Introduction
Clear cell renal cell carcinoma (ccRCC) is the most
common histological subtype of RCC, accounting for >70% of RCC
(1). At the time of diagnosis,
~30% of patients have metastatic disease (2). Although surgical resection can
effectively resolve ccRCC, 20-40% of patients continue to develop
local recurrence or distinct metastasis following surgery (3,4).
Molecular-targeted agents repressing the vascular endothelial
growth factor (VEGF) or mammalian target of rapamycin
(mTOR) genes have been routinely administered to patients
with metastatic or recurrent RCC. Among these drugs, sunitinib is a
common molecular-targeted agent that is recommended as a first-line
therapy for patients with advanced RCC. Unfortunately, most
patients treated with these drugs eventually suffer from
progressive disease due to intrinsic or acquired resistance
(5).
In a previous study, metabolic reprogramming was
observed in sunitinib-resistant RCC cells, resulting in the
acquisition of sunitinib resistance (6). In recent years, novel drugs have been
developed as second-line treatments for advanced RCC. Nivolumab is
an IgG4 antibody that causes immune checkpoint blockade by
decreasing inhibitory signaling via the programmed death ligand-1
pathway (7). Nivolumab increases
the overall survival (OS) time and is associated with decreased
toxicity in comparison with everolimus according to the CheckMate
025 study (8). However, due to the
high cost of nivolumab, it is necessary to define its usefulness
from the viewpoint of efficacy as well as cost (9). Indeed, the phase 3 CheckMate 025
study demonstrated longer OS times with nivolumab compared with
everolimus, but not significantly so. Additionally, the objective
response rate of nivolumab-treated patients was only 25% (8). Therefore, it is necessary to identify
novel therapeutic modalities to defeat sunitinib resistance.
MicroRNAs (miRNAs/miRs) are a class of small
noncoding RNAs (~22 nucleotides) that have roles in the inhibition
or degradation of target RNA transcripts in a sequence-dependent
manner (10). Numerous miRNAs have
tissue-specific expression (11),
and >2,000 different miRNAs have been identified in humans
(12). miRNAs are abnormally
expressed in several human cancer types, and certain miRNAs are
frequently downregulated in numerous types of cancer (13-15),
suggesting that they function as tumor suppressors by targeting
multiple oncogenes. Several studies have demonstrated that
modulating miRNA expression levels can increase the efficacy of
chemotherapy (16,17). Furthermore, silencing multiple
genes using a single miRNA can simultaneously control several
signaling pathways and minimize compensatory mechanisms that cause
therapeutic resistance (18).
miRNAs have also been reported to be associated with sunitinib
resistance. For example, miR-144-3p mediates sunitinib resistance
by targeting the AT-rich interactive domain 1A gene in ccRCC
(19). Therefore, miRNAs may
represent promising candidates for the treatment of RCC in patients
with intrinsic or acquired resistance to sunitinib.
Accordingly, the aim of the present study was to
investigate the functional importance of miR-99a-3p and to discover
the molecular targets that are regulated by this miRNA in
sunitinib-resistant RCC. Gain-of-function studies were performed in
miR-99a-3p transfectants and novel miR-99a-3p-mediated molecular
targets and pathways were investigated through in silico
analysis and RNA sequencing. The discovery that miR-99a-3p
regulates targets and pathways may provide novel insights into the
mechanisms of intrinsic or acquired resistance to sunitinib.
Materials and methods
Clinical tissues and human RCC cell
lines
ccRCC and normal adjacent kidney tissues were
collected from 40 patients who sequentially underwent radical or
partial nephrectomy at Kagoshima University Hospital (Kagoshima,
Japan) between 2005 and 2010 (Table
I). The stage and grade of the samples were determined
according to the American Joint Committee on Cancer/International
Union Against Cancer classification and histologically graded
(20) at the Department of
Veterinary Histopathology of Kagoshima University. The samples were
kept in RNAlater™ (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at −20°C until RNA extraction. The present study was approved
by the Bioethics Committee of Kagoshima University, and written
informed consent and was obtained from all patients.
 | Table IPatient characteristics (n=40). |
Table I
Patient characteristics (n=40).
| Characteristic | Value |
|---|
| Median age (range),
years | 66.5 (41-89) |
| Sex, n (%) | |
| Male | 28 (70.0) |
| Female | 12 (30.0) |
| Pathological tumor
stagea, n (%) | |
| pT1a | 20 (50.0) |
| pT1b | 13 (32.5) |
| pT2 | 0 (0.0) |
| pT3a | 4 (10.0) |
| pT3b | 3 (7.5) |
| pT4 | 0 (0.0) |
| Tumor gradea, n (%) | |
| G1 | 3 (7.5) |
| G2 | 30 (75.0) |
| G3 | 6 (15.0) |
| N/A | 1 (2.5) |
| Metastasisa, n (%) | |
| M 0 | 36 (90.0) |
| M 1 | 2 (5.0) |
| N/A | 2 (5.0) |
| Venous
invasiona, n (%) | |
| v 1 | 25 (62.5) |
| v 0 | 15 (37.5) |
Human RCC cells (786-o, A498, ACHN, Caki1, and Caki2
cells) were purchased from the American Type Culture Collection
(ATCC, Manassas, VA, USA). The sunitinib-resistant 786-o
(SU-R-786-o) cell line was previously established by administration
of sunitinib to mice (6).
Cell culture and RNA extraction
Cells were cultured in RPMI-1640 medium (Invitrogen;
Thermo Fisher Scientific, Inc.) with 10% fetal bovine serum and
kept in a humidified incubator (5% CO2) at 37°C. Routine
tests for mycoplasma infection were negative. Total RNA, including
the miRNA and the mRNA fractions, was extracted using a mir-Vana
miRNA Isolation kit (Thermo Fisher Scientific, Inc.) according to
the manufacturer’s protocol. The quality of the RNA was tested
using an RNA 6000 Nano assay kit and a 2100 Bioanalyzer (both
Agilent Technologies, Inc., Santa Clara, CA, USA). Human kidney
total RNA (cat. no. AM7976; Thermo Fisher Scientific, Inc.) was
used as normal kidney control RNA.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Stem-loop RT-qPCR (TaqMan MicroRNA Assays; Assay ID:
002141 for miR-99a-3p; Applied Biosystems; Thermo Fisher
Scientific, Inc.) was employed to quantify miRNA following the
manufacturer’s protocol. Human RNU48 (P/N: 001006; Applied
Biosystems; Thermo Fisher Scientific, Inc.) was used as an internal
control, and the 2−ΔΔCq method was used to calculate the
relative changes (21). For
ribonucleotide reductase regulatory subunit-M2 (RRM2),
SYBR-Green qPCR was performed, and the primer sequences are listed
in Table II. Briefly, 500 ng
total RNA was reverse transcribed into cDNA using the High Capacity
cDNA Reverse Transcription kit (Thermo Fisher Scientific, Inc.)
under the incubation conditions of 25°C for 10 min, 37°C for 120
min and 85°C for 5 min. qPCR was performed using a Power SYBR Green
Master Mix (cat. no. 4367659) on a 7300 Real-time PCR System (both
Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling protocol used was as follows: Initial activation step
at 95°C for 10 min, followed by 40 cycles of a denaturation step at
95°C for 15 sec and an annealing/extension step at 60°C for 1 min.
The amplification specificity was confirmed by monitoring the
dissociation curve of the amplified product. All expression data
were normalized to the glucuronidase β gene, and the
2−ΔΔCq method was employed to calculate the relative
changes.
| Table IISequences of the primers used in the
present study. |
Table II
Sequences of the primers used in the
present study.
| Gene | Forward
(3′-5′) | Reverse
(3′-5′) |
|---|
| GUSB |
CGTCCCACCTAGAATCTGCT |
TTGCTCACAAAGGTCACAGG |
| RRM2 |
CACGGAGCCGAAAACTAAAGC |
TCTGCCTTCTTATACATCTGCCA |
| MKI67 |
ACGCCTGGTTACTATCAAAAGG |
CAGACCCATTTACTTGTGTTGGA |
| PPP6R1 |
TGACCTGCACACAAGCTCG |
GGTTGACGACCTTGCACTC |
| PLXNA1 |
ACCCACCTAGTGGTGCACTC |
CGGTTAGCGGCATAGTCCA |
Transfection with miRNA mimic and small
interfering (si)RNA into RCC and SU-R-786-o cells
As described previously (22), ACHN, 786-o and SU-R-786-o cells
were transfected using Lipofectamine™ RNAiMAX transfection reagent
and Opti-MEM (both Thermo Fisher Scientific, Inc.) containing 10 nM
mature miRNA or RRM2 siRNA. Mature miRNAs and pre-miR miRNA
precursors (hsa-miR-99a-3p; product ID, PM12983; negative
control miRNA product ID, AM17111) were employed for the
gain-of-function experiments, whereas RRM2 siRNA (product ID,
HSS109390 and HSS109392) and negative control siRNA (product ID,
D-001810-10) (all Thermo Fisher Scientific, Inc.) were employed for
the loss-of-function experiments. The sequences of all miRNA mimics
and siRNAs are listed in Tables
III and IV. Different
negative controls (miRNA/siRNA) were used for each cancer cell line
to prevent off-target effects. The optimization of the transfection
efficacy of the microRNA precursors in the RCC cell lines was based
on the downregulation of PTK9 mRNA by over-expression of
miR-1, as recommended by the manufacturer (Thermo Fisher
Scientific, Inc.). The transfection efficiency of all miRNA mimics
was evaluated accordingly. In order to establish the RRM2 siRNA
transfection efficacy, RT-qPCR and western blot analyses were
performed to confirm the downregulation of RRM2 mRNA and
protein levels.
| Table IIISequences of the miRNA mimics used in
the present study. |
Table III
Sequences of the miRNA mimics used in
the present study.
