Introduction
Lung cancer is one of the leading causes of
mortality worldwide. Even though numerous anticancer drugs have
been developed, it remains a challenge to effectively treat lung
cancer without increasing chemoresistance. Non-small cell lung
cancer (NSCLC) is a type of lung cancer that is further classified
into three histopathological subtypes: Adenocarcinoma, squamous
cell carcinoma and large-cell carcinoma (LCC) (1). NSCLC accounts for 80% of lung cancer
cases, and of these, ~10% are LCC (2). LCC occurs in the lungs and is
characterized by poor differentiation, rapid growth and early
metastasis (3,4). These cancers also have an aggressive
pattern and are associated with a poorer prognosis. The 5-year
survival rate for patients with stage III/IV LCC is <10%
(5). Although LCC of the lung is a
relatively uncommon type of lung cancer, a better understanding of
the molecular and cellular biology of LCC, the mechanisms
underlying LCC drug resistance and the possible molecular pathway
associated with autophagy-mediated drug resistance may identify
more effective NSCLC therapeutic strategies and targets. Therefore,
the present study used two LCC cell lines, H1299 and H460.
Camptothecin (CPT), which is a natural compound
originally derived from the Asian tree Camptotheca
acuminata, was synthesized by Wall and Wani in 1966 (6). CPT is able to form a stable tertiary
structure with DNA and topoisomerase I, thus resulting in formation
of the topoisomerase I-CPT complex, which inhibits topoisomerase I
and results in DNA damage and cell death (7). Recent studies have revealed that CPT
and its derivatives exert broad antitumor activity against various
tumors cells in vitro and in vivo. CPT and its
derivatives have been reported to exhibit anticancer activities
against various types of cancer cells, including lung cancer
(8), gastric cancer (9), esophageal cancer (10), colorectal cancer (11) and breast cancer (12). Oral topotecan has been used as a
second-line medication for the treatment of metastatic ovarian
cancer (13) and small-cell lung
cancer (14). Furthermore,
irinotecan-based chemotherapy improves overall response rate,
overall survival and progression-free survival, and has been
recommended as a first-line treatment in Asian patients with stage
IIIB/IV NSCLC (15).
Although the clinical use of CPT derivatives has
exhibited efficacy in the treatment of the aforementioned types of
cancer, de novo and developed clinical resistance to these drugs is
common. The mechanism underlying the resistance of cancer cells to
CPT-based anticancer drugs remains to be fully elucidated, since
the resistance and selectivity towards cancer cells is
multifactorial. Sugimoto et al revealed that quantitative
reduction of topoisomerase I content contributes to the most
frequently occurring events in the development of resistance to CPT
in various tumor cell lines (16).
In addition, a previous study indicated that the increased
expression of ATP-dependent drug transporters, such as ATP-binding
cassette subfamily C member 4 and ATP binding cassette subfamily G
member 2 (ABCG2), is sufficient to confer resistance of lung cancer
cells to the CPT-based anticancer drugs irinotecan and topotecan
(17). Furthermore, it has been
reported that breast cancer induces resistance to topotecan and
irinotecan via regulation of the cell cycle and DNA repair activity
(18). Although numerous novel
therapies have been developed, the prognosis of patients has not
significantly improved, and chemoresistance is one of the main
reasons for the low survival of patients with lung cancer. CPT
derivatives are also affected by chemoresistance during the
treatment of lung cancer. For example, an increase in ABCG2
expression is often correlated with irinotecan and topotecan
resistance, and may result in clinical failure in patients with
advanced NSCLC (19). Therefore,
the present study applied CPT as a model to determine the
mechanisms underlying chemoresistance in NSCLC cells.
Autophagy is a cellular degradation response to
various types of stress, including starvation, hypoxia, reactive
oxygen species (ROS) and DNA damage (20,21).
Membrane receptors receive signals, which are communicated to the
cell interior, thus resulting in activation of autophagy, which
degrades dysfunctional proteins and organelles, in order to yield
more energy for adaptation to adverse environments and avoid cell
apoptosis (22). Therefore, cell
fate depends on the association between apoptosis and autophagy.
According to previous studies, when cells are treated with
chemotherapy, autophagy serves a major role in chemoresistance
(23). For example, celecoxib is
able to suppress autophagic flux by preventing lysosome function,
and strengthens the cytotoxicity of imatinib in imatinib-resistant
myeloid leukemia cells (24). In
addition, human epidermal growth factor receptor 2-overexpressing
breast cancer cells exposed to trastuzumab exhibit increased
autophagy, and protect breast cancer cells from the inhibitory
effects of trastuzumab. Conversely, the blockade of autophagosome
formation/function significantly enhances the growth inhibitory
activity of trastuzumab in trastuzumabrefractory breast cancer
cells (25). These findings
suggest that inhibiting autophagy may be a novel target for
increasing drug effects.
The present study aimed to examine the effects of
CPT on cell viability, migration, apoptosis and autophagy in the
H1299 NSCLC cell line. The results demonstrated that CPT exerted
limited cytotoxic and anti-metastatic effects on H1299 cells. In
addition, apoptosis and DNA damage were not increased following CPT
dose accumulation. However, CPT induced the increased formation of
autophagosomes in the H1299 NSCLC cell line in a dose-dependent
manner. Furthermore, the present study revealed that the autophagy
inhibitor, 3-methyladenine (3-MA), was able to suppress CPT-induced
autophagy. The results demonstrated that 3-MA enhanced the
cytotoxicity of CPT in CPT-resistant H1299 cells. Accordingly, 3-MA
may serve as a novel agent to enhance the antitumor activity of
conventional therapeutic agents in CPT-resistant H1299 cells.
Materials and methods
Cell culture
Human NSCLC cell lines H1299 and H460 were
generously provided by Dr K. Fang (National Taiwan Normal
University, Taipei, Taiwan). H1299 and H460 cells were grown in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100
µg/ml streptomycin and 100 U/ml penicillin (Gibco; Thermo
Fisher Scientific, Inc.), at 37°C in a humidified incubator
containing 5% CO2. Cell morphology was observed under an
inverted light microscope.
Source of CPT and 3-MA, and half maximal
inhibitory concentration (IC50) values of CPT
CPT and 3-MA were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). CPT was dissolved in dimethyl
sulfoxide (DMSO) to form a stock concentration of 5 mM, and 3-MA
was freshly dissolved in ddH2O at 20 mM. Cells were
treated with 0.5, 1, 2 and 5 µM CPT, and co-treated with
0.1, 0.5, 1 and 5 mM 3-MA for 24 h at at 37°C in a humidified
incubator containing 5% CO2. To determine the
IC50 values of CPT, 3×103 H1299 or H460
cells/well in 96-well plates were used. After 48 h of plating,
cells were treated with various doses of CPT (0.01, 0.1, 0.5, 1,
10, 50, 100 or 200 µM) for 24 h. The cells were incubated
for 4 h with DMEM containing 0.5% FBS and 0.5 mg/ml MTT at 37°C in
an atmosphere containing 5% CO2 to determine the number
of surviving cells. After removing the medium containing MTT and
replacing with 100 µl DMSO, absorbance was recorded at 570
nm using a Multiskan Ascent microplate reader (Thermo Fisher
Scientific, Inc.). The IC50 values of each agent were
determined using Ascent Software, version 2.6 (Thermo Fisher
Scientific, Inc.).
Cell viability assay
A cell viability assay was performed as described
previously with slight modifications (26). Briefly, 5×104 H1299 and
H460 cells/well were seeded in 12-well plates, and were treated
with DMSO as a vehicle or with various concentrations of CPT for 24
h, respectively. Subsequently, the cells were suspended in 0.2%
trypan blue and counted using a Countess automated cell counter
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer’s protocol.
Cell migration assays
The migration of H1299 and H460 cells was determined
using wound-healing assays as previously described (27). Briefly, for wound-healing assays,
3×105 H1299 and H460 cells/well were seeded in 12-well
plates, and were grown overnight until they reached ~95%
confluence, after which wound gaps were generated using a sterile
pipette tip. Cellular debris was removed with PBS and cells were
incubated in medium containing 0.5, 1, 2, and 5 µM CPT. The
wound-healing ability of the cells was documented after 16 h using
the Nikon Eclipse TE2000U microscope (Nikon Corporation, Melville,
NY, USA). The migration distance was assessed using TScratch
software, version 1.0 (MathWorks Inc., Natick, MA, USA). The
migration rate was calculated according to the relative cell
migration area for each treatment.
Annexin V staining
H1299 and H460 cells (1×105 per well in a
6-well plate) were collected following incubation with CPT and/or
3-MA for 24 h. The cells were washed with PBS and were then
resuspended in Annexin V binding buffer [140 mM NaCl, 10 mM
HEPES-NaOH (pH 7.4) and 2.5 mM CaCl2]. Following 300 × g
centrifugation for 5 min, the cells were incubated with Annexin V
binding buffer containing 1.25 ml fluorescein isothiocyanate
(FITC)-conjugated Annexin V and PI (BD Pharmingen; BD Biosciences,
San Jose, CA, USA) at room temperature for 15 min in the dark. Data
acquisition and analysis were performed using the BD Accuri™ C6
flow cytometer with BD Accuri™ C6 software version 1.0.264.21 (both
from BD Biosciences).
Western blot analysis
A total of 1×106 cells per 10 cm dish
were treated with the indicated concentrations of CPT and/or 3-MA
for 24 h. Cells were lysed with radioimmunoprecipitation assay
lysis buffer (cat. no. 20-188; EMD Millipore, Billerica, MA, USA)
containing 0.05 M Tris-HCl (pH 7.4), 0.15 M NaCl, 0.25% deoxycholic
acid, 1% NP-40, 1 mM EDTA and a protease inhibitor mixture
(Sigma-Aldrich; Merck KGaA). After one freeze-thaw cycle, cell
lysates were centrifuged at 14,000 × g for 30 min at 4°C. Proteins
were collected and concentration was determined using the Pierce™
Bicinchoninic Acid Protein Assay kit (Thermo Fisher Scientific,
Inc.) before boiling with the electrophoresis sample buffer for 5
min. The 25 µg sample lysate was mixed with the reducing
sample buffer and resolved by 10% SDS-PAGE. Proteins were then
blotted on polyvinylidene fluoride membranes (Pall Corporation,
Port Washington, NY, USA) and blocked for 1 h in blocking solution
(5% non-fat dry milk, 1% Tween-20 in PBS) at room temperature.
Membranes were washed with Tris-buffered saline with 0.1% Tween-20
(TBS-T). Proteins were determined using primary antibodies against
human caspase-3 (1:1,000, cat. no. AS-55041; Anaspec Inc., Fremont,
CA, USA), human Bax (1:1,000, cat. no. ab32503; Abcam, Cambridge,
MA, USA), human PARP (1:1,000, cat. no. 9541; Cell Signaling
Technology, Inc., Danvers, MA, USA), human caspase-9 (1:1,000, cat.
no. 9501; Cell Signaling Technology, Inc.), phosphorylated-H2A
histone family member X (Ser139) (γH2AX; 1:500, cat. no.
sc-101696; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), human
autophagy-related 3 (Atg3; 1:1,000, cat. no. AP1807c; Abgent, Inc.,
San Diego, CA, USA), human microtubule-associated proteins 1A/1B
light chain 3B (LC3B; 1:1,000, cat. no. 2775; Cell Signaling
Technology, Inc.), huaman sequestome-1 (SQSTM)/P62 (1:1,000, cat.
no. GTX100685; GeneTex, Inc., Irvine, CA, USA), human
lysosomal-associated membrane protein 2 (LAMP2; 1:1,000, cat. no.
