Introduction
Liver kinase B1 (LKB1), also referred to as serine
threonine kinase 11, plays diverse roles in cellular proliferation,
energy metabolism, apoptosis and polarity, by regulating a variety
of substrates. LKB1 activates at least 14 adenosine monophosphate
(AMP)-activated protein kinase (AMPK)-associated kinases, and the
most extensively investigated substrate is AMPK (1). LKB1/AMPK is activated when the
AMP/ATP ratio is high under energy stress conditions, and restores
intracellular ATP levels by stimulating catabolic and inhibiting
anabolic pathways (2). Studies on
LKB1 in cancer have demonstrated its role as a master tumor
suppressor in the majority of human cancer types, including
melanoma (3), non-small-cell lung
carcinoma (4) and other epithelial
cancer types (5). However, recent
studies have reported that LKB1 acts as a proto-oncogene in certain
types of cancer. Bardeesy et al (6) indicated that LKB1−/− mouse
embryonic fibroblasts were resistant to transformation by activated
Ha-Ras, either alone or with immortalizing genes. Jeon et al
(7) reported that knockdown of
LKB1 and AMPK in breast cancer cells attenuated tumor development
due to failure to inhibit acetyl-CoA carboxylase activity and to
maintain intracellular NADPH levels. Furthermore, Martinez-Lopez
et al (8) reported that
glycine N-methyltransferase (GNMT) knockout mice may develop
hepatocellular carcinoma (HCC). Reduced expression of GNMT in mouse
and human HCC cells increased the activity of LKB1 and RAS. Lee
et al (9) demonstrated that
the stabilization and activation of LKB1/STE20-related kinase
adaptor α (STRADA)/scaffolding mouse 25 (MO25) complex by S-phase
kinase-associated protein 2-dependent ubiquitination was crucial
for cell survival under energy stress conditions. They also
indicated that LKB1 was highly expressed in late-stage HCC and its
overexpression predicts poor survival outcomes. Furthermore, a
study by Huang et al (10)
suggested that the expression of LKB1 was decreased in HCC
patients, and that low LKB1 expression predicted poor survival. Due
to these contradicting results, the aim of the present study was to
elucidate the role of LKB1 in HCC.
Materials and methods
Patients, specimens and follow-up
In the present study, two independent cohorts of
patients who underwent curative resection at the Hepatic Surgery
Center of Tongji Hospital of Huazhong University of Science and
Technology (Wuhan, China) between January 2004 and January 2014
were enrolled. For cohort 1, a total of 229 HCC tissues and matched
surrounding analogous non-cancerous tissues (ANT) were collected
for immunohistochemical (IHC) analysis; these patients were
diagnosed with liver tumors, hepatectomy was performed and
pathological analysis confirmed the diagnosis of HCC. Complete
clinicopathological data and follow-up results were acquired for
this cohort. Cohort 2, lacking follow-up data, included 60 HCC
samples and matched ANT for western blot analysis of LKB1
expression. Furthermore, the level of phosphorylated (p)-AMPK
(Thr172) was measured to elucidate the activation of downstream
signaling in 10 pairs of ANT and HCC samples. The preoperative
diagnosis of HCC was performed according to the diagnostic criteria
of the American Association for the Study of Liver Diseases
(11). All the patients were
followed-up until October 2014, with a median survival time of
23.30±0.97 months. Overall survival was defined as the time
interval between the date of surgery and the date of death or the
last follow-up. Disease-free survival was defined as the time
interval between the date of surgery and the date of recurrence
confirmed by abdominal ultrasound examinations and serum
α-fetoprotein levels. If no recurrence was diagnosed, patients were
censored on the date of death or the last follow-up. The median
disease-free survival time was 17.02±0.98 months. The present study
was approved by the Ethics Committee of Tongji Hospital, Huazhong
University of Science and Technology (Wuhan, China). The study
protocol conformed to the principles outlined in the Declaration of
Helsinki and written informed consent was obtained from each
patient.
IHC
Formalin-fixed, paraffin-embedded tissues were
sectioned at 2 μm, deparaffinized in xylene and rehydrated
through a graded series of ethanol. Antigen retrieval was performed
by microwave heating in 10 mM Tris base and 1 mM EDTA (pH 9.0).
Endogenous peroxidase was blocked with 3%
H2O2 in methanol. The sections were then
incubated with primary antibody at 4°C overnight (Table I). A Dako EnVision kit (Dako,
Glostrup, Denmark) was used for incubation with the secondary
antibody (Table I) and detection
of peroxidase activity. Hematoxylin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used to counterstain the nuclei for 5 min
at room temperature. IHC scores were obtained by multiplying the
percentage score with the intensity score of positively stained
cells, as described previously (12). Scoring was performed by two
certified pathologists independently, who were blinded to the
patients’ clinical and demographic information. The expression
status is represented by the mean of several independent readings.
An overall score of >6 and ≤6 was considered to indicate high
and low expression, respectively. The Edmondson-Steiner, Barcelona
Clinic Liver Cancer (BCLC) and tumor-node-metastasis (TNM) stages
were also determined (13,14).
 | Table IAntibodies used in this study. |
Table I
Antibodies used in this study.
| Antigen | Catalogue no.,
manufacturer | Dilution and
application |
|---|
| LKB1 | AP7239A, Abgent,
San Diego, CA, USA | 1:100 for IHC |
| LKB1 | 3050, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| p-AMPKThr172 | 50081S, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| AMPKα | 5831, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| GAPDH | KC-5G4, KangChen
Bio-tech, Shanghai, China | 1:50,000 for
WB |
| β-actin | sc-47778, Santa
Cruz Biotechnology, Santa Cruz, CA, USA | 1:10,000 for
WB |
|
p21Cip1 | 2947, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
|
p27Kip1 | 3686, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| c-Myc | 1472-1, Epitomics,
Burlingame, CA, USA | 1:2,000 for WB |
| PARP | 9532, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| Cleaved-PARP | 5625, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| Bcl-2 | 3498, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| Bax | 5023, Cell
Signaling Technology, Beverly, MA, USA | 1:1,000 for WB |
| HRP-conjugated
anti-rabbit IgG | Jackson
ImmunoResearch Laboratories, Inc. West Grove, PA, USA | 1:3,000 for WB |
| HRP-conjugated
anti-mouse IgG | Jackson
ImmunoResearch Laboratories, Inc. West Grove, PA, USA | 1:4,000 for WB |
| Secondary
antibody | Envision kit (HRP,
rabbit/mouse, DAB+), Dako | Ready-to-use for
IHC |
Cell lines and culture
The HCC cell lines MHCC97L, MHCC97H and HCCLM3 were
obtained from the Liver Cancer Institute of Zhongshan Hospital
(Fudan University, Shanghai, China). The HCC cell lines HLE and HLF
were kindly provided by Shanshan Wang and Gang Li (Department of
Molecular Biology, Peking University Health Science Center,
Beijing, China). The hepatoblastoma cell line HepG2, and the HCC
cell lines Hep3B, Huh7 and SK-Hep1 were purchased from the China
Center for Type Culture Collection (Wuhan, China). The HCC cell
line PLC/PRF-5 was purchased from the cell bank of the Chinese
Academy of Sciences (Shanghai, China). All cell lines were
maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere with 5% CO2 at 37°C.
Western blot analysis
HCC cell lines and samples were lysed in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Haimen, China) with proteinase and phosphatase
inhibitor cocktail (Hoffmann-La Roche Ltd., Basel, Switzerland),
and the protein concentration was determined by using Pierce™ BCA
Protein Assay Kit (Thermo Fisher Scientific, Inc.). A total of 20
μg of each protein was separated by 10% SDS-PAGE (Boster
Biotechnology, Wuhan, China) and transferred to a polyvinylidene
difluoride membrane (Hoffmann-La Roche Ltd.). The membrane was
blocked with 5% skimmed milk dissolved by 1X Tris-buffered saline
containing Tween-20 and incubated with specific primary antibodies
at 4°C overnight (Table I),
followed by incubation with a horseradish peroxidase
(HRP)-conjugated anti-mouse or anti-rabbit secondary antibody
(Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) at
37°C for 2 h (Table I). Detection
was performed using a ChemiDoc™ Imaging System (Bio-Rad
Laboratories Inc., Hercules, CA, USA).
Lentivirus production, transfection and
establishment of stable cell clones
The pLKO.1-TRC cloning vector (cat. no. 10878) was
from Addgene, Inc. (Cambridge, MA, USA). Small hairpin (sh)RNA
specific for LKB1 (shLKB1) oligos were synthesized by Tsingke
Technology (Wuhan, China) and were inserted into the pLKO.1 vector,
which was then transfected into 293 cells with psPAX2 and pMD2.G
(cat. nos. 12260 and 12259, respectively; Addgene, Inc.) using
X-tremeGENE™ HP DNA Transfection Reagent (Sigma-Aldrich; Merck
KGaA). After 48 h of incubation, the virus-containing supernatant
was collected and filtered through a 0.45-μm filter (PALL,
Port Washington, NY, USA) (15).
