Introduction
Colorectal cancer (CRC) is one of the most commonly
diagnosed malignancies and is a leading cause of cancer-related
mortality worldwide (1,2). Although CRC diagnosis and treatment
strategies have improved over the past decades, CRC is still
considered a major public health concern (3). Despite the development of novel
target agents for CRC therapy, the survival rate still remains
unsatisfactory (4,5).
Tumor necrosis factor (TNF)-related
apoptosis-inducing ligand (TRAIL) is a member of the tumor necrosis
factor family of cytokines. Due to their cancer cell specificity
and marked anticancer effects in a variety of pre-clinical trials,
TRAIL is considered an attractive anticancer agent (6). TRAIL-induced apoptosis is mediated by
binding to cell surface death receptor (DR)5 (also known TRAIL-R2)
or DR4 (also known TRAIL-R1) (7).
The therapeutic potential of soluble TRAIL (sTRAIL) and of
monoclonal antibodies directed against DRs have been extensively
exploited in phase I/II clinical trials (8-10).
However, despite promising preclinical data and favorable toxicity
profiles, these attempts have displayed poor anticancer activity
and limitations in use due to the frequent incidence of primary or
acquired tumor cell resistance, which can occur in CRC (6). Among strategies which can be used to
overcome TRAIL resistance, combined treatment is an attractive
method that enhances TRAIL sensitivity. In our previous study, we
demonstrated that the potent NF-κB inhibitor, parthenolide,
sensitized resistant CRC cells to TRAIL, and combined treatment
with parthenolide represented a possible novel therapeutic strategy
for CRC treatment (11). For
better clinical outcomes, an in-depth analysis of the resistance
mechanisms and the identification of markers to predict response to
TRAIL are warranted.
Lipocalin 2 (LCN2), also known as oncogene 24p3
neutrophil gelatinase-associated lipocalin (NGAL), siderocalin, or
uterocalin, is a 25 kDa secreted glycoprotein that belongs to the
lipocalin superfamily (12). LCN2
is an effective carrier of small, hydrophobic ligands and exerts
bacteriostatic effects by sequestering the siderophoreiron complex
(13,14). In addition to these immune defense
functions, LNC2 is involved in acute phase response, kidney cell
differentiation, erythropoiesis and iron metabolism (13,15-17).
Furthermore, LCN2 is involved in cancer biology. LCN2 has been
identified in colonic epithelial cells under various inflammatory
conditions, inflammatory bowel diseases and colorectal cancers
(15). In vitro
experimental evidence has indicated the inverse association between
LCN2 and cell motility, and LCN2 has been shown to suppress CRC
progression and metastasis (18).
Our previous study demonstrated that LCN2 was negatively associated
with an advanced stage and metastasis (19). Thus, LCN2 may play a differential
role in early and late stage disease. However, a deep understanding
of the role of LCN2 as a molecular target is lacking. Moreover, if
a new perspective of LCN2 as a target for chemoresistance is
suggested, this may provide new insight into the role of LCN2 in
cancer.
The present study was undertaken to investigate the
potential of LCN2 as a target molecule for predicting TRAIL
sensitivity and to explore the molecular mechanisms that regulate
TRAIL resistance by LCN2. Our findings may provide new insight into
TRAIL resistance in CRC and may lead to the development of novel
therapeutic targets for the disease.
Materials and methods
Chemicals and reagents
TRAIL, which was purchased from Pepprotech (Rocky
Hill, NJ, USA), was dissolved in 1X PBS to a concentration of 100
ng/μl. Annexin V-FITC and propidium iodide (PI) were
purchased from Thermo Fisher Scientific (Waltham, MA, USA). The
caspase inhibitor, z-VAD-FMK, and the mitogen-activated protein
kinase (MAPK) inhibitor, SB203580, were obtained from Sigma-Aldrich
(St. Louis, MO, USA). The protein levels of DR4 and DR5 were
analyzed using phycoerythrin (PE)-conjugated DR4 and DR5 obtained
from ProSci Inc. (San Diego, CA, USA).
Patients and tissue specimens
A total of 71 CRC tissues were obtained through the
Biobank of Chonbuk National University Hospital, a member of the
National Biobank of Korea. All patients had a pathological
diagnosis of CRC. The samples were frozen in liquid nitrogen and
stored at -80°C. This study consisted of 28 (39.4%) females and 43
(60.5%) males with a mean age of 63.1 years. All patients provided
written informed consent prior to collecting the tissue samples.
The study protocol was approved by the Institutional Review Boards
of Chonbuk National University Hospital (IRB no. 2016-04-018).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells and human
normal tissue/matched tumor samples using TRIzol reagent (Thermo
Fisher Scientific). Reverse transcription was performed using M-MLV
Reverse Transcriptase (Promega, Madison, WI, USA) according to the
manufacturer’s instructions. qPCR was performed using an ABI 7500
real-time PCR system (Applied Biosystems, Foster City, CA, USA). In
brief, 20 μl of master mix were prepared on ice with 10
μl of 2X SYBR-Green, 1 μl of primers, 2 μl of
DNA and 7 μl of nuclease-free water. The master mix was
initially denatured at 95°C for 10 min followed by 40 cycles of
denaturation at 95°C for 15 sec, annealing and extension at 60°C
for 30 sec. The geometric average Cq value was used to calculate
the relative expression of LCN2 using the 2-ΔΔCq method,
which was normalized to beta-2-microglobulin (B2M) (20). The primers used in this experiment
were as follows: 5′-TCACCTCCGTCCTGTTTAGG-3′ (forward) and
5′-CGAAGTCAGCTCCTTGGTTC-3′ (reverse) for LCN2;
‘5-CACAGCAATGGGAACATAGC-3′ (forward) an ‘5-CAG GGACTTCTCTCTTCTTC-3′
(reverse) for DR4; 5′-CTGAAA GGCATCTGCTCAGGTG-3′ (forward) and
5′-CAGAGTCT GCA TTAC CTTCTAG-3′ (reverse) for DR5; 5′-CCTGAATTG
CTATGTGTCTGGG-3′ (forward) and 5′-TGATGCTGCTT ACATGTCTCGA-3′
(reverse) for B2M. The LCN2 expression level was subdivided into he
low and high groups according to the mean value.
Cell culture
The human colorectal cancer cell lines, HT-29,
DLD-1, SW480, HCT116 and SW620, were purchased from the American
Type Culture Collection (ATCC, Manassas, VA, USA). The cells were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (FBS), 100 units penicillin and 100 units streptomycin in a
humidified 5% CO2 atmosphere at 37°C.
Small interfering RNA (siRNA) for the
inhibition of gene expression
The siRNA sequences used for the targeted silencing
of the LCN2 gene (NCBI Ref Seq NM_005564.4) were from Ambion
(Austin, TX, USA), and those for the DR5 gene (NCBI Ref Seq
NM_003842) were from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). LCN2 siRNA, DR5 siRNA and scrambled siRNA as the negative
control (Ambion) were transfected into the HT-29 and DLD-1 cells
using TransiT-X2® transfection reagent (Mirus Bio,
Madison, WI, USA) according to the manufacturer’s instructions.
Reverse transcription-PCR (RT-PCR)
Total RNA was isolated from the cultured cells using
TRIzol reagent (Invitrogen/ Thermo Fisher Scientific), and cDNA was
synthesized with Super Script II reverse-transcriptase
(Invitrogen/Thermo Fisher Scientific) according to the
manufacturer’s instructions. GAPDH expression was used as an
internal control. The following primer sequences were used:
5′-TCACCTCCGTCC TGTTTAGG-3′ (forward) and 5′-CGAAGTCAGCTCCTTGG
TTC-3′ (reverse) for LCN2, and 5′-CAGGTGTCAACATGTT GTCC-3′
(forward) and 5′-ATCGAAGCACTGTCTCAGAG-3′ (reverse) for DR5, which
generate 242 and 136 bp products, respectively. Following initial
denaturation at 95°C for 1 min, PCR was performed for various
cycles (30 sec at 94°C, 1 min at annealing temperature and 2 min at
72°C) using Taq polymerase. The reaction products (10 μl)
were separated on a 2% agarose gel and stained with Redsafe™
(Intron, Daejeon, Korea). The DNA band intensity was analyzed by
densitometry using an NαBI imager (Neogene Science, Suwon,
Korea).