| miRNA | Mature accession
no.a | Sequence
(5′-3′) |
|---|
| let-7c-5p | MIMAT0000064 |
UGAGGUAGUAGGUUGUAUGGUU |
| miR-1-3p | MIMAT0000416 |
UGGAAUGUAAAGAAGUAUGUAU |
| miR-135-5p | MIMAT0000428 |
UAUGGCUUUUUAUUCCUAUGUGA |
| miR-144-3p | MIMAT0000436 |
UACAGUAUAGAUGAUGUACU |
| miR-204-5p | MIMAT0000265 |
UUCCCUUUGUCAUCCUAUGCCU |
| miR-23b-3p | MIMAT0000418 |
AUCACAUUGCCAGGGAUUACCAC |
| miR-26b-5p | MIMAT0000083 |
UUCAAGUAAUUCAGGAUAGGU |
| miR-27b-3p | MIMAT0000419 |
UUCACAGUGGCUAAGUUCUGC |
| miR-29b-3p | MIMAT0000100 |
UAGCACCAUUUGAAAUCAGUGUU |
| miR-29c-3p | MIMAT0000681 |
UAGCACCAUUUGAAAUCGGUUA |
| miR-30a-5p | MIMAT0000087 |
UGUAAACAUCCUCGACUGGAAG |
| miR-31-3p | MIMAT0004504 |
UGCUAUGCCAACAUAUUGCCAU |
| miR-429 | MIMAT0001536 |
UAAUACUGUCUGGUAAAACCGU |
| miR-766-3p | MIMAT0003888 |
ACUCCAGCCCCACAGCCUCAGC |
| miR-99a-3p | MIMAT0024017 |
ACCCACCTAGTGGTGCACTC |
| Table IVSequences of the siRNAs used in the
present study. |
Table IV
Sequences of the siRNAs used in the
present study.
| siRNA | Cat. no. | Directionality | Sequence
(5′-3′) |
|---|
| si-RRM2_1 | 10620318 | Sense |
GCCUGAUGUUCAAACACCUGGUACA |
| 10620319 | Antisense |
UGUACCAGGUGUUUGAACAUCAGGC |
| si-RRM2_2 | 10620318 | Sense |
ACCAUGAUAUCUGGCAGAUGUAUAA |
| 10620319 | Antisense |
UUAUACAUCUGCCAGAUAUCAUGGU |
Cell proliferation, colony formation,
apoptosis and cell cycle assays, and determination of half maximal
inhibitory concentration (IC50) values
To investigate the functional importance of
miR-99a-3p and RRM2, cell proliferation, colony formation
and apoptosis assays were performed using ACHN, 786-o, and
SU-R-786-o cells. Didox (3,4-dihydroxybenzohydroxamic acid; Cayman
Chemical Company, Ann Arbor, MI, USA) was used as an RRM2
inhibitor. The cell proliferation was examined 72 h after
transfection using XTT assays (Roche Applied Science, Penzberg,
Germany), according to the manufacturer’s instructions. For the
colony formation assays, 1,000 cells were plated into 10-cm dishes
following transfection for 10 days to confirm optimal colony
formation, followed by staining with 0.04% crystal violet (Nacalai
Tesque, Inc., Kyoto, Japan) at room temperature for 10 min. The
cell cycle and apoptosis assays were performed by flow cytometry
(CytoFLEX Analyzer; Beckman Coulter, Inc., Brea, CA, USA) using a
Cycletest PLUS DNA Reagent kit and FITC Annexin V Apoptosis
Detection kit (both BD Biosciences, San Jose, CA, USA),
respectively, following the manufacturer’s protocols (23). Cell viability was assessed using an
XTT cell proliferation assay kit. The IC50 values of
sunitinib were assessed in accordance with the relative survival
curve.
Plasmid construction and dual-luciferase
reporter assay
Partial wild-type sequences of the 3′-untranslated
region (UTR) of RRM2 or those with a deleted miR-99a-3p target site
were inserted between the XhoI and PmeI restriction
sites in the 3′-UTR of the hRluc gene in a psiCHECK-2 vector
(C8021; Promega Corporation, Madison, WI, USA). ACHN, 786-o, and
SU-R-786-o cells were transfected with 50 ng vector and 10 nM
miR-99a-3p. The activities of firefly and Renilla luciferases in
cell lysates were recorded. The procedure for the dual-luciferase
reporter assays was described previously (24).
Western blotting
The cells were harvested 72 h after transfection and
total protein lysate was prepared with a radioimmunoprecipitation
assay buffer (Thermo Fisher Scientific, Inc.) containing a protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
The protein concentrations were determined using the Bradford assay
(25). Protein lysates (50
µg) were separated on NuPAGE 4-12% Bistris gels (Invitrogen;
Thermo Fisher Scientific, Inc.) and transferred to polyvinylidene
difluoride membranes. Immunoblotting was performed with diluted
rabbit polyclonal anti-RRM2 antibodies (1:500; cat. no. 11661-1-AP;
Proteintech Group, Inc., Chicago, IL, USA), rabbit polyclonal
anti-poly(ADP-ribose) polymerase (PARP) antibodies (1:500; cat. no.
9542), rabbit monoclonal anti-cleaved PARP antibodies (1:500; cat.
no. 5625) (both Cell Signaling Technology, Inc., Danvers, MA, USA),
and rabbit polyclonal anti-β-actin antibodies (1:5,000; cat. no.
bs-0061R; Bioss, Beijing, China). Specific complexes were
visualized using an echochemiluminescence detection system (GE
Healthcare Life Sciences, Little Chalfont, UK) as described
previously (26).
In silico analysis for identifying genes
regulated by miR-99a-3p
In silico analysis was used to identify genes
targeted by miR-99a-3p. To obtain candidate target genes regulated
by miR-99a-3p, TargetScan database Release 7.1 (http://www.targetscan.org) was used. Additionally, the
Gene Expression Omnibus (GEO) database (accession nos. GSE36895 and
GSE22541; https://www.ncbi.nlm.nih.gov/geo/) was employed to
identify upregulated genes in ccRCC tissues.
Bioinformatics analysis
In order to evaluate the clinical relevance, The
Cancer Genome Atlas (TCGA) cohort database of 534 patients with
ccRCC was used. Full sequencing and clinical information were
obtained through University of California Santa Cruz Xena
(http://xena.ucsc.edu/), cBioPortal for Cancer
Genomics (http://www.cbioportal.org/public-portal/), and TCGA
(https://tcga-data.nci.nih.gov/tcga/).
The present study met the criteria for the publication guidelines
provided by TCGA (http://cancergenome.nih.gov/publications/publicationguidelines).
Statistical analysis
The statistical comparisons between two or three
variables and numerical values were analyzed by Mann-Whitney U
tests and Bonferroni-adjusted Mann-Whitney U tests, respectively.
All experiments were performed in triplicate. Spearman’s rank tests
were used to evaluate the correlation between the expression of
miR-99a-3p and RRM2. Kaplan-Meier and log-rank methods were
used to analyze the associations between miR-99a-3p and candidate
target genes, including RRM2, and OS time by using the
OncoLnc dataset (http://www.oncolnc.org/), which contains survival data
for 8,647 patients from 21 cancer studies performed by TCGA.
OncoLnc is a useful tool for exploring survival correlations, and
for downloading clinical data coupled to expression data for mRNAs,
miRNAs or long noncoding RNAs as previously described (27). All analyses were performed on
Expert StatView software version 5.0 (SAS Institute, Inc., Cary,
NC, USA). P<0.05 was considered to indicate statistically
significant differences.
Results
Identification of miRNAs that exhibit
decreased expression in sunitinib-resistant RCC
Initially, 15 miRNAs that exhibited decreased
expression and had not been previously analyzed in
sunitinib-resistant RCC were selected (28-31)
(Table III). XTT assays were
performed using 786-o and SU-R-786-o cells transfected with these
15 miRNAs in order to select candidate miRNAs (Fig. 1A). The results revealed that 5
miRNA transfectants (miR-1-3p, miR-29c-3p, miR-429, miR-766-3p and
miR-99a-3p) inhibited cell proliferation in comparison with
miR-control. Additionally, among these 5 miRNAs, the OncoLnc
analysis revealed a trend towards longer OS times in the patients
with high miR-99a-3p expression (n=253) compared with those in the
patients with low expression (n=253) in the TCGA ccRCC cohort, but
this was not statistically significant (P=0.0546; Fig. 1B). Therefore, miR-99a-3p was chosen
for further analyses.
