
that elevated miR-552 levels promote the oncogenic properties of
CRC cells. Based on these data, we predicted that miR-552 inhibits
the expression of tumor suppressor genes and aimed to identify a
potential target of this miRNA relevant in our experimental
setting. The use of miRNA binding site prediction algorithms
(TargetScan version 7.1) revealed that the p53 tumor
suppressor gene has a potential binding site within its 3′-UTR. The
putative binding site was found only in humans and chimps (Fig. 4A), suggesting that the interaction
between these two genetic elements came late in the evolution. To
examine whether miR-552 directly binds to the potential binding
site in the p53 3′-UTR, we cloned p53 3′UTR in the wild-type or
mutant configuration in the psiCHECK-2 luciferase vector and
reporter assay was performed (Fig.
4B). The luciferase activity of the wild-type construct was
efficiently repressed by miR-552. However, miR-552 did not affect
luciferase reporter activity when the binding site was mutated,
suggesting miR-552 directly interacts with its binding site in the
p53 3′-UTR. Next, to investigate whether miR-552 regulates the p53
levels, we generated HCT116 and DLD-1 CRC cells expressing either
MSCV-puro or MSCV-puro-miR-552 and examined the p53 levels by
western blot analysis (Fig. 4C).
The levels of p53 increased in the control cells in a
time-dependent manner. By contrast, the cells expressing miR-552
did not exhibit as much of an increment as the control cells,
suggesting that miR-552 suppressed p53 expression. Intriguingly,
DLD-1 CRC cells are known to express mutant p53, but our sequencing
results indicated that the miR-552 binding site in p53 3′-UTR was
intact (Fig. 4D). As expected, the
downregulation of p53 by miR-552 was observed in the DLD-1 cells
(Fig. 4C). Taken together, these
data suggest that miR-552 negatively modulates p53 tumor suppressor
in CRC, which is through a direct interaction between miR-552 and
its binding site in the p53 3′-UTR.
Discussion
Cancer is a complex disease involving multiple
genetic and epigenetic aberrations (1,2). The
functional loss of APC, thereby aberrant Wnt signaling, has been
found in >80% of CRC samples (3) and it is recognized as one of the
earliest genetic events during the development of this type of
tumor (37). Another common
genetic abnormality observed in CRC is the inactivation of the
tumor suppressor p53 signals (16,17).
However, the inter-relationship between the Wnt/β-catenin pathway
and p53 tumor-suppressive signaling has yet to be fully understood.
The data of this study suggest that the activation of Wnt signaling
downregulates the expression of p53 via the upregulation of
miR-552, a p53-targeting miRNA. This is intriguing as the
hyperactivation of Wnt occurs in the early stages of CRC
development and may eventually decrease p53 levels, which may help
progression to later stages by driving cell proliferation and
survival. This offers a unique opportunity where the development of
CRC can be interrupted by restoring miR-552 expression. Previous
studies have provided clear evidence that miR-34 links p53 tumor
suppressor signals to the Wnt pathway (38,39).
The expression of miR-34 is increased by p53 (40); miR-34 directly targets and
decreases the expression of some activators of canonical Wnt
signaling, such as WNT1, WNT3, β-catenin, LEF1, Axin2 and the Wnt
coreceptor LDL receptor-related protein (LRP) 6, which leads to the
suppression of Wnt signaling (39); the loss of p53 functions or miR-34
promote CRC progression, which is dependent on the hyperactivation
of the Wnt pathway (39). These
results revealed that miR-34 is a mediator that transduces the
inactivation of p53 tumor suppressor functions to heightened Wnt
signaling in cancers. To the best of our knowledge, miR-552 is the
first onco-miRNA connecting the hyperactive Wnt pathway with
down-modulation of p53 levels, which may contribute to colon
carcinogenesis. Previous studies have demonstrated that the loss of
p53 function plays a decisive role in carcinogenesis and, indeed,
inactivating mutations in p53 have been described in approximately
40-50% of CRCs (16,17). In this regard, the normalization of
miR-552 can restore the level of p53 in CRC cells to that in normal
cells and may be a feasible therapeutic option for treating
patients with CRC driven by high Wnt signaling. The regulation of
p53 and miR-552 by the Wnt signals using CRC clinical samples
warrants further investigation.