LKB1 overexpression lentivirus was purchased from Genecreate
Technology (Wuhan, China). HCC cells were transfected with
lentiviral particles in the presence of 8 μg/ml polybrene
(Sigma-Aldrich; Merck KGaA) with a multiplicity of infection (MOI)
ranging from 50 to 100. At 72 h after transfection, cells were
selected with 5 μg/ml puromycin (Merck Calbiochem,
Darmstadt, Germany) for 2 weeks. Selected pools of LKB1-knockdown
or overexpressing cells were used for the subsequent experiments.
The shRNA sequences are listed in Table II. HCC-LM3 shLKB1 and Huh7 shLKB1
refer to the HCC cell lines transfected with shLKB1 (LKB1
knockdown), whereas HLF LKB1 refers to the cell lines transfected
with LKB1 overexpression virus. Control cells were transfected with
empty vector.
| Table IIshRNA sequences used in this
study. |
Table II
shRNA sequences used in this
study.
| Identifier | Sequence | Reference |
|---|
| shLKB1#2 sense |
CCGGGCCAACGTGAAGAAGGAAATT | Broad Institute
http://portals.broadinstitute.org/gpp/public/gene/search |
|
CTCGAGAATTTCCTTCTTCACGTTGGCTTTTTG |
| shLKB1#2
antisense |
AATTCAAAAAGCCAACGTGAAGAA |
|
GGAAATTCTCGAGAATTTCCTTCTTCACGTTGGC |
| shLKB1#3 sense |
CCGGGATCCTCAAGAAGAAGAAGTT |
|
CTCGAGAACTTCTTCTTCTTGAGGATCTTTTTG |
| shLKB1#3
antisense |
AATTCAAAAAGATCCTCAAGAAGAA |
|
GAAGTTCTCGAGAACTTCTTCTTCTTGAGGATC |
Cell proliferation assay
HCC-LM3 shLKB1, HLF-LKB1 cells (1×103
cells/well) or Huh-7 shLKB1 cells (3×103 cells/well) and
the same amount of control cells were seeded into 96-well plates.
At the indicated time points, Cell Counting Kit-8 reagent (Beyotime
Institute of Biotechnology) was added, followed by incubation for 1
h at 37°C. The plate was read using an ELISA plate reader (Elx 800;
Bio-Tek, Winooski, VT, USA) at a wavelength of 450 nm. Experiments
were repeated three times.
Colony formation assay
Transfected or control cells were seeded into 6-well
plates at 500 cells/well, and the medium was changed every 3 days.
After 10 days of incubation, the cells were fixed with 4% formalin
and stained with 0.1% crystal violet solution (ServiceBio
Technology, Wuhan, China). The numbers of colonies >100
μm in diameter were quantified with a ChemiDoc™ Imaging
System (Bio-Rad Laboratories, Inc.). The experiments were repeated
three times.
Apoptosis assay
Huh-7 shLKB1, HCC-LM3 shLKB1 or the corresponding
control cells were seeded into 6-well plates at 5×105
cells/well. After the cells were attached to the culture dish and
had entered the logarithmic proliferation phase, they were
thoroughly trypsinized, suspended, washed with ice-cold
phosphate-buffered saline 3 times, re-suspended with 1X binding
buffer, incubated with Annexin V and 7-aminoactinomycin D (BD
Biosciences, Franklin Lakes, NJ, USA) at 37°C for 15 min, and
analyzed with a BD FACSCalibur (BD Biosciences). Experiments were
repeated three times.
In vivo tumorigenicity assay
HCC-LM3 shLKB1 cells (2×106), Huh-7
shLKB1 cells (5×106), HLF-LKB1 cells (1×106)
and equal amounts of the corresponding control cells were suspended
in 100 μl DMEM and subcutaneously injected into the flank of
5-week-old male nude mice (weight, 18-19 g). All the experimental
mice were purchased from HFK Technology (Beijing, China) and kept
under specific pathogen-free conditions with free access to food
and water. The experimental mice were routinely monitored and
sacrificed at the indicated time points. The length and width of
the tumors was manually monitored using a Vernier caliper. Tumor
volume (V) was calculated according to the following equation: V
(mm3) = 0.5 × L × W2, where L is the length
and W the width in mm (16). The
animal experiments were approved by the Ethics Committee of Tongji
Hospital, Huazhong University of Science and Technology (Wuhan,
China).
Statistical analysis
Statistical analysis was performed using SPSS 19.0
(IBM Corp., Armonk, NY, USA) or Prism 6.0 (GraphPad Software, Inc.,
La Jolla, CA, USA) software. Comparison between groups was
performed by a two-tailed Student’s t-test, analysis of variance
with Bonferroni’s post hoc test, Chi-squared test, Spearman’s
correlation coefficient test or a non-parametric test, including
the Wilcoxon’s signed-rank test. Kaplan-Meier analysis and the
log-rank test were used to compare the survival between subgroups.
A Cox proportional hazards model was used for univariate and
multivariate analyses to determine the factors independently
associated with survival and recurrence. P<0.05 was considered
to indicate a statistically significant difference.
Results
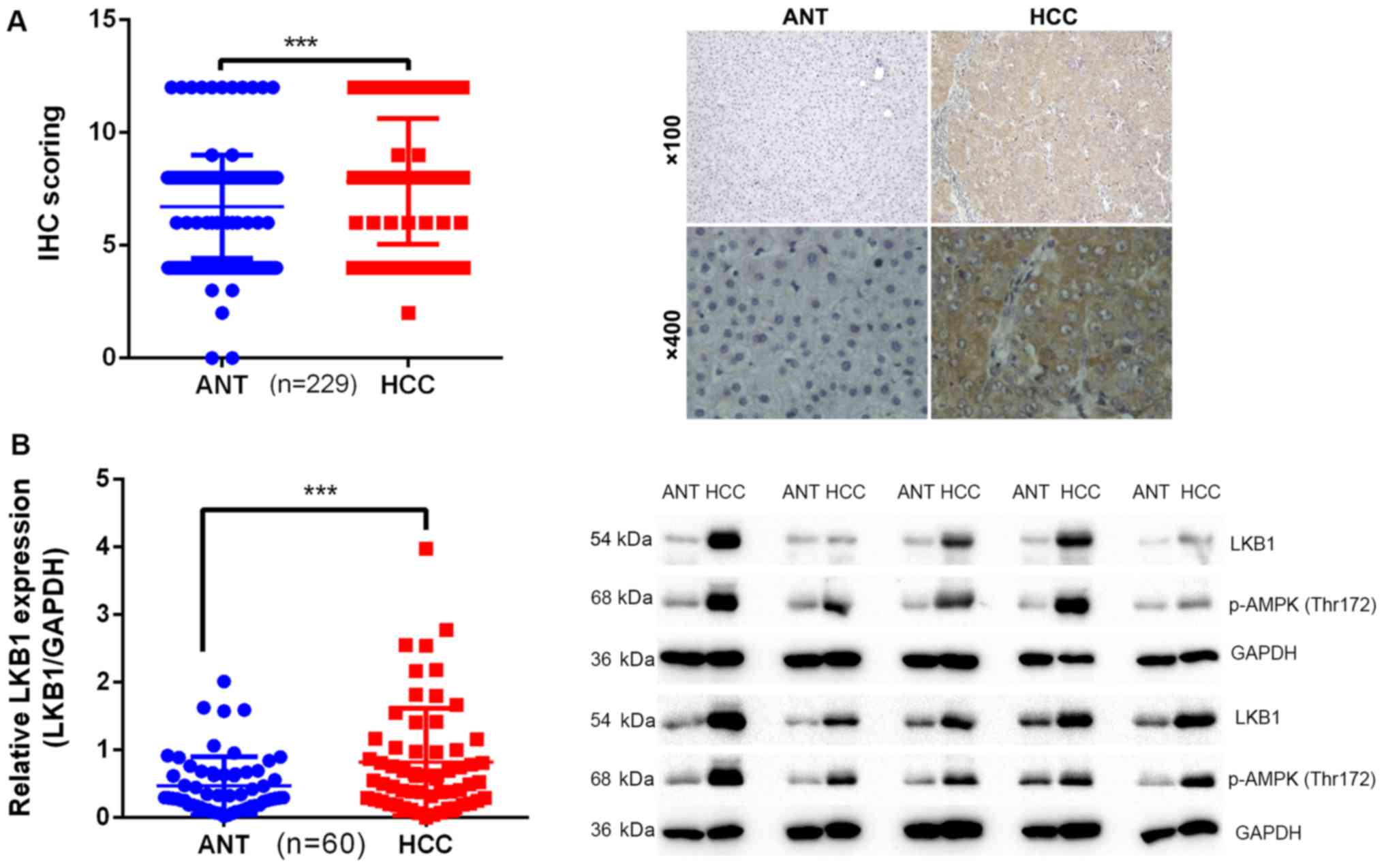
LKB1 expression is upregulated in
HCC
To determine the clinical significance of LKB1 in
the development of HCC, the expression pattern of LKB1 was examined
in two cohorts of patients. Cohort 1 included 229 patients
(Table III) and cohort 2
included 60 patients. The expression of LKB1 was examined by IHC in
matched pairs of HCC and ANT specimens in cohort 1. The results
indicated that the expression levels of LKB1 were significantly
higher in HCC tissues (8.288±2.922) compared with those in ANT
tissues (6.716±2.293; Fig. 1A).