AM1851b; Abgent, Inc.), human mTOR (1:1,000, cat. no. 1612-S;
Epitomics; Abcam), human β-actin (1:8,000, cat. no. 612656; BD
Biosciences) and human GAPDH (1:8,000, cat. no. YH80536; Yao-Hong
Biotechnology, Inc., New Taipei City, Taiwan). Each membrane was
incubated with appropriate primary antibodies at 4°C overnight and
washed with PBS containing Tween. After washing with PBS with 0.5%
Tween, blots were incubated with a 1:10,000 dilution of horseradish
peroxidase (HRP)-conjugated goat anti-mouse (cat. no. #20102;
Leadgene Biomedical, Inc., Tainan, Taiwan) or goat anti-rabbit IgG
(cat. no. #20202; Leadgene Biomedical, Inc.) antibodies for 1.5 h
at room temperature. The signals of specific proteins were detected
using a Western Bright ECL HRP substrate kit (Advansta, Inc., Menlo
Park, CA, USA)
Treatment with a pan-caspase
inhibitor
To determine whether caspase-mediated apoptosis
affected 3-MA-enhanced reductions in the proliferation of
CPT-treated cells, the pan-caspase inhibitor Z-VAD-FMK (cat. no.
S7023; Selleck Chemicals, Houston, TX, USA) was dissolved in DMSO.
Briefly, 5×104 NSCLC H1299 cells/well were seeded in a
12-well plate and were pretreated with 50 µM Z-VAD-FMK for 2
h at 37°C. Subsequently, the cells were treated with CPT/3-MA for
24 h. Cellular viability was determined using the trypan blue
exclusion assay, as described previously (28).
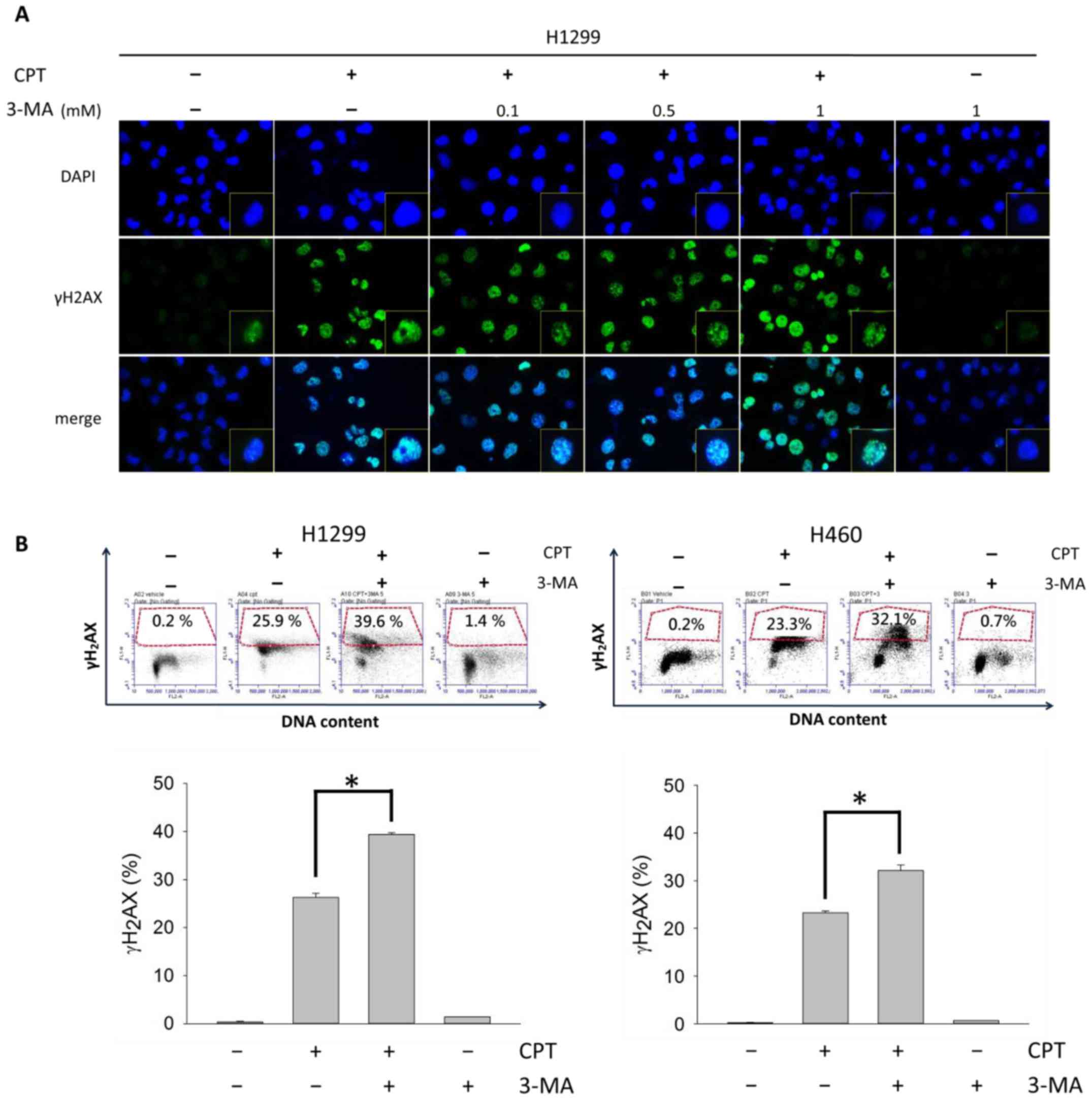
Flow cytometric detcetion of γH2AX
Briefly, cells were collected and fixed in 70%
ethanol at −20°C overnight. Prior to flow cytometry, the ethanol
was aspirated, and cells were rinsed with PBS and incubated with
anti-γH2AX (1:100; cat. no. sc-101696; Santa Cruz Biotechnology,
Inc.) dissolved in PBS containing 1% bovine serum albumin (BSA; cat
.no. 101-9048-46-8; MDBio, Inc., Taipei, Taiwan) and 0.5% Triton
(BSA-T-PBS) at 4°C overnight. The cells were then incubated with
FITC-conjugated anti-mouse immunoglobulin G (1:500, cat. no.
GTX26816; Genetex, Inc.) dissolved in BSA-T-PBS for 1 h at 4°C in
the dark. The cells were then rinsed and suspended in 1 µl
20 µg/ml propidium iodide (PI) containing RNase for 20 min
at 37°C, and FITC (γH2AX) and PI (DNA content) were quantified
using BD Accuri™ C6 flow cytometer with BD Accuri™ C6 software
(both BD Biosciences).
Immunofluorescence detection of γH2AX
nuclear foci
H1299 cells (3×104/well) grown on a
24-well plate were exposed to various concentrations of 3-MA in the
presence of 0.5 µM CPT for 24 h at 37°C. The cells were then
washed with PBS and fixed in 4% paraformaldehydefor 20 min at room
temperature. The fixed cells were washed twice with PBS,
permeabilized with 0.5% Triton-PBS, blocked with 1% BSA-PBS and
incubated for 2 h with anti-γH2AX mouse monoclonal antibodies (cat.
no. sc-101696; Santa Cruz Biotechnology, Inc.). The antibodies were
then washed off and cells were rinsed three times in 0.5%
Triton-PBS and incubated for 1 h in the dark with a FITC-conjugated
antimouse IgG secondary antibody (1:500, cat. no. GTX26816;
Genetex, Inc.). Cells were then rinsed twice in 0.5% Triton-PBS and
the cells were stained with 0.5 µg/ml DAPI (cat. no. 32670;
Sigma-Aldrich; Merck KGaA) for 5 min. Images were captured using a
fluorescence microscope (TE2000-U; Nikon Corporation).
Fluorescence microscopy
The green fluorescent protein (GFP)
fluorescence-tagged LC3 (LC3-GFP) construct was kindly provided by
Dr Wei-Pang Huang (Department of Life Science, National Taiwan
University, Taipei, Taiwan) (29).
H1299 and H460 cells were seeded onto 6-well microplates and
transfected with 1 µg wild-type or mutant pEGFP-LC3
constructs using Lipofectamine® and PLUS™ reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to a
previous study with minor modifications (29). A total of 48 h post-transfection,
the medium was replaced with seletive medium containing 0.4 mg/ml
(for H460 cells) and 0.8 mg/ml (for H1299 cells) Geneticin
selective antibiotic (G418 Sulfate; cat. no 10131035; Gibco; Thermo
Fisher Scientic, Inc.). Surviving colonies were picked 2 weeks
later and amplified. Stable transfectants were examined for LC3-GFP
expression and used in relevant experiments. Cells were seeded onto
a cover glass slide chamber (Nunc™ Lab-Tek™; Thermo Fisher
Scientific, Inc.), and after the designated treatments, the cells
were examined under a Nikon Eclipse TE2000U fluorescence microscope
(Nikon Corporation). The GFP-LC3 puncta were quantified by counting
the number of cells as described previously (29,30).
Briefly, the GFP-LC3 puncta in a single cell were manually counted
under a fluorescence microscope. For each group, 50 cells were
randomly selected to determine the average number of GFP-LC3
puncta/cells. The presented results were selected from experiments
performed at least three times.
Statistical analysis
All experiments were performed at least in
triplicate. Data were analyzed by multivariate analysis of
variance. If a significant difference was found, a Holm-Sidak
multiple comparison test was used to identify significant groups.
Statistical analyses were conducted using SigmaPlot software
version 12.0 (Systat Software, Inc., San Jose, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
IC50 values in NSCLC cell
lines
The present study was conducted using two LCC cell
lines, H1299 (p53-deleted, p53null) and H460
(wild-type p53-expressing, p53wt), since CPT is a
recommended therapeutic treatment for advanced NSCLC (31). The IC50 values of CPT
were initially determined in H1299 and H460 cells prior to
examining the antitumor effects of CPT and the CPT-resistant
effects of H1299 and H460 cells. The determined IC50 for
CPT was 1 µM in H1299 cells and 1 µM in H460 cells
(data not shown).
CPT failed to induce a cytotoxic effect
on drug-resistant H1299 and H460 cell lines in a dose-responsive
manner
Since CPT is able to inhibit tumor cell viability
and metastasis of lung carcinoma cells (32) the present study aimed to determine
its molecular/cellular mechanism. H1299 and H460 lung carcinoma
cells were incubated for 24 h in the presence of various
concentrations of CPT. As shown in Fig. 1A, the trypan blue assay
demonstrated that CPT damaged cell viability of H1299 and H460
cells. In H1299 cells, the percentage of viable cells decreased
from 100±1.8% in the untreated group to 55.8±2.3, 49.2±3.9,
47.3±11.4 and 39.2±4.6% in the presence of 0.5, 1, 2 and 5
µM CPT, respectively. In H460 cells, the percentage of
viable cells decreased from 100±0.0% in the untreated group to
47.4±4.9, 44.46±4.5, 47.07±3.3 and 39.9±0.5% in the presence of
0.5, 1, 2 and 5 µM CPT, respectively; therefore, the
susceptibility of the two lung carcinoma cells to CPT was
considered similar.