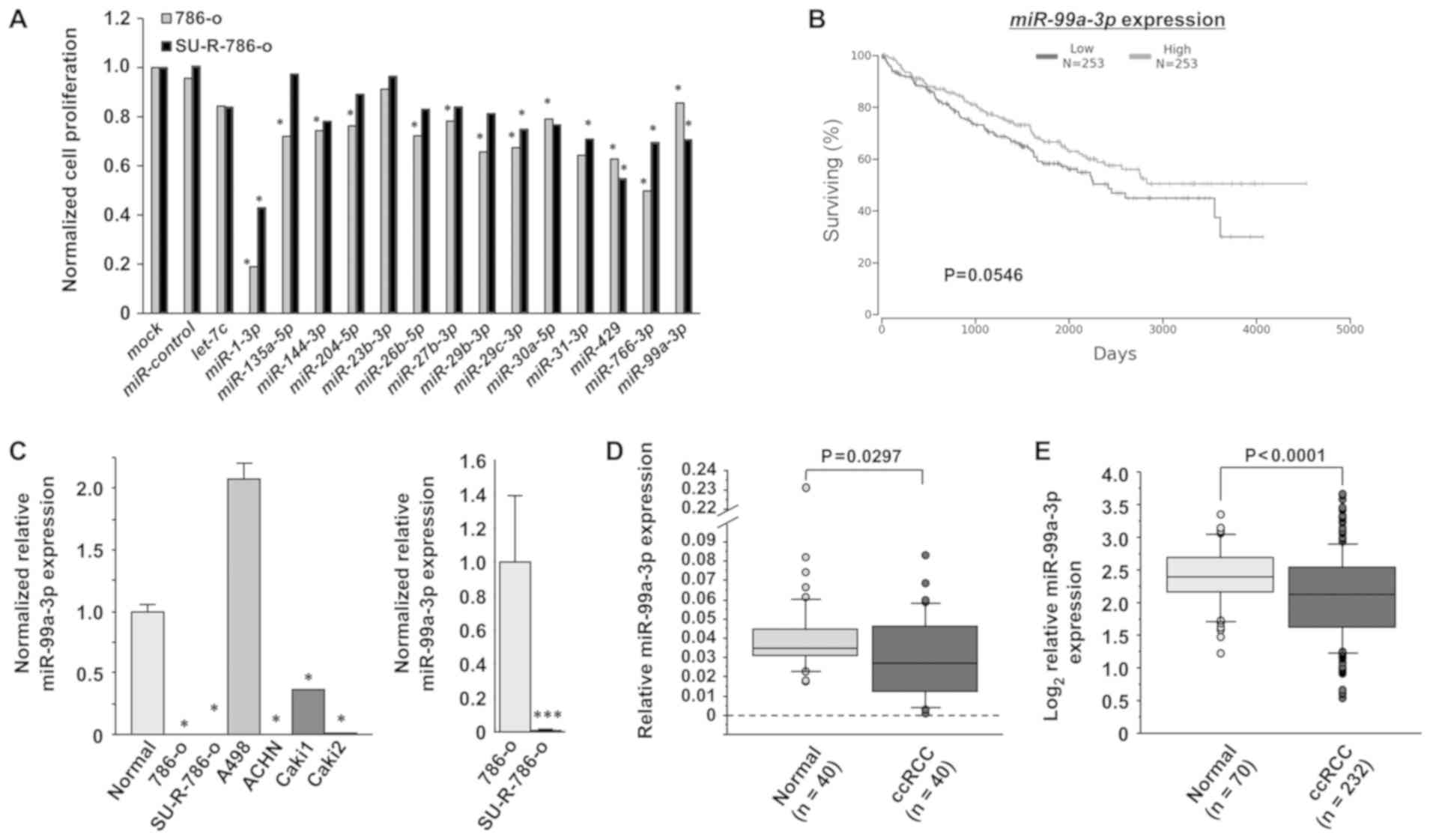 | Figure 1Clinical significance and expression
levels of miR-99a-3p in RCC. (A) Cell proliferation was examined by
XTT assays 72 h after transfection with 10 nM miRNAs in 786-o and
SU-R-786-o cells in order to select candidate miRNAs. Significant
cell proliferation inhibition was observed in the two cell types
transfected with the following miRNAs: miR-1-3p, miR-29c-3p,
miR-429, miR-766-3p and miR-99a-3p. *P<0.0001 versus
miR-control. (B) Analysis of a ccRCC cohort from TCGA in OncoLnc
revealed longer OS times in the patients with high miR-99a-3p
expression (n=253) in comparison with those with low expression
(n=253), but the difference was not statistically significant
(P=0.0546). (C) The expression levels of miR-99a-3p were
significantly lower in 4 RCC cell lines (786-o, ACHN, Caki1 and
Caki2) and in SU-R-786-o cells than those in normal kidney cells.
The expression levels of miR-99a-3p in SU-R-786-o cells were lower
than those in 786-o cells. *P<0.0001 vs. normal;
***P<0.05 vs. 786-o. (D) The miR-99a-3p levels were
lower in clinical ccRCC tissues (n=40) compared with their adjacent
noncancerous tissues (n=40) (P=0.0297). (E) In a dataset obtained
from TCGA, miR-99a-3p expression was significantly downregulated in
ccRCC samples (n=232) compared with that in normal samples (n=70)
(P<0.001). RCC, renal cell carcinoma; ccRCC, clear cell RCC;
miR/ miRNA, microRNA; TCGA, The Cancer Genome Atlas. |
miR-99a-3p expression in RCC cell lines,
SU-R-786-o cells, and ccRCC clinical tissues
The expression levels of miR-99a-3p were examined in
clinical ccRCC tissues (n=40), their adjacent noncancerous tissues
(n=40), RCC cell lines and SU-R-786-o cells by RT-qPCR. The
miR-99a-3p levels of were significantly lower in four of the RCC
cell lines (786-o, ACHN, Caki1 and Caki2) and in SU-R-786-o cells
than in normal kidney cells (P<0.0001; Fig. 1C). Notably, the expression levels
in the SU-R-786-o cells were lower than those in the parental 786-o
cells (P=0.0495). Furthermore, miR-99a-3p revealed lower expression
in the clinical ccRCC specimens compared with their adjacent
noncancerous tissues (P=0.0297; Fig.
1D). The clinicopathological information of the patients is
listed in Table I. No significant
associations were observed between any of the clinicopathological
parameters and miR-99a-3p expression in this cohort (data not
shown). In addition, within the ccRCC dataset from TCGA, the
expression level of miR-99a-3p was significantly downregulated in
patients with ccRCC (n=232) compared with that in healthy patients
(n=70; P<0.0001; Fig. 1E).
These data imply that miR-99a-3p may be a potential therapeutic
target in RCC and sunitinib-resistant RCC cells.
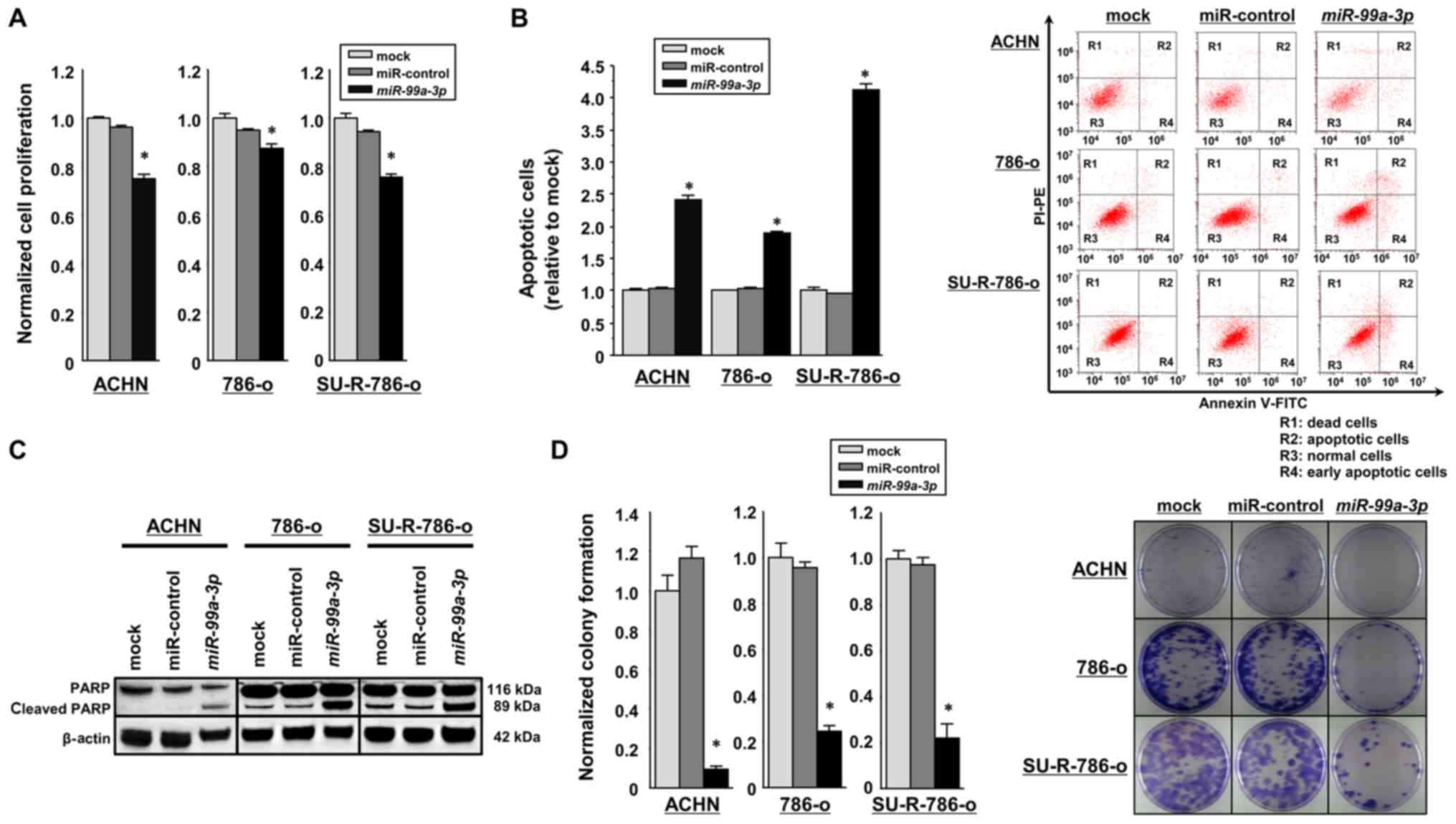
Effects of restoring miR-99a-3p
expression on cell proliferation, apoptosis, cell cycle and colony
formation in RCC cell lines and SU-R-786-o cells
In order to investigate the functional roles of
miR-99a-3p, gain-of-function studies were performed using
miRNA-transfected ACHN, 786-o and SU-R-786-o cells. Using XTT
assays, miR-99a-3p overexpression was revealed to significantly
suppress cell proliferation in comparison with the mock or
miR-control transfectants (P<0.0001; Fig. 2A). As miR-99a-3p transfection
significantly inhibited cell proliferation in SU-R-786-o and other
RCC cells, it was hypothesized that this miRNA may induce cell
apoptosis. Hence, flow cytometric analyses were performed to count
the number of apoptotic cells following the restoration of
miR-99a-3p expression. The number of apoptotic cells (apoptotic and
early apoptotic cells) was significantly higher in
miR-99a-3p-transfected SU-R-786-o cells than in the mock or
miR-control transfectants (P<0.0001; Fig. 2B). Similarly, in the ACHN and 786-O
cells, the miR-99a-3p transfectants exhibited increased apoptosis
in comparison with the controls (P<0.0001; Fig. 2B). Western blot analyses
demonstrated that the expression of cleaved PARP was markedly
increased in the miR-99a-3p transfectants compared with that in the
controls (Fig. 2C). The cell cycle
effects were also investigated using miR-99a-3p-transfected 786-o
and SU-R-786-o cells. Overexpression of miR-99-3p induced S-phase
arrest in the two cell types (Fig.
S1). In addition, colony formation assays using SU-R-786-o,
ACHN, and 786-o cells revealed significantly decreased colony
numbers in miR-99a-3p transfectants compared with those in the mock
or miR-control transfectants (Fig.