This was further confirmed by western blot analysis in specimens
from cohort 2: The intensity ratio (LKB1/GAPDH) in HCC tissues
(0.8236±0.7931) was significantly higher compared with that in ANT
tissues (0.4727±0.4279; Fig. 1B).
p-AMPK (Thr172) was also upregulated in samples with high
expression of LKB1 (Fig. 1B).
These results suggested that LKB1 may play a protooncogenic role in
HCC.
| Table IIICorrelation between LKB1 expression
and clinicopathological characteristics in 229 HCC patients. |
Table III
Correlation between LKB1 expression
and clinicopathological characteristics in 229 HCC patients.
| Clinicopathological
variables | Low expression | High
expression | Percentage (%) | P-value |
|---|
| Sex |
| Male | 45 | 161 | 89.96 | 0.643a |
| Female | 6 | 17 | 10.04 | |
| Age, years |
| ≤50 | 28 | 86 | 49.78 | 0.297b |
| >50 | 23 | 92 | 50.22 | |
| BMI,
kg/m2 |
| <25 | 37 | 124 | 70.31 | 0.763b |
| ≥25 | 14 | 53 | 29.26 | |
| Missing | | 1 | 0.44 | |
| Alcohol intake |
| Yes | 14 | 61 | 32.75 | 0.469 |
| No | 37 | 115 | 66.38 | |
| Missing | | 2 | 0.87 | |
| Smoking |
| Current, past | 12 | 81 | 40.61 | 0.015 |
| Never | 39 | 96 | 58.95 | |
| Missing | | 1 | 0.44 | |
| HBV |
| Negative | 3 | 16 | 8.30 | 0.466 |
| Positive | 48 | 160 | 90.83 | |
| Missing | | 2 | 0.87 | |
| Cirrhosis |
| Absent | 10 | 39 | 21.40 | 0.724 |
| Present | 41 | 139 | 78.60 | |
| Tumor number |
| Single | 44 | 137 | 79.04 | 0.150 |
| Multiple | 7 | 41 | 20.96 | |
| Tumor size, cm |
| ≤5 | 25 | 64 | 38.86 | 0.005b |
| >5 | 26 | 112 | 60.26 | |
| Missing | | 2 | 0.87 | |
| Tumor
encapsulation |
| None | 14 | 91 | 45.85 | 0.009 |
| Complete | 37 | 86 | 53.71 | |
| Missing | | 1 | 0.44 | |
| Vascular
invasion |
| Unidentified | 44 | 113 | 68.56 | 0.002 |
| Identified | 7 | 65 | 31.44 | |
| PVTT |
| Unidentified | 46 | 132 | 77.73 | 0.015 |
| Identified | 5 | 46 | 22.27 | |
| Local invasion |
| Unidentified | 49 | 166 | 93.88 | 0.738a |
| Identified | 2 | 11 | 5.68 | |
| Missing | | 1 | 0.44 | |
| Distant
metastasis |
| Absent | 51 | 168 | 95.63 | 0.214a |
| Present | 0 | 9 | 3.93 | |
| Missing | | 1 | 0.44 | |
|
Differentiation |
| Poor | 17 | 64 | 35.371 | 0.02b |
| Moderate | 23 | 86 | 47.598 | |
| High | 11 | 28 | 17.031 | |
| Edmondson-Steiner
grade |
| I-II | 22 | 63 | 37.12 | 0.313 |
| III-IV | 29 | 115 | 62.88 | |
| Child-Pugh
stage |
| A | 47 | 135 | 79.47 | 0.016a |
| B | 4 | 42 | 20.09 | |
| Missing | | 1 | 0.44 | |
| TNM stage |
| I–II | 46 | 134 | 78.60 | 0.021a |
| III–IV | 5 | 44 | 21.40 | |
| BCLC stage |
| 0-A | 37 | 80 | 51.09 | <0.001b |
| B | 4 | 17 | | |
| C | 10 | 81 | | |
| Fasting glucose
level, mM |
| ≤6.1 | 34 | 132 | 72.49 | 0.934b |
| >6.1 | 12 | 25 | 16.16 | |
| Missing | 5 | 21 | 11.35 | |
| Diabetes |
| Yes | 15 | 23 | 16.59 | 0.005 |
| No | 36 | 155 | 83.41 | |
| ALT, U/l |
| ≤40 | 31 | 110 | 61.57 | 0.845b |
| >40 | 19 | 63 | 35.81 | |
| Missing | 1 | 5 | 2.62 | |
| AST, U/l |
| ≤40 | 32 | 104 | 59.39 | 0.264b |
| >40 | 17 | 66 | 36.24 | |
| Missing | 2 | 8 | 4.37 | |
| TBIL,
μμ |
| ≤17.1 | 41 | 128 | 73.80 | 0.181b |
| >17.1 | 9 | 43 | 22.71 | |
| Missing | 1 | 7 | 3.49 | |
| γ-GGT, U/l |