The role of miRNAs in normal cellular physiology has
been studied extensively, and the notion that they are
dysregu-lated in a wide range of human diseases, including cancer,
has been convincing (41). A
previous study demonstrated that miR-552 was highly expressed in
CRC samples (36). We thus
hypothesized that the expression of miR-552 may be affected by LINE
L1 as their sequences overlapped. The present study indicated that
this LINE was not active, and thus miR-552 was not expressed as a
part of it and that the expression of this miRNA may be controlled
by its own promoter (Fig. 2). Our
results shed light into the underlying mechanisms of the
dysregulation of miR-552 in CRC, i.e., the β-catenin/TCF4 complex
and Myc, a major downstream target of the Wnt pathway, directly
bind to their corresponding binding sites in the miR-552 promoter
region to enhance its expression. Taken together with the results
from the previous section, these data suggest that the Wnt-Myc axis
can increased the miR-552 levels in HCT116 CRC cells.
Among the identified targets of the miR-552 are the
cell fate determination factor dachshund family transcription
factor 1 (DACH1) (30) and a
disintegrin and metalloprotease (ADAM) family member 28
(ADAM28) (31). The
inhibition of the expression of these genes by miR-552 increased
the oncogenic properties of CRC cells, such as cell proliferation,
migration and clonogenicity (30,31)
and our study suggests that aberrant Wnt signals may also
downregulate these genes by upregulating miR-552. Notably, DACH1 is
a negative regulator of the Wnt pathway and the downregulation of
DACH1 by miR-552 results in increased Wnt/β-catenin signaling
(30). This and the results of the
present study indicate the presence of a feedforward loop that may
play a critical role in promoting the survival of CRC cells:
miR-552 elevates the activity of the Wnt pathway, which in turn
increases the expression of miR-552. Given the critical role of the
Wnt signaling in normal cellular processes, such as development,
this miRNA may be involved in the regulation of normal cell
physiology. One of the key findings of this study is the regulation
of p53 levels by the Wnt signaling, which is mediated by miR552.
Together with previous findings (30), our study indicates the presence of
a potential positive feedback loop between the Wnt signals and the
levels of p53 tumor suppressor.
In conclusion, the current study demonstrates that
miR-552 acts as an oncogene in CRC. Mechanistically, the
Wnt/β-catenin signals reduce the expression of p53 tumor suppressor
via the upregulation of this miRNA. Previous observations that
miR-552 expression was higher in patients with pancreatic cancer
and that Wnt signaling was necessary for pancreatic carcinogenesis
(42) suggest that this miRNA may
be a potential therapeutic target in these types of cancer.
Acknowledgments
The pcDNA-Myc and pcDNA control vector were gifts
from Dr D.C. Kang at Hallym University (Chuncheon, Korea), and
non-targeting shRNA control vector and pSUPER vectors expressing
shRNA for β-catenin and TCF4 were kindly provided by Dr H.S. Kang
at Pusan National University (Pusan, Korea).
Funding
This study was supported by grants from the Basic
Science Research Program through the National Research Foundation
of Korea (NRF) funded by the Ministry of Science, ICT and Future
Planning (NRF-2013R1A1A2008838 and NRF-2016R1A2B4011758) to
SWK.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BK designed and performed the experiments, analyzed
the data, and wrote the manuscript; DUK performed the experiments;
TOK analyzed the data; HSK supervised the study and analyzed the
data; SWK designed and supervised the study, planned experiments,
analyzed the data and wrote the manuscript. All the authors have
edited and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Grothey A, Sargent D, Goldberg RM and
Schmoll HJ: Survival of patients with advanced colorectal cancer
improves with the availability of fluorouracil-leucovorin,
irinotecan, and oxaliplatin in the course of treatment. J Clin
Oncol. 22:1209–1214. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fransén K, Klintenäs M, Osterström A,
Dimberg J, Monstein HJ and Söderkvist P: Mutation analysis of the
BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas.