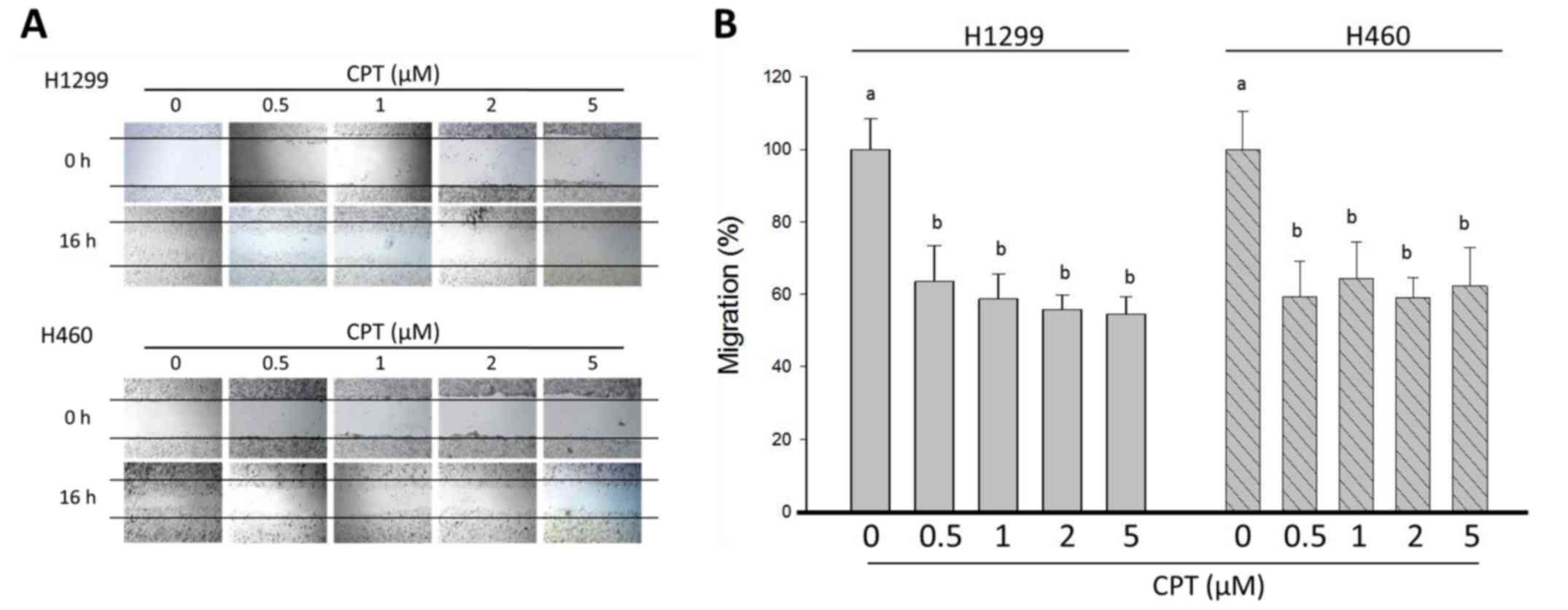
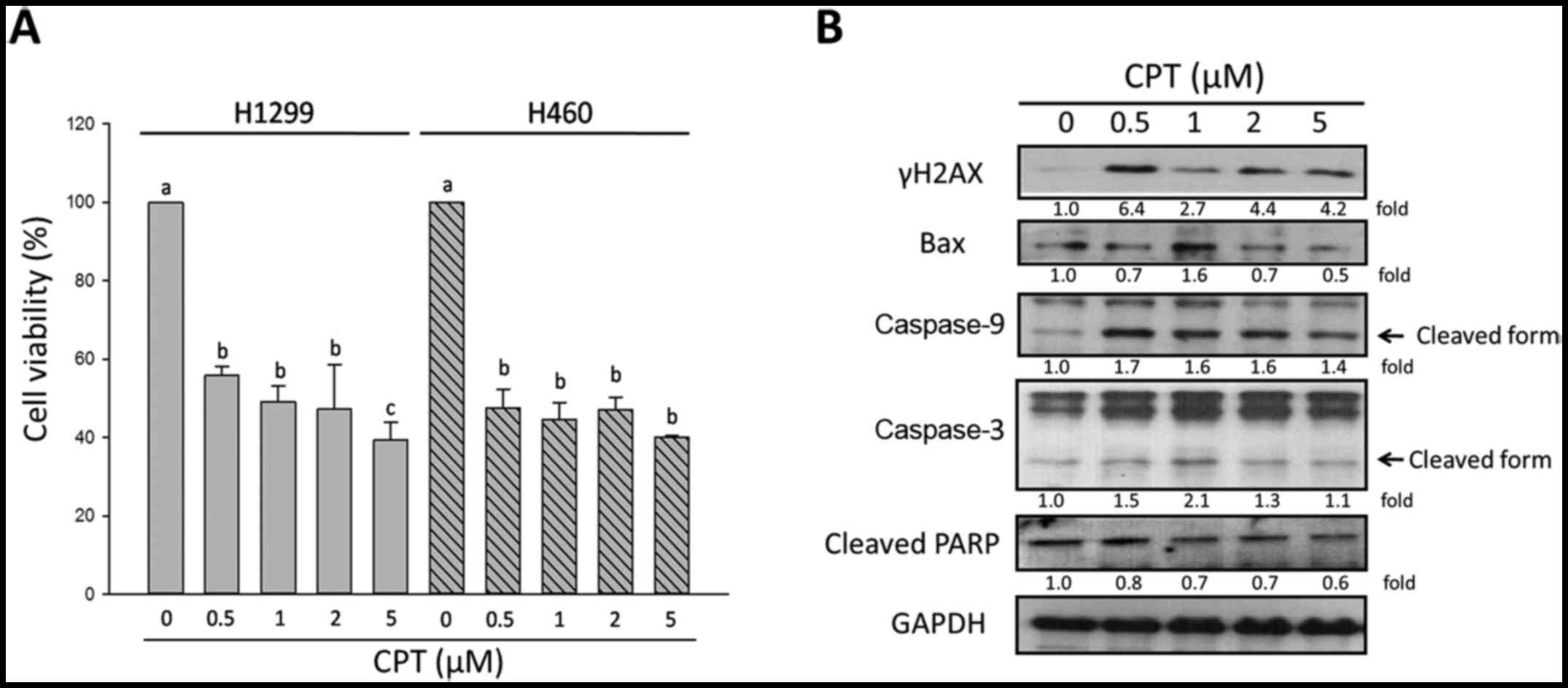 | Figure 1Effects of CPT on cell viability and
apoptosis of NSCLC cells. (A) Viability of two non-small cell lung
cancer cell lines, H1299 and H460, was determined by trypan blue
exclusion assay. (B) Alterations in the expression levels of
apoptosis-associated proteins and the DNA damage-sensor γH2AX in
H1299 cells, as determined by western blotting. GAPDH was used as
an internal control to ensure equal loading. Data are presented as
the means ± standard deviation of at least three independent
experiments. a vs. a, P>0.05; a vs. b, P<0.05; a vs. c,
P<0.001. γH2AX, phosphorylated-H2A histone family, member X
(Ser139); Bax, B-cell lymphoma 2-associated X protein;
CPT, camptothecin; PARP, poly (ADP-ribose) polymerase. |
Most types of cancer harbor significant patterns of
relapse following treatment due to evolved resistance. It has been
reported that cancer cells exist due to their resistance to cell
death and the loss of their ability to undergo apoptosis-induced
death, thus leading to uncontrolled proliferation (33). Evasion of apoptosis may contribute
to tumor development and progression, and to treatment resistance,
since the majority of anticancer therapies that are currently
available, including CPT-based chemotherapy, primarily act by
activating the apoptotic pathway in NSCLC (8). A better understanding of the pathway
underlying tumor resistance to apoptotic cell death is required, in
order to provide understanding of the molecular mechanisms
underlying development of resistance to targeted therapy.
Therefore, the present study detected apoptosis and
DNA-damage-related protein expression in H1299 cells treated with
various concentrations of CPT. The phosphorylation of H2AX at
Ser139, resulting in the formation of γH2AX puncta in
the nuclei, is an early event in the cellular response to DNA
damage. The expression levels of γH2AX were detected in order to
evaluate DNA damage of H1299 cells. As shown in Fig. 1B, the results of western blotting
indicated that, in the presence of 0.5 µM CPT, the
expression levels of the DNA damage biomarker γH2AX were increased,
as were the expression levels of cleaved caspase-9 and caspase-3.
Furthermore, 1 µM CPT increased B-cell lymphoma 2
(Bcl-2)-associated X protein (Bax) and cleaved caspase-3 expression
(Fig. 1B). These results indicated
that low dosages of CPT induced apoptosis and DNA damage of H1299
cells; however, the protein expression levels of γH2AX, Bax,
cleaved caspase-9, cleaved caspase-3 and cleaved poly (ADP-ribose)
polymerase (PARP) were not markedly increased by CPT in a
dose-dependent manner. These observations suggested that H1299
NSCLC cells may eventually become resistant in response to higher
concentrations of CPT. Therefore, 0.5 µM CPT may be
considered a reasonable dose due to its significant effect on cell
viability; cells were treated with 0.5 µM CPT in subsequent
experiments.
The present study also determined the effects of
various concentrations of CPT on the migration of H1299 and H460
cells using wound-healing assays. In the control group, the cells
migrated into the wound area and the wound edges became
indistinguishable; however, following the addition of CPT, cells
exhibited slower wound healing (Fig.
2A). In response to various concentrations of CPT, H1299 cell
migration was inhibited from 100.0±8.4% in the control group, to
63.5±9.9, 58.7±6.8, 55.85±4.0 and 54.6±4.6% in response to 0, 0.5,
1, 2 and 5 µM CPT, respectively (Fig. 2B). The percentage of cell viability
and wound healing was significantly decreased in H1299 cells in the
presence of 0.5-5 µM CPT compared with in the control group
(P<0.05). However, there was no alteration in the percentage of
cell viability and wound healing following CPT dosage accumulation,
thus indicating that treatment did not affect growth and
metastasis; in addition, the inhibitory effects of CPT on H1299
cells reached a plateau. Consistent with the results in H1299
cells, H460 cells were treated with the indicated concentrations of
CPT for 24 h, after which the migration rate of cells was
significantly reduced compared with in the control group; however,
no change in migration was detected as the CPT dosage accumulated
(Fig. 2). These observations
supported the finding that higher concentrations of CPT (1, 2 and 5
µM) exhibited no further inhibitory effect on cell viability
and metastasis compared with the lower CPT concentration (0.5
µM) on H1299 and H460 NSCLC cells.