Quantification of DR expression on the
cell surface
To quantify the cell surface expression of DR4 and
DR5, the cells were harvested by trypsinization, washed in PBS, and
incubated for 30 min at 4°C with 500 ng/test of phyco-erythrin
(PE)-conjugated monoclonal anti-human DR4 (cat. no. 12-6644-41) and
DR5 (cat. no. 12-9908-42) antibodies (eBiosciences, San Diego, CA,
USA). Non-immune mouse IgG was used as the negative control.
Fluorescence was measured using a BD LSR flow cytometer and
processed with CellQuest software for analysis.
Cell viability assay
The DLD-1 and HT-29 cells were plated at a density
of 1.0×104 cells per well in 96-well plates. Following
transfection with scrambled siRNA or LCN2 siRNA, the medium was
removed, and 200 μl of fresh medium, including various
concentrations of TRAIL (10, 50, 100, 300 and 500 ng/ml) were
added. Subsequently, 20 μl of 3-(4,5-dimethylthiazol-2yl)-2,
5-diphenyltetrazolium bromide (MTT, 2.5 mg dissolved in 50
μl of DMSO; Sigma-Aldrich) were added to each well.
Following incubation for 4 h at 37°C, the culture medium containing
MTT was removed and 200 μl of DMSO were added. This was
followed by shaking until the crystals were dissolved. Viable cells
were detected by measuring the absorbance at 570 nm using a
microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Detection of apoptosis
Apoptotic cell death was determined by staining the
cells with Annexin V-FITC (Ex/Em, 488/519 nm). In brief,
1×105 cells in a 60-mm culture dish were transfected
with scrambled siRNA or LCN2 siRNA. After 48 h, the cells were
pre-treated with Z-VAD-FMK (10 nM) or SB203580 (50 μM) for 1
h, and 50 ng/ml of TRAIL was then added to the transfected cells
for 24 h. The cells were washed twice with cold PBS and then
resuspended in 500 μl of binding buffer (10 mM HEPES/NaOH pH
7.4, 140 mM NaCl, and 2.5 mM CaCl2) at a concentration
of 1×106 cells/ml. Annexin V-FITC (5 μl) and PI
(1 μg/ml) was then added, and the cells were analyzed with a
BD FACSCalibur™ (BD Biosciences, San Jose, CA, USA).
Protein extraction and western blot
analysis
The DLD-1 and HT-29 cells were harvested by
resolving in RIPA buffer (50 mM Tris-HCl,150 mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS and protease inhibitors)
and were centrifuged at 1,3870 × g at 4°C for 30 min. Following
centrifugation, supernatants were used as whole cell extracts. The
protein concentration in the cell lysates or tissue lysates was
measured using a Protein Quantification kit from Bio-Rad (Hercules,
CA, USA). Subsequently, 50 μg of protein or 30 μg of
protein per lane were loaded onto 8-15% SDS-polyacrylamide gels.
After transferring and blocking using 3% of bovine albumin serum,
the polyvinylidene difluoride (PVDF) membranes were probed with
various antibodies [anti-LCN2 (1:1,000; AF1757; R&D
Systems,Minneapolis, MN, USA), anti-DR4 (1:1,000; #1167; ProSci Ψ™,
Poway, CA, USA), anti-DR5 (1:1,000; #2019; ProSci Ψ™),
anti-Fas-associated death-domain-like IL-1β-converting enzyme
(FLICE)-inhibitory protein (FLIP, 1:1,000; #8510; Cell Signaling
Technology, Danvers, MA, USA), anti-Bid (1:1,000; SC-11423; Santa
Cruz Biotechnology), anti-caspase-8 (1:1,000; SC-73526; Santa Cruz
Biotechnology), anti-caspase-3 (1:1,000; SC-7148; Santa Cruz
Biotechnology), anti-cleaved caspase-3 (1:1,000; #9661; Cell
Signaling Technology), anti-poly(ADP-ribose) polymerase (PARP,
1:1,000, SC-7150; Santa Cruz Biotechnology), anti-Bcl-xL (1:1,000;
SC-8392; Santa Cruz Biotechnology), anti-Bcl-2 (1:1,000; SC-783;
Santa Cruz Biotechnology), anti-cytochrome c (1:1,000,
SC-65396; Santa Cruz Biotechnology), anti-caspase-9 (1:1,000;
#9502S; Cell Signaling Technology), anti-phospho-ERK (1:1,000;
#9106; Cell Signaling Technology), anti-phospho-p38 (1:1,000,
#4511; Cell Signaling Technology), anti-phospho-JNK (1:1,000;
#4668; Cell Signaling Technology), anti-C/EBP homologous protein
(CHOP, 1:1,000; #2895; Cell Signaling Technology) and anti-actin
(1:2,000; A2066; Sigma-Aldrich) antibodies] in 4°C for overnight.
HRP-conjugated goat anti-rabbit IgG (SC-2004; Santa Cruz
Biotechnology), goat anti-mouse (SC-2005; Santa Cruz Biotechnology)
and mouse anti-goat (SC-2354; Santa Cruz Biotechnology) secondary
antibodies were used at a concentration of 1:3,000 for 1 h at room
temperature. Binding of the antibody to the antigen was detected
using enhanced ECL prime (GE Healthcare, NJ, USA) and was captured
and analyzed by the Las-3000 luminescent Image Analyzer (Fuji Film,
Tokyo, Japan).
Statistical analysis
The association between the LCN2 and DR4/5 levels in
the human specimens was analyzed using the Chi-square
(χ2) test. The Student’s t-test (for differences between
2 groups) or one-way analysis of variance (ANOVA) with Tukey’s test
were used to analyze differences between more than 2 groups. The
data are presented as the means ± SD of at least 3 independent
experiments. All data were entered into Microsoft Excel 5.0, and
GraphPad Prism 5.0 was used. A probability (P)-value <0.05 was
considered to indicate a statistically significant difference.
Results
LCN2 and DR5 have a negative association
in human CRC tissues and cells
To identify the association between LCN2 and DRs in
patients with CRC, the expression levels of LCN2, DR4 and DR5 in
the specimens from patients with CRC and CRC cell lines were
examined. First, the mRNA levels of LCN2, DR4 and DR5 in 71 frozen
tissues of patients with CRC was measured by RT-qPCR, and the
association of the mRNA expression of each of the DRs with mRNA
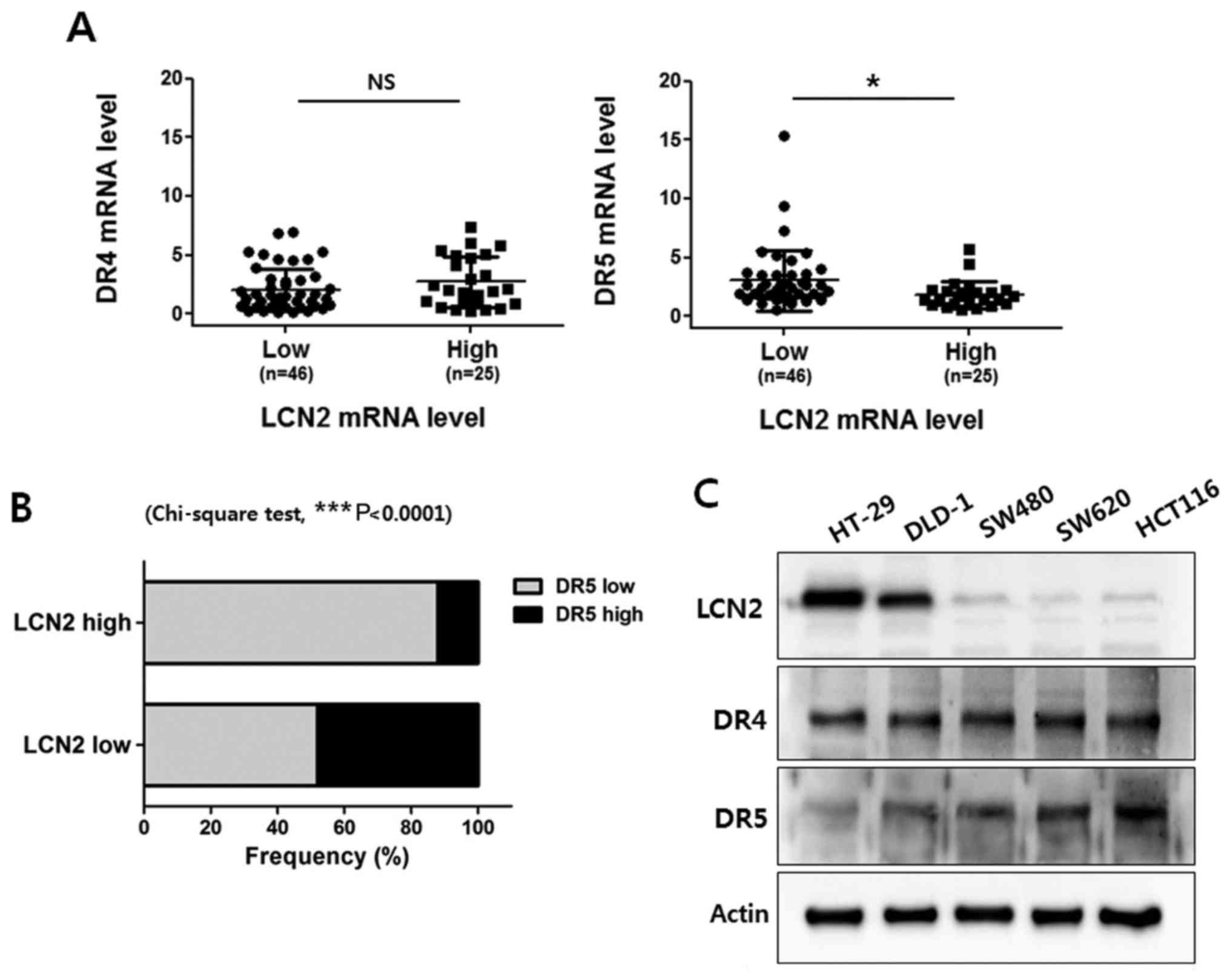
expression of LCN2 was analyzed. As shown in Fig. 1A, we did not observe any
association between the DR4 and LCN2 mRNA levels in the CRC
tissues. Of note, the LCN2 level was found to be negatively
associated with the DR5 expression level in the CRC tissue samples.