2D). Furthermore, cell viability assays were performed using
SU-R-786-o cells treated with various concentrations of sunitinib,
and the viability of the cells was assessed with XTT assays
(Fig. 2E). Notably, sunitinib
sensitivity was restored in miR-99a-3p-transfected SU-R-786-o
cells; the sunitinib IC50 values were 3.04, 1.72 and
1.38 µM in the SU-R-786-o cells transfected with
miR-control, those transfected with miR-99a-3p, and the parental
786-o cells, respectively. These results suggest that miR-99a-3p
may function as a tumor suppressor in SU-R-786-o and other RCC
cells.
Identification of the RRM2 gene as a
target for miR-99a-3p in SU-R-786-o cells
In order to gain further insights into the molecular
mechanisms and pathways associated with the tumor-suppressing
functions of miR-99a-3p in SU-R-786-o cells, a combination of in
silico analyses and RNA sequencing were performed on SU-R-786-o
cells. Fig. 3A indicates the
method of narrowing down the genes targeted by miR-99a-3p. The
candidate target genes were identified using in silico
analyses including TargetScan database Release 7.1 and the GEO
database (accession nos. GSE36895 and GSE22541). Overall, 1,592
candidate target genes were selected that had ≥1 target sites.
Additionally, from the GEO database, 12,831 genes were
significantly upregulated in clinical ccRCC tissues in comparison
with normal kidney tissues. RNA sequencing expression analysis was
applied to identify the genes significantly upregulated in
SU-R-786-o cells compared with the parental 786-o cells, and 16
candidate target genes were selected. Among these, 4 genes
[RRM2, proliferation marker Ki-67 (MKI67),
serine/threonine-protein phosphatase 6 regulatory subunit 1
(PPP6R1) and plexin-A1 (PLXNA1)] were chosen that
were associated with significant differences in OS time, as
revealed by Kaplan-Meier analysis of TCGA ccRCC cohort using the
OncoLnc dataset (Figs. 3B and
S2). Of these 4 candidate genes,
RRM2 was investigated due to its knockdown efficiency being
higher than that of the other 3 genes in miR-99a-3p-transfected
SU-R-786-o cells than in the mock or miR-control transfectants
(Fig. S3). Furthermore, the
RRM2 expression levels in RCC cell lines were examined by
RT-qPCR. RRM2 was revealed to be significantly upregulated in all
tested RCC cell lines compared with RNA from normal kidneys
(Fig. 3C). Notably, RRM2
expression in SU-R-786-o cells was significantly upregulated in
comparison with that in parental 786-o cells (P<0.0001).
Additionally, it was upregulated in patients with ccRCC (n=534)
compared with healthy individuals (n=72) in the ccRCC cohort from
TCGA database (P<0.0001; Fig.
3D).
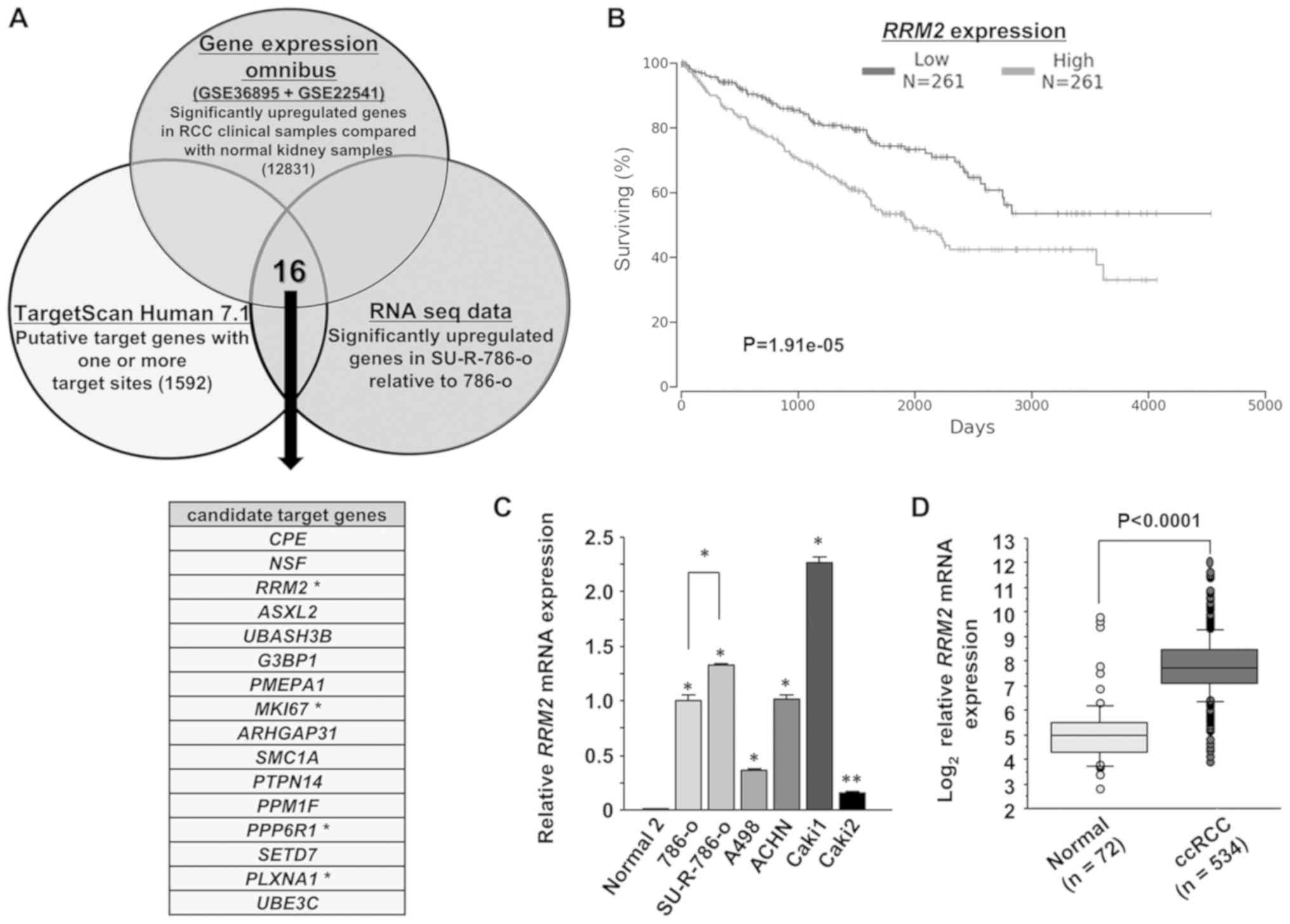 | Figure 3Identification of RRM2 as a
candidate miR-99a-3p target gene. (A) Venn diagram of the results
from RNA sequencing and in silico analyses indicated 16
putative candidate target genes as key factors in SU-R-786-o cells.
Four genes, RRM2, MKI67, PPP6R1 and
PLXNA1, were linked to significant differences in OS rates
by Kaplan-Meier analysis of ccRCC cohort from TCGA. (B)
Kaplan-Meier analysis demonstrated that the group of patients with
high RRM2 expression (n=261) exhibited lower OS rates
compared with those in the low expression group in the OncoLnc
dataset (n=261) (P<0.0001). (C) The mRNA expression levels of
RRM2 were examined in RCC cell lines by reverse
transcription-quantitative polymerase chain reaction. The
expression levels were significantly upregulated in RCC cells in
comparison with those in normal kidney cells. RRM2
expression in SU-R-786-o cells was significantly higher than that
in 786-o cells. *P<0.0001 and
**P<0.001. (D) The mRNA levels of RRM2 were
significantly upregulated in the ccRCC samples of TCGA dataset
(n=534) compared with those in the normal samples (n=72).
(P<0.0001). RRM2, ribonucleotide reductase regulatory
subunit-M2; MKI67; proliferation marker Ki-67; PPP6R1,
serine/threonine-protein phosphatase 6 regulatory subunit 1;
PLXNA1, plexin-A1; miR, microRNA; OS, overall survival; TCGA, The
Cancer Genome Atlas. |
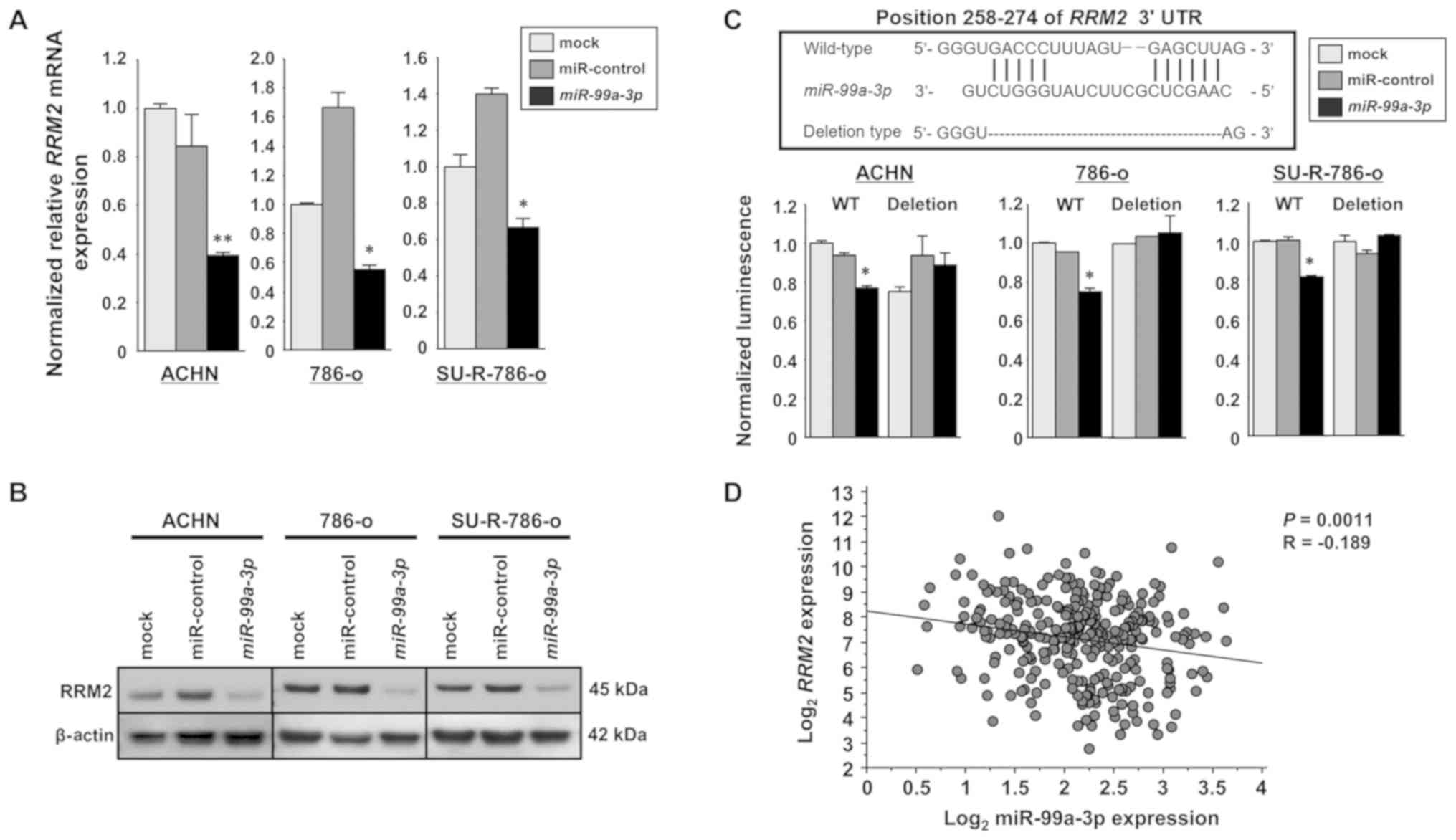
RRM2 is directly targeted by miR-99a-3p
in SU-R-786-o cells
RT-qPCR and western blot analyses were performed to
confirm that overexpression of miR-99a-3p resulted in
downregulation of RRM2 in ACHN, 786-o and SU-R-786-o cells.