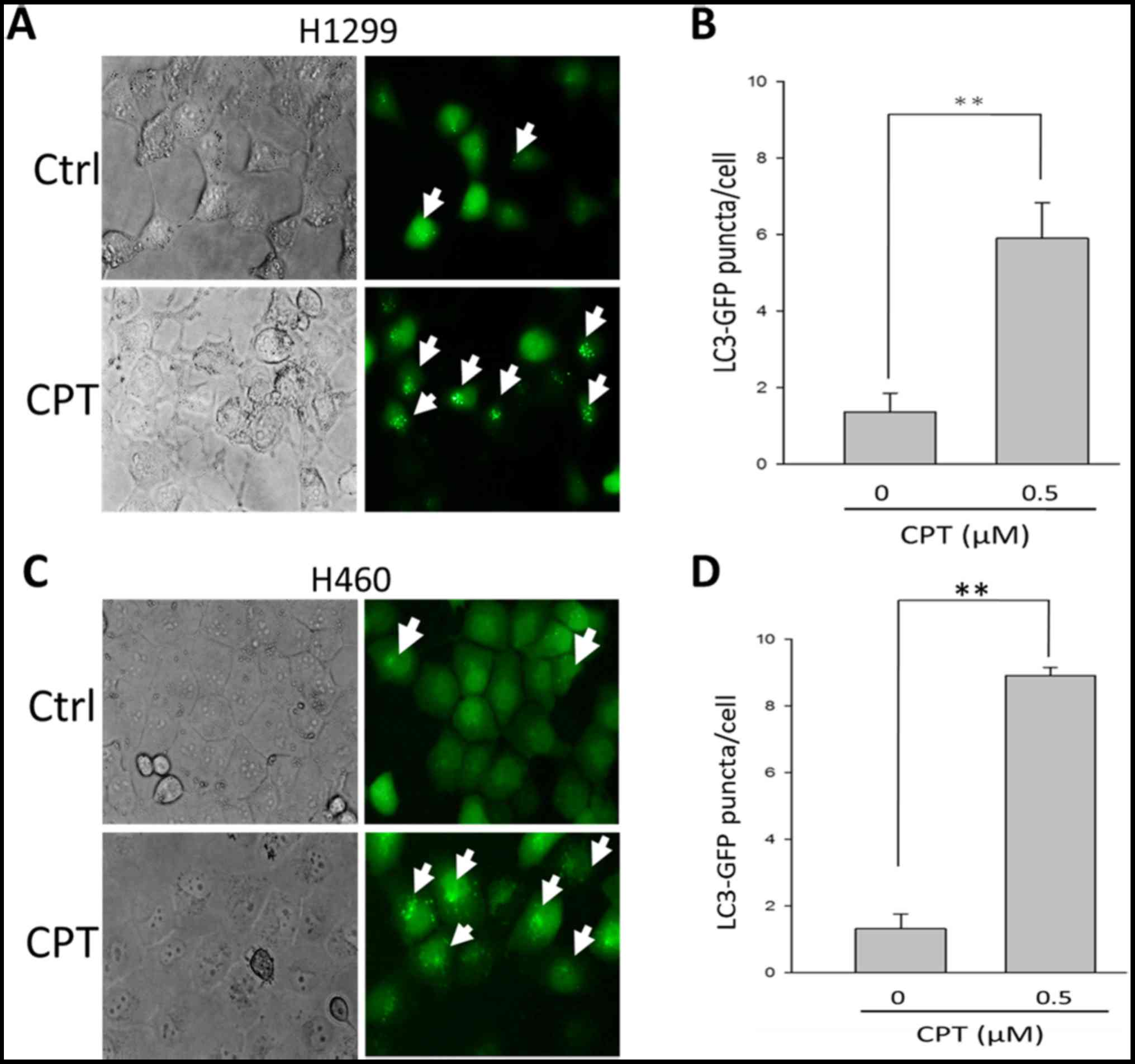
A recent study suggested that when treated with
chemotherapeutic agents, certain tumor cells fail to undergo
apoptosis and instead undergo autophagy followed by delayed cell
death (34). To determine whether
autophagy is associated with the suppression of CPT-induced
apoptotic cell death, the autophagic marker
phosphatidylethanolamine-LC3 (LC3-II) was detected in H1299 cells
in the presence of 0.5 µM CPT. As shown in Fig. 3A and B, 1.4±0.5 and 6.0±0.9 GFP-LC3
puncta were detected in H1299 cells in the presence of 0 and 0.5
µM CPT, respectively. These findings indicated that
supplementation with 0.5 µM CPT may lead to an enrichment of
GPF-LC3 protein in H1299 cells (P<0.05), thus suggesting that
H1299 cells respond to CPT by activating autophagy. Similar effects
were detected on H460 NSCLC cells (Fig. 3C and D).
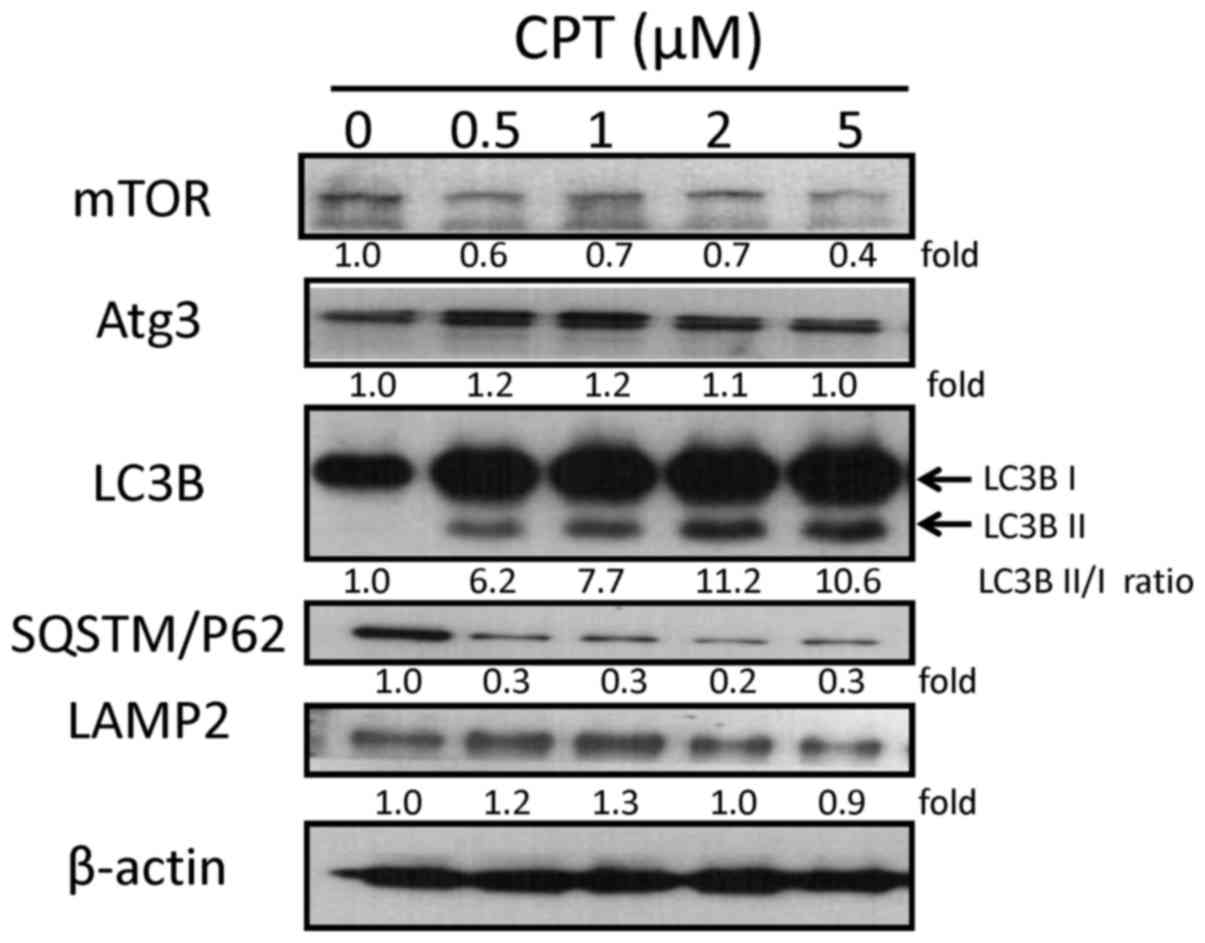
Since the present study demonstrated that CPT
increased autophagy in H1299 cells, it was hypothesized that
alterations in cell survival may be due to autophagy. The protein
expression levels of mammalian target of rapamycin (mTOR), Atg3,
LC3B, SQSTM/p62 and LAMP2, which are involved in the autophagic
process, were also examined by western blot analysis (Fig. 4). The expression levels of Atg3
were slightly increased in response to 0.5, 1 and 2 µM CPT,
and appeared the same in the the control and 5 µM CPT
groups. Decreases in the expression levels of mTOR and p62 were
detected in response to various CPT concentrations. LAMP2 was
increased in response to 0.5 and 1 µM CPT, and was only
slightly decreased in response to 5 µM CPT. Furthermore, the
expression levels of the protein LC3B-I (an unprocessed form of
LC3) and the cleaved protein LC3B-II (lipidated and
autophagosome-associated form of LC3) were markedly increased in
H1299 cells following CPT treatment at various concentrations
compared with in the control group (Fig. 4). These findings were consistent
with detection of the increased formation of autophagosomes, and
suggested that CPT may enhance autophagy in an H1299 NSCLC cell
lines in a dose- dependent manner.