| ≤50 | 19 | 55 | 32.31 | 0.007b |
| >50 | 28 | 109 | 59.83 | |
| Missing | 4 | 14 | 7.86 | |
| AFP,
μg/l |
| ≤20 | 18 | 53 | 31.0 | 0.442b |
| >20 | 29 | 117 | 63.76 | |
| Missing | 4 | 8 | 5.24 | |
| CEA (ng/ml) |
| ≤5.9 | 46 | 138 | 80.35 | 0.744b |
| >5.9 | 1 | 10 | 4.80 | |
| Missing | 4 | 30 | 14.85 | |
| CA19-9, U/ml |
| ≤40 | 41 | 138 | 78.17 | 0.501b |
| >40 | 5 | 12 | 7.42 | |
| Missing | 5 | 28 | 14.41 | |
| Adjuvant TACE |
| Yes | 4 | 12 | 6.99 | 0.760a |
| No | 47 | 166 | 93.01 | |
| Entecavir
therapy |
| Yes | 26 | 98 | 54.15 | 0.607 |
| No | 25 | 80 | 45.85 | |
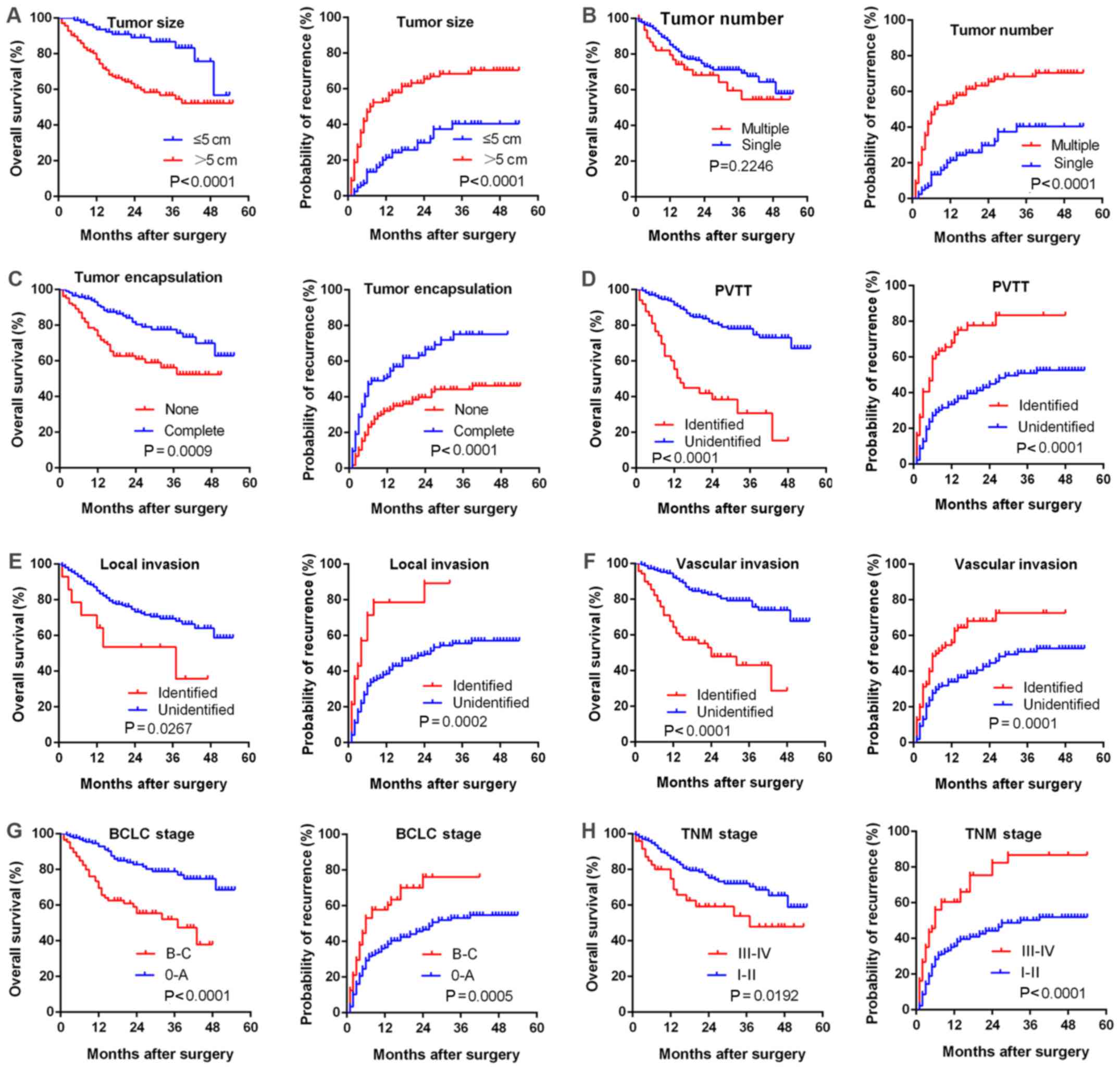
Upregulated LKB1 expression is correlated
with numerous malignant characteristics and poor prognosis
Due to the evidence supporting the possible
proto-oncogenic role of LKB1 in HCC, the present study then aimed
to further elucidate the correlation between LKB1 expression and
clinical characteristics. Upregulation of LKB1 was significantly
(P<0.001) correlated with several clinicopathological
characteristics associated with aggressive biological behavior of
cancer cells, including higher number of tumor foci, larger tumor
size, incomplete tumor encapsulation, vascular invasion, portal
vein tumor thrombus (PVTT), poor differentiation, advanced
Edmondson-Steiner grade, advanced BCLC grade and TNM stage
(Fig. 2A–H). Most importantly,
upregulated LKB1 expression was correlated with a shorter overall
survival (P=0.0158), shorter disease-free survival (P=0.0461) and
higher early recurrence (P=0.0372; Fig. 2J).
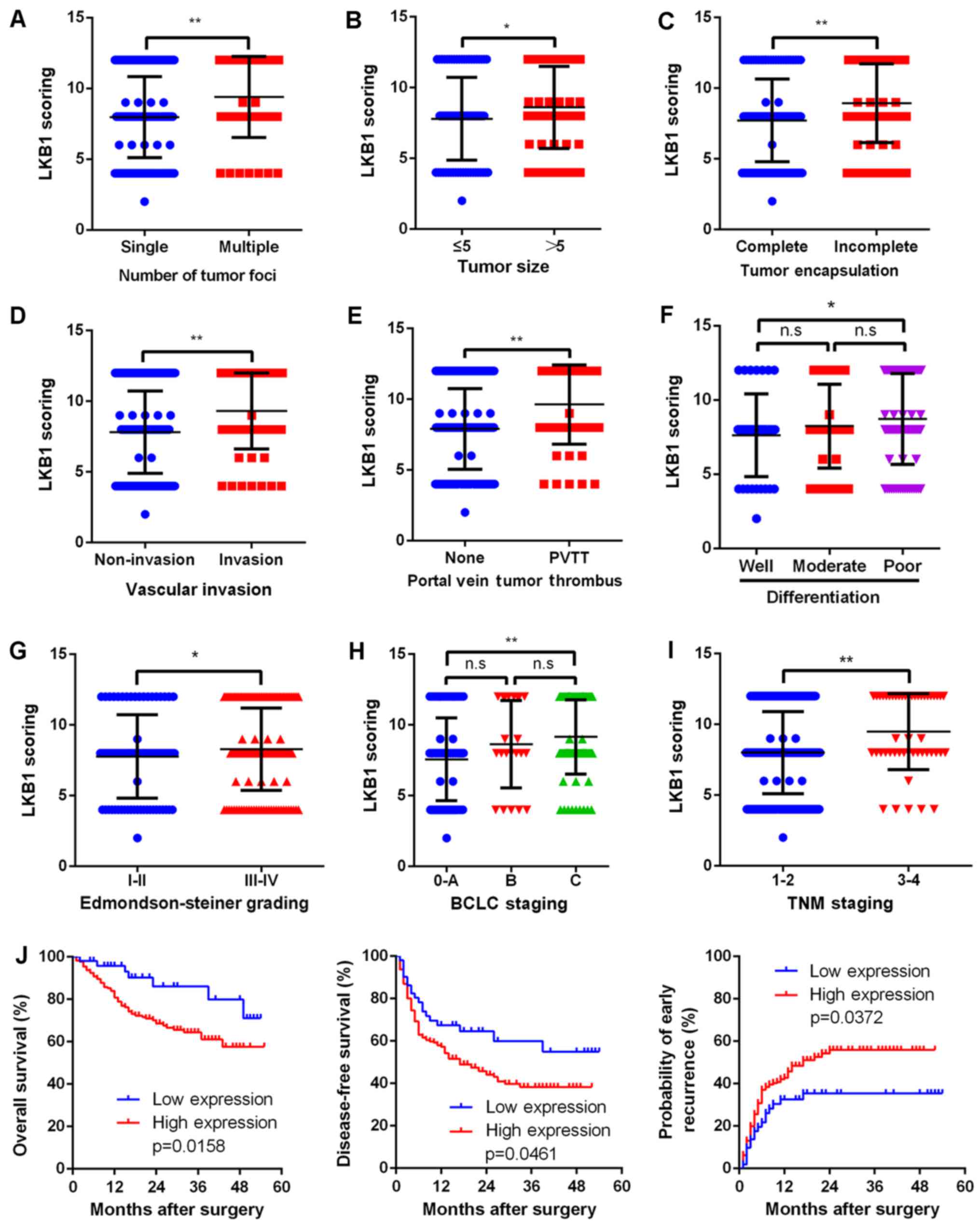 | Figure 2Upregulated LKB1 expression predicts
aggressive clinicopathological characteristics and poor prognosis
in HCC patients. Relative expression scores of LKB1 in 229 human
HCC samples divided by (A) number of tumor foci, (B) tumor size,
(C) tumor encapsulation, (D) vascular invasion, (E) PVTT, (F) tumor
differentiation, (G) Edmondson-Steiner grade, (H) BCLC stage and
(I) TNM stage. All values are presented as dot plots, with the
middle bars representing the median and vertical bars representing
the range of data. (J) Kaplan-Meier analysis of the correlation
between LKB1 expression and the OS and DFS of HCC patients as well
as early recurrence. **P<0.01, *P<0.05;
n.s, non-significant. HCC, hepatocellular carcinoma; LKB1, liver
kinase B1; PVTT, portal vein tumor thrombus; BCLC, Barcelona Clinic
Liver Cancer; TNM, tumor-node-metastasis. |
Furthermore, a large tumor size, multiple tumor
foci, incomplete tumor encapsulation, PVTT, local invasion,
vascular invasion, advanced BCLC or TNM stage were correlated with
poorer survival (Fig. 3).
Univariate and multivariate analyses revealed that high LKB1
expression in HCC patients may serve as an independent prognostic
marker for overall survival (P=0.018 and 0.046 for uni- and
multivariate analysis, respectively), whereas it had no significant
predictive value regarding recurrence (P=0.054 and 0.383,
respectively; Table IV).
| Table IVUnivariate and multivariate analyses
of prognostic factors in overall survival and recurrence. |
Table IV
Univariate and multivariate analyses
of prognostic factors in overall survival and recurrence.
