Introduction
Osteosarcoma (OS) is a highly malignant and
aggressive bone tumor that mostly occurs in children and
adolescents. OS is characterized by early lung metastasis and a
poor prognosis (1). The 5-year
survival rate of patients with localized OS has remained stable at
60-70% in recent years due to advances in chemotherapy. However,
the development of metastasis reduces the survival rate to 20%
(2).
Epigenetic processes play a key role in the
regulation of gene expression by affecting chromatin structure. DNA
methylation and histone modification are important epigenetic
mediators of transcriptional suppression, and they are essential
for biological processes (3).
Aberrant epigenetic regulation, such as DNA hypermethylation and
histone deacetylation, is a frequent event within the promoters of
tumor suppressor genes in cancer cells (4) and contributes to cancer development,
progression and metastasis (5).
DNA methyltransferase (DNMT) and histone deacetylase (HDAC)
inhibitors synergistically reactivate epigenetically silenced tumor
suppressor genes and induce growth inhibition and apoptosis of
tumor cells (6). Recently, DNMT
and HDAC inhibitors were shown to directly inhibit endothelial cell
growth and tumor angiogenesis (7,8).
Angiogenesis is characterized by the formation of new blood vessels
and is required for fundamental physiological processes, including
embryonic development and tissue repair (9); it also has the potential to promote
tumor progression and the development of metastasis (10). Tumor cells secrete pro-angiogenic
factors, such as vascular endothelial growth factor (VEGF). VEGF,
also referred to as VEGF-A, belongs to a large family of proteins.
VEGFs and VEGF-Rs are important for vessel formation in healthy
individuals, as well as tumor angiogenesis, and the interaction
between receptors and ligands mediates the survival and
proliferation of malignant cells (11).
Vascular endothelial growth inhibitor (VEGI), also
referred to as TL1A or TNFSF15, is a member of the tumor necrosis
factor (TNF) superfamily (12).
VEGI is found in endothelial cells and acts as an endogenous
angiogenic inhibitor. VEGI in endothelial cells inhibits cell
growth and migration (13). VEGI
is known to operate via two receptors: Death receptor-3 (DR3) and
decoy receptor-3 (DcR3). DR3 is a member of the TNF receptor
superfamily and is also known as TNFSF25; it contains a death
domain in the cytoplasm that is associated with the induction of
apoptosis and nuclear factor-κB activation (14). DcR3 is a decoy receptor for VEGI
and is a secreted soluble protein (14-16)
that acts as an antagonist to VEGI/DR3 interaction. DcR3 is
expressed in a wide range of human tissues and is overexpressed in
several tumors (17).
We previously demonstrated that VPA and Trichostatin
A (TSA), which were histone deacetylase inhibitors, increased the
expression of VEGI while exerting little effect on its receptor,
DR3, and sensitized both OS and human microvascular endothelial
(HMVE) cells to apoptosis via the VEGI/DR3 autocrine and paracrine
pathways (18). The aim of the
present study was to further investigate the effect of the
combination of DNMT and HDAC inhibitors on VEGI and DR3. We
observed that the combination of the DNA methylation inhibitor Hy
with VPA increased the expression of VEGI and markedly increased
the expression of DR3 in OS and HMVE cells compared with
monotherapy, and also enhanced the production of soluble VEGI
without enhancing the production of DcR3. Combination treatment of
OS cell culture media significantly inhibited vascular tube
formation. Furthermore, the physical interaction of VEGI and VEGF-A
was observed by immunoprecipitation. These findings may provide
evidence of additional VEGI-mediated anti-angiogenic machinery.
Materials and methods
Cell culture and drugs
U-2 OS and SaOS-2 human OS cells were purchased from
the American Type Culture Collection and Riken BRC Cell Bank,
respectively. The U-2 OS and SaOS-2 cells were cultured in McCoy's
5A modified medium (Invitrogen; Thermo Fisher Scientific, Inc.).
Primary normal HMVE cells were purchased from the Cell Systems
Corporation and cultured using a CS-C medium kit (DS Pharma
Biomedical Co., Ltd.). All media contained 10% fetal bovine serum
(FBS) (MP Biomedical), penicillin (100 U/ml) and streptomycin (100
µg/ml). All the cells were cultured in a humidified
atmosphere of 5% CO2/95% air at 37°C. VPA was purchased
from Wako Pure Chemical Industries, Ltd., and Hy was purchased from
Sigma-Aldrich; Merck KGaA.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
U-2 OS, SaOS-2 cells and HMVE cells were cultured
with or without 20 µM Hy and 1.0 mM VPA. The culture medium
was changed on day 3. Total RNA was isolated from each cell culture
dish on days 3 and 7 using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), and 2.0 µg was reverse-transcribed
using a High-Capacity RNA-to-cDNA kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. qPCR using TaqMan Gene Expression assays (Applied
Biosystems; Thermo Fisher Scientific, Inc.) was performed to detect
VEGI, DR3 and DcR3 mRNA expression. The primer sets were
Hs00270802_ml for VEGI mRNA, Hs00600930_ml for DR3 mRNA, and
Hs00187070_ml for DcR3 mRNA (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The amount of GAPDH mRNA (as an internal
reference) was estimated using human GAPDH endogenous control
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The relative
gene expression was analyzed and calculated via the
2−ΔΔCq method (19),
and the mRNA expression levels of VEGI, DR3 and DcR3 were
normalized to GAPDH.
Chromatin immunoprecipitation (ChIP)
assay
The ChIP assay was performed using the EZ-Magna
ChIP™ A kit (EMD Millipore; cat. no. 17-408) according to the
manufacturer's instructions. The ChIP samples were obtained from
SaOS-2 cells cultured with or without 20 µM Hy and 1.0 mM
VPA for 3 days. In brief, nuclear lysate containing protein-DNA
complexes was prepared from cross-linking proteins bound to DNA
after formaldehyde treatment. An aliquot of the nuclear lysate was
used to immunoprecipitate acetylated histone-DNA complexes with an
anti-acetyl-histone H3 rabbit polyclonal antibody (kit supplied by
EMD Millipore; cat. no. 06-599B). DNA was extracted from the
precipitated acetylated histone-DNA complexes. The VEGI gene
promoter sequence was amplified by a PCR using Takara Ex Taq™ DNA
polymerase (Takara Bio, Inc.) and the promoter region from −1515 to
−953 with the following primers: Sense, 5′-GTTCCAACAC CACCTCTTTC-3′
and antisense, 5′-AGTTCTAAATCACG GCTTGG-3′, for the VEGI promoter.