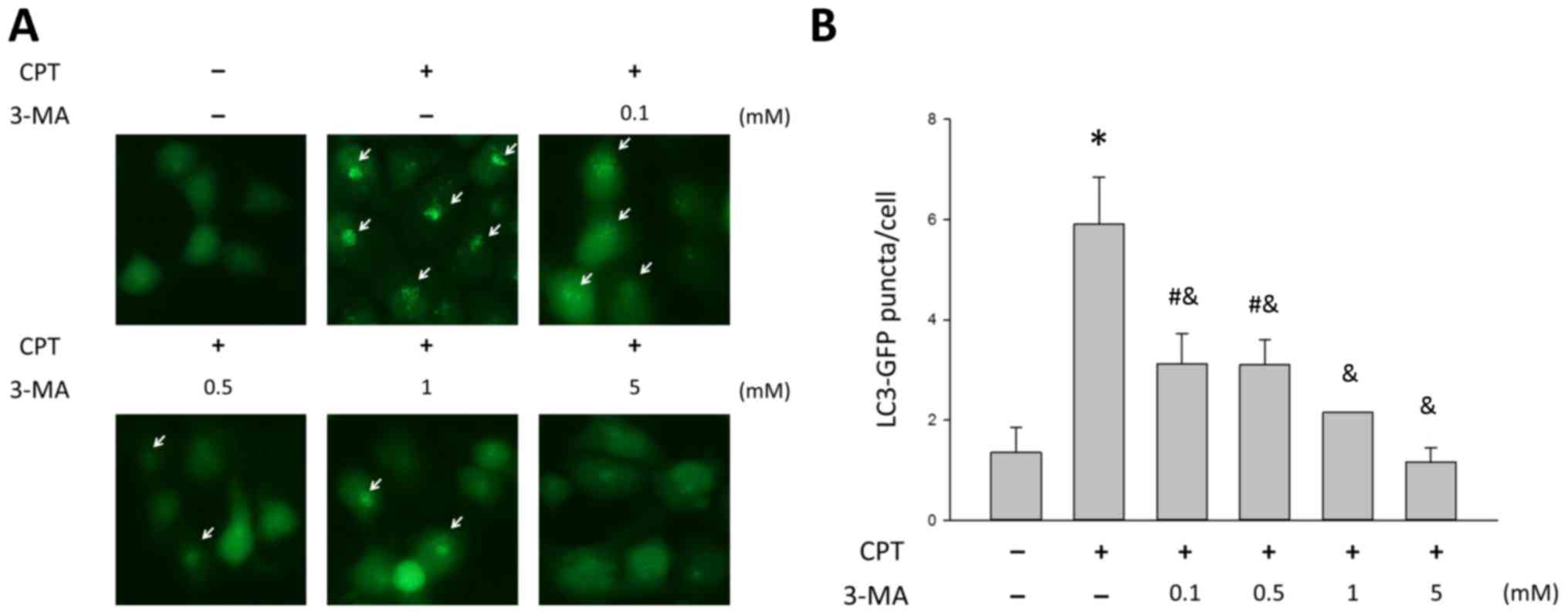
3-MA blocks CPT-induced autophagy in
H1299 lung cancer cells in a dose-dependent manner
To determine the role of autophagy in CPT-treated
NSCLC, the present study cotreated H1299 cells with various
concentrations of the autophagy inhibitor 3-MA and 0.5 µM
CPT, in order to determine whether 3-MA blocked CPT-induced
autophagy. As shown in Fig. 5,
3-MA had an effect on CPT-induced autophagy; treatment with CPT
alone resulted in 5.91±0.9 GFP-LC3 puncta, whereas 3-MA induced a
significant dose-dependent decrease in autophagy in H1299 cells
(P<0.05). To further validate the inhibitory effects of 3-MA on
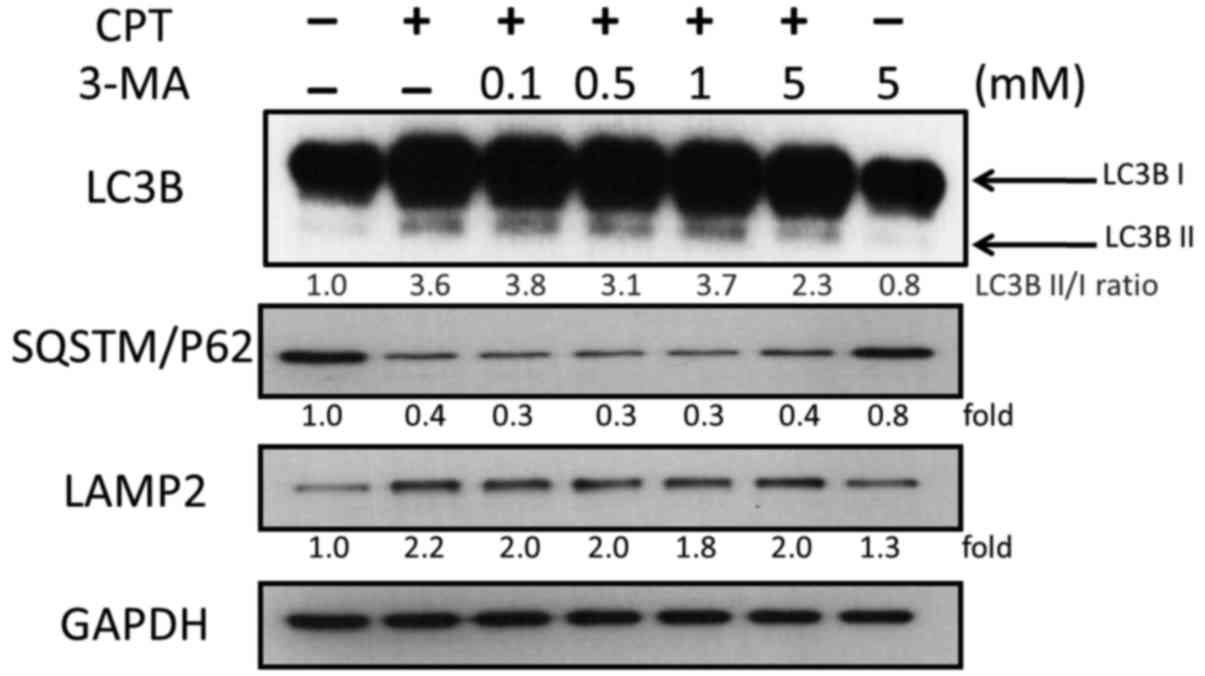
CPT-induced autophagy, activation of LC3B and the expression of
autophagic proteins were analyzed by western blotting. As shown in
Fig. 6, 3-MA treatment reduced the
accumulation of LC3B induced by 0.5 µM CPT, but exhibited no
marked effects on SQSTM/P62 and LAMP2 (Fig. 6). The ratio of LC3BII/I was
decreased in a dose-dependent manner; the ratio of LC3BII/I was 3.8
and 2.3 in response to 0.1 and 5 mM 3-MA, respectively. These
findings indicated that 3-MA blocked CPT-induced autophagy in H1299
lung carcinoma cells.
3-MA enhances the anticancer effect of
CPT via autophagy inhibition
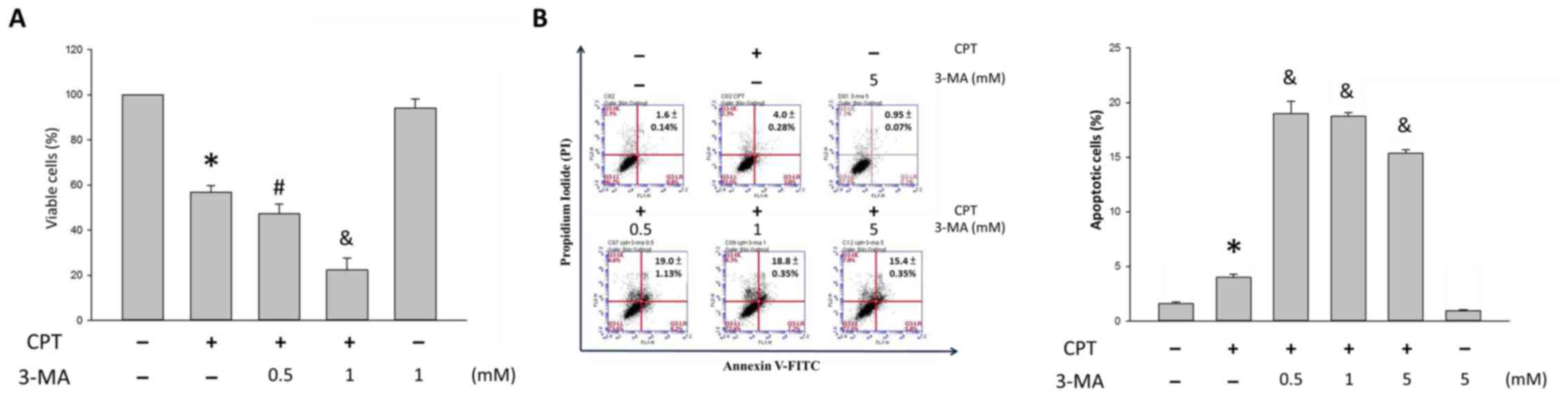
As shown in Fig.
7A, the percentage of viable H1299 cells was 56.8±3.1, 47.3±4.3
and 22.3±5.3% following treatment with 0, 0.5 and 1 mM 3-MA, in the
presence of 0.5 µM CPT, respectively, as opposed to
100.0±0.2% in the control group and 94.2±4.0 % in the 1 mM 3-MA
group. Once CPT-induced autophagy was blocked, the cytotoxic
effects of CPT were enhanced in H1299 cells. Furthermore,
suppressing autophagy increased apoptosis, as determined using the
Annexin V/PI staining assay. Following treatment with vehicle
control, or 0, 0.5, 1 and 5 mM 3-MA in the presence of 0.5
µM CPT for 24 h, apoptosis was detected (Fig. 7B). In response to CPT, the
percentage of apoptotic H1299 cells was 4.0±0.1%; however, in
response to CPT and 3-MA cotreatment, the percentage of apoptotic
cells was 19.0±1.13, 18.8±0.35 and 15.4±0.35% following 3-MA
accumulation (Fig. 7B).
The present study indicated that there was a slight
increase in apoptosis in the CPT group; however, in cells receiving
CPT and 3-MA cotreatment apoptosis was significantly increased
compared with in those receiving CPT only (P<0.05; Fig. 7B). These results suggested that,
following cotreatment with 3-MA, CPT may efficiently induce
apoptosis of CPT-resistant H1299 cells and CPT-induced autophagy
may be suppressed. Since the major mechanism underlying CPT-induced
cellular cytotoxicity and apoptosis is induction of DNA damage, the
present study examined whether blocking autophagy affected the
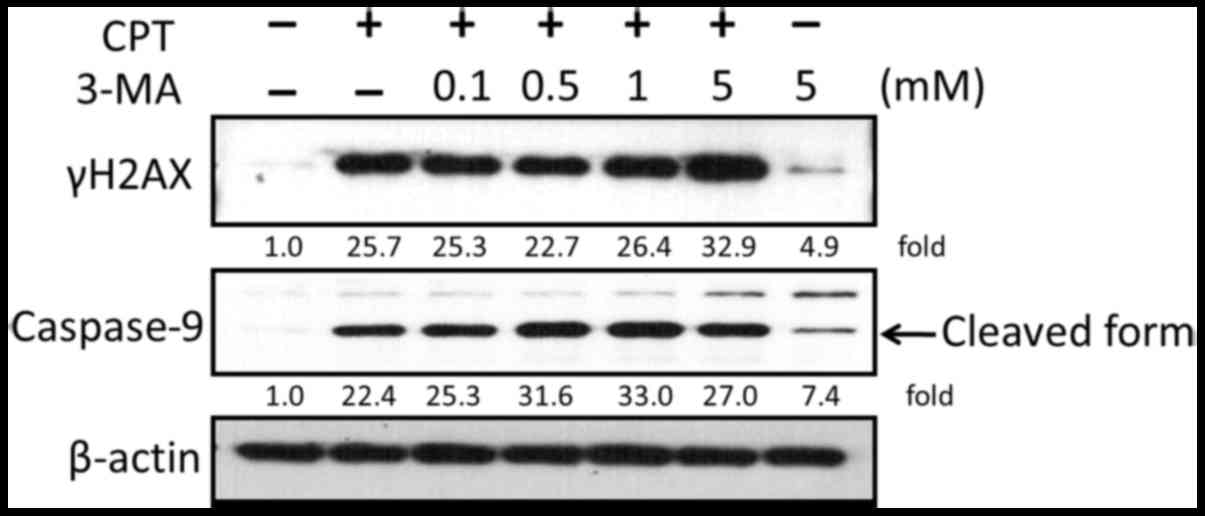
anti-lung cancer effects of CPT. As shown in Fig. 8, the expression levels of γH2AX
were increased in response to CPT and 3-MA cotreatment compared
with in cells treated with CPT or 3-MA alone. In addition, the
expression levels of the apoptosis-associated protein caspase-9
were detected by western blotting. The results indicated that
activation of caspase-9 was enhanced in response to the suppression
of CPT-induced autophagy by 3-MA.