A, Overall survival
|
|---|
| Factors | Univariate analysis
| Multivariate
analysis
|
|---|
| HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| Age (>50 vs. ≤50
years) | 0.990 | 0.775-1.265 | 0.938 | | | |
| Sex (male vs.
female) | 1.528 | 1.115-2.092 | 0.008 | 1.155 | 0.290-4.604 | 0.302 |
| Diabetes mellitus
(yes vs. no) | 1.57 | 0.677-3.641 | 0.293 | | | |
| Blood glucose
(>6.1 vs. ≤6.1 mM) | 0.923 | 0.453-1.882 | 0.826 | | | |
| Tumor foci
(multiple vs. single) | 0.850 | 0.641-1.128 | 0.26 | | | |
| Tumor size (>5
vs. ≤5 cm) | 3.270 | 1.779-6.011 | <0.001 | 4.161 | 1.587-10.910 | 0.004 |
| Tumor encapsulation
(none vs. complete) | 0.432 | 0.263-0.711 | 0.001 | 1.403 | 0.708-2.779 | 0.332 |
| Differentiation
(poor vs. high + moderate) | 2.224 | 1.246-3.970 | 0.007 | 0.397 | 0.110 -1.430 | 0.158 |
| Edmondson-Steiner
grade (III+IV vs. I+II) | 0.832 | 0.639-1.804 | 0.173 | | | |
| TNM stage (III+IV
vs. I+II) | 0.732 | 0.557-0.961 | 0.025 | 0.591 | 0.227-1.538 | 0.281 |
| Child-Pugh stage (B
vs. A) | 1.387 | 0.786-2.448 | 0.259 | | | |
| BCLC stage (B+C vs.
0+A) | 0.596 | 0.460-0.772 | <0.001 | 1.772 | 0.541-5.803 | 0.344 |
| PVTT (identified
vs. unidentified) | 2.314 | 1.407-3.805 | 0.001 | 2.291 | 0.778-6.750 | 0.133 |
| Vascular invasion
(identified vs. unidentified) | 0.514 | 0.401-0.685 | <0.001 | 1.155 | 0.290-4.604 | 0.838 |
| Local invasion
(identified vs. unidentified) | 2.661 | 1.211-5.847 | 0.015 | 3.126 | 0.880-11.102 | 0.078 |
| Distant metastasis
(identified vs. unidentified) | 1.446 | 0.453-4.613 | 0.533 | | | |
| HBV (positive vs.
negative) | 0.939 | 0.405-2.176 | 0.883 | | | |
| Cirrhosis (present
vs. absent) | 0.795 | 0.567-1.114 | 0.182 | | | |
| AST (>40 vs.
≤40) | 2.168 | 1.135-3.574 | 0.002 | 1.295 | 0.660 -2.540 | 0.452 |
| γ-GGT (>50 vs.
≤50) | 1.994 | 1.090-3.648 | 0.025 | 1.204 | 0.507 -2.862 | 0.674 |
| AFP (≥20 vs.
<20) | 2.196 | 1.170-4.124 | 0.014 | 1.605 | 0.781-3.297 | 0.198 |
| CA199 (>40 vs.
≤40) | 2.752 | 1.344-5.634 | 0.006 | 7.273 | 3.079-17.177 | <0.001 |
| LKB1 expression
(high vs. low) | 2.617 | 1.179-5.808 | 0.018 | 2.372 | 1.014-5.550 | 0.046 |
B, Recurrence
|
| Factors | Univariate analysis
| Multivariate
analysis
|
| HR | 95% CI | P-value | HR | 95% CI | P-value |
|
| Age (>50 vs ≤50
years) | 0.855 | 0.595-1.23 | 0.399 | | | |
| Sex (male vs.
female) | 0.912 | 0.502-1.657 | 0.762 | | | |
| Diabetes mellitus
(yes vs. no) | 0.569 | 0.313-1.035 | 0.065 | | | |
| Blood glucose
(>6.1 vs. ≤6.1 mM) | 0.828 | 0.492-1.392 | 0.476 | | | |
| Tumor foci
(multiple vs. single) | 1.988 | 1.337-2.956 | 0.001 | 1.295 | 0.610-2.749 | 0.500 |
| Tumor size (>5
vs. ≤5 cm) | 2.983 | 1.937-4.595 | <0.001 | 2.599 | 1.539-4.391 | <0.001 |
| Tumor encapsulation
(none vs. complete) | 2.220 | 1.533-3.215 | <0.001 | 1.520 | 0.969-2.383 | 0.068 |
| Differentiation
(poor vs. high + moderate) | 0.046 | 0.218-0.755 | 0.004 | 1.212 | 0.867-1.692 | 0.260 |
| Edmondson-Steiner
grade (III+IV vs. I+II) | 1.325 | 0.902-1.948 | 0.152 | | | |
| TNM stage (III+IV
vs. I+II) | 2.510 | 1.694-3.719 | <0.001 | 0.994 | 0.751-1.315 | 0.964 |
| Child-Pugh stage (B
vs. A) | 1.146 | 0.736-1.785 | 0.546 | | | |
| BCLC stage (B+C vs.
0+A) | 2.385 | 1.637-3.475 | <0.001 | 0.825 | 0.587-1.160 | 0.269 |
| PVTT (identified
vs. unidentified) | 0.369 | 0.249-0.547 | <0.001 | 0.673 | 0.456-0.994 | 0.047 |
| Vascular invasion
(identified vs. unidentified) | 2.045 | 1.407-2.971 | <0.001 | 1.256 | 0.827-1.909 | 0.285 |
| Local invasion
(identified vs. unidentified) | 3.619 | 1.975-6.632 | <0.001 | 2.367 | 1.072-5.228 | 0.033 |
| Distant metastasis
(identified vs. unidentified) | 1.593 | 0.699-3.632 | 0.268 | | | |
| HBV (positive vs.
negative) | 0.848 | 0.466-1.540 | 0.588 | | | |
| Cirrhosis
(identified vs. unidentified) | 1.234 | 0.776-1.963 | 0.374 | | | |
| AST (>40 vs.
≤40) | 1.593 | 1.097-2.313 | 0.014 | 1.363 | 0.862-2.155 | 0.185 |
| γ-GGT (>50 vs.
≤50) | 1.861 | 1.203-2.800 | 0.005 | 1.706 | 0.946-3.076 | 0.076 |
| AFP (≥20 vs.
<20) | 1.352 | 0.897-2.038 | 0.150 | | | |
| CA199 (>40 vs.
≤40) | 1.644 | 0.877-3.082 | 0.121 | | | |
| LKB1 expression
(high vs. low) | 1.622 | 0.992-2.652 | 0.054 | 1.263 | 0.747-2.133 | 0.383 |
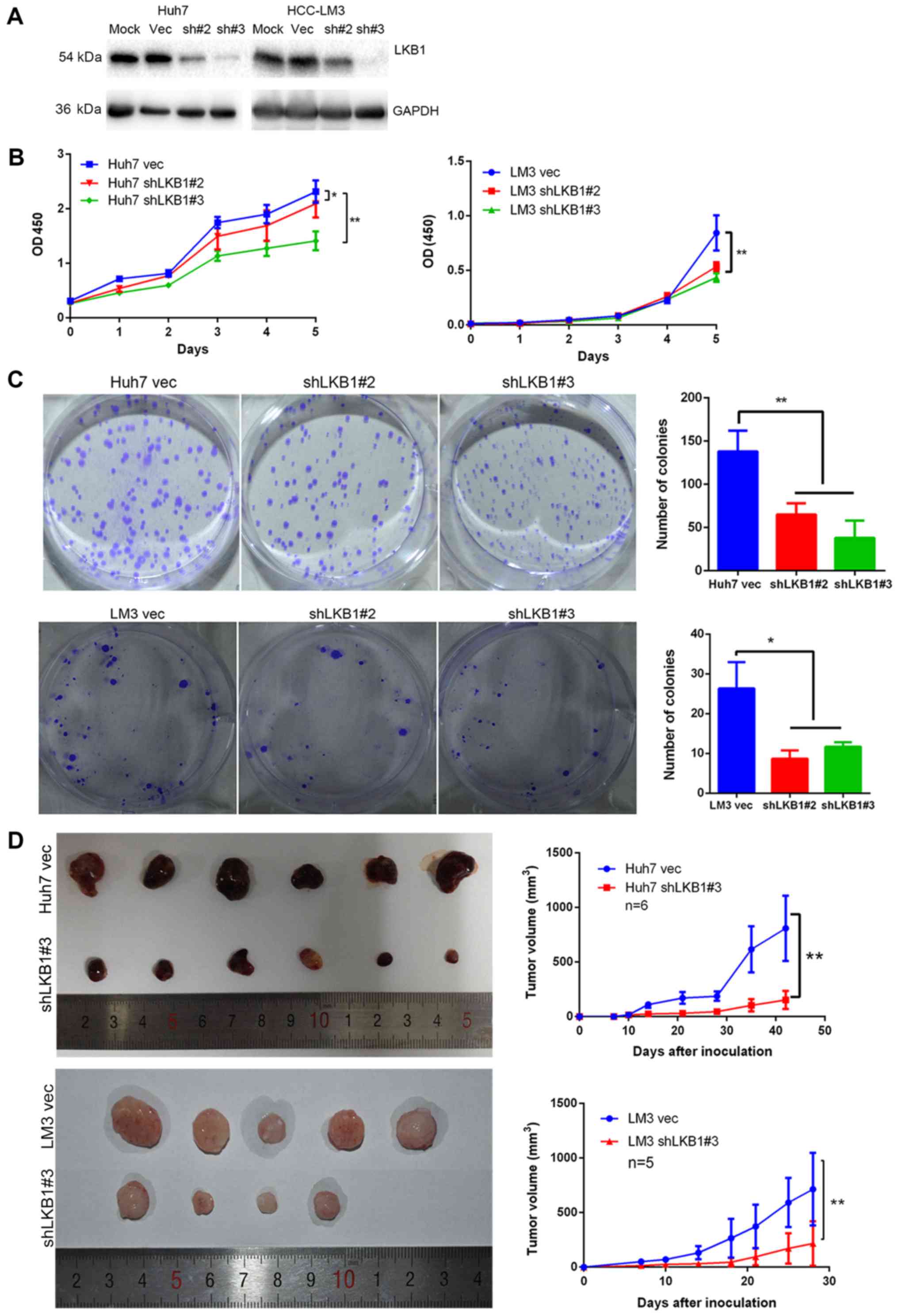
Knockdown of LKB1 expression inhibits HCC
cell proliferation
In order to examine the role of LKB1 in HCC cell
lines, LKB1 expression was knocked down in Huh7 and HCC-LM3 cells
(Fig. 4A), which exhibit high and
moderate endogenous LKB1 expression, respectively (data not shown).