Carcinogenesis. 25:527–533. 2004. View Article : Google Scholar
|
|
3
|
Basu S, Haase G and Ben-Ze'ev A: Wnt
signaling in cancer stem cells and colon cancer metastasis.
F1000Res. 2016, 5:https://doi.org/10.12688/f1000research.7579.1.
View Article : Google Scholar
|
|
4
|
Kang YJ, Park HJ, Chung HJ, Min HY, Park
EJ, Lee MA, Shin Y and Lee SK: Wnt/β-catenin signaling mediates the
antitumor activity of magnolol in colorectal cancer cells. Mol
Pharmacol. 82:168–177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu Y, Xie S, Zhang W, Zhang C, Gao C, Sun
Q, Cai Y, Xu Z, Xiao M, Xu Y, et al: Twa1/Gid8 is a beta-catenin
nuclear retention factor in Wnt signaling and colorectal
tumorigenesis. Cell Res. 27:1422–1440. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Najdi R, Holcombe RF and Waterman ML: Wnt
signaling and colon carcinogenesis: Beyond APC. J Carcinog.
10:52011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
You Z, Saims D, Chen S, Zhang Z, Guttridge
DC, Guan KL, MacDougald OA, Brown AM, Evan G, Kitajewski J, et al:
Wnt signaling promotes oncogenic transformation by inhibiting
c-Myc-induced apoptosis. J Cell Biol. 157:429–440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morin PJ: beta-catenin signaling and
cancer. Bioessays. 21:1021–1030. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rivlin N, Brosh R, Oren M and Rotter V:
Mutations in the p53 tumor suppressor gene: Important milestones at
the various steps of tumorigenesis. Genes Cancer. 2:466–474. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marine JC, Francoz S, Maetens M, Wahl G,
Toledo F and Lozano G: Keeping p53 in check: Essential and
synergistic functions of Mdm2 and Mdm4. Cell Death Differ.
13:927–934. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meek DW: The p53 response to DNA damage.
DNA Repair (Amst). 3:1049–1056. 2004. View Article : Google Scholar
|
|
13
|
Liebermann DA, Hoffman B and Vesely D: p53
induced growth arrest versus apoptosis and its modulation by
survival cytokines. Cell Cycle. 6:166–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Giaccia AJ and Kastan MB: The complexity
of p53 modulation: Emerging patterns from divergent signals. Genes
Dev. 12:2973–2983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moll UM and Petrenko O: The MDM2-p53
interaction. Mol Cancer Res. 1:1001–1008. 2003.
|
|
16
|
Al-Kuraya KS: KRAS and TP53 mutations in
colorectal carcinoma. Saudi J Gastroenterol. 15:217–219. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iacopetta B: TP53 mutation in colorectal
cancer. Hum Mutat. 21:271–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sadot E, Geiger B, Oren M and Ben-Ze'ev A:
Down-regulation of beta-catenin by activated p53. Mol Cell Biol.
21:6768–6781. 2011. View Article : Google Scholar
|
|
19
|
Riascos-Bernal DF, Chinnasamy P, Cao LL,
Dunaway CM, Valenta T, Basler K and Sibinga NE: β-catenin
C-terminal signals suppress p53 and are essential for artery
formation. Nat Commun. 7:123892016. View Article : Google Scholar
|
|
20
|
Griffiths-Jones S: The microRNA registry.
Nucleic Acids Res. 32(Database issue): D109–D111. 2004. View Article : Google Scholar :
|
|
21
|
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee
DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al:
Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic
Acids Res. 33:e1792005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek
SH and Kim VN: MicroRNA genes are transcribed by RNA polymerase II.
EMBO J. 23:4051–4060. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Piriyapongsa J and Jordan IK: A family of
human microRNA genes from miniature inverted-repeat transposable
elements. PLoS One. 2:e2032007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Piriyapongsa J, Marino-Ramirez L and
Jordan IK: Origin and evolution of human microRNAs from
transposable elements. Genetics. 176:1323–1337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Finnegan DJ: Retrotransposons. Curr Biol.