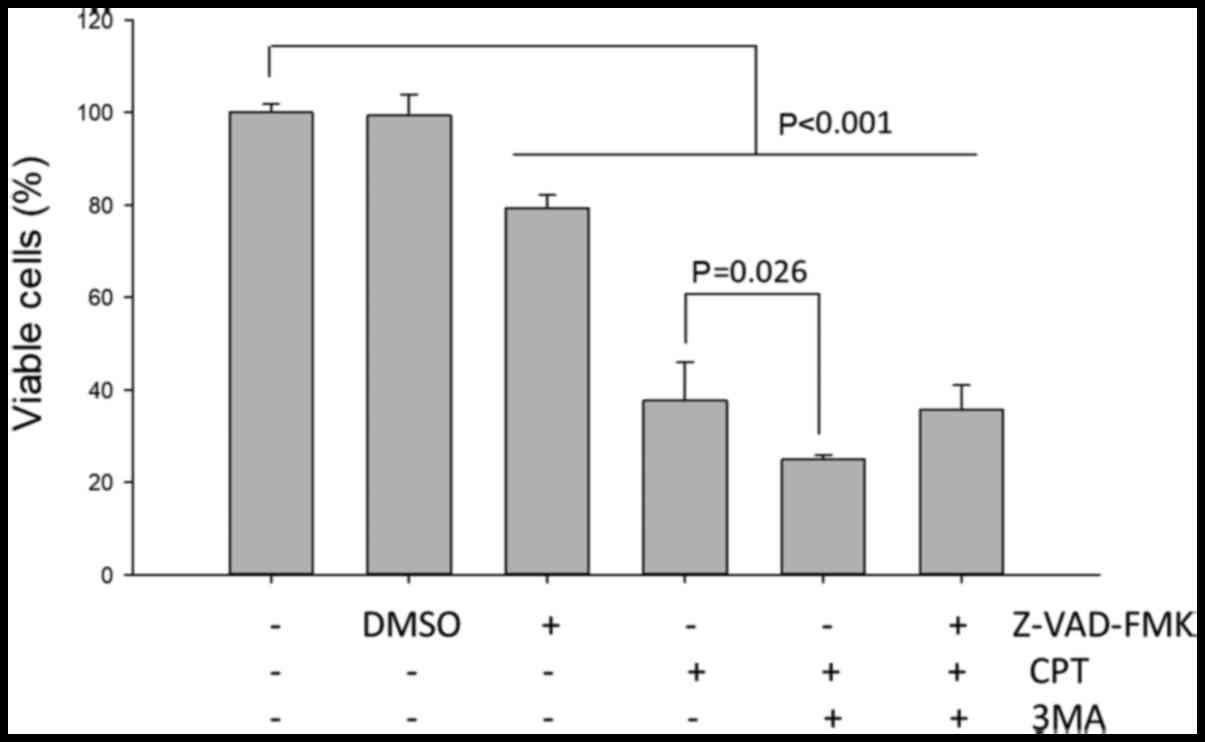
Effects of a pan-caspase inhibitor
H1299 NSCLC cells were pretreated with or without
the pan-caspase inhibitor Z-VAD-FMK for 2 h prior to treatment with
CPT/3-MA for 24 h. Subsequently, cell viability was analyzed; the
results indicated that pretreatment with the caspase inhibitor did
not significantly affect the 3-MA-enhanced cytotoxic effects on
CPT-treated H1299 cells (Fig. 9).
These results indicated that 3-MA-enhanced cell death of NSCLC cell
lines may not predominantly occur via a caspase-dependent
pathway.
CPT-induced DNA damage is increased
following autophagy blockade
To further demonstrate that the observed
DNA-damaging effects were caused by blocking CPT-induced autophagy,
γH2AX foci formation was analyzed in H1299 cells (Fig. 10A), and a γH2AX/PI double staining
assay was conducted in H1299 and H460 cells (Fig. 10B). γH2AX foci are generally
regarded as markers of DNA double-strand breaks (35); therefore, increases in γH2AX foci
indicate CPT-induced DNA damage in H1299. Notably, DNA damage was
enhanced in H1299 and H460 cells in response to CPT and 3-MA
cotreatment, and the effects of 3-MA on DNA damage were
concentration- dependent.
Discussion
Drug resistance or chemoresistance is a phenomenon
whereby cells undergo adaptive mutation under environmental stress
(36). Chemoresistance disrupts
anticancer drug actions and renders therapy ineffective.
Chemoresistance is induced by numerous factors, including
inhibition of drug absorption (37), epigenetic modifications (38), cell cycle alterations (39) and inhibition of apoptosis (40). Accordingly, one of the critical
factors mediating chemoresistance is inhibition of cell apoptosis.
To understand whether inhibition of apoptosis is attributed to a
plateau of the cytotoxic effects of CPT, apoptotic factor
expression was detected by western blot analysis. Notably, since
low doses CPT increased caspase activation, decreases in the
activation of caspase-3 and caspase-9 were detected in cells
following treatment with high doses of CPT. In addition, other
apoptotic factors, and the DNA damage marker γH2AX, were also
similarly reduced following treatment with increasing doses of CPT.
These data indicated that when lung cancer H1299 cells were treated
with a relatively high concentration of CPT, the inhibition of
apoptosis may enhance resistance of H1299 cells to antitumor CPT
treatment. Despite the initial high responsiveness of cells to CPT
and its derivatives (8-14), cancer cells often develop acquired
resistance following treatment (41), which significantly limits the
therapeutic efficacy of CPT. Methods for reversing CPT resistance
are currently lacking; therefore, a better understanding of the
mechanisms underlying such resistance is essential. A reduction in
apoptotic cell death may be one of the factors that mediate
chemoresistance, and the present study revealed that high
concentrations of CPT may diminish apoptotic death and DNA damage
in H1299 and H460 NSCLC cell lines.
In our previous studies, it was demonstrated that
some chemosensitizers could reduce antagonism and sensitize cancer
cells to CPT, thus enhancing CPT-induced anticancer effects under
the IC50 concentration (0.5 µM) (8,42).
In the present study, there was no marked alteration in H1299 and
H460 cell viability, wound closure, DNA damage and apoptotic cell
death between 0.5 and 1 µM CPT treatment. Conversely,
CPT-induced formation of autophagosomes was increased and
regulation of the expression of autophagy marker proteins was
affected in H1299 and H460 lung carcinoma cells. As indicated by
the reduction in apoptotic protein expression and the increase in
autophagy at higher CPT concentrations, it was suggested that H1299
and H460 cells may eventually became less sensitive to higher
concentrations of CPT. Since higher concentrations of CPT
(>IC50: 1 µM) did not exhibit an increased
anticancer effect, a lower dose of CPT (0.5 µM) was
considered a reasonable dosage for subsequent experiments.
Numerous studies have reported that autophagy
induces chemoresistance against anticancer drugs by inhibiting
apoptosis of cancer cells (23,43).
One suggested strategy to overcome acquired resistance to CPT and
its derivatives is the inhibition of autophagosome formation
(44,45). A recent study demonstrated that
defective autophagosome formation in p53null
colorectal cancer may enhance drug-induced apoptosis (46). In the present study, the antitumor
effects of CPT were detected on both H460 p53wt
and H1299 p53null lung cancer cell lines. A
similar pattern was detected in p53null H1299 and
p53wt H460 cells with regards to cell death, cell
migration and autophagy in the presence of CPT; therefore, it may
be suggested that CPT-mediated antitumor effects occur
independently of p53 expression. On the basis of these results,
further studies are required to verify the essential role of
defective autophagosome formation in CPT-induced apoptosis in other
NSCLC lines, such as p53wt A549, and p53-mutated
CL1-0 and CL1-5 lung adenocarcinoma cells.
To understand the role of autophagy in CPT-treated
NSCLC cells, in the present study H1299 cells were cotreated with
various concentrations of 3-MA and 0.5 µM CPT, and the
effects of 3-MA on the suppression of CPT-induced autophagy were
confirmed. 3-MA is an inhibitor of phosphatidylinositol 3-kinase
(PI3K), which serves an essential role in controlling the
activation of mTOR and the regulation of autophagy. The group of
PI3K inhibitors includes 3-MA, wortmannin and LY294002. Among them,
wortmannin is able to suppress autophagy regardless of nutrient
status; however, 3-MA has been revealed to promote autophagy flux
when used under nutrient-rich conditions for a prolonged period of
time, whereas it is still capable of suppressing starvation-induced
autophagy (47). Due to the dual
roles of 3-MA in autophagy, it has been widely used as an autophagy
inhibitor in various types of cancer therapy (48-50).
The present study confirmed that 3-MA significantly inhibited
cytoprotective autophagy in H1299 cells in a dose-dependent manner.
The results of western blotting further revealed a marked decrease
in LC3B expression in response to 3-MA, which may be associated
with the reduction in autophagosome formation.
Since CPT-induced autophagy was suppressed by 3-MA,
inhibition of H1299 cells was enhanced; in particular, apoptosis of
H1299 cells was increased, as determined by trypan blue dye
exclusion assay and Annexin V/PI staining. To further confirm that
the observed apoptotic effects were produced by blocking
CPT-induced autophagy, the expression levels of mitochondrial
apoptotic proteins, including Bax, Bcl-2, caspase-3, caspase-9 and
PARP, were detected via western blotting. The results demonstrated
that activation of caspase-9 was enhanced in response to inhibition
of CPT-induced autophagy by 3-MA. However, there was no significant
alteration in the expression levels of Bax, caspase-3 and PARP in
response to cotreatment with CPT and 3-MA (data not shown).
Conversely, a marked decrease in the protein expression levels of
Bcl-2 was detected following 3-MA dosage accumulation in the
presence of 0.5 µM CPT (data not shown). Therefore, the
elevated Bax/Bcl-2 ratio indicated that 3-MA enhanced the
susceptibility of H1299 cells to autophagy-inhibited apoptosis.
These results suggested a significant improvement in CPT
sensitivity following 3-MA cotreatment; this effect is most likely
due to the suppression of autophagy and enhanced apoptosis of H1299
NSCLC cells.
Caspases are cysteine proteases that have critical
roles in apoptosis (51,52). The present study demonstrated that
treatment with CPT and 3-MA activated caspase-9. However,
pretreatment with a pan-caspase inhibitor did not significantly
suppress CPT/3-MA-induced cell death, thus suggesting that
CPT/3-MA-induced apoptosis was not associated with
caspase-dependent pathways; this finding differs from previous
reports. It has previously been reported that the apoptotic effects
of CPT/3-MA are usually associated with the activation of
caspase-3, caspase-8 and caspase-9 in colon, liver (53) and lung cancer cells (54). The different pathways associated
with CPT/3-MA-induced apoptosis may be due to various cell types,
treatment duration and CPT/3-MA concentration.
Although it is widely believed that anticancer
agents inhibit the viability of cancer cells through inducing
apoptosis, accumulating evidence has revealed that other
apoptosis-independent cell death modalities, such as autophagic
cell death, may also contribute to cancer cell death, thus
resulting in the inhibitory effects of anticancer drugs on cancer
cells. For example, voacamine, which is a bisindolic alkaloid,
induces apoptosis-independent autophagic cell death of the
multidrug- resistant human osteosarcoma cell line U-2 OS (55). Similarly, autosis, a newly
identified non-apoptotic form of cell death, may contribute to cell
death during autophagy (56). In
the present study, 3-MA significantly enhanced CPT-induced cell
death and increased its inhibitory effect on the viability of NSCLC
cells; furthermore, apoptosis was markedly increased in
CPT/3-MA-treated cells compared with in cells treated with CPT
alone, thus confirming that 3-MA increased apoptosis of CPT-treated
cells. Pretreatment with the pan-caspase inhibitor Z-VAD-FMK did
not significantly affect the viability of NSCLC cells, thus
suggesting that apoptosis may not be fully responsible for NSCLC
cell death induced by CPT and 3-MA cotreatment. Additionally,
non-apoptotic cell death, such as autosis or other forms of
autophagic cell death, may contribute to the enhancing effects of
3-MA on CPT-induced proliferation inhibition and death of NSCLC
cells.
Accumulating evidence has revealed that autophagy is
capable of attenuating DNA damage by decreasing generation of ROS
and modulating DNA repair activity (57,58).