RRM2 mRNA and protein levels were significantly decreased in
miR-99a-3p transfectants compared with those in the mock or
miR-control transfectants (Fig. 4A and
B). Dual luciferase reporter assays were performed to examine
whether the RRM2 gene was regulated through direct
interaction by miR-99a-3p. The TargetScan database predicted a
binding site for miR-99a-3p in the 3′-UTR of RRM2 (positions
258-274). Vectors encoding the partial wild-type sequence of the
3′-UTR of RRM2 were employed, including the predicted
miR-99a-3p target sites. The luminescence intensity was
significantly diminished in the case of co-transfection with
miR-99a-3p and the vector carrying the wild-type 3′-UTR. In
contrast, no decrease in luminescence was observed following
transfection with the binding site deletion vector (P<0.0001;
Fig. 4C). Furthermore, the
relationship between miR-99a-3p and RRM2 expression levels
in clinical ccRCC tissues was investigated using TCGA database. A
significant negative correlation was revealed between miR-99a-3p
and RRM2 mRNA expression according to Spearman’s rank test
(P=0.0011, R=-0.189; Fig. 4D).
These results suggest that miR-99a-3p directly binds to specific
sites at positions 258-274 of the RRM2 3′-UTR.
Effects of RRM2 knockdown on cell
proliferation, apoptosis, cell cycle and colony formation in
SU-R-786-o cells
In order to investigate the functional role of
RRM2 in SU-R-786-o cells, loss-of-function assays were
conducted using si-RRM2. The knockdown efficacies of si-RRM2
transfection were examined in ACHN, 786-o and SU-R-786-o cells. In
the present study, two siRNAs targeting RRM2 were employed
(si-RRM2_1 and si-RRM_2). RT-qPCR and western blot analyses
indicated that these siRNAs effectively downregulated RRM2
mRNA and protein expression in ACHN, 786-o and SU-R-786-o cells
(P<0.0001; Fig. 5A and B). XTT
assays demonstrated that cell proliferation was inhibited in the
si-RRM2 transfectants in comparison with that in the mock or
si-control transfectants (Fig.
5C). In the apoptosis assays, the number of apoptotic cells was
significantly greater in the si-RRM2 transfectants than in the
controls (Fig. 5D). Western blot
analyses demonstrated that the levels of cleaved PARP were markedly
increased when RRM2 was silenced (Fig. 5E). The cell cycle assays revealed
that S-phase arrest was induced in the si-RRM2 transfected 786-o
cells, whereas RRM2 knockdown in the SU-R-786-o cells
increased the fraction of cells in the G0/G1
phase (Fig. S1). Furthermore,
colony formation assays confirmed that the development of colonies
was significantly suppressed in the RRM2-knockdown RCC
cells, including SU-R-786-o cells, compared with that in the
controls (Fig. 5F). These results
indicated that high expression of RRM2 is associated with
oncogenic effects in ACHN, 786-o and SU-R-786-o cells.
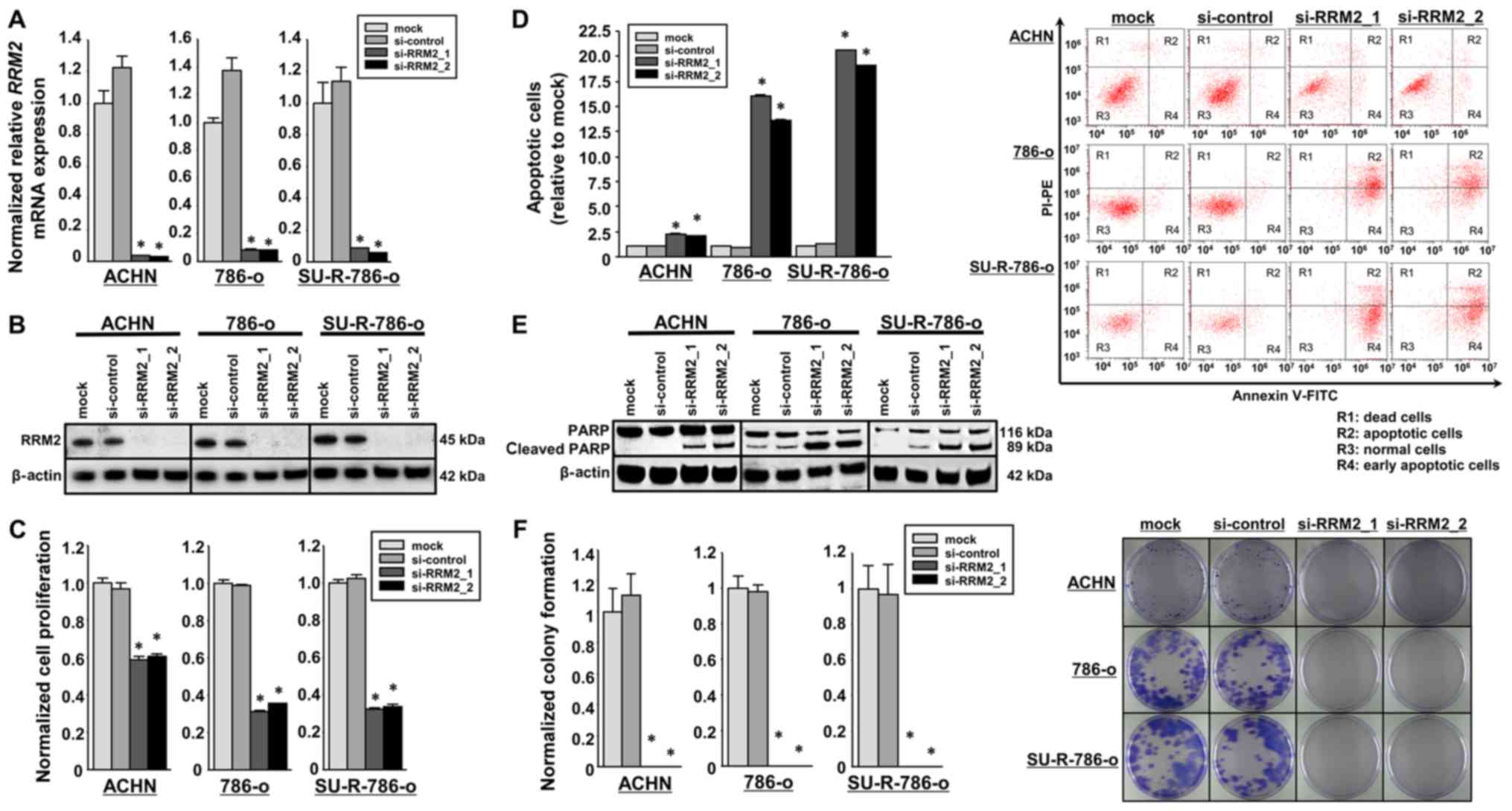 | Figure 5Effects of si-RRM2 transfection on
SU-R-786-o, ACHN and 786-o cells. (A) The expression of RRM2
mRNA was significantly inhibited in cells transfected with si-RRM2
compared with that in the mock and si-control groups. GUSB
was employed as an internal control. *P<0.0001 versus
mock and si-control groups. (B) The expression of RRM2 protein, as
observed by western blot analysis, was markedly inhibited in cells
with si-RRM2 compared with that in the mock or si-control groups.