The initial denaturation and final extension of the PCR were
performed at 95°C for 5 min and at 72°C for 7 min, respectively,
and the PCR conditions were 95°C for 1 min, 55°C for 1 min and 72°C
for 1 min for 35 cycles. The amplified fragments were resolved by
electrophoresis in a 1.5% agarose gel with ethidium bromide
staining.
Methylation-specific polymerase chain
reaction (MSP)
Bisulfite-converted genomic DNA was obtained from
SaOS-2 cells after 3 days of Hy and/or VPA treatment using a
Cells-to-CpG™ bisulfite conversion kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The bisulfite-converted genomic DNA was used as a
template for MSP using a GC-rich PCR system (Roche Diagnostics).
The primer pairs for the amplified methylated DNA for the region
from -55 to +143 were as follows: Sense,
5′-TTACGACGGGTAGAGAGTACG-3′ and antisense, 5′-ACT
TAAAATAAAAACGCGCCC-3′. The primer pairs for the amplified
unmethylated DNA for the region from −51 to +150 were as follows:
Sense, 5′-GGAATTATGATGGGTAGAGAGT ATG-3′ and antisense,
5′-CAATAAAACTTAAAATAAAAAC ACACCC-3′. These primers were designed
with the MethPrimer software program (available at www.urogene.org/methprimer2). The initial
denaturation and final extension steps of the PCR were performed at
95°C for 5 min and at 72°C for 10 min, respectively. The
amplification conditions of the PCR were 35 cycles at 95°C for 1
min, at 57°C for 1 min, and at 72°C for 2 min. The amplified
fragments were resolved by electrophoresis on 1.5% agarose gels
with ethidium bromide staining.
Enzyme-linked immunosorbent assay
(ELISA)
U-2 OS and SaOS-2 cells were seeded at
2.0×105 cells/dish in 10-cm tissue culture dishes
containing 5 ml of medium per dish. HMVE cells were seeded at
1.0×104 cells/dish in 6-cm tissue culture dishes
containing 2 ml of medium per dish. After 24 h (Day 0), 20
µM Hy and/or 1.0 mM VPA were added to the medium and
cultured for 7 days. The medium was changed after 3 days (Day 3),
and the remaining dishes were cultured for a further 4 days (until
Day 7). To detect soluble VEGI, 96-well plates were coated with the
capture antibody (anti-human VEGI mouse monoclonal antibody, 2.0
µg/ml; Santa Cruz Biotechnology, Inc.; cat. no. sc-53975)
overnight at room temperature, and then washed three times with
washing buffer (PBS containing 0.05% Tween-20). Standard protein,
recombinant human TL1A/TNFSF15 (R&D Systems, Inc.; cat. no.
1319-TL-010) and samples were incubated in each well for 2 h at
room temperature. After washing, biotin-conjugated anti-human VEGI
rabbit polyclonal antibody (0.5 µg/ml, Abcam; cat. no.
ab84233) was added to the wells and incubated for 2 h at room
temperature. Following 20 min incubation at room temperature with
horseradish peroxidase-conjugated streptavidin, substrate solution
(both from R&D Systems, Inc.) was added to the wells and
incubated for 20 min at room temperature. The detection of soluble
DcR3 was performed using an ELISA development system (R&D
Systems, Inc.) according to the manufacturer's instructions. To
determine the optical density of each well, the absorbance at 450
nm was measured against a reference wavelength of 570 nm using a
microplate reader (Bio-Rad Laboratories, Inc.). The effect of Hy or
VPA was evaluated by determining the mean value of the soluble
forms per 104 viable cells in the treated cultures, and
was expressed as the ratio of the mean value in the untreated
control cultures.
Plasmid construction and
transfection
VEGI cDNA plasmid (pVEGI) was obtained from RT-PCR
fragments amplified from U-2 OS cell total RNA (18). The siRNA designed for VEGI mRNA was
5′-ACCUGACAGUUGUGAGACAtt-3′ (sense strand) and was synthesized by
Applied Biosystems; Thermo Fisher Scientific, Inc. For
transfection, U-2 OS and SaOS-2 cells were seeded at
1×105 cells/well in 6-well tissue culture plates and
cultured in 2 ml of medium for 24 h. The culture medium was changed
to Opti-MEM (Invitrogen; Thermo Fisher Scientific, Inc.), and the
cells were transfected with 2.0 µg of plasmid DNA or 20 nM
of siRNA using Lipofectamine™ 2000 and RNAiMAX (both from
Invitrogen; Thermo Fisher Scientific, Inc.), respectively,
according to the manufacturer's instructions. Whole-cell lysate and
cell culture medium were collected 48 h after transfection.
Whole-cell lysis was performed and the findings were analyzed by an
immunoprecipitation assay. In addition, the cell culture medium was
subjected to an in vitro tube formation assay.
Western blot analyses
SaOS-2 and HMVE cells with or without Hy and VPA
treatment and pVEGI-transfected OS cells were washed twice with PBS
and lysed in radioimmunoprecipitation assay buffer [50 mM Tris-HCl
(pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1%
SDS] supplemented with a complete protease inhibitor cocktail
(Roche Diagnostics). In brief, the cell lysates were first
incubated on ice for 30 min and then sonicated three times (5 sec
each time) prior to centrifugation at 7,700 × g for 10 min at 4°C.