Of the 71 patients with CRC, the DR5 level in the LCN2 low
expression group was 0.558-15.380 (3.015±2.57) and the DR5 level in
the LCN2 high expression group was 0.529-5.679 (1.823±1.168).
Moreover, our analysis indicated that LCN2 expression was
negatively regulated along with DR5 expression in the CRC specimens
(Fig. 1B).
Subsequently, western blot analysis was performed to
determine the protein expression levels of DR4, DR5 and LCN2 in 5
CRC cell lines (HT-29, DLD-1 SW480, SW620 and HCT116) (Fig. 1C). The protein levels of LCN2 were
higher in the HT-29 and DLD-1 cell lines than in the SW480, HCT116
and SW620 cell lines. However, the protein levels of DR5 were
higher in the SW480, HCT116 and SW620 cell lines than in the HT-29
and DLD-1 cell lines. Conversely, all the cell lines exhibited
similar levels of DR4. Taken together, these findings suggest that
LCN2 is negatively associated with DR5 in CRC.
Downregulation of LCN2 enhances DR5
expression and sensitizes the cells to TRAIL-induced apoptosis
To define the role of LCN2 in the regulation of DR5,
the DLD-1/ HT-29 and HCT116 cells were employed as representatives
of DR5-deficient cells and DR5-enriched cells, respectively. As
shown in Fig. 2A, the DR5 protein
and mRNA levels were increased by the silencing of LCN2 in the
DLD-1/HT-29 cells. On the contrary, the DR4 protein level was not
affected by transfection with LCN2 siRNA. Of note, the knockdown of
DR5 in the HCT116 cells did not affect the regulation of LCN2
expression (Fig. 2B). These
results were confirmed by flow cytometry. The expression of DR4 on
the cell surface was not altered by transfection with LCN2 siRNA
compared to transfection with scrambled siRNA; however, DR5
expression was markedly increased by the silencing of LCN2 in the
DLD-1 and HT-29 cells (Fig. 2C).
These results indicate that the inhibition of LCN2 promotes the
expression of DR5, and that LCN2 regulates DR5 as an upstream
factor from DR5 in TRAIL-induced apoptosis.
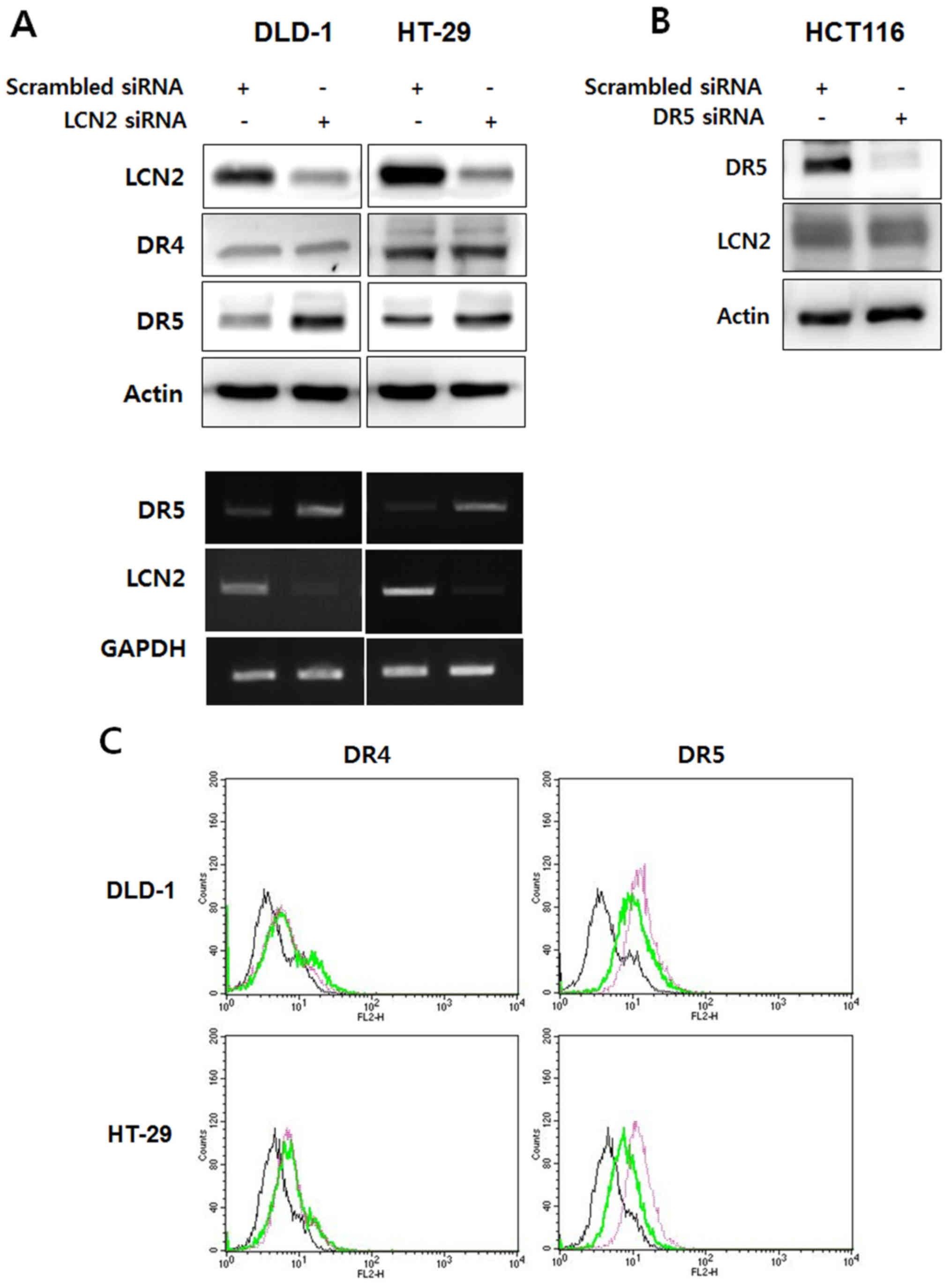 | Figure 2Knockdown of LCN2 can lead to the
upregulation of DR5 in TRAIL-resistant CRC cells. (A) DLD-1 and
HT-29 cells, which have TRAIL resistant characteristics, were
transfected with LCN2 siRNA and the endogenous levels of DR4/5 and
LCN2 were then detected by western blot analysis and RT-PCR. (B)
HCT116 cells with TRAIL sensitive characteristics were transfected
with DR5 siRNA. LCN2 and DR5 levels were also evaluated by western
blot analysis. (C) The cell surface expression of DR4 and DR5 was
determined by flow cytometry using PE-conjugated DR4/5 antibody. In
the histograms the live cell populations are presented as follows:
Left panels (DR4): black line, scramble siRNA + IgG antibody; green
line, scramble siRNA + DR4 antibody; pink line, LNC2 siRNA + DR4
antibody. Right panels (DR5): black line, scramble siRNA + IgG
antibody; green line, scramble siRNA + DR5 antibody; pink line,
LNC2 siRNA + DR5 antibody. LCN2, lipocalin 2; DR, death receptor;
TRAIL, TNF-related apoptosis-inducing ligand. |
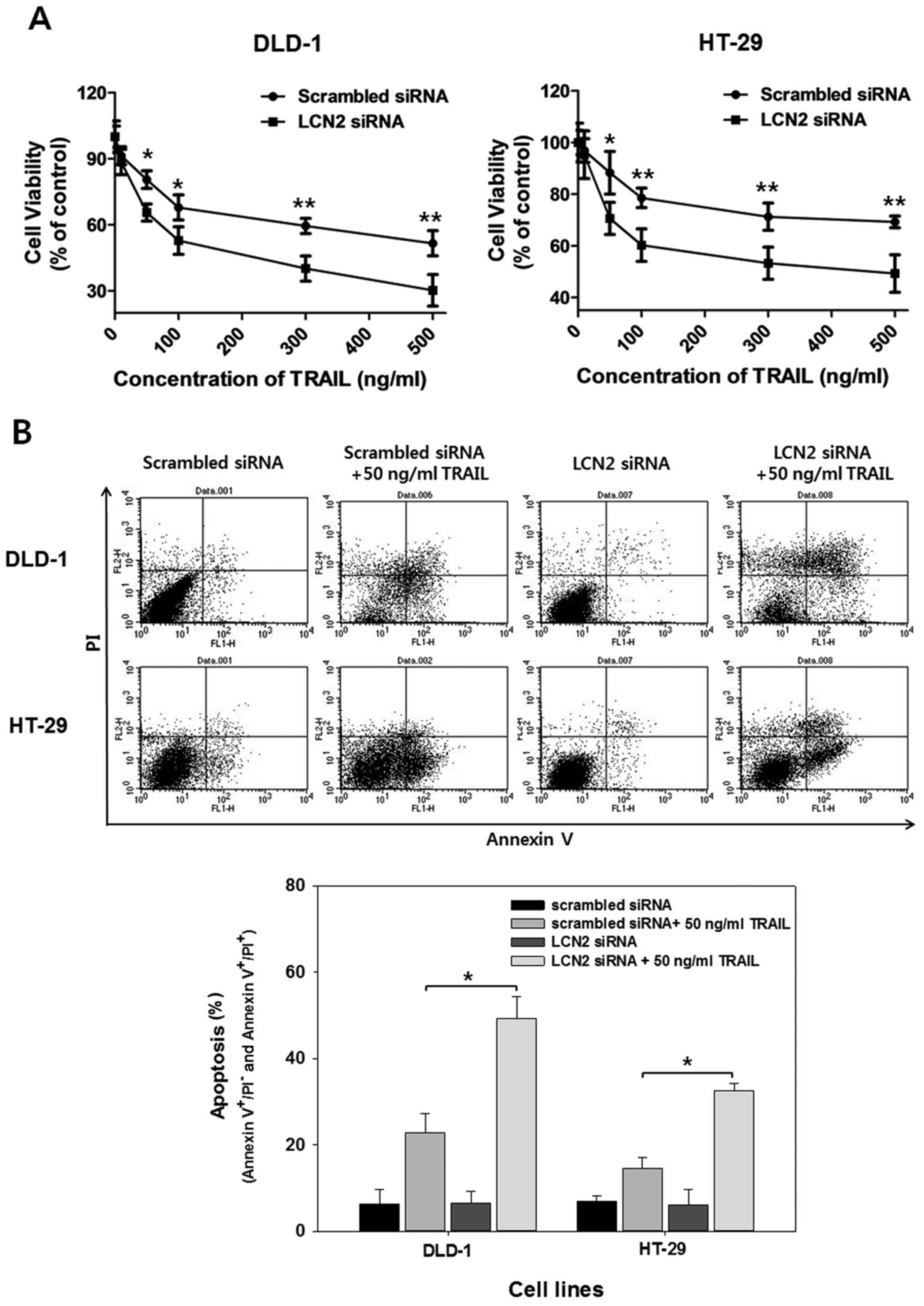
We also identified whether DR5 upregulation by LCN2
silencing affects the TRAIL-induced apoptosis of DR5-deficient
cells. Cell viability assay was performed following treatment with
various concentrations of TRAIL (50, 100, 300 and 500 ng/ml) for 24
h followed by transfection with scrambled siRNA or LCN2 siRNA. As
shown by the results of MTT assay, TRAIL-induced cell death was
increased by the silencing of LCN2 compared to the cells
transfected with scrambled siRNA in both cell lines (Fig. 3A).
To ascertain the above-mentioned observations,
apoptotic cell death analysis was performed using Annexin V/PI
staining. As shown in Fig. 3B, the
cells transfected with LCN2 siRNA exhibited greater apoptotic cell
death when treated with 50 ng/ml of TRAIL compared to the cells
transfected with scrambled siRNA. Moreover, the quantification of
apoptotic cell death was carried out by counting cells in the lower
right and upper right quadrants. The results revealed that
TRAIL-induced apoptosis was significantly increased by the
silencing of LCN2 from 22.8 to 49.2% in the DLD-1 cells and from
14.6 to 32.5% in the HT-29 cells. These results indicate that the
downregulation of LCN2 enhances DR5 expression and promotes the
TRAIL-induced apoptosis of TRAIL-resistant CRC cells.