The CCK-8 and colony formation assays indicated that knockdown of
LKB1 significantly inhibited cell proliferation (Fig. 4B and C). Furthermore, LKB1 was
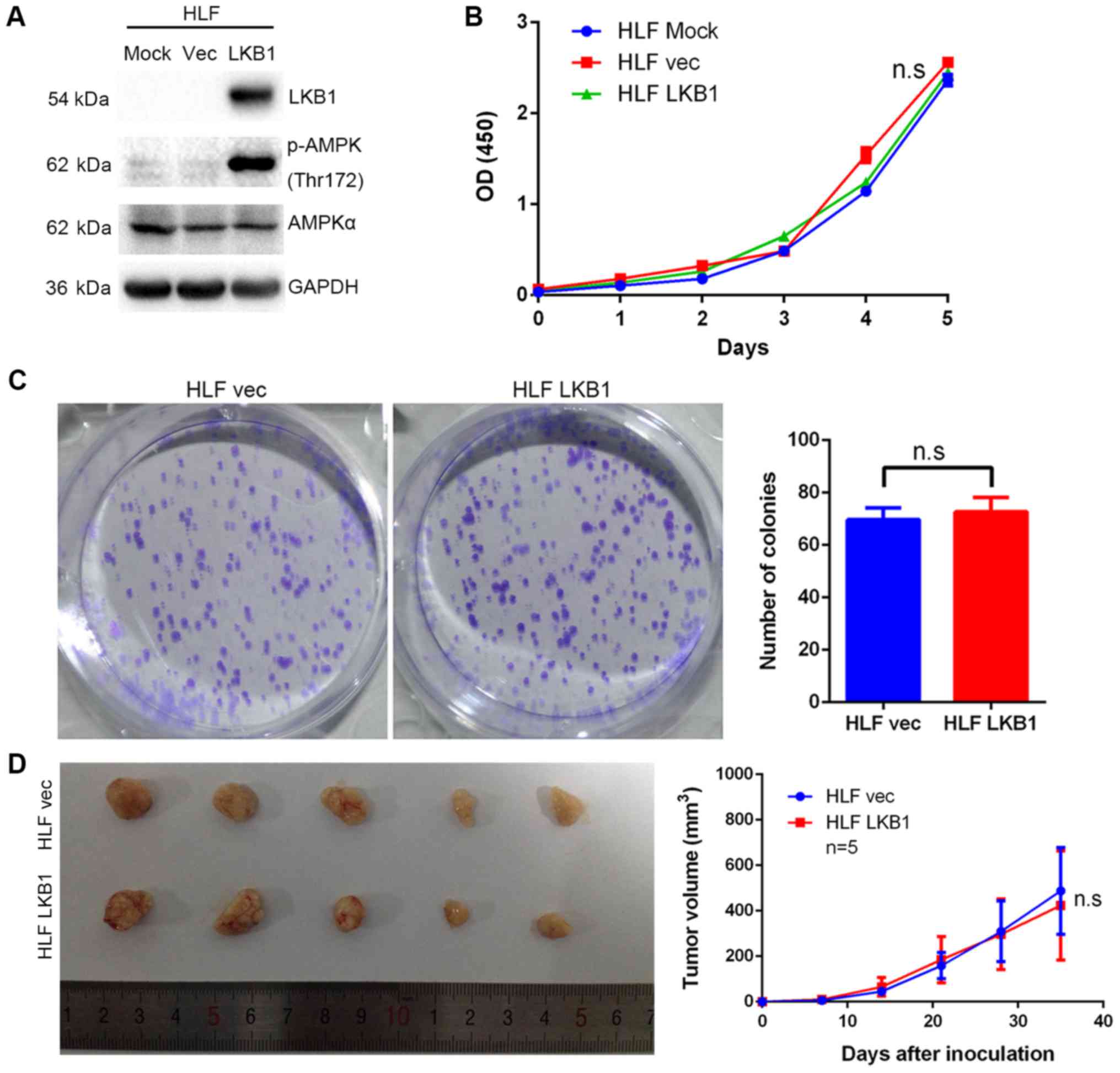
found to be ectopically overexpressed in HLF cells (Fig. 5A), which have no detectable LKB1
expression (data not shown). However, overexpression of LKB1
exerted no effect on the growth of HLF cells (Fig. 5B and C). The in vivo
tumorigenicity assay indicated that the volume of tumors grown from
subcutaneously injected cells was smaller in the LKB1 knockdown
groups compared with that in the control groups (Fig. 4D). However, no significant
difference in volume was observed between the tumors derived from
LKB1-overexpressing HLF and those from control cells (Fig. 5D).
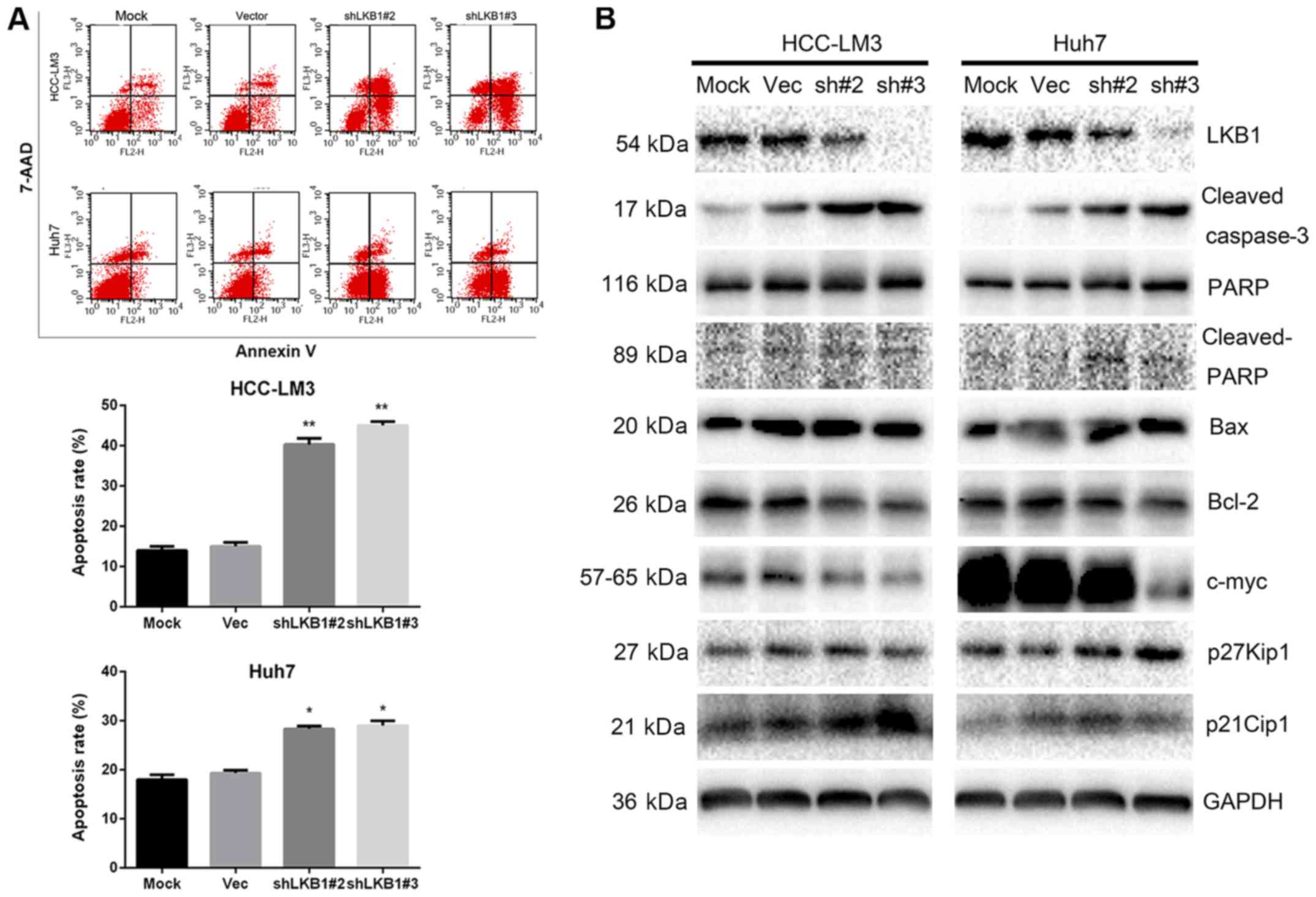
LKB1 knockdown inhibits tumor cell
proliferation by promoting cell apoptosis
Since knockdown of LKB1 inhibited Huh7 and HCC-LM3
cell proliferation, flow cytometric analysis was performed to
determine whether this antiproliferative effect was due to cell
cycle arrest. No significant differences in the distribution of
cells in each phase of the cell cycle were observed (data not
shown). However, the cell apoptosis assay indicated that, in
LKB1-knockdown cells, the apoptotic rate was higher compared with
that in the control cells (Fig.