The supernatant was collected and the protein concentration was
measured using NanoDrop (Thermo Fisher Scientific, Inc.). An
aliquot of the supernatant (equivalent to 30 µg protein) was
mixed with a 3-fold volume of SDS sample buffer (BioLab) containing
10% β-mercaptoethanol, heated to 95°C for 10 min, and
electrophoresed on a 4-12% Bis-Tris gel with the MES SDS Running
Buffer (Invitrogen; Thermo Fisher Scientific, Inc.) before being
transferred onto a nitrocellulose membrane. The membrane was
blocked for 30 min at room temperature in blocking buffer
containing 5% skimmed milk in Tris-buffered saline with Tween-20
[TBST; 10 mM Tris, 150 mM NaCl (pH 7.4), 1% Tween-20] and then
incubated for 90 min at room temperature with anti-human VEGI mouse
monoclonal antibody (1:200 dilution; Santa Cruz Biotechnology,
Inc., cat. no. sc-53975) or anti-human DR3 mouse monoclonal
antibody (1:500 dilution; Santa Cruz Biotechnology, Inc.; cat. no.
sc-374203) in TBST buffer, after which time the membrane was washed
with TBST three times (10 min each time). The membrane was then
incubated for 90 min at room temperature with horseradish
peroxidase-conjugated goat anti-mouse IgG antibody for VEGI and DR3
(1:5,000 dilution; Santa Cruz Biotechnology, Inc.; cat. no.
sc-2005) and was detected and visualized with SuperSignal™ West
Pico PLUS chemiluminescence substrate (Thermo Fisher Scientific,
Inc.). The same membrane was then stripped using stripping buffer
(Thermo Fisher Scientific, Inc.) and re-probed using actin, which
was detected with anti-actin rabbit polyclonal antibody
(Sigma-Aldrich; Merck KGaA; cat. no. A2066) at a 1:200 dilution in
TBST buffer.
HMVE cell proliferation assay
HMVE cells (1×103 cells per well) were
seeded onto 96-well tissue culture plates containing 100 µl
of CS-C complete medium in each well. After 24 h (Day 0), 20
µM Hy and 1.0 mM VPA were added to the medium and cultured
for 7 days. The medium was changed after 3 days (Day 3) and the
remaining dishes were cultured for a further 4 days (until Day 7).
The number of viable cells in each well was estimated using a
CellTiter 96® AQueous One solution cell proliferation
assay kit (Promega Corporation) according to the manufacturer's
instructions, and the findings are presented as the ratio to the
mean level of optical density in control cultures.
In vitro tube formation assay
HMVE cells were subjected to an in vitro tube
formation assay using a Cultrex® in vitro angiogenesis assay
tube formation kit (Trevigen Inc.), according to the manufacturer's
instructions. In brief, 1.0×104 HMVE cells were seeded
onto BME gel pre-coated 96-well plates and cultured with HMVE cell
culture media in the absence of angiogenesis mediators and FBS.
After 1 h, Hy and/or VPA was added, or the medium was changed to
Hy- and/or VPA-treated OS cell culture media after centrifugation
for 1 min at 800 × g at 4°C, and then incubation was extended for
another 16 h. Calcein AM staining was performed after washing twice
with 1X PBS by gentle pipetting. The endothelial cells and tubes
were examined using a fluorescence microscope (Nikon Corporation).
The ratio of the mean value was estimated by counting the number of
complete tubular shapes in four independent experiments by four
independent researchers.
Immunoprecipitation assay
A total of 200 µg cell lysate obtained from
U-2 OS and SaOS-2 cells (1×106 cells) treated with VPA
or transfected with pVEGI was pre-cleaned with 20 µl of
protein G-Sepharose beads (Santa Cruz Biotechnology, Inc.) for 3 h
at 4°C and washed twice with 1X immunoprecipitation (IP) buffer [50
mM Tris-HCl (pH 7.5), 120 mM NaCl, 0.2 mM NaF, 0.2 mM
Na3VO4, 1 mM PMSF and 0.1% NP-40]. Following
centrifugation at 11,100 × g for 1 min at 4°C, 100 µl
supernatant was collected and incubated with protein G-Sepharose
beads (Bio-Rad Laboratories, Inc.) coated with 20 µg
anti-VEGF-165 mouse monoclonal antibody (BioLegend; cat. no.
662702) in 500 µl 1X IP buffer overnight at 4°C. The beads
were then gently washed four times with 1X IP buffer, and the
proteins bound to the beads were eluted with 3X SDS sample buffer
containing 10% β-mercaptoethanol. The protein samples obtained from
IP were subjected to western blotting to detect anti-V5-Tag
horseradish peroxidase-conjugated antibody (Invitrogen; Thermo
Fisher Scientific, Inc.; cat. no. R961-25), as described above.
Opposite detection was performed with protein G-Sepharose beads
coated with 20 µg anti-TL1A rabbit polyclonal antibody
(Abcam; cat. no. ab85566) and was detected by western blotting
using anti-VEGF-165A mouse monoclonal antibody (Abcam; cat. no.
ab69479. The membrane was then incubated for 90 min at room
temperature with anti-mouse IgG TrueBlot® ULTRA (1:1,000
dilution; Rockland Immunochemicals, Inc.; cat. no. 18-8817-31) as a
secondary antibody.
Statistical analysis
Data are presented as the mean ± standard error. The
data of three or more groups were analyzed by one-way ANOVA
followed by the Newman-Keuls test for multiple comparisons.
P-values <0.05 were considered to indicate statistically
significant differences.
Results
Effects of Hy and VPA on VEGI mRNA and
protein expression in OS cells
U-2 OS and SaOS-2 cells were cultured with or
without 20 µM Hy and 1.0 mM VPA for 3 or 7 days, and the
degree of VEGI mRNA transcription was quantitated by qPCR. The VEGI
mRNA expression in both cell lines was increased ~2.5- to 4.0-fold
by VPA treatment (P<0.001) and 2.5- to 4.3-fold by combination
treatment with Hy and VPA (P<0.001). Hy treatment also increased
VEGI expression 1.5- to 2.0-fold. However, there was no significant
difference compared with no treatment as a control (P>0.05)
(Fig. 1A). The protein translation
of VEGI under these conditions was confirmed by western blotting
(Fig. 1B). These results suggest
that the VEGI expression was more prominently enhanced by exposure
to VPA rather than to Hy. SaOS-2 cells were examined to confirm the
effect of VPA on the VEGI expression by a ChIP assay. The result
demonstrated that acetylated histone bound to the VEGI promoters
after VPA treatment and combination treatment (Fig. 1C).