Mitochondria, which are the major source of ROS, cause ROS
production accompanied with DNA damage (59). Autophagy inhibits ROS production,
in order to protect cells from DNA damage (60,61);
therefore, blocking autophagy may induce an excess generation of
ROS, consequently leading to more severe DNA damage (62). When cells encounter DNA damage,
they initiate DNA repair mechanisms to reduce it. Notably,
autophagy is also involved in the process of DNA repair; autophagy
induces the generation of ATP and recycles dNTP to improve DNA
repair activity, whereas suppression of autophagy leads to
decreased levels of checkpoint kinase 1 and a markedy diminished
ability to repair DNA double-strand breaks (63).
According to previous results, the present study
aimed to determine whether inhibiting autophagy affected the degree
of DNA damage. Following inhibition of CPT-induced autophagy, the
present study detected γH2AX through immunofluorescence and flow
cytometry. The present study demonstrated that suppression of
CPT-induced autophagy significantly increased γH2AX foci formation.
These findings suggested that inhibition of autophagy increased DNA
damage and γH2AX expression. In addition, DNA damage was only
slightly increased in response to treatment with 3-MA alone.
Therefore, it may be suggested that inhibition of autophagy by 3-MA
promotes ROS generation leading to DNA damage; this may explain why
γH2AX is upregulated in response to treatment with 3-MA alone.
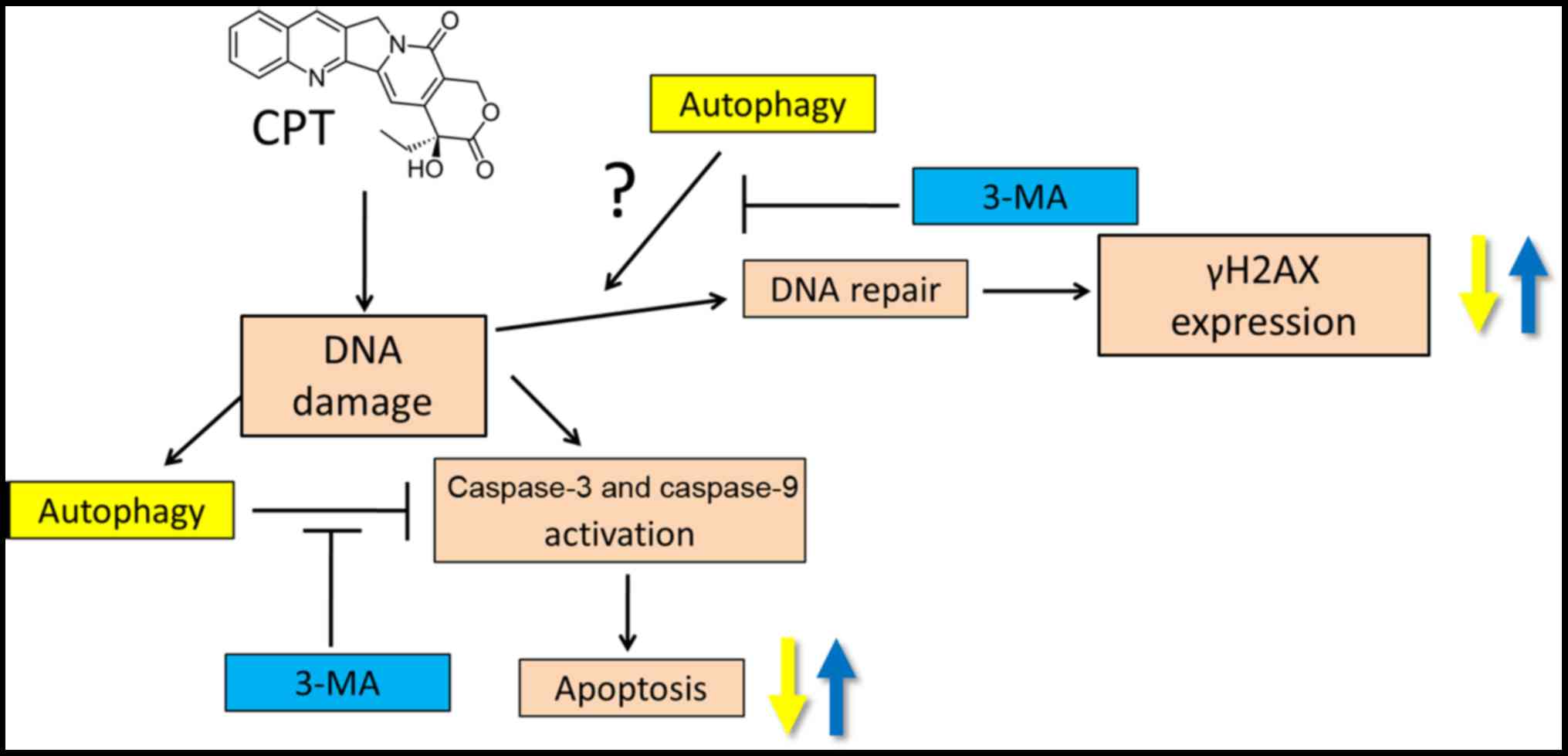
In conclusion, the present study provided detailed
insights into the CPT-resistant mechanisms of NSCLC cells. The
results demonstrated that CPT failed to further induce cell
cytotoxicity, metastasis-inhibiting effects, DNA damage and
apoptotic death following dosage accumulation, and the inhibitory
effects of CPT on H1299 and H460 cells reached an early plateau.
Conversely, CPT increased the formation of autophagosomes in NSCLC
H1299 cells in a dose-dependent manner, and the present study
indicated that CPT-induced autophagy may serve a protective role in
NSCLC with regards to DNA damage and apoptosis. Cotreatment with
the autophagy inhibitor 3-MA blocked CPT-induced autophagy, and
activated caspase-9 and γH2AX, thereby enhancing induction of
apoptosis and DNA damage in NSCLC cells. Taken together, 3-MA may
serve as a promising clinical adjuvant to enhance CPT-based
chemotherapies for the future treatment of lung cancer (Fig. 11).
Funding
The present study was supported by grants from the
Ministry of Science and Technology (MOST), Taiwan (grant nos.
MOST105-2311-B-037-001, MOST106-2320-B-037-012,
MOST107-2314-B-037-028-MY3 and 107-2320-B-037-023), the NSYSU-KMU
Joint Research Project (grant nos. NSYSU K M U10 6 - P 015, NSYSU K
M U10 6 - P 019, NSYSUKMU107-P02 and NSYSUKMU107-P013), and the Aim
for the Top Universities grants from Kaohsiung Medical University
(grant nos. KMU-TP105A03 and KMU-M107002).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
CCC initated the work. YHC, SHH and CCC designed
experiments. HWH, KCH and WL performed most of the assays. CYW,
WPH, JYFC and BHC helped to acquire data and conduct statistical
analysis. WPH helped conduct the transftection assay and analysed
the autophagic puncta. YHC and CCC wrote the manuscript. All
authors have read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors of the present study would like to thank
the Center for Research Resources and Development at Kaohsiung
Medical University (Kaohsiung, Taiwan) for support with
instrumentation, including the confocal microscope and flow
cytometer.
References
|
1
|
Kayser G, Csanadi A, Kakanou S, Prasse A,
Kassem A, Stickeler E, Passlick B and Zur Hausen A: Downregulation
of MTSS1 expression is an independent prognosticator in squamous
cell carcinoma of the lung. Br J Cancer. 112:866–873. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang A: Chemotherapy, chemoresistance and
the changing treatment landscape for NSCLC. Lung Cancer. 71:3–10.
2011. View Article : Google Scholar
|
|
3
|
Ling DJ, Chen ZS, Liao QD, Feng JX, Zhang
XY and Yin TY: Differential effects of MTSS1 on invasion and
proliferation in subtypes of non-small cell lung cancer cells. Exp
Ther Med. 12:1225–1231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McNeill PM, Wagman LD and Neifeld JP:
Small bowel metastases from primary carcinoma of the lung. Cancer.
59:1486–1489. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Downey RJ, Asakura S, Deschamps C and
Colby TV: Large cell carcinoma of the lung: Results of resection
for a cure. J Thorac Cardiovasc Surg. 117:599–604. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wall ME, Wani MC, Cook CE, Palmer KH,
McPhail AT and Sim GA: Plant antitumor agents. 1. The isolation and
structure of camptothecin, a novel alkaloidal leukemia and tumor
inhibitor from Camptotheca acuminata. J Am Chem Soc. 88:3888–3890.
1966. View Article : Google Scholar
|
|
7
|
Wadkins RM, Bearss D, Manikumar G, Wani
MC, Wall ME and Von Hoff DD: Topoisomerase I-DNA complex stability
induced by camptothecins and its role in drug activity. Curr Med
Chem Anticancer Agents. 4:327–334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chou HL, Fong Y, Wei CK, Tsai EM, Chen JY,
Chang WT, Wu CY, Huang HW and Chiu CC: A quinone-containing
compound enhances camptothecin-induced apoptosis of lung cancer
through modulating endogenous ROS and ERK signaling. Arch Immunol
Ther Exp (Warsz). 65:241–252. 2017. View Article : Google Scholar
|
|
9
|
Kawakami T, Machida N, Yasui H, Kawahira
M, Kawai S, Kito Y, Yoshida Y, Hamauchi S, Tsushima T, Todaka A, et
al: Efficacy and safety of irinotecan monotherapy as third-line
treatment for advanced gastric cancer. Cancer Chemother Pharmacol.
78:809–814. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim M, Keam B, Kim TM, Kim HG, Kim JS, Lee
SS, Shin SH, Kim MK, Park KU, Kim DW, et al: Phase II study of
irinotecan and cisplatin combination chemotherapy in metastatic,
unresectable esophageal cancer. Cancer Res Treat. 49:416–422. 2017.
View Article : Google Scholar :
|
|
11
|
Zhang B, Wang T, Yang S, Xiao Y, Song Y,
Zhang N and Garg S: Development and evaluation of oxaliplatin and
irinotecan co-loaded liposomes for enhanced colorectal cancer
therapy. J Control Release. 238:10–21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Keyvani-Ghamsari S, Rabbani-Chadegani A,
Sargolzaei J and Shahhoseini M: Effect of irinotecan on HMGB1, MMP9
expression, cell cycle, and cell growth in breast cancer (MCF-7)
cells. Tumour Biol. 39:10104283176983542017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bruchim I, Ben-Harim Z, Piura E, Haran G
and Fishman A: Analysis of two topotecan treatment schedules in
patients with recurrent ovarian cancer. J Chemother. 28:129–134.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kubo T, Fujiwara K, Hotta K, Okada T,
Kuyama S, Harita S, Ninomiya T, Kamei H, Hosokawa S, Bessho A, et
al: A phase II study of topotecan and cisplatin with sequential
thoracic radiotherapy in elderly patients with small-cell lung
cancer: Okayama Lung Cancer Study Group 0102. Cancer Chemother
Pharmacol. 78:769–774. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang XQ, Li CY, Xu MF, Zhao H and Wang D:
Comparison of first-line chemotherapy based on irinotecan or other
drugs to treat non-small cell lung cancer in stage IIIB/IV: A
systematic review and meta-analysis. BMC Cancer. 15:9492015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugimoto Y, Tsukahara S, Oh-hara T, Isoe T
and Tsuruo T: Decreased expression of DNA topoisomerase I in
camptothecin- resistant tumor cell lines as determined by a
monoclonal antibody. Cancer Res. 50:6925–6930. 1990.PubMed/NCBI
|
|
17
|
Kawabata S, Oka M, Shiozawa K, Tsukamoto
K, Nakatomi K, Soda H, Fukuda M, Ikegami Y, Sugahara K, Yamada Y,
et al: Breast cancer resistance protein directly confers SN-38
resistance of lung cancer cells. Biochem Biophys Res Commun.