6A). Western blot analysis further confirmed an increased
amount of cleaved caspase-3 and PARP in the knockdown group
(Fig. 6B). In addition, reduced
c-Myc expression and elevated expression of p21Cip1 and
p27Kip1 were observed in LKB1-knockdown cells,
suggesting that p21Cip1 and p27Kip1 affect
cell proliferation via other mechanisms (Fig. 6B).
Discussion
LKB1 has been reported to act as a tumor suppressor
in the majority of published studies. LKB1 suppresses cell growth
and viability through the LKB1/AMPK/mammalian target of rapamycin
signaling pathway (17). However,
certain studies suggested that LKB1 exerts a proto-oncogenic effect
through modulating cellular metabolism and resistance to oncogenic
transformation (8,9). Therefore, it is of paramount
importance to elucidate the function of LKB1 in different types of
cancer. In the present study, the expression pattern of LKB1 was
detected in two cohorts of HCC and paired ANT specimens. The
results demonstrated that LKB1 was frequently upregulated in HCC
tissues, and the high expression of LKB1 was correlated with
numerous malignant characteristics, shorter overall survival and
earlier recurrence. It was also revealed that a large tumor size,
multiple tumor foci, incomplete tumor encapsulation, PVTT, local
invasion, vascular invasion, and advanced BCLC or TNM stage were
associated with a worse prognosis. Knockdown of LKB1 inhibited cell
proliferation by promoting apoptosis and regulating
proliferation-associated genes, but overexpression of LKB1 exerted
no effect on the proliferation of HCC cells. It is well-known that,
under quiescent conditions, LKB1 is localized to the nucleus and
activation of LKB1 requires translocation from the nucleus to the
cytoplasm by forming a heterotrimer with the proteins STRADA and
MO25 (18,19). It may be hypothesized that enhanced
LKB1 expression in HCC cells does not affect STRADA and MO25 and,
accordingly, LKB1 translocation to the cytoplasm remains
unchanged.
HCC is one of the leading causes of
cancer-associated mortality worldwide, and its incidence is
increasing (20). The Asia-Pacific
area is the region with the highest prevalence of HCC (21,22),
and a large number of patients are first diagnosed with HCC at an
advanced stage. Therefore, the therapeutic efficacy is not optimal,
and mortality due to cancer recurrence or metastasis is common. In
the present study, the protooncogenic role of LKB1 in HCC was
demonstrated. Whether and how LKB1 affects HCC metastasis, and the
possible therapeutic approaches based on LKB1, remain to be further
investigated in future studies.
Germline mutation of LKB1 is responsible for a
precancerous condition referred to as Peutz-Jeghers syndrome, which
is characterized by the development of benign hamartomatous polyps
in the gastrointestinal tract and hyperpigmented macules on the
lips and oral mucosa. Patients with Peutz-Jeghers syndrome develop
gastrointestinal hamartomas and have a markedly increased risk for
developing gastrointestinal, breast and gynecological cancers
(23). Dahmani et al
(24) reported a novel LKB1
isoform, which lacks the N-terminal region and a portion of the
kinase domain, named ΔN-LKB1. This enhances the metabolic activity
of AMPK in HeLa cells and NCI-H460 lung cancer cells and has
intrinsic oncogenic properties. In order to explore the possibility
of mutated LKB1 in HCC tissues and cell lines used in the present
study, the literature on LKB1 mutation in HCC was reviewed. Kim
et al (25) collected 80
HCC samples and 7 dysplastic nodules to investigate potential
mutations in all 9 exons of LKB1. The results revealed the presence
of only one missense mutation of CCG→CTG (Pro→Leu) among the 80 HCC
cases, whereas no mutation was identified among the 7 dysplastic
nodules. Pineau et al (26)
collected 57 hepatobiliary cancer cell lines for detection of
homozygeous deletions, and no homozygous deletion of LKB1 was
detected in the HCC cell lines used in their study. Therefore, the
effect of LKB1 observed in the present study was likely exerted by
a non-mutated protein.
Activation of LKB1 by phosphorylation at the Ser428,
Ser307 and Ser399 sites is required for translocation from the
nucleus to the cytoplasm (27-29).
It has been reported that LKB1 regulates glucose metabolism and
suppresses gluconeogenesis in the normal liver (30), and knockout of LKB1 in mouse livers
leads to the inability to use glucose, resulting in severe
hyperglycemia (31). Apoptosis is
a type of programmed cell death under various types of stress
(32,33). It is reasonable to hypothesize that
LKB1-knockdown cells underwent apoptosis due to inability to use
glucose. LKB1 may be used as a potential therapeutic target in HCC
treatment by agents suppressing phosphorylation at Ser428, Ser307
and Ser399, thereby inhibiting nuclear export of LKB1.
The in vivo tumor inhibitory effect of LKB1
was previously investigated by knockout of LKB1 in mice (34,35),
and the most recent study indicated that LKB1 acts as a master
gatekeeper of liver regeneration (36). Another previous study indicated
that LKB1 was downregulated in HCC and that low expression is
correlated with poor prognosis (10). This conclusion was made based on
IHC staining analysis of 70 cases. In the present study, in which
the scale of samples included was enlarged, different conclusions
were reached. Along with the results of previous studies (8,9,37),
the present study suggests that LKB1 plays a proto-oncogenic role
in HCC. It is suggested that the function of LKB1 varies between
different cancer types and pathological conditions. Therefore, the
heterogeneity of cancers should be taken into consideration in
cancer therapy.
Funding
The present study was supported by the National
Natural Science Fund (grant nos. 31671348, 81572427 and
81572855).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article. The authors declare
that materials described in the manuscript, including all relevant
raw data, will be freely available to any scientist wishing to use
them for non-commercial purposes, without breaching participant
confidentiality.
Authors’ contributions
XT and LC designed the experiments, XT and ZL
performed the experiments. ZL collected clinicopathological data.
XT, BZ, XC and LC analyzed the results. XT and ZL generated the
data, prepared the panels and assembled the figures and tables. XT
and LC wrote the manuscript. All authors have reviewed and approved
the final version of the manuscript.
Ethics approval and consent to
participate
The protocol of the present study, involving both
human clinical samples and animal experimentation, was approved by
the Ethics Committee of Tongji Hospital, Huazhong University of
Science and Technology. Written informed consent was obtained from
all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Changshu Ke and Jing
Xiong (Department of Pathology, Tongji Hospital) for their
assistance with the IHC scoring, Zhanguo Zhang (Hepatic Surgery
Center, Tongji Hospital) for their assistance with the shRNA
lentivirus production, Xiaolan Li (Public Experimental Platform,
Tongji Hospital) for conducting the flow cytometry, Lanping Ding
(Institute of Organ Transplantation, Tongji Hospital) and Shunchang
Zhou (Department of Experimental Zoology, Tongji Medical College)
for animal care, and Wei Dong (Hepatic Surgery Center, Tongji
Hospital) for insightful discussion.