β-actin was employed as a loading control. (C) Cell proliferation
was examined using XTT assays in cells with RRM2 knockdown,
revealing a significant inhibition compared with the control
groups. *P<0.0001. (D) Apoptosis assays using flow
cytometry indicated that the number of apoptotic cells was
significantly greater in si-RRM2 transfectants than in the mock or
siRNA-control transfection groups. *P<0.0001. (E)
Western blot analysis of apoptotic marker cleaved PARP in ACHN
786-o, and SU-R-786-o cells demonstrated a significant difference
in cleaved PARP levels between cells with and without RRM2
silencing. β-actin was employed as a loading control. (F) Colony
formation assays demonstrated that colony growth was repressed in
cells with RRM2 knockdown compared with that in the mock or
si-control groups. *P<0.0001. si-, small interfering
RNA; RRM2, ribonucleotide reductase regulatory subunit-M2; GUSB,
glucuronidase β; PARP, poly(ADP-ribose) polymerase; FITC,
fluorescein isothiocyanate. |
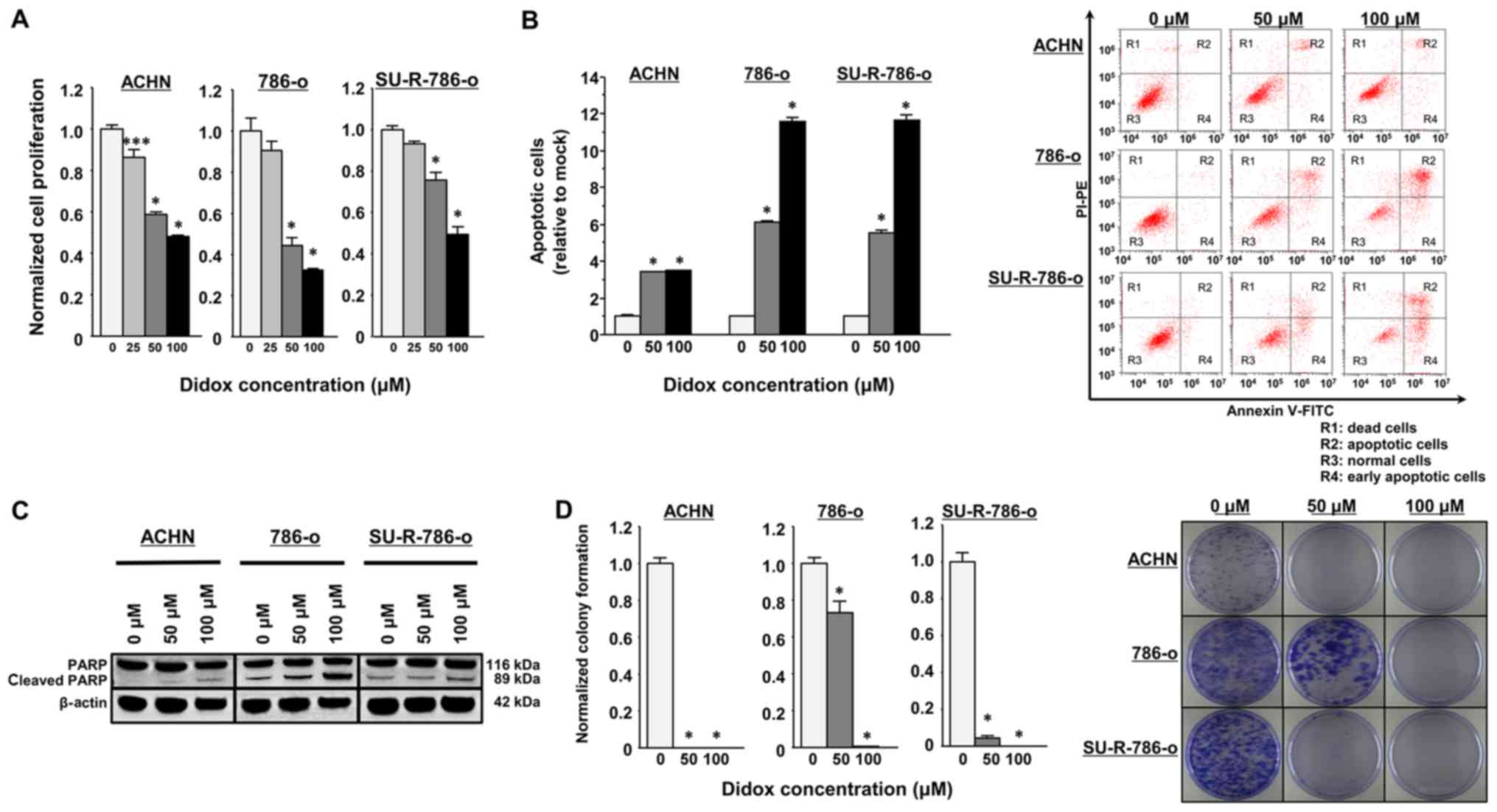
Effects of the RRM2 inhibitor Didox on
cell proliferation, apoptosis and colony formation in SU-R-786-o
cells
The above findings imply that RRM2 inhibitors may be
a promising anticancer agent for repressing cell proliferation and
colony formation by enhancing apoptosis. Didox has strong
inhibitory effects against ribonucleotide reductase that are
associated with DNA synthesis and repair by blocking the synthesis
of deoxyribonucleotides; this inhibitor has demonstrated strong
antitumor effects (32-35). In preliminary analyses, the
inhibition of RRM2 by Didox (50 or 100 µM) was demonstrated
to significantly decrease the proliferation of SU-R-786-o, ACHN and
parental 786-o cells (Fig. 6A).
Apoptosis assays revealed that Didox had significant apoptotic
effects in these cells (P<0.0001; Fig. 6B). Western blot analyses also
revealed that the levels of cleaved PARP were markedly increased in
cells treated with Didox in a concentration-dependent manner
(Fig. 6C). In addition, 50 or 100
µM Didox led to significant inhibition of colony formation
of the cells in a concentration-dependent manner (P<0.0001;
Fig. 6D). These data suggest that
the RRM2 inhibitor Didox may lead to anticancer effects by
inhibiting cancer cell growth, promoting apoptosis and modulating
the colony formation ability in SU-R-786-o and other RCC cells.
Discussion
The guide-strand RNA derived from double-stranded
miRNA is maintained for direct binding of the RNA-induced silencing
complex to target mRNAs, whereas the passenger-strand RNA is
degraded (10,36). Although a previous study performed
functional analyses of miR-99a (guide-strand) in RCC (37), miR-99a-3p (passenger-strand) was
selected as a candidate miRNA and therapeutic target in RCC and
SU-R-786-o cells in the present study. Recently, the two strands of
pre-miR-145, miR-145-5p (guide-strand) and miR-145-3p
(passenger-strand), have been reported to act as antitumor miRNAs
in bladder cancer cells by regulating the gene encoding
ubiquitin-like with PHD and ring finger domains 1 (23). In addition, the passenger strand
miR-21-3p has been reported to mediate cisplatin resistance in
ovarian cancer (38). Therefore,
passenger-strand miRNAs may also be associated with resistance to
chemotherapeutic agents. In fact, several other studies have
reported on miRNAs involved in resistance to molecular-targeted
agents in various cancer types (39,40).
Yumioka et al (41)
demonstrated that restoring miR-194-5p expression in
sunitinib-resistant ccRCC cells sensitized to sunitinib by
downregulating lysosome-associated membrane protein 2. On the other
hand, Kishikawa et al (42)
reported that decreased miR-122 expression may be involved in
sorafenib sensitivity through the upregulation of solute carrier
family 7 expression in hepatocellular carcinoma. The present study
focused on miR-99a-3p due to the observation that this miRNA
strongly inhibited the viability of 786-o and SU-R-786-o cells.
Additionally, a trend towards shorter OS times in patients with low
expression levels of miR-99a-3p was observed, but this was not
statistically significant. Furthermore, cell function assays
demonstrated that apoptosis was induced and colony formation was
inhibited in ACHN, 786-o and SU-R-786-o cells transfected with this
miRNA. To the best of our knowledge, no studies have revealed that
passenger-strand miR-99a-3p is associated with tumorigenesis in
RCC. In other cancer types, miR-99a-3p has been reported to act as
a tumor suppressor in naïve and castration-resistant prostate
cancer (43). Additionally,
miR-99a-3p was validated as a predictor of response to standard
fluoropyrimidine-based chemotherapy in patients with metastatic
colorectal cancer (44).
Therefore, the present finding that miR-99a-3p acts as a tumor
suppressor in RCC cells, including the SU-R-786-o, is reasonable,
even though SU-R-786-o cells were the only sunitinib-resistant cell
line used in this study. Therefore, additional studies are
necessary to investigate the functional roles of miR-99a-3p and
RRM2 in more sunitinib-resistant RCC cell lines. In terms of
the regulatory mechanisms of miR-99a-3p, this study attempted to
explore how miR-99a-3p downregulation occurred in normal and
sunitinib-resistant RCC. Based on the TCGA ccRCC cohort using the
cBioPortal, genetic copy number alterations involving miR-99a-3p
were revealed in only 0.2% of cases (1 out of 528 sequenced cases).
Notably, no reports have suggested that miR-99a-3p is regulated
epigenetically by DNA methylation, histone modification or
noncoding RNAs in RCC or other cancer types. Future studies are
necessary to elucidate the mechanisms of miR-99a-3p downregulation
in normal and sunitinib-resistant RCC. In this study, the
expression levels of miR-99a-3p were significantly lower than in
normal kidney cell RNA, with the exception of the A498 cells. It is
possible that the variation in the expression levels of miR-99a-3p
in the various cell lines are due to their different clinical
origins.
The RRM2 protein is one of two subunits of the
ribonucleotide reductase complex, catalyzing the formation of
deoxyribonucleotides from ribonucleotides. Oncogenic roles of
RRM2 have been reported in several cancer types, including
adrenocortical cancer, gastric adenocarcinoma, breast cancer and
melanoma (45-48). In colorectal cancer and non-small
cell lung carcinoma, RRM2 upregulation was revealed to be
associated with shorter survival time (49,50).
Overexpression of RRM2 has been reported to enhance the
potential of cellular transformation by various oncogenes and to
increase the malignant potential of transformed cells (51). However, to the best of our
knowledge, this is the first study demonstrating the oncogenic role
of RRM2 in RCC as well as sunitinib-resistant RCC cells.
Previous studies have demonstrated that RRM2 knockdown causes
S-phase arrest with no particular enrichment of the
G1-phase population in ccRCC cell lines (52). However, no reports have described
the effects of miR-99a-3p expression on the cell cycle. In the
present study, cell cycle arrest was evaluated in miR-99a-3p- and
si-RRM2-transfectants using flow cytometry and demonstrated that
S-phase arrest was induced in the transfected 786-o cells. By
contrast, in the SU-R-786-o cells, miR-99a-3p transfection caused
S-phase arrest, whereas RRM2 knockdown caused
G0/G1-phase arrest. These results suggest
that there are additional complex mechanisms mediated by
RRM2 that affect cell cycle arrest in sunitinib-resistant
cells. Further studies are necessary to elucidate these
mechanisms.