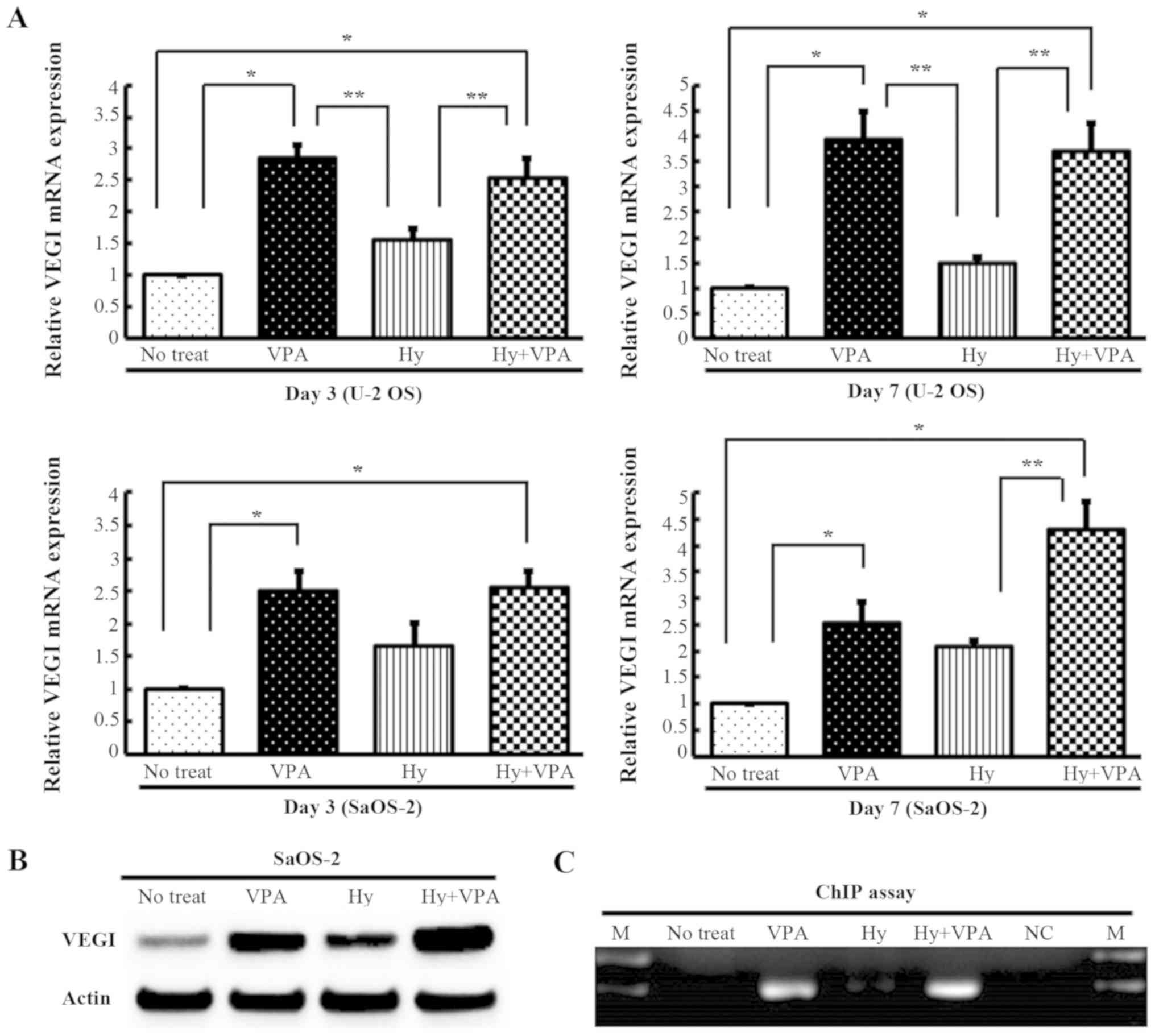 | Figure 1Effects of Hy and VPA on the
expression of VEGI and the analysis of the acetylation status of
VEGI gene promoters in OS cell lines. U-2 OS and SaOS-2 cells were
treated with 20 µM Hy and/or 1.0 mM VPA for 3 (Day 3) or 7
days (Day 7). The medium was changed on Day 3. (A) VEGI mRNAs was
quantitated using qPCR analysis. The values are expressed as the
ratio to the average value in the no treat as a control. Each bar
indicates the mean ± SE of values from four independent experiments
of each samples, in three sets of condition cultures.
*P<0.001, significant difference in comparison to no
treat. **P<0.01, significant difference in comparison to Hy. (B)
The VEGI protein expression was examined by western blotting. (C) A
ChIP assay was performed with 20 µM Hy and/or 1.0 mM VPA
treatment on Day 3 in SaOS-2 cells using anti-acetyl histone H3
antibodies. DNA bound to acetylated histones was amplified using
specific primers for the VEGI promoter region. M, molecular marker;
NC, negative control; Hy, hydralazine; VPA, sodium valproate; VEGI,
vascular endothelial growth inhibitor; OS, osteosarcoma; qPCR,
quantitative polymerase chain reaction; SE, standard error; ChIP,
chromatin immunoprecipitation. |
Effects of Hy and VPA on the mRNA and
protein expression of the VEGI-related receptor DR3 in OS
cells
U-2 OS and SaOS-2 cells were cultured with or
without 20 µM Hy and 1.0 mM VPA for 3 or 7 days, and the
mRNA transcription of DR3 was quantitated by qPCR. The DR3 mRNA
expression in both cell lines was increased 1.6- to 1.9-fold by VPA
treatment (P<0.01) and 1.7- to 2.5-fold by Hy treatment
(P<0.01). Combination treatment with Hy and VPA increased the
expression 2.1- to 4.2-fold (P<0.01) (Fig. 2A). The translation of DR3 under
these conditions was confirmed in SaOS-2 cells by western blotting
(Fig. 2B). These results indicate
that the expression of DR3 was more prominently enhanced by
exposure to Hy rather than to VPA. SaOS-2 cells were examined by an
MSP assay to confirm the effects of Hy on DR3 expression.