280:1216–1223. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feeney GP, Errington RJ, Wiltshire M,
Marquez N, Chappell SC and Smith PJ: Tracking the cell cycle
origins for escape from topotecan action by breast cancer cells. Br
J Cancer. 88:1310–1317. 2003. View Article : Google Scholar
|
|
19
|
Han JY, Lim HS, Yoo YK, Shin ES, Park YH,
Lee SY, Lee JE, Lee DH, Kim HT and Lee JS: Associations of ABCB1,
ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and
clinical outcome in patients with advanced non-small cell lung
cancer. Cancer. 110:138–147. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Czarny P, Pawlowska E, Bialkowska-Warzecha
J, Kaarniranta K and Blasiak J: Autophagy in DNA damage response.
Int J Mol Sci. 16:2641–2662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song X, Narzt MS, Nagelreiter IM,
Hohensinner P, Terlecki-Zaniewicz L, Tschachler E, Grillari J and
Gruber F: Autophagy deficient keratinocytes display increased DNA
damage, senescence and aberrant lipid composition after oxidative
stress in vitro and in vivo. Redox Biol. 11:219–230. 2017.
View Article : Google Scholar :
|
|
22
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang B, Liu X, Liu Y, Kong D, Liu X,
Zhong R and Ma S: Inhibition of autophagy sensitizes MDR-phenotype
ovarian cancer SKVCR cells to chemotherapy. Biomed Pharmacother.
82:98–105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu Y, Liu LL, Liu SS, Fang ZG, Zou Y, Deng
XB, Long ZJ, Liu Q and Lin DJ: Celecoxib suppresses autophagy and
enhances cytotoxicity of imatinib in imatinib-resistant chronic
myeloid leukemia cells. J Transl Med. 14:2702016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vazquez-Martin A, Oliveras-Ferraros C and
Menendez JA: Autophagy facilitates the development of breast cancer
resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS
One. 4:e62512009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chiu CC, Chou HL, Chen BH, Chang KF, Tseng
CH, Fong Y, Fu TF, Chang HW, Wu CY, Tsai EM, et al: BPIQ, a novel
synthetic quinoline derivative, inhibits growth and induces
mitochondrial apoptosis of lung cancer cells in vitro and in
zebrafish xenograft model. BMC Cancer. 15:9622015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiu CC, Liu PL, Huang KJ, Wang HM, Chang
KF, Chou CK, Chang FR, Chong IW, Fang K, Chen JS, et al:
Goniothalamin inhibits growth of human lung cancer cells through
DNA damage, apoptosis, and reduced migration ability. J Agric Food
Chem. 59:4288–4293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsai JR, Chong IW, Chen YH, Hwang JJ, Yin
WH, Chen HL, Chou SH, Chiu CC and Liu PL: Magnolol induces
apoptosis via caspase-independent pathways in non-small cell lung
cancer cells. Arch Pharm Res. 37:548–557. 2014. View Article : Google Scholar
|
|
29
|
Tung YT, Hsu WM, Lee H, Huang WP and Liao
YF: The evolutionarily conserved interaction between LC3 and p62
selectively mediates autophagy-dependent degradation of mutant
huntingtin. Cell Mol Neurobiol. 30:795–806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ciechomska IA and Tolkovsky AM:
Non-autophagic GFP-LC3 puncta induced by saponin and other
detergents. Autophagy. 3:586–590. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Saeki T, Mhashilkar A, Chada S, Branch C,
Roth JA and Ramesh R: Tumor-suppressive effects by
adenovirus-mediated mda-7 gene transfer in non-small cell lung
cancer cell in vitro. Gene Ther. 7:2051–2057. 2000. View Article : Google Scholar
|
|
32
|
Ramnath N, Yu J, Khushalani NI, Gottlieb
RH, Schwarz JK, Iyer RV, Rustum YM and Creaven PJ: Scheduled
administration of low dose irinotecan before gemcitabine in the
second line therapy of non-small cell lung cancer: A phase II
study. Anticancer Drugs. 19:749–752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holohan C, Van Schaeybroeck S, Longley DB
and Johnston PG: Cancer drug resistance: An evolving paradigm. Nat
Rev Cancer. 13:714–726. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fukuda T, Oda K, Wada-Hiraike O, Sone K,
Inaba K, Ikeda Y, Makii C, Miyasaka A, Kashiyama T, Tanikawa M, et
al: Autophagy inhibition augments resveratrol-induced apoptosis in
Ishikawa endometrial cancer cells. Oncol Lett. 12:2560–2566. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sánchez-Flores M, Pásaro E, Bonassi S,
Laffon B and Valdiglesias V: γH2AX assay as DNA damage biomarker
for human population studies: Defining experimental conditions.
Toxicol Sci. 144:406–413. 2015. View Article : Google Scholar
|
|
36
|
Galhardo RS, Hastings PJ and Rosenberg SM:
Mutation as a stress response and the regulation of evolvability.
Crit Rev Biochem Mol Biol. 42:399–435. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamagata T, Kusuhara H, Morishita M,
Takayama K, Benameur H and Sugiyama Y: Improvement of the oral drug
absorption of topotecan through the inhibition of intestinal
xenobiotic efflux transporter, breast cancer resistance protein, by
excipients. Drug Metab Dispos. 35:1142–1148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Furukawa Y and Kikuchi J: Epigenetic
mechanisms of cell adhesion-mediated drug resistance in multiple
myeloma. Int J Hematol. 104:281–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yan LH, Wang XT, Yang J, Lian C, Kong FB,
Wei WY, Luo W, Xiao Q and Xie YB: Reversal of multidrug resistance
in gastric cancer cells by CDX2 downregulation. World J
Gastroenterol. 19:4155–4165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Owen HC, Appiah S, Hasan N, Ghali L,
Elayat G and Bell C: Phytochemical modulation of apoptosis and
autophagy: Strategies to overcome chemoresistance in leukemic stem
cells in the bone marrow microenvironment. Int Rev Neurobiol.
135:249–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Beretta GL, Perego P and Zunino F:
Mechanisms of cellular resistance to camptothecins. Curr Med Chem.
13:3291–3305. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chou HL, Fong Y, Lin HH, Tsai EM, Chen JY,
Chang WT, Wu CY, David Wang HM, Huang HW and Chiu CC: An acetamide
derivative as a camptothecin sensitizer for human non-small-cell
lung cancer cells through increased oxidative stress and JNK
activation. Oxid Med Cell Longev. 2016:91281022016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu HM, Shao LJ, Jiang ZF and Liu RY:
Gemcitabine-induced autophagy protects human lung cancer cells from
apoptotic death. Hai. 194:959–966. 2016.
|
|
44
|
Zhang JW, Zhang SS, Song JR, Sun K, Zong
C, Zhao QD, Liu WT, Li R, Wu MC and Wei LX: Autophagy inhibition
switches low-dose camptothecin-induced premature senescence to
apoptosis in human colorectal cancer cells. Biochem Pharmacol.
90:265–275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li DD, Sun T, Wu XQ, Chen SP, Deng R,
Jiang S, Feng GK, Pan JX, Zhang XS, Zeng YX, et al: The inhibition
of autophagy sensitises colon cancer cells with wild-type p53 but
not mutant p53 to topotecan treatment. PLoS One. 7:e450582012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Amin A, Bajbouj K, Koch A, Gandesiri M and
Schneider-Stock R: Defective autophagosome formation in p53-null
colorectal cancer reinforces crocin-induced apoptosis. Int J Mol
Sci. 16:1544–1561. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q,
Wenk MR, Ong CN, Codogno P and Shen HM: Dual role of
3-methyladenine in modulation of autophagy via different temporal
patterns of inhibition on class I and III phosphoinositide
3-kinase. J Biol Chem. 285:10850–10861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fiorini C, Menegazzi M, Padroni C, Dando
I, Dalla Pozza E, Gregorelli A, Costanzo C, Palmieri M and
Donadelli M: Autophagy induced by p53-reactivating molecules
protects pancreatic cancer cells from apoptosis. Apoptosis.
18:337–346. 2013. View Article : Google Scholar
|
|
49
|
Sun Y, Liu Z, Zou X, Lan Y, Sun X, Wang X,
Zhao S, Jiang C and Liu H: Mechanisms underlying
3-bromopyruvate-induced cell death in colon cancer. J Bioenerg
Biomembr. 47:319–329. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Shin D, Kim EH, Lee J and Roh JL: RITA
plus 3-MA overcomes chemoresistance of head and neck cancer cells
via dual inhibition of autophagy and antioxidant systems. Redox
Biol. 13:219–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Joseph B, Ekedahl J, Lewensohn R,
Marchetti P, Formstecher P and Zhivotovsky B: Defective caspase-3
relocalization in non-small cell lung carcinoma. Oncogene.
20:2877–2888. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zakeri Z and Lockshin RA: Cell death:
History and future. Adv Exp Med Biol. 615:1–11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lin SY, Chang YT, Liu JD, Yu CH, Ho YS,
Lee YH and Lee WS: Molecular mechanisms of apoptosis induced by
magnolol in colon and liver cancer cells. Mol Carcinog. 32:73–83.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Seo JU, Kim MH, Kim HM and Jeong HJ:
Anticancer potential of magnolol for lung cancer treatment. Arch
Pharm Res. 34:625–633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Meschini S, Condello M, Calcabrini A,
Marra M, Formisano G, Lista P, De Milito A, Federici E and Arancia
G: The plant alkaloid voacamine induces apoptosis-independent
autophagic cell death on both sensitive and multidrug resistant
human osteosarcoma cells. Autophagy. 4:1020–1033. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu Y and Levine B: Autosis and autophagic
cell death: The dark side of autophagy. Cell Death Differ.
22:367–376. 2015. View Article : Google Scholar :
|
|
57
|
Shao J, Yang X, Liu T, Zhang T, Xie QR and
Xia W: Autophagy induction by SIRT6 is involved in oxidative
stress-induced neuronal damage. Protein Cell. 7:281–290. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lin W, Yuan N, Wang Z, Cao Y, Fang Y, Li
X, Xu F, Song L, Wang J, Zhang H, et al: Autophagy confers DNA
damage repair pathways to protect the hematopoietic system from
nuclear radiation injury. Sci Rep. 5:123622015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rouschop KM, Ramaekers CH, Schaaf MB,
Keulers TG, Savelkouls KG, Lambin P, Koritzinsky M and Wouters BG:
Autophagy is required during cycling hypoxia to lower production of
reactive oxygen species. Radiother Oncol. 92:411–416. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kaushik S and Cuervo AM: Autophagy as a
cell-repair mechanism: Activation of chaperone-mediated autophagy
during oxidative stress. Mol Aspects Med. 27:444–454. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tokarz P, Piastowska-Ciesielska AW,
Kaarniranta K and Blasiak J: All-trans retinoic acid modulates DNA
damage response and the expression of the VEGF-A and MKI67 genes in
ARPE-19 cells subjected to oxidative stress. Int J Mol Sci.
17:8982016. View Article : Google Scholar :
|
|
63
|
Liu EY, Xu N, O’Prey J, Lao LY, Joshi S,
Long JS, O’Prey M, Croft DR, Beaumatin F, Baudot AD, et al: Loss of
autophagy causes a synthetic lethal deficiency in DNA repair. Proc
Natl Acad Sci USA. 112:773–778. 2015. View Article : Google Scholar : PubMed/NCBI
|