22:R432–R437. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li M, Marin-Muller C, Bharadwaj U, Chow
KH, Yao Q and Chen C: MicroRNAs: Control and loss of control in
human physiology and disease. World J Surg. 33:667–684. 2009.
View Article : Google Scholar
|
|
27
|
Lytle JR, Yario TA and Steitz JA: Target
mRNAs are repressed as efficiently by microRNA-binding sites in the
5′ UTR as in the 3′UTR. Proc Natl Acad Sci USA. 104:9667–9672.
2007. View Article : Google Scholar
|
|
28
|
Jansson MD and Lund AH: MicroRNA and
cancer. Mol Oncol. 6:590–610. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu WK, Law PT, Lee CW, Cho CH, Fan D, Wu
K, Yu J and Sung JJ: MicroRNA in colorectal cancer: From benchtop
to bedside. Carcinogenesis. 32:247–253. 2011. View Article : Google Scholar
|
|
30
|
Cao J, Yan XR, Liu T, Han XB, Yu JJ, Liu
SH and Wang LB: MicroRNA-552 promotes tumor cell proliferation and
migration by directly targeting DACH1 via the Wnt/beta-catenin
signaling pathway in colorectal cancer. Oncol Lett. 14:3795–802.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang J, Li H, Wang Y, Wang L, Yan X, Zhang
D, Ma X, Du Y, Liu X and Yang Y: MicroRNA-552 enhances metastatic
capacity of colorectal cancer cells by targeting a disintegrin and
metalloprotease 28. Oncotarget. 7:70194–7210. 2016.PubMed/NCBI
|
|
32
|
Jeong D, Kim J, Nam J, Sun H, Lee YH, Lee
TJ, Aguiar RC and Kim SW: MicroRNA-124 links p53 to the NF-κB
pathway in B-cell lymphomas. Leukemia. 29:1868–1874. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
34
|
Dalby B, Cates S, Harris A, Ohki EC,
Tilkins ML, Price PJ and Ciccarone VC: Advanced transfection with
Lipofectamine 2000 reagent: Primary neurons, siRNA, and
high-throughput applications. Methods. 33:95–103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cho Y, Song SH, Lee JJ, Choi N, Kim CG,
Dean A and Kim A: The role of transcriptional activator GATA-1 at
human beta-globin HS2. Nucleic Acids Res. 36:4521–4528. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang N and Liu W: Increased expression of
miR-552 acts as a potential predictor biomarker for poor prognosis
of colorectal cancer. Eur Rev Med Pharmacol Sci. 22:412–416.
2018.PubMed/NCBI
|
|
37
|
Rowan AJ, Lamlum H, Ilyas M, Wheeler J,
Straub J, Papadopoulou A, Bicknell D, Bodmer WF and Tomlinson IP:
APC mutations in sporadic colorectal tumors: A mutational 'hotspot'
and interdependence of the 'two hits'. Proc Natl Acad Sci USA.
97:3352–3357. 2000. View Article : Google Scholar
|
|
38
|
Kim NH, Cha YH, Kang SE, Lee Y, Lee I, Cha
SY, Ryu JK, Na JM, Park C, Yoon HG, et al: p53 regulates nuclear
GSK-3 levels through miR-34-mediated Axin2 suppression in
colorectal cancer cells. Cell Cycle. 12:1578–1587. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li
XY, Kang SE, Cha SY, Ryu JK, Na JM, et al: p53 and microRNA-34 are
suppressors of canonical Wnt signaling. Sci Signal. 4:ra712011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hermeking H: p53 enters the microRNA
world. Cancer Cell. 12:414–418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ha TY: The role of microRNAs in regulatory
T cells and in the immune response. Immune Netw. 11:11–41. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schultz NA, Werner J, Willenbrock H,
Roslind A, Giese N, Horn T, Wøjdemann M and Johansen JS: MicroRNA
expression profiles associated with pancreatic adenocarcinoma and
ampullary adenocarcinoma. Mod Pathol. 25:1609–1622. 2012.
View Article : Google Scholar : PubMed/NCBI
|