Demethylation of the promoter region of DR3 was verified by the
conversion of cytosine to uracil following Hy treatment and
combination treatment (Fig.
2C).
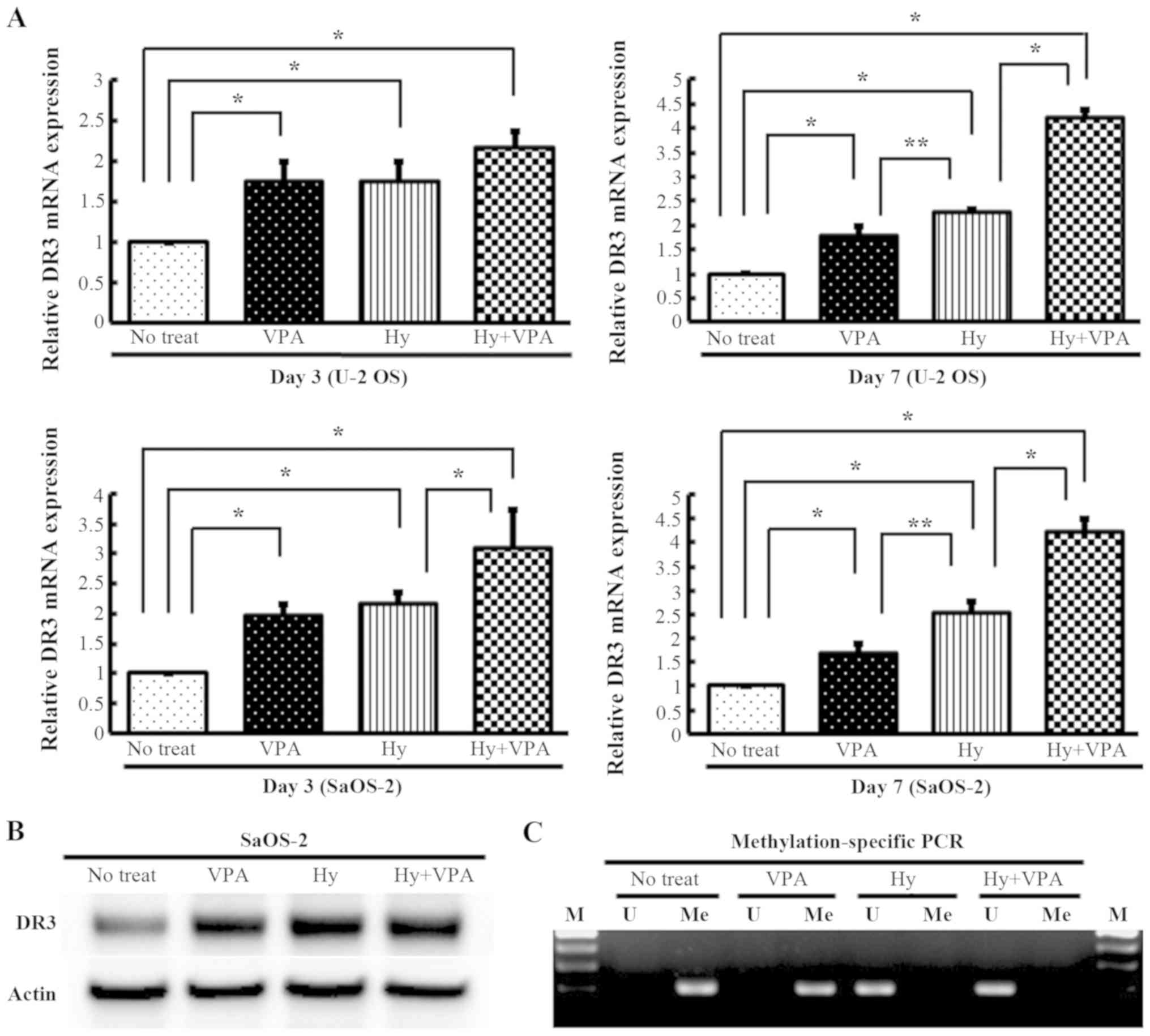 | Figure 2Effects of Hy and VPA on the DR3 and
analysis of the methylation status on the DR3 gene promoter in OS
cell lines. U-2 OS and SaOS-2 cells were treated with 20 µM
Hy and/or 1.0 mM VPA for 3 (Day 3) or 7 days (Day 7). The medium
was changed on day 3. (A) DR3 mRNAs was quantitated using qPCR
analysis. The values are expressed as the ratio to the average
value in the no treat as a control. Each bar indicates the mean ±
SE of values from four independent experiments for each sample, in
three sets of culture conditions. *P<0.01,
significant difference in comparison to no treat and to Hy.
**P<0.05, significant difference in comparison to Hy.
(B) The DR3 protein expression was examined by western blotting.