Downregulation of LCN2 enhances
TRAIL-mediated apoptotic signaling by engaging the extrinsic
pathway
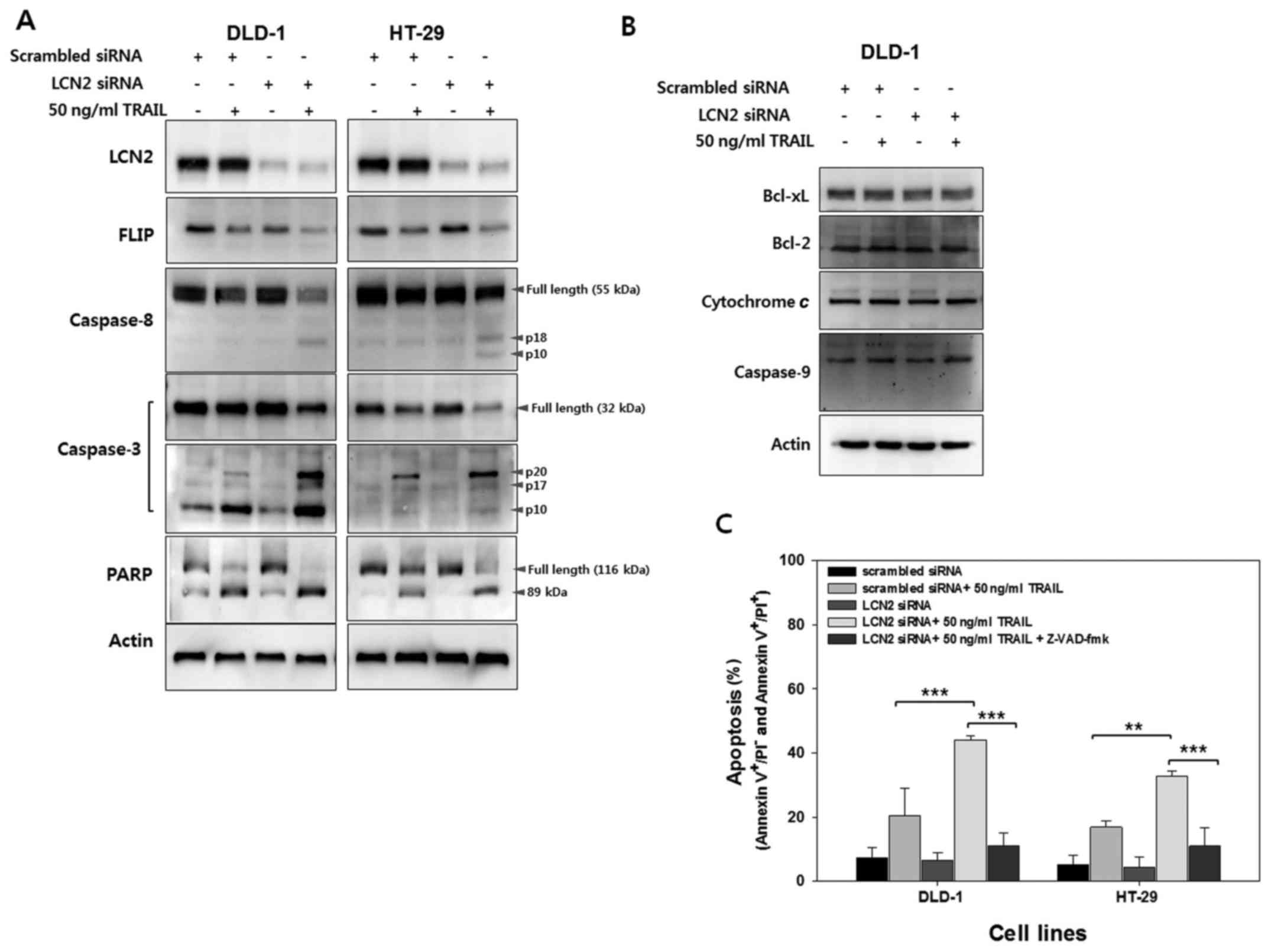
Having shown LCN2-related sensitization to
TRAIL-induced apoptosis, we then investigated the regulation of the
molecules involved in death signaling by western blot analysis
(Fig. 4A). FLIPs are
anti-apoptotic proteins that can be recruited to the death-inducing
signaling complex (DISC) (21). As
FLIP contains two death effector domains (DED) and an inactive
caspase domain, FLIP can inhibit death receptor-mediated apoptosis
by binding to Fas-associated death domain (FADD) or caspase-8
(21,22). As shown in Fig. 4A, lower levels of FLIP were
observed following treatment with TRAIL in the cells in which LCN2
was silenced (LCN2 siRNA + 50 ng/ ml TRAIL) compared to the control
cells (scrambled siRNA + 50 ng/ml TRAIL). These results suggest
that LCN2 affects FLIP to inhibit death receptor-mediated cell
death (Fig. 4A, 2nd panel from the top). Thus, the decreased FLIP
expression by treatment with TRAIL following the silencing of LCN2
can induce the processing of caspase-8 activation. As expected, the
cleavage of caspase-8 was much higher in the TRAIL-treated cells in
which LCN2 was silenced than in the TRAIL-treated cells transfected
with the scrambled siRNA (Fig. 4A, 3rd panel from the top). To
identify the next caspase cascade, the expression level of
caspase-3 was analyzed using full-length caspase-3 and cleaved
caspase-3 antibody, respectively. During the activation of
caspase-3, the cleaved form of caspase-3 increases, while the full
length decreases. As shown in the 4th and 5th panels (from the top)
in Fig. 4A, caspase-3 activation
was markedly induced in the TRAIL-treated cells in which LCN2 was
silenced. These results indicate that the silencing of LCN2
enhances the TRAIL-induced cleavage of caspase-8 and caspase-3 in
their active forms in TRAIL-resistant CRC cells. Following the
activation of caspases during apoptosis, the cleavage of PARP
occurs by several caspases, including caspase-3, which cleaves
113-kDa PARP into 89- and 24-kDa polypeptides (23,24).
Thus, in this study, to confirm the effects of LCN2 regulation on
PARP cleavage, the cells were treated with 50 ng/ml of TRAIL