Malignant cells often exhibit a shift in cellular
metabolism from oxidative phosphorylation to glycolysis, known as
the Warburg effect (53,54). As the Warburg effect is considered
a fundamental property of neoplasia, targeting glycolysis may be a
therapeutically relevant strategy for cancer treatment (55). In addition, a previous study
demonstrated that the Warburg effect contributes to resistance to
molecular-targeted agents in various cancer types (56). Previous studies have indicated that
ccRCC exhibits increased glucose utilization as a result of
overexpression of the genes encoding glucose transporter protein
type 1 and hexokinase-2, known as glycolytic enzymes (57,24).
In addition, it has also been demonstrated that metabolic
reprogramming and chromatin remodeling occur in sunitinib-resistant
cells (6). Notably, previous
reports on cervical and breast cancer confirmed that RRM2
overexpression specifically upregulates hypoxia-inducible factor
(HIF)-1α-associated proliferation and differentiation pathways and
VEGF expression via the activation of the extracellular
signal-regulated kinase 1/2 signaling pathway (58,59).
As continuous HIF activation is thought to be critical for RCC
progression and acquired resistance to tyrosine kinase inhibitors
and mTOR inhibitors (60), the
finding that RRM2 was upregulated via miR-99a-3p
downregulation in sunitinib-resistant cells may represent another
mechanism through which RCC cells acquire sunitinib resistance.
Indeed, several reports have confirmed that the upregulation of
RRM2 contributes to resistance to chemotherapeutic agents in
various types of cancer (61-63).
Therefore, further studies using in vivo models are required
to elucidate the associations between angiogenesis and sunitinib
resistance.
In summary, the present study demonstrated that
miR-99a-3p was downregulated in several RCC cell lines and
SU-R-786-o cells. Additionally, this miRNA was demonstrated to act
as a tumor suppressor through regulating oncogenic RRM2. To
the best of our knowledge, this is the first report to demonstrate
that tumor-suppressive miR-99a-3p directly targets RRM2. The
identification of novel molecular pathways and targets regulated by
the miR-99a-3p/RRM2 axis may improve our understanding of
sunitinib-resistant RCC.
Supplementary Materials
Funding
This study was supported by a Grant-in-Aid for
Scientific Research from the Japan Society for the Promotion of
Science, Tokyo, Japan (nos. 16H05464, 17H04332 and 16K11015).
Availability of data and materials
All datasets used in this study are already provided
as a part of the submitted article.
Authors’ contributions
YO, HY, MY, HE and MN conceived of the study and
designed the experiments. YO, HY, TS and SS performed the
experiments. YO, HY and HE drafted the manuscript. All authors
reviewed the manuscript and approved the final version.
Ethics approval and consent to
participate
The present study was approved by the Bioethics
Committee of Kagoshima University, and written informed consent and
was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Acknowledgments
The authors thank Ms. Mutsumi Miyazaki and Ms. Keiko
Yoshitomi, Department of Urology, Graduate School of Medical and
Dental Sciences, Kagoshima University (Kagoshima, Japan), for their
excellent laboratory assistance.
References
|
1
|
López JI: Renal tumors with clear cells. A
review Pathol Res Pract. 209:137–146. 2013.
|
|
2
|
Ohba K, Miyata Y, Yasuda T, Asai A,
Mitsunari K, Matsuo T, Mochizuki Y, Matsunaga N and Sakai H:
Efficacy and safety of sunitinib alternate day regimen in patients
with metastatic renal cell carcinoma in Japan: Comparison with
standard 4/2 schedule. Asia Pac J Clin Oncol. 14:153–158. 2018.
|
|
3
|
Yang F, Zhou X, Du S, Zhao Y, Ren W, Deng
Q, Wang F and Yuan J: LIM and SH3 domain protein 1 (LASP-1)
overexpression was associated with aggressive phenotype and poor
prognosis in clear cell renal cell cancer. PLoS One.
9:e1005572014.
|
|
4
|
Teng J, Gao Y, Chen M, Wang K, Cui X, Liu
Y and Xu D: Prognostic value of clinical and pathological factors
for surgically treated localized clear cell renal cell carcinoma.
Chin Med J (Engl). 127:1640–1644. 2014.
|
|
5
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008.
|
|
6
|
Yoshino H, Nohata N, Miyamoto K, Yonemori
M, Sakaguchi T, Sugita S, Itesako T, Kofuji S, Nakagawa M, Dahiya
R, et al: PHGDH as a key enzyme for serine biosynthesis in
HIF2alpha targeting therapy for renal cell carcinoma. Cancer Res.
77:6321–6329. 2017.
|
|
7
|
Rajan A, Kim C, Heery CR, Guha U and
Gulley JL: Nivolumab, anti-programmed death-1 (PD-1) monoclonal
antibody immunotherapy: Role in advanced cancers. Hum Vaccin
Immunother. 12:2219–2231. 2016.
|
|
8
|
Motzer RJ, Escudier B, McDermott DF,
George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G,
Plimack ER, et al CheckMate 025 Investigators: Nivolumab versus
Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med.
373:1803–1813. 2015.
|
|
9
|
Sarfaty M, Leshno M, Gordon N, Moore A,
Neiman V, Rosenbaum E and Goldstein DA: Cost Effectiveness of
Nivolumab in Advanced Renal Cell Carcinoma. Eur Urol. 73:628–634.
2018.
|
|
10
|
Carthew RW and Sontheimer EJ: Origins and
Mechanisms of miRNAs and siRNAs. Cell. 136:642–655. 2009.
|
|
11
|
Landgraf P, Rusu M, Sheridan R, Sewer A,
Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M,
et al: A mammalian microRNA expression atlas based on small RNA
library sequencing. Cell. 129:1401–1414. 2007.
|
|
12
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39(Database): D152–D157. 2011.
|
|
13
|
Kawakami K, Enokida H, Chiyomaru T,
Tatarano S, Yoshino H, Kagara I, Gotanda T, Tachiwada T, Nishiyama
K, Nohata N, et al: The functional significance of miR-1 and
miR-133a in renal cell carcinoma. Eur J Cancer. 48:827–836.
2012.
|
|
14
|
Xie M, Dart DA, Guo T, Xing XF, Cheng XJ,
Du H, Jiang WG, Wen XZ and Ji JF: MicroRNA-1 acts as a tumor
suppressor microRNA by inhibiting angiogenesis-related growth
factors in human gastric cancer. Gastric cancer. 21:41–54.
2018.
|
|
15
|
Xu W, Zhang Z, Zou K, Cheng Y, Yang M,
Chen H, Wang H, Zhao J, Chen P, He L, et al: MiR-1 suppresses tumor
cell proliferation in colorectal cancer by inhibition of
Smad3-mediated tumor glycolysis. Cell Death Dis. 8:e27612017.
|
|
16
|
Xi Y: MicroRNA: A New Player for Cancer
Chemoprevention. J Integr Oncol. 2:pii: 1052013.
|
|
17
|
Yi B, Piazza GA, Su X and Xi Y: MicroRNA
and cancer chemo-prevention. Cancer Prev Res (Phila). 6:401–409.
2013.
|
|
18
|
Lin HM, Nikolic I, Yang J, Castillo L,
Deng N, Chan CL, Yeung NK, Dodson E, Elsworth B, Spielman C, et al:
MicroRNAs as potential therapeutics to enhance chemosensitivity in
advanced prostate cancer. Sci Rep. 8:78202018.
|
|
19
|
Xiao W, Lou N, Ruan H, Bao L, Xiong Z,
Yuan C, Tong J, Xu G, Zhou Y, Qu Y, et al: Mir-144-3p Promotes Cell
Proliferation, Metastasis, Sunitinib Resistance in Clear Cell Renal
Cell Carcinoma by Downregulating ARID1A. Cell Physiol Biochem.
43:2420–2433. 2017.
|
|
20
|
Sobin LH and Compton CC: TNM seventh
edition: what’s new, what’s changed: communication from the
International Union Against Cancer and the American Joint Committee
on Cancer. Cancer. 116:5336–5339. 2010.
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
22
|
Ichimi T, Enokida H, Okuno Y, Kunimoto R,
Chiyomaru T, Kawamoto K, Kawahara K, Toki K, Kawakami K, Nishiyama
K, et al: Identification of novel microRNA targets based on
microRNA signatures in bladder cancer. Int J Cancer. 125:345–352.
2009.
|
|
23
|
Matsushita R, Yoshino H, Enokida H, Goto
Y, Miyamoto K, Yonemori M, Inoguchi S, Nakagawa M and Seki N:
Regulation of UHRF1 by dual-strand tumor-suppressor microRNA-145
(miR-145-5p and miR-145-3p): Inhibition of bladder cancer cell
aggressiveness. Oncotarget. 7:28460–28487. 2016.
|
|
24
|
Yoshino H, Enokida H, Itesako T, Kojima S,
Kinoshita T, Tatarano S, Chiyomaru T, Nakagawa M and Seki N:
Tumor-suppressive microRNA-143/145 cluster targets hexo-kinase-2 in
renal cell carcinoma. Cancer Sci. 104:1567–1574. 2013.
|
|
25
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976.
|
|
26
|
Yoshino H, Chiyomaru T, Enokida H,
Kawakami K, Tatarano S, Nishiyama K, Nohata N, Seki N and Nakagawa
M: The tumour-suppressive function of miR-1 and miR-133a targeting
TAGLN2 in bladder cancer. Br J Cancer. 104:808–818. 2011.
|
|
27
|
Anaya J: OncoLnc: linking TCGA survival
data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science.