(C) Methylation-specific PCR (MSP) was performed with 20 µM
Hy and/or 1.0 mM VPA treatment on day 3 in SaOS-2 cells. Bisulfited
genomic DNA amplified by unmethylated-specific (U) and
methylated-specific (Me) primers designed in the CpG island of the
DR3 promoter region. M, molecular marker. Hy, hydralazine; VPA,
sodium valproate; DR3, death receptor-3; OS, osteosarcoma; qPCR,
quantitative polymerase chain reaction; SE, standard error. |
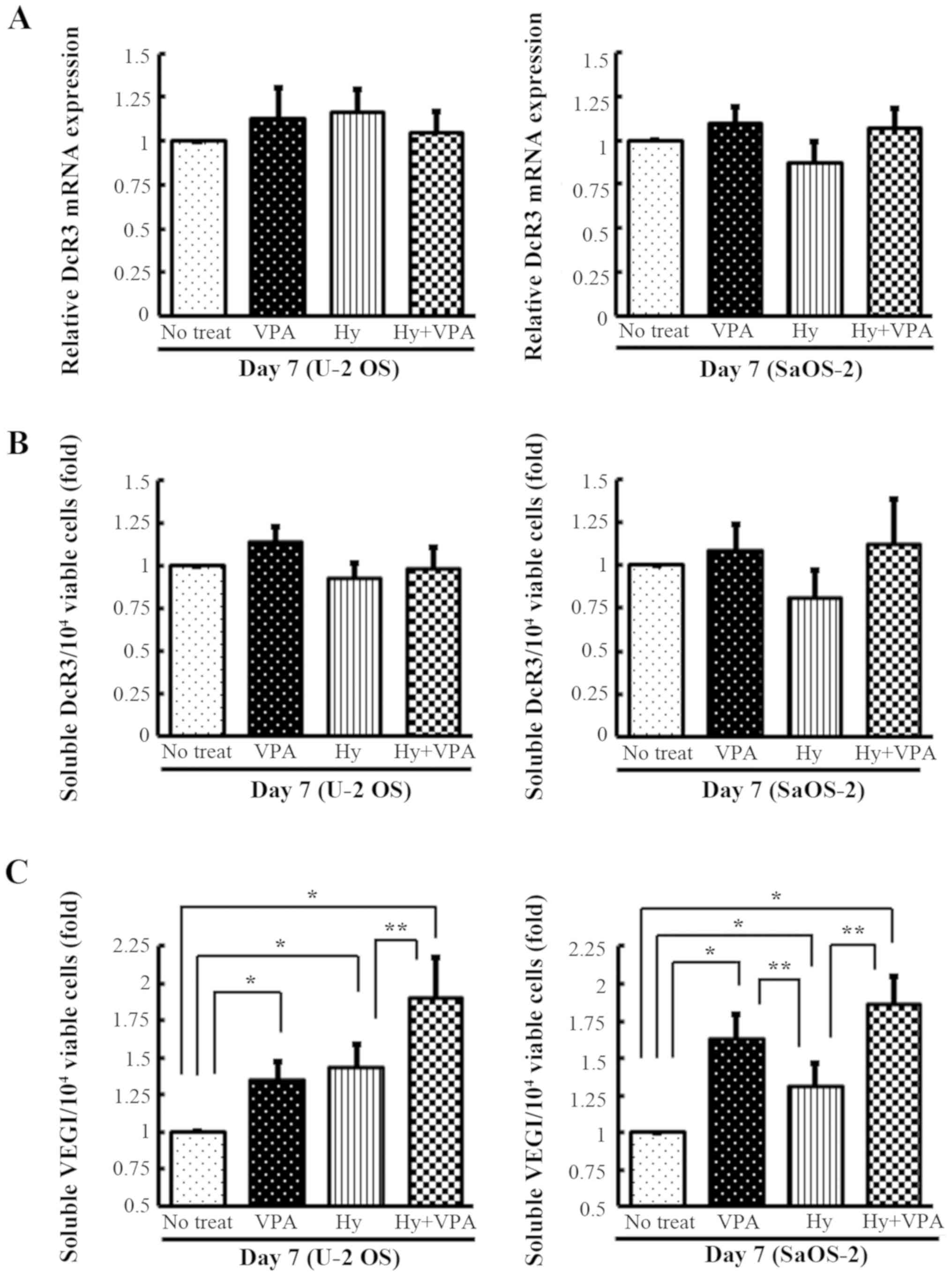
Effects of Hy and VPA on the DcR3
expression levels and the expression of soluble forms of VEGI in OS
cells
To determine the effects of Hy and VPA on DcR3
expression and the production of soluble VEGI, U-2 OS and SaOS-2
cells were cultured with and without 20 µM Hy and 1.0 mM VPA
for 7 days. The DcR3 mRNA transcription was quantitated by qPCR.
The DcR3 mRNA expression was unchanged in both cell lines after 7
days of culture (Fig. 3A). The
secreted DcR3 in the culture medium was analyzed by ELISA and found
to be essentially unchanged compared with no treatment (Fig. 3B). The accumulation of soluble VEGI
was increased 1.3- to 1.6-fold by VPA treatment (P<0.001). A
statistically significant 2.0-fold increase in soluble VEGI was
observed following combination treatment in both types of OS cells
on day 7 (P<0.001) (Fig.
3C).
Effects of Hy and VPA on the
transcription of VEGI, its related receptors and their soluble
forms in HMVE cells
qPCR analysis of HMVE cells revealed that VEGI mRNA
transcription was increased ~2.5-fold following VPA treatment
(P<0.01) (Fig. 4A), while that
of DR3 was increased ~1.9-fold by Hy treatment (P<0.001)
(Fig. 4C). The combination of Hy
and VPA increased the VEGI and DR3 gene expression ~2.8-fold
(P<0.001) (Fig. 4A and C). The
protein expression profiles reflected the trends in gene expression
(Fig. 4B and D). A 1.5-fold
increase in soluble VEGI levels was observed after treatment with
1.0 mM VPA (P<0.001), while combination treatment resulted in a
2.0-fold increase (P<0.001) (Fig.
4G). However, the DcR3 gene expression and secretion did not
differ to a statistically significant extent from no treatment
(control; P>0.05) (Fig. 4E and
F).