following transfection with scrambled or LCN2 siRNA. As shown in
the 6th panel (from the top) in Fig.
4A, a higher level of cleaved PARP following treatment with
TRAIL was observed in the cells in which LCN2 was silenced compared
with the scrambled siRNA-transfected cells treated with TRAIL.
To determine whether the TRAIL-induced apoptosis
resulting from LCN2 silencing is dependent on the initiation of the
extrinsic or intrinsic pathways, molecules involved in the
intrinsic pathway were examined by western blot analysis (Fig. 4B). The expression of Bid, which
links the extrinsic and intrinsic pathways, was not altered by LCN2
regulation with or without TRAIL treatment. Moreover,
mitochondria-related anti-(Bcl-2 and Bcl-xL) or pro-apoptotic
(cytochrome c and caspase-9) molecules also exhibited
similar levels with or without LCN2 regulation. Collectively, these
data provide evidence that the downregulation of LCN2 enhances
TRAIL-induced apoptosis through the extrinsic apoptotic
pathway.
To further evaluate the findings shown in Fig. 4A, the cells in which LCN2 was
silenced were pre-treated with Z-VAD-FMK, a general caspase
inhibitor, and apoptotic cell death was evaluated by Annexin V
assay. As shown in Fig. 4C, the
apoptotic cell death induced by TRAIL treatment of the cells in
which LCN2 was silenced decreased following pretreatment with
Z-VAD-FMK. These results suggest that the downregulation of LCN2
sensitizes TRAIL-resistant CRC cells to TRAIL and induces
TRAIL-mediated apoptosis related to the extrinsic pathway.
p38 MAPK and CHOP are involved in
LCN2-related TRAIL sensitivity
Several studies have demonstrated that the
regulation of DR5 and TRAIL sensitivity is mediated by MAPK
signaling (25-28). Thus, in this study, to investigate
the possible role of MAPK signaling in the enhancement of the
sensitivity of CRC cells to TRAIL by LCN2 silencing, LCN2 was
downregulated by transfection with targeting siRNA in
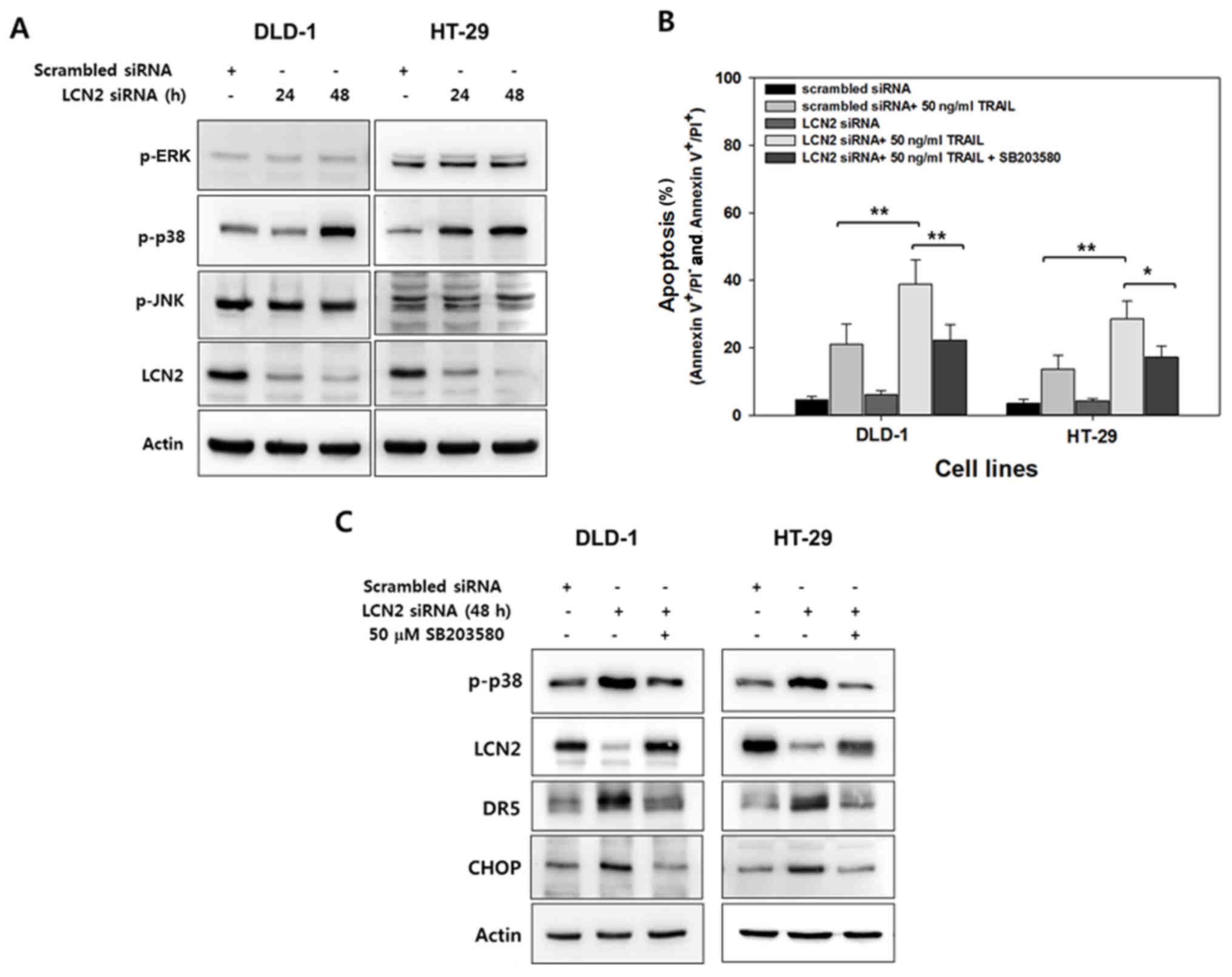
TRAIL-resistant cells in a time-dependent manner. The active form
of MAPKs was then determined by western blot analysis with
antibodies that recognize the phosphorylated forms of these
kinases. As shown in Fig. 5A, only
the levels of p-p38 MAPK exhibited a marked increase following the
silencing of LCN2 in both cell lines.