2:e672016.
|
|
28
|
Berkers J, Govaere O, Wolter P, Beuselinck
B, Schöffski P, van Kempen LC, Albersen M, Van den Oord J, Roskams
T, Swinnen J, et al: A possible role for microRNA-141
down-regulation in sunitinib resistant metastatic clear cell renal
cell carcinoma through induction of epithelial-to-mesenchymal
transition and hypoxia resistance. J Urol. 189:1930–1938. 2013.
|
|
29
|
Papadopoulos EI, Yousef GM and Scorilas A:
Cytotoxic activity of sunitinib and everolimus in Caki-1 renal
cancer cells is accompanied by modulations in the expression of
apoptosis-related microRNA clusters and BCL2 family genes. Biomed
Pharmacother. 70:33–40. 2015.
|
|
30
|
Goto Y, Kurozumi A, Nohata N, Kojima S,
Matsushita R, Yoshino H, Yamazaki K, Ishida Y, Ichikawa T, Naya Y,
et al: The microRNA signature of patients with sunitinib failure:
Regulation of UHRF1 pathways by microRNA-101 in renal cell
carcinoma. Oncotarget. 7:59070–59086. 2016.
|
|
31
|
Merhautova J, Hezova R, Poprach A,
Kovarikova A, Radova L, Svoboda M, Vyzula R, Demlova R and Slaby O:
miR-155 and miR-484 Are Associated with Time to Progression in
Metastatic Renal Cell Carcinoma Treated with Sunitinib. BioMed Res
Int. 2015:9419802015.
|
|
32
|
Elford HL, Freese M, Passamani E and
Morris HP: Ribonucleotide reductase and cell proliferation I
Variations of ribonucleotide reductase activity with tumor growth
rate in a series of rat hepatomas. J Biol Chem. 245:5228–5233.
1970.
|
|
33
|
Elford HL, Wampler GL and van’t Riet B:
New ribonucleotide reductase inhibitors with antineoplastic
activity. Cancer Res. 39:844–851. 1979.
|
|
34
|
Elford HL, Van’t Riet B, Wampler GL, Lin
AL and Elford RM: Regulation of ribonucleotide reductase in
mammalian cells by chemotherapeutic agents. Adv Enzyme Regul.
19:151–168. 1980.
|
|
35
|
van’t Riet B, Wampler GL and Elford HL:
Synthesis of hydroxy- and amino-substituted benzohydroxamic acids:
Inhibition of ribonucleotide reductase and antitumor activity. J
Med Chem. 22:589–592. 1979.
|
|
36
|
Chendrimada TP, Finn KJ, Ji X, Baillat D,
Gregory RI, Liebhaber SA, Pasquinelli AE and Shiekhattar R:
MicroRNA silencing through RISC recruitment of eIF6. Nature.
447:823–828. 2007.
|
|
37
|
Cui L, Zhou H, Zhao H, Zhou Y, Xu R, Xu X,
Zheng L, Xue Z, Xia W, Zhang B, et al: MicroRNA-99a induces
G1-phase cell cycle arrest and suppresses tumorigenicity in renal
cell carcinoma. BMC Cancer. 12:5462012.
|
|
38
|
Pink RC, Samuel P, Massa D, Caley DP,
Brooks SA and Carter DR: The passenger strand, miR-21-3p, plays a
role in mediating cisplatin resistance in ovarian cancer cells.
Gynecol Oncol. 137:143–151. 2015.
|
|
39
|
Ayers D and Vandesompele J: Influence of
microRNAs and Long Non-Coding RNAs in Cancer Chemoresistance. Genes
(Basel). 8:82017.
|
|
40
|
An X, Sarmiento C, Tan T and Zhu H:
Regulation of multidrug resistance by microRNAs in anti-cancer
therapy. Acta Pharm Sin B. 7:38–51. 2017.
|
|
41
|
Yumioka T, Osaki M, Sasaki R, Yamaguchi N,
Onuma K, Iwamoto H, Morizane S, Honda M, Takenaka A and Okada F:
Lysosome-associated membrane protein 2 (LAMP-2) expression induced
by miR-194-5p downregulation contributes to sunitinib resistance in
human renal cell carcinoma cells. Oncol Lett. 15:893–900. 2018.
|
|
42
|
Kishikawa T, Otsuka M, Tan PS, Ohno M, Sun
X, Yoshikawa T, Shibata C, Takata A, Kojima K, Takehana K, et al:
Decreased miR122 in hepatocellular carcinoma leads to
chemoresistance with increased arginine. Oncotarget. 6:8339–8352.
2015.
|
|
43
|
Arai T, Okato A, Yamada Y, Sugawara S,
Kurozumi A, Kojima S, Yamazaki K, Naya Y, Ichikawa T and Seki N:
Regulation of NCAPG by miR-99a-3p (passenger strand) inhibits
cancer cell aggressiveness and is involved in CRPC. Cancer Med.
7:1988–2002. 2018.
|
|
44
|
Molina-Pinelo S, Carnero A, Rivera F,
Estevez-Garcia P, Bozada JM, Limon ML, Benavent M, Gomez J, Pastor
MD, Chaves M, et al: MiR-107 and miR-99a-3p predict chemotherapy
response in patients with advanced colorectal cancer. BMC Cancer.
14:6562014.
|
|
45
|
Grolmusz VK, Karászi K, Micsik T, Tóth EA,
Mészáros K, Karvaly G, Barna G, Szabó PM, Baghy K, Matkó J, et al:
Cell cycle dependent RRM2 may serve as proliferation marker and
pharmaceutical target in adrenocortical cancer. Am J Cancer Res.
6:2041–2053. 2016.
|
|
46
|
Kang W, Tong JH, Chan AW, Zhao J, Wang S,
Dong Y, Sin FM, Yeung S, Cheng AS, Yu J, et al: Targeting
ribonucleotide reductase M2 subunit by small interfering RNA exerts
anti-oncogenic effects in gastric adenocarcinoma. Oncol Rep.
31:2579–2586. 2014.
|
|
47
|
Shah KN, Mehta KR, Peterson D, Evangelista
M, Livesey JC and Faridi JS: AKT-induced tamoxifen resistance is
overturned by RRM2 inhibition. Mol Cancer Res. 12:394–407.
2014.
|
|
48
|
Zuckerman JE, Hsueh T, Koya RC, Davis ME
and Ribas A: siRNA knockdown of ribonucleotide reductase inhibits
melanoma cell line proliferation alone or synergistically with
temozolomide. J Invest Dermatol. 131:453–460. 2011.
|
|
49
|
Liu X, Zhang H, Lai L, Wang X, Loera S,
Xue L, He H, Zhang K, Hu S, Huang Y, et al: Ribonucleotide
reductase small subunit M2 serves as a prognostic biomarker and
predicts poor survival of colorectal cancers. Clin Sci (Lond).
124:567–578. 2013.
|
|
50
|
Mah V, Alavi M, Márquez-Garbán DC, Maresh
EL, Kim SR, Horvath S, Bagryanova L, Huerta-Yepez S, Chia D,
Pietras R, et al: Ribonucleotide reductase subunit M2 predicts
survival in subgroups of patients with non-small cell lung
carcinoma: Effects of gender and smoking status. PLoS One.
10:e01276002015.
|
|
51
|
Fan H, Villegas C and Wright JA:
Ribonucleotide reductase R2 component is a novel malignancy
determinant that cooperates with activated oncogenes to determine
transformation and malignant potential. Proc Natl Acad Sci USA.
93:14036–14040. 1996.
|
|
52
|
Avolio TM, Lee Y, Feng N, Xiong K, Jin H,
Wang M, Vassilakos A, Wright J and Young A: RNA interference
targeting the R2 subunit of ribonucleotide reductase inhibits
growth of tumor cells in vitro and in vivo. Anticancer Drugs.
18:377–388. 2007.
|
|
53
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer’s Achilles’ heel. Cancer Cell. 13:472–482.
2008.
|
|
54
|
Vander Heiden MG: Targeting cancer
metabolism: A therapeutic window opens. Nat Rev Drug Discov.
10:671–684. 2011.
|
|
55
|
Lai IL, Chou CC, Lai PT, Fang CS, Shirley
LA, Yan R, Mo X, Bloomston M, Kulp SK, Bekaii-Saab T, et al:
Targeting the Warburg effect with a novel glucose transporter
inhibitor to overcome gemcitabine resistance in pancreatic cancer
cells. Carcinogenesis. 35:2203–2213. 2014.
|
|
56
|
Liu J, Pan C, Guo L, Wu M, Guo J, Peng S,
Wu Q and Zuo Q: A new mechanism of trastuzumab resistance in
gastric cancer: MACC1 promotes the Warburg effect via activation of
the PI3K/AKT signaling pathway. J Hematol Oncol. 9:762016.
|
|
57
|
Yamasaki T, Seki N, Yoshino H, Itesako T,
Yamada Y, Tatarano S, Hidaka H, Yonezawa T, Nakagawa M and Enokida
H: Tumor-suppressive microRNA-1291 directly regulates glucose
transporter 1 in renal cell carcinoma. Cancer Sci. 104:1411–1419.
2013.
|
|
58
|
Wang N, Zhan T, Ke T, Huang X, Ke D, Wang
Q and Li H: Increased expression of RRM2 by human papillomavirus E7
oncoprotein promotes angiogenesis in cervical cancer. Br J Cancer.
110:1034–1044. 2014.
|
|
59
|
Shah KN, Wilson EA, Malla R, Elford HL and
Faridi JS: Targeting Ribonucleotide Reductase M2 and NF-κB
Activation with Didox to Circumvent Tamoxifen Resistance in Breast
Cancer. Mol Cancer Ther. 14:2411–2421. 2015.
|
|
60
|
Rini BI and Atkins MB: Resistance to
targeted therapy in renal-cell carcinoma. Lancet Oncol.
10:992–1000. 2009.
|
|
61
|
Putluri N, Maity S, Kommagani R, Creighton
CJ, Putluri V, Chen F, Nanda S, Bhowmik SK, Terunuma A, Dorsey T,
et al: Pathway-centric integrative analysis identifies RRM2 as a
prognostic marker in breast cancer associated with poor survival
and tamoxifen resistance. Neoplasia. 16:390–402. 2014.
|
|
62
|
Zhou B, Su L, Hu S, Hu W, Yip ML, Wu J,
Gaur S, Smith DL, Yuan YC, Synold TW, et al: A small-molecule
blocking ribo-nucleotide reductase holoenzyme formation inhibits
cancer cell growth and overcomes drug resistance. Cancer Res.
73:6484–6493. 2013.
|
|
63
|
Zhang M, Wang J, Yao R and Wang L: Small
interfering RNA (siRNA)-mediated silencing of the M2 subunit of
ribonucleotide reductase: a novel therapeutic strategy in ovarian
cancer. Int G Gynecol Cancer. 23:659–666. 2013.
|