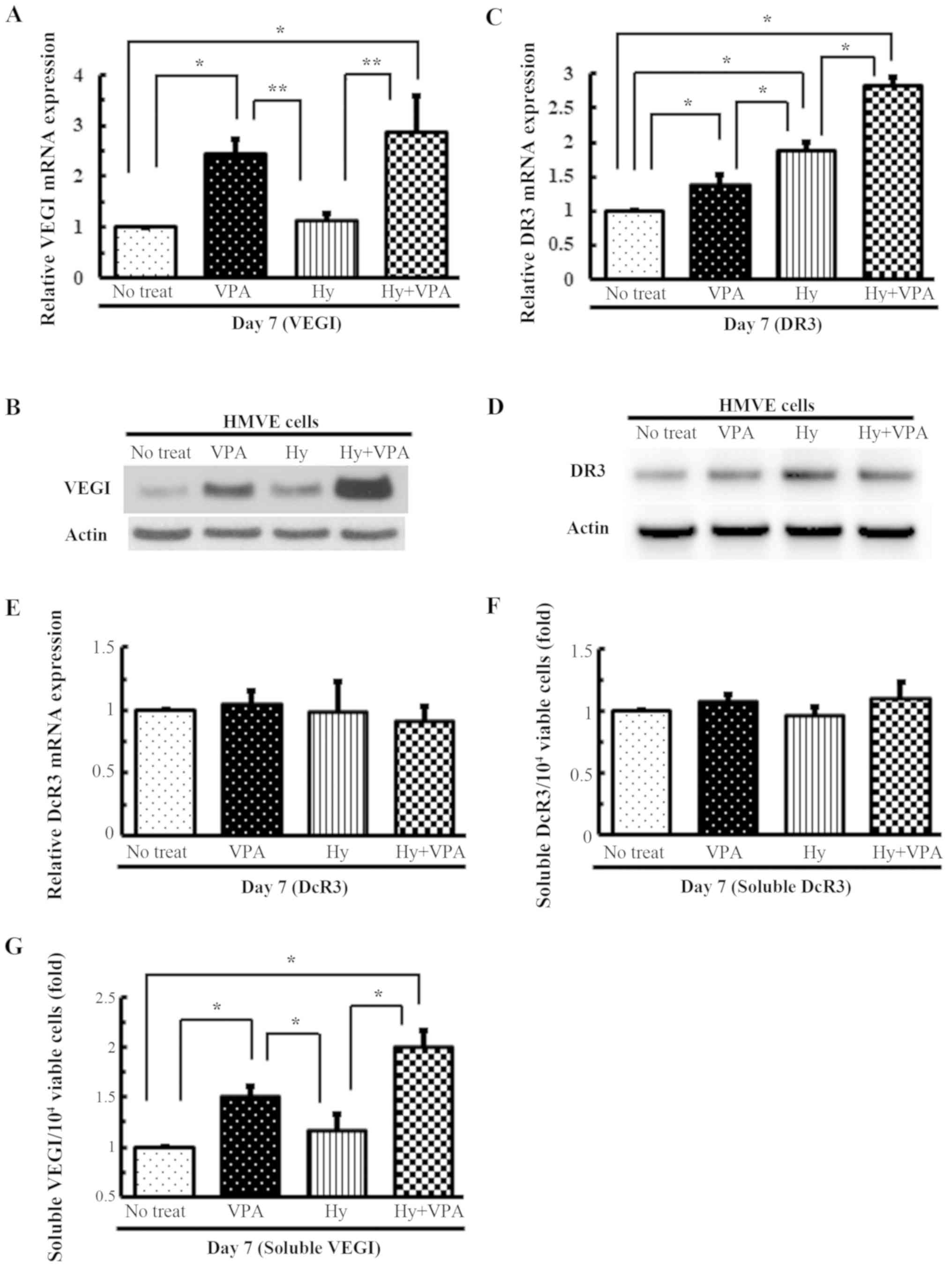 | Figure 4Effects of Hy and VPA on VEGI and its
related receptors on transcription and the production of their
soluble forms in the HMVE cells. HMVE cells were cultured in medium
with 20 µM Hy and/or 1.0 mM VPA for 7 days (Day 7). The
medium was changed on day 3. The gene expression levels of (A)
VEGI, (C) DR3 and (E) DcR3 were quantitated using qPCR. The values
are expressed as the ratio to the mean value in the no treat as a
control. Each bar indicates the mean ± SE of values obtained from
four independent experiments for each sample, under three sets of
culture conditions. *P<0.01, significant difference
in comparison to no treat and to Hy. **P<0.05,
significant difference in comparison to Hy. The (B) VEGI and (D)
DR3 protein expression levels were examined using western blotting.
The amount of (F) soluble DcR3 and (G) VEGI in the medium was
determined on Day 7. The values are indicated as the ratio of the
mean amounts of soluble DcR3 and VEGI per 104 viable
cells in no treat is expressed as 1.0. Each bar indicates the mean
± SE of values obtained from four independent experiments for each
sample, under three sets of culture conditions.
*P<0.001, significant difference in comparison to no
treat and to Hy. Hy, hydralazine; VPA, sodium valproate; VEGI,
vascular endothelial growth inhibitor; HMVE, human microvascular
endothelial; DR3, death receptor-3; DcR3, decoy receptor-3; qPCR,
quantitative polymerase chain reaction; SE, standard error. |
Effects of Hy and VPA on cell
proliferation and effects of Hy and VPA inhibitor treatment of OS
cell culture media on vascular tube formation
An analysis of the HMVE cell growth revealed that 20
µM Hy and 1.0 mM VPA treatment was not associated with
notable changes compared with no treatment as a control. However,
their combination markedly inhibited HMVE cell growth (P<0.001)
(Fig. 5A-a and -b). We previously
demonstrated that VEGI from VPA-treated OS culture medium markedly
inhibited HMVE tube formation (18). To confirm whether soluble VEGI in
the culture medium of OS cells treated with Hy alone or combination
treatment with VPA and Hy can inhibit neovascularization, we
performed the same experiment as described in our previous study
(18). The results demonstrated
that treatment with Hy and/or VPA alone slightly affected tube
formation (P<0.001) (Fig. 5B-d and
-e), whereas combination treatment and the treatment of OS cell
medium markedly inhibited tube formation in HMVE cells (P<0.001)
(Fig. 5B-f, B-i and C).