References
|
1
|
Alessi DR, Sakamoto K and Bayascas JR:
LKB1-dependent signaling pathways. Annu Rev Biochem. 75:137–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hardie DG: AMP-activated/SNF1 protein
kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu W, Monahan KB, Pfefferle AD, Shimamura
T, Sorrentino J, Chan KT, Roadcap DW, Ollila DW, Thomas NE,
Castrillon DH, et al: LKB1/STK11 inactivation leads to expansion of
a prometastatic tumor subpopulation in melanoma. Cancer Cell.
21:751–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han Li F, Li X, Wang F, Wang R, Gao H,
Wang Y, Fang X, Zhang Z, Yao WS, et al: LKB1 Inactivation elicits a
redox imbalance to modulate non-small cell lung cancer plasticity
and therapeutic response. Cancer Cell. 27:698–711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Herrmann JL, Byekova Y, Elmets CA and
Athar M: Liver kinase B1 (LKB1) in the pathogenesis of epithelial
cancers. Cancer Lett. 306:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bardeesy N, Sinha M, Hezel AF, Signoretti
S, Hathaway NA, Sharpless NE, Loda M, Carrasco DR and DePinho RA:
Loss of the Lkb1 tumour suppressor provokes intestinal polyposis
but resistance to transformation. Nature. 419:162–167. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeon SM, Chandel NS and Hay N: AMPK
regulates NADPH homeostasis to promote tumour cell survival during
energy stress. Nature. 485:661–665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martinez-Lopez N, Garcia-Rodriguez JL,
Varela-Rey M, Gutiérrez V, Fernández-Ramos D, Beraza N, Aransay AM,
Schlangen K, Lozano JJ, Aspichueta P, et al: Hepatoma cells from
mice deficient in glycine N-methyltransferase have increased RAS
signaling and activation of liver kinase B1. Gastroenterology.
143:787–798.e13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SW, Li CF, Jin G, Cai Z, Han F, Chan
CH, Yang WL, Li BK, Rezaeian AH, Li HY, et al: Skp2-dependent
ubiquitination and activation of LKB1 is essential for cancer cell
survival under energy stress. Mol Cell. 57:1022–1033. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang Y-H, Chen Z-K, Huang K-T, Li P, He
B, Guo X, Zhong JQ, Zhang QY, Shi HQ, Song QT, et al: Decreased
expression of LKB1 correlates with poor prognosis in hepatocellular
carcinoma patients undergoing hepatectomy. Asian Pac J Cancer Prev.
14:1985–1988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases: Management of
hepatocellular carcinoma: An update. Hepatology. 53:pp. 1020–1022.
2011, View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Li JC, Sun XR, Xu HX, Zhou Y, Qiu J,
Ke SJ, Cui AW, Wang YH, Wang ZJWM, et al: Up-regulation of
Kruppel-like factor 8 promotes tumor invasion and indicates poor
prognosis for hepatocellular carcinoma. Gastroenterology.
139:2146–2157.e12. 2010. View Article : Google Scholar
|
|
13
|
Edmondson HA and Steiner PE: Primary
carcinoma of the liver: A study of 100 cases among 48,900
necropsies. Cancer. 7:462–503. 1954. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou L, Rui J-A, Ye D-X, Wang S-B, Chen
S-G and Qu Q: Edmondson-Steiner grading increases the predictive
efficiency of TNM staging for long-term survival of patients with
hepatocellular carcinoma after curative resection. World J Surg.
32:1748–1756. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moffat J, Grueneberg DA, Yang X, Kim SY,
Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK,
et al: A lentiviral RNAi library for human and mouse genes applied
to an arrayed viral high-content screen. Cell. 124:1283–1298. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang B, Halder SK, Kashikar ND, Cho YJ,
Datta A, Gorden DL and Datta PK: Antimetastatic role of Smad4
signaling in colorectal cancer. Gastroenterology. 138:969–980.e1–3.
2010. View Article : Google Scholar
|
|
17
|
Han D, Li SJ, Zhu YT, Liu L and Li MX:
LKB1/AMPK/mTOR signaling pathway in non-small-cell lung cancer.
Asian Pac J Cancer Prev. 14:4033–4039. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baas AF, Boudeau J, Sapkota GP, Smit L,
Medema R, Morrice NA, Alessi DR and Clevers HC: Activation of the
tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD.
EMBO J. 22:3062–3072. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boudeau J, Baas AF, Deak M, Morrice NA,
Kieloch A, Schutkowski M, Prescott AR, Clevers HC and Alessi DR:
MO25alpha/beta interact with STRADalpha/beta enhancing their
ability to bind, activate and localize LKB1 in the cytoplasm. EMBO
J. 22:5102–5114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
22
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Korsse SE, Peppelenbosch MP and van Veelen
W: Targeting LKB1 signaling in cancer. Biochim Biophys Acta.
1835.194–210. 2013.
|
|
24
|
Dahmani R, Just PA, Delay A, Canal F,
Finzi L, Prip-Buus C, Lambert M, Sujobert P, Buchet-Poyau K, Miller
E, et al: A novel LKB1 isoform enhances AMPK metabolic activity and
displays oncogenic properties. Oncogene. 34:2337–2346. 2015.
View Article : Google Scholar
|
|
25
|
Kim CJ, Cho YG, Park JY, Kim TY, Lee JH,
Kim HS, Lee JW, Song yH, Nam SW, Lee sH, et al: Genetic analysis of
the LKB1/STK11 gene in hepatocellular carcinomas. Eur J Cancer.
40:136–141. 2004. View Article : Google Scholar
|
|
26
|
Pineau P, Marchio A, Nagamori S, Seki S,
Tiollais P and Dejean A: Homozygous deletion scanning in
hepatobiliary tumor cell lines reveals alternative pathways for
liver carcinogenesis. Hepatology. 37:852–861. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie Z, Dong Y, Scholz R, Neumann D and Zou
MH: Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta
is required for metformin-enhanced activation of the AMP-activated
protein kinase in endothelial cells. Circulation. 117:952–962.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie Z, Dong Y, Zhang J, Scholz R, Neumann
D and Zou MH: Identification of the serine 307 of LKB1 as a novel
phosphorylation site essential for its nucleocytoplasmic transport
and endothelial cell angiogenesis. Mol Cell Biol. 29:3582–3596.
2009. View Article : Google Scholar :
|
|
29
|
Zhu H, Moriasi CM, Zhang M, Zhao Y and Zou
MH: Phosphorylation of serine 399 in LKB1 protein short form by
protein kinase Cζ is required for its nucleocytoplasmic transport
and consequent AMP-activated protein kinase (AMPK) activation. J
Biol Chem. 288:16495–16505. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Patel K, Foretz M, Marion A, Campbell DG,
Gourlay R, Boudaba N, Tournier E, Titchenell P, Peggie M, Deak M,
et al: The LKB1-salt-inducible kinase pathway functions as a key
gluconeogenic suppressor in the liver. Nat Commun. 5:45352014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shaw RJ, Lamia KA, Vasquez D, Koo SH,
Bardeesy N, Depinho RA, Montminy M and Cantley LC: The kinase LKB1
mediates glucose homeostasis in liver and therapeutic effects of
metformin. Science. 310:1642–1646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Evan G and Littlewood T: A matter of life
and cell death. Science. 281:1317–1322. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lowe SW, Cepero E and Evan G: Intrinsic
tumour suppression. Nature. 432:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyoshi H, Deguchi A, Nakau M, Kojima Y,
Mori A, Oshima M, Aoki M and Taketo MM: Hepatocellular carcinoma
development induced by conditional beta-catenin activation in
Lkb1+/− mice. Cancer Sci. 100:2046–2053. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nakau M, Miyoshi H, Seldin MF, Imamura M,
Oshima M and Taketo MM: Hepatocellular carcinoma caused by loss of
heterozygosity in Lkb1 gene knockout mice. Cancer Res.
62:4549–4553. 2002.PubMed/NCBI
|
|
36
|
Maillet V, Boussetta N, Leclerc J, Fauveau
V, Foretz M, Viollet B, Couty JP, Celton-Morizur S, Perret C and
Desdouets C: LKB1 as a gatekeeper of hepatocyte proliferation and
genomic integrity during liver regeneration. Cell Reports.
22:1994–2005. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martínez-López N, Varela-Rey M,
Fernández-Ramos D, Woodhoo A, Vázquez-Chantada M, Embade N,
Espinosa-Hevia L, Bustamante FJ, Parada LA, Rodriguez MS, et al:
Activation of LKB1-Akt pathway independent of phosphoinositide
3-kinase plays a critical role in the proliferation of
hepatocellular carcinoma from nonalcoholic steatohepatitis.
Hepatology. 52:1621–1631. 2010. View Article : Google Scholar : PubMed/NCBI
|