To confirm the association between p38 MAPK with the
alteration of TRAIL sensitivity by LCN2 regulation, the cells were
pre-treated with SB203580, a p38 MAPK specific inhibitor, and
apoptosis was evaluated by Annexin V/PI (Fig. 5B). The percentage of LCN2-silenced
cells treated with TRAIL that underwent apoptotic death was
approximately 38% in the DLD-1 cells and 27% in the HT-29 cells. Of
note, following treatment with SB203580, these percentages
significantly decreased down to 22% in the DLD-1 cells and 17% in
the HT-29 cells (Fig. 5B). Western
blot analysis was also performed to examine the association between
p38 MAPK activity and LCN2 as regards TRAIL sensitivity (Fig. 5C). The levels of phosphorylated p38
MAPK were induced by the silencing of LCN2, and they were also
reduced by pre-treatment with SB203580. Conversely, downregulated
LCN2 by targeted siRNA was recovered by p38 specific inhibitor in
both cell lines. The level of DR5 was also altered by LCN2
silencing and pretreatment with SB203580. These results suggest
that LCN2 is responsible for TRAIL sensitivity and is crosslinked
with p38 MAPK.
CHOP, a member of the C/EBP family of transcription
factors, serves as a substrate for p38 MAPK and is a key regulator
of DR5 (29,30). We thus hypothesized that CHOP is
regulated by LCN2 silencing and p38 MAPK inhibition. To verify
this, when LCN2 was downregulated by targeting siRNA, the level of
CHOP substantially increased. Conversely, CHOP expression decreased
by pre-treatment with the selective inhibitor of p38 MAPK in the
TRAIL-resistant CRC cells (Fig.
5C). Overall, these data demonstrate that LCN2 silencing
enhances TRAIL sensitivity through the regulation of p38
MAPK/CHOP/DR5 signaling and the proposed molecular mechanisms of
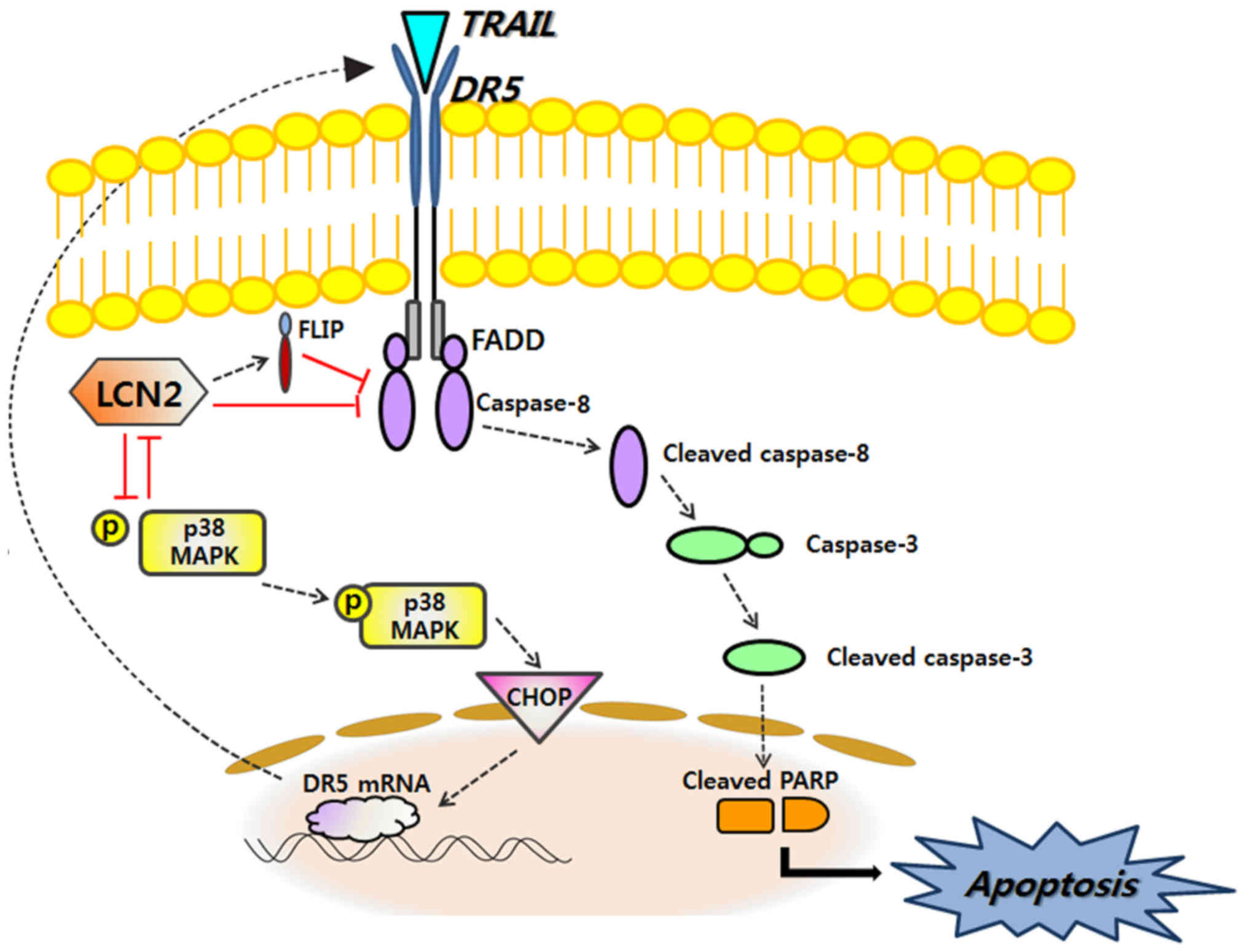
LCN2 on TRAIL resistance are illustrated in Fig. 6.
Discussion
TRAIL has been identified as a cytotoxic cancer cell
specific ligand with no effect on normal cells. However, the
clinical utility of TRAIL has been limited due to multiple
mechanisms of TRAIL resistance (31). A substantial number of cancer cells
are resistant to TRAIL, particularly in highly malignant tumors,
such as pancreatic cancer, melanoma, neuroblastoma and CRC
(32). Moreover, the repeated
application of TRAIL to susceptible cancer cells results in the
selection and expansion of TRAIL-resistant cells with acquired
resistance (33,34). Thus, identifying the mechanisms
responsible for TRAIL resistance will not only provide insight
regarding transduction of the death signal, but is also essential
for designing strategies to overcome resistance to TRAIL for future
clinical applications.
Several antibodies targeting DR4/5 and recombinant
human TRAIL have been developed and tested for clinical use in
cancer therapy. In particular, dulanermin (recombinant human
TRAIL), mapatumumab (DR4 targeting monoclonal antibody), drozitumab
(DR5 targeting monoclonal antibody) and conatumumab (DR5 targeting
monoclonal antibody) have generally been well tolerated and appear
to be safe in patients with CRC (35-38).
However, the efficacy results available thus far have been
disappointing. For this reason, agonists of DR4/5 have been applied
for combined treatment with other chemotherapeutic agents, although
these attempts have not demonstrated significantly improved
efficacy over what is expected (35,38).
Therefore, the identification of a biomarker that can predict the
sensitivity of TRAIL and increase the understanding of the TRAIL
resistance mechanisms may aid in the development of targeted
therapies. In this study, we provide evidence that LCN2 has
potential for use as a prediction marker of TRAIL sensitivity. In
addition, we demonstrate that LCN2 is a key mediator of resistance
to TRAIL in CRC.
The expression levels of DR4 and DR5 determine the
cell fate in response to TRAIL treatment. Several studies have
indicated that the upregulation of DRs can sensitize cells to
TRAIL-induced cell death (26,27,39,40).
In this study, we found that the silencing of LCN2 promoted
TRAIL-induced apoptosis through the upregulation of DR5 at both the
mRNA and protein level. Moreover, we demonstrated that
LCN2-dependent DR5 regulation contributed to the sensitizing
effects of TRAIL-induced apoptosis and was an upstream event that
negatively affected DR5 expression.
Numerous mechanisms have been proposed to explain
the induction of DR5, including p53 induction, reactive oxygen
species (ROS) regeneration and MAPK activation, which are largely
dependent on cell type (30,41).
In particular, MAPK activation has been suggested to play a key
role in DR5 induction (42,43).
Thus, in this study, we investigated whether MAPK plays a role in
DR5 regulation by LCN2. Our findings indicated that only p38 MAPK
was involved in DR5 upregulation by LCN2 silencing in
TRAIL-resistant CRC cells. In addition, we provide evidence that
the inhibition of p38 MAPK with the specific inhibitor, SB203580,
can counteract the increased expression of LCN2, and a similar
result was also observed in the analysis of apoptotic cell death
using Annexin V/PI staining. These results clearly identify a high
degree of crosstalk between LCN2 and p38 MAPK. Through these
observations, we provide new insight into the role of LCN2 in TRAIL
resistance.
CHOP is a 29 kDa protein composed of an N-terminal
transcriptional activation domain and a C-terminal basicleucine
zipper (bZIP) domain. In 1996, Wang and Ron demonstrated that CHOP
serves as a link between a specific stress-activated protein
kinase, p38, and cellular growth and differentiation (29). Moreover, Bruhat et al
reported that p38 MAPK regulated the expression of CHOP at the
post-transcriptional level (44).
For that reason, CHOP expression has been considered to play a
crucial role in p38 MAPK-regulated DR5 and TRAIL sensitivity. In
the present study, we identified that the activation of p38 MAPK by
LCN2 silencing induced CHOP expression. Our observations are
supported by those of earlier study showing the role of CHOP in
upregulating DR5 and enhancing the effects of TRAIL by p38 MAPK
activation (29,44). Inversely, the selective inhibition
of p38 MAPK by SB203580 recovered the LCN2 level and abolished CHOP
expression. Taken together, these results indicate that CHOP is
regulated by p38 MAPK as a transcription factor. CHOP was also
negatively regulated by LCN2 due to the crosstalk between LCN2 and
p38 MAPK.
LCN2 was originally considered a biomarker of
inflammation, ischemia, infection and kidney damage due to its
function to capture bacterial siderophores (45). Moreover, the mRNA and protein
levels of LCN2 have been shown to be elevated in various types of
cancer, including CRC, demonstrating that LCN2 may serve as a
cancer biomarker (46). In our
previous study, we suggested the possibility of using LCN2 as a
diagnostic and prognostic marker by classifying the expression
pattern by CRC stage and metastasis using patient specimens
(19). In addition, we identified
that LCN2 negatively regulates cell proliferation and the EMT
process through glycolysis using human CRC cells (19). Although the functional role of LCN2
in tumorigenesis and progression in cancer has been investigated,
studies linking LCN2 to various cancer pathologies, such as
chemoresistance are lacking. In this study, we confirmed that LCN2
is a key mediator of cancer resistance to TRAIL. Using human
specimens and CRC cell lines, the level of LCN2 was found to be
inversely associated with DR5 and cellular susceptibility to
TRAIL-induced apoptosis. Furthermore, we demonstrate that
LCN2-related resistance to TRAIL signaling appears to directly
regulate the extrinsic apoptotic pathway and crosstalk with p38
MAPK, which is responsible for DR5-mediated apoptosis (Fig. 6). Thus, in this study, for the
first time, at least to the best of our knowledge, we provide a new
algorithm of LCN2 as a potential biomarker for the prediction of
tumor resistance to TRAIL or the DR5 targeted therapies.
To our knowledge, this study is the first study to
identify the possibility of using LCN2 as a prediction marker of
TRAIL sensitivity in CRC. Using in vitro experiments, we
observed that LCN2 negatively regulates p38 MAPK-mediated DR5 and
mediates TRAIL-induced apoptosis via the extrinsic apoptotic
pathway. Accordingly, we demonstrate a new role of LCN2 as a sensor
for TRAIL response in CRC. These findings provide new insight into
the role of LCN2 and the underlying molecular mechanisms of TRAIL
resistance in CRC, indicating LCN2 as a potential prediction marker
and therapeutic target.
Funding
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Science, ICT and Future Planning
(NRF-2015R1C1A2A01055803) and by the Fund of Biomedical Research
Institute, Chonbuk National University Hospital.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
SLK and SWK conceived and planned the experiments.
ISM and YRP analyzed the mRNA levels in the human CRC samples. SLK
and ISM carried out all the in vitro experiments. STL and
SWK contributed to the interpretation of the results. SLK and ISM
took the lead in writing the manuscript. All authors provided
critical feedback and helped shape the research, analysis and
manuscript. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Institutional
Review Boards of Chonbuk National University Hospital (IRB no.
2016-04-018). All patients provided written informed consent prior
to collecting the tissue samples.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
3
|
Hara K, Beppu T, Kimura M, Fujita Y,
Takata T, Nishio K and Ono N: Influence of novel supramolecular
substance, [2] rotaxane, on the caspase signaling pathway in
melanoma and colon cancer cells in vitro. J Pharmacol Sci.
122:153–157. 2013. View Article : Google Scholar
|
|
4
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng L, Ren W, Xie L, Li M, Liu J, Hu J,
Liu BR and Qian XP: Anti-EGFR MoAb treatment in colorectal cancer:
Limitations, controversies, and contradictories. Cancer Chemother
Pharmacol. 74:1–13. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hellwig CT and Rehm M: TRAIL signaling and
synergy mechanisms used in TRAIL-based combination therapies. Mol
Cancer Ther. 11:3–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han B, Yao W, Oh YT, Tong JS, Li S, Deng
J, Yue P, Khuri FR and Sun SY: The novel proteasome inhibitor
carfilzomib activates and enhances extrinsic apoptosis involving
stabilization of death receptor 5. Oncotarget. 6:17532–17542. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashkenazi A, Holland P and Eckhardt SG:
Ligand-based targeting of apoptosis in cancer: The potential of
recombinant human apoptosis ligand 2/Tumor necrosis factor-related
apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol.
26:3621–3630. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koehler BC, Jäger D and Schulze-Bergkamen
H: Targeting cell death signaling in colorectal cancer: Current
strategies and future perspectives. World J Gastroenterol.
20:1923–1934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kelley SK, Harris LA, Xie D, Deforge L,
Totpal K, Bussiere J and Fox JA: Preclinical studies to predict the
disposition of Apo2L/tumor necrosis factor-related
apoptosis-inducing ligand in humans: Characterization of in vivo
efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther.
299:31–38. 2001.PubMed/NCBI
|
|
11
|
Kim SL, Liu YC, Park YR, Seo SY, Kim SH,
Kim IH, Lee SO, Lee ST, Kim DG and Kim SW: Parthenolide enhances
sensitivity of colorectal cancer cells to TRAIL by inducing death
receptor 5 and promotes TRAIL-induced apoptosis. Int J Oncol.
46:1121–1130. 2015. View Article : Google Scholar
|
|
12
|
Skerra A: Lipocalins as a scaffold.
Biochim Biophys Acta. 1482:337–350. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang J, Goetz D, Li JY, Wang W, Mori K,
Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, et al: An
iron delivery pathway mediated by a lipocalin. Mol Cell.
10:1045–1056. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Devireddy LR, Gazin C, Zhu X and Green MR:
A cell-surface receptor for lipocalin 24p3 selectively mediates
apoptosis and iron uptake. Cell. 123:1293–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nielsen BS, Borregaard N, Bundgaard JR,
Timshel S, Sehested M and Kjeldsen L: Induction of NGAL synthesis
in epithelial cells of human colorectal neoplasia and inflammatory
bowel diseases. Gut. 38:414–420. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanjeevani S, Pruthi S, Kalra S, Goel A
and Kalra OP: Role of neutrophil gelatinase-associated lipocalin
for early detection of acute kidney injury. Int J Crit Illn Inj
Sci. 4:223–228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan C, Yuanjie T, Zhengqun X, Jiayan C and
Kongdan L: Neutrophil gelatinase-associated lipocalin attenuates
ischemia/ reperfusion injury in an in vitro model via autophagy
activation. Med Sci Monit. 24:479–485. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee HJ, Lee EK, Lee KJ, Hong SW, Yoon Y
and Kim JS: Ectopic expression of neutrophil gelatinase-associated
lipocalin suppresses the invasion and liver metastasis of colon
cancer cells. Int J Cancer. 118:2490–2497. 2006. View Article : Google Scholar
|
|
19
|
Kim SL, Lee ST, Min IS, Park YR, Lee JH,
Kim DG and Kim SW: Lipocalin 2 negatively regulates cell
proliferation and epithelial to mesenchymal transition through
changing metabolic gene expression in colorectal cancer. Cancer
Sci. 108:2176–2186. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Irmler M, Thome M, Hahne M, Schneider P,
Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C,
et al: Inhibition of death receptor signals by cellular FLIP.
Nature. 388:190–195. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Scaffidi C, Schmitz I, Zha J, Korsmeyer
SJ, Krammer PH and Peter ME: Differential modulation of apoptosis
sensitivity in CD95 type I and type II cells. J Biol Chem.
274:22532–22538. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tewari M, Quan LT, O’Rourke K, Desnoyers
S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS and Dixit VM:
Yama/CPP32 beta, a mammalian homolog of CED-3, is a
CrmA-inhibitable protease that cleaves the death substrate
poly(ADP-ribose) polymerase. Cell. 81:801–809. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim EY, Ryu JH and Kim AK: CAPE promotes
TRAIL-induced apoptosis through the upregulation of TRAIL receptors
via activation of p38 and suppression of JNK in SK-Hep1
hepatocellular carcinoma cells. Int J Oncol. 43:1291–1300. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lepage C, Léger DY, Bertrand J, Martin F,
Beneytout JL and Liagre B: Diosgenin induces death receptor-5
through activation of p38 pathway and promotes TRAIL-induced
apoptosis in colon cancer cells. Cancer Lett. 301:193–202. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ohtsuka T, Buchsbaum D, Oliver P, Makhija
S, Kimberly R and Zhou T: Synergistic induction of tumor cell
apoptosis by death receptor antibody and chemotherapy agent through
JNK/p38 and mitochondrial death pathway. Oncogene. 22:2034–2044.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pennati M, Sbarra S, De Cesare M,
Lopergolo A, Locatelli SL, Campi E, Daidone MG, Carlo-Stella C,
Gianni AM and Zaffaroni N: YM155 sensitizes triple-negative breast
cancer to membrane-bound TRAIL through p38 MAPK- and CHOP-mediated
DR5 upregulation. Int J Cancer. 136:299–309. 2015. View Article : Google Scholar
|
|
29
|
Wang XZ and Ron D: Stress-induced
phosphorylation and activation of the transcription factor CHOP
(GADD153) by p38 MAP Kinase. Science. 272:1347–1349. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yamaguchi H and Wang HG: CHOP is involved
in endoplasmic reticulum stress-induced apoptosis by enhancing DR5
expression in human carcinoma cells. J Biol Chem. 279:45495–45502.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ozören N and El-Deiry WS: Cell surface
Death Receptor signaling in normal and cancer cells. Semin Cancer
Biol. 13:135–147. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
den Hollander MW, Gietema JA, de Jong S,
Walenkamp AM, Reyners AK, Oldenhuis CN and de Vries EG: Translating
TRAIL-receptor targeting agents to the clinic. Cancer Lett.
332:194–201. 2013. View Article : Google Scholar
|
|
33
|
Yoshida T, Zhang Y, Rivera Rosado LA and
Zhang B: Repeated treatment with subtoxic doses of TRAIL induces
resistance to apoptosis through its death receptors in MDA-MB-231
breast cancer cells. Mol Cancer Res. 7:1835–1844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jin Z, McDonald ER III, Dicker DT and
El-Deiry WS: Deficient tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) death receptor transport to the
cell surface in human colon cancer cells selected for resistance to
TRAIL-induced apoptosis. J Biol Chem. 279:35829–35839. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wainberg ZA, Messersmith WA, Peddi PF,
Kapp AV, Ashkenazi A, Royer-Joo S, Portera CC and Kozloff MF: A
phase 1B study of dulanermin in combination with modified FOLFOX6
plus bevacizumab in patients with metastatic colorectal cancer.
Clin Colorectal Cancer. 12:248–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trarbach T, Moehler M, Heinemann V, Köhne
CH, Przyborek M, Schulz C, Sneller V, Gallant G and Kanzler S:
Phase II trial of mapatumumab, a fully human agonistic monoclonal
antibody that targets and activates the tumour necrosis factor
apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with
refractory colorectal cancer. Br J Cancer. 102:506–512. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rocha Lima CM, Bayraktar S, Flores AM,
MacIntyre J, Montero A, Baranda JC, Wallmark J, Portera C, Raja R,
Stern H, et al: Phase Ib study of drozitumab combined with
first-line mFOLFOX6 plus bevacizumab in patients with metastatic
colorectal cancer. Cancer Invest. 30:727–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fuchs CS, Fakih M, Schwartzberg L, Cohn
AL, Yee L, Dreisbach L, Kozloff MF, Hei YJ, Galimi F, Pan Y, et al:
TRAIL receptor agonist conatumumab with modified FOLFOX6 plus
bevacizumab for first-line treatment of metastatic colorectal
cancer: A randomized phase 1b/2 trial. Cancer. 119:4290–4298. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Takeda K, Stagg J, Yagita H, Okumura K and
Smyth MJ: Targeting death-inducing receptors in cancer therapy.
Oncogene. 26:3745–3757. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Do MT, Na M, Kim HG, Khanal T, Choi JH,
Jin SW, Oh SH, Hwang IH, Chung YC, Kim HS, et al: Ilimaquinone
induces death receptor expression and sensitizes human colon cancer
cells to TRAIL-induced apoptosis through activation of ROS-ERK/p38
MAPK-CHOP signaling pathways. Food Chem Toxicol. 71:51–59. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu GS, Burns TF, McDonald ER III, Jiang W,
Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, et al:
KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor
gene. Nat Genet. 17:141–143. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ohtsuka T and Zhou T: Bisindolylmaleimide
VIII enhances DR5-mediated apoptosis through the MKK4/JNK/p38
kinase and the mitochondrial pathways. J Biol Chem.
277:29294–29303. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shenoy K, Wu Y and Pervaiz S: LY303511
enhances TRAIL sensitivity of SHEP-1 neuroblastoma cells via
hydrogen peroxide-mediated mitogen-activated protein kinase
activation and up-regulation of death receptors. Cancer Res.
69:1941–1950. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bruhat A, Jousse C, Wang XZ, Ron D,
Ferrara M and Fafournoux P: Amino acid limitation induces
expression of CHOP, a CCAAT/enhancer binding protein-related gene,
at both transcriptional and post-transcriptional levels. J Biol
Chem. 272:17588–17593. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Flo TH, Smith KD, Sato S, Rodriguez DJ,
Holmes MA, Strong RK, Akira S and Aderem A: Lipocalin 2 mediates an
innate immune response to bacterial infection by sequestrating
iron. Nature. 432:917–921. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Candido S, Maestro R, Polesel J, Catania
A, Maira F, Signorelli SS, McCubrey JA and Libra M: Roles of
neutrophil gelatinase-associated lipocalin (NGAL) in human cancer.
Oncotarget. 5:1576–1594. 2014. View Article : Google Scholar : PubMed/NCBI
|