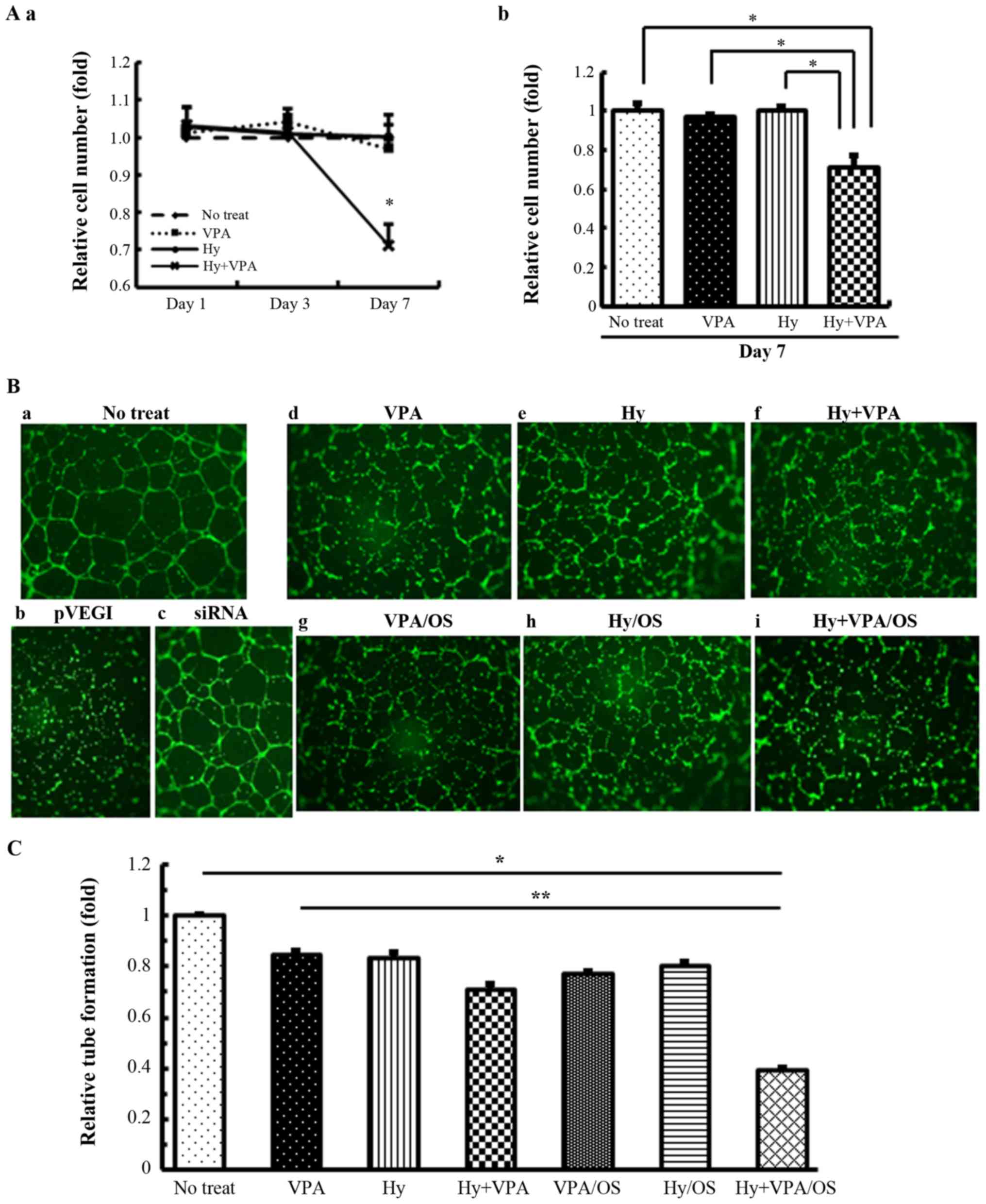 | Figure 5Effects of Hy and VPA on HMVE cell
proliferations and the effects of Hy and VPA-treated OS cell
culture media on vascular tube formation. (A) The growth of HMVE
cells was analyzed with 20 µM Hy and/or 1.0 mM VPA on the
indicated days. (a) Cell growth on Day 1, 3 and 7 of treatment is
indicated on the polygonal line graph. (b) Cell growth on Day 7 of
treatment with Hy and/or VPA. The values are the indicated ratio,
with the mean optical density of no treat set at 1. Each bar
indicates the mean ± SE of 8 wells of separate experiments,
performed in triplicate. *P<0.001, significant
difference in comparison to Hy + VPA. (B) HMVE cells were cultured
or harvested under each condition: (a) No treat (control); (b)
pVEGI (OS culture media transfected plasmid VEGI); (c) siRNA (OS
culture media transfected siRNA for VEGI); (d) 1.0 mM VPA; (e) 20
µM Hy; (f) 20 µM Hy and 1.0 mM VPA; (g) VPA/OS (1.0
mM VPA-treated OS cell culture medium); (h) Hy/OS (20 µM
Hy-treated OS cell culture medium); (i) Hy + VPA/OS (20 µM
Hy and 1.0 mM VPA-treated OS cell culture medium). OS culture media
treated with Hy and/or VPA was obtained from U-2 OS cells after 7
days of culturing in media. The pVEGI and siRNA medium was obtained
from plasmid VEGI- or the siRNA of VEGI-transfected U-2 OS cells
cultured for 48 h. The endothelial cell tube formation was examined
by fluorescence microscopy. (C) Tube formation was counted based on
the identification of a complete tubular shape in four independent
experiments. Counting was performed by four independent
researchers. The values are the indicated ratio of the mean amounts
of complete tubular shape in no treat is expressed as 1.0. Each bar
indicates the mean ± SE of values. *P<0.001,
significant difference in comparison to no treat.
**P<0.001, significant difference in comparison to Hy
+ VPA/OS. Hy, hydralazine; VPA, sodium valproate; HMVE, human
microvascular endothelial; OS, osteosarcoma; SE, standard error;
VEGI, vascular endothelial growth inhibitor; SE, standard
error. |
VEGI directly interferes with VEGF-A in
vitro
U-2 OS and SaOS-2 cells were cultured with or
without 20 µM Hy and 1.0 mM VPA for 3 or 7 days, and the
VEGF-A mRNA transcription was quantitated by qPCR. The VEGF-A mRNA
expression of both cell lines was significantly decreased under all
treatment conditions (P<0.05) (Fig.
6A). To determine the mechanism underlying the VEGI-mediated
anti-angiogenesis without VEGI/DR3-induced apoptosis, a
VEGI/V5-His-tag expression vector was transfected into OS cells,
and cell lysis was performed via an immunoprecipitation assay. The
results demonstrated that the V5-His-tagged VEGI bound to
VEGF-A-coated beads and precipitated the VEGF-A/VEGI immune complex
(Fig. 6B-a). OS cell lysis after
VPA treatment also created VEGI/VEGF-A immune complexes (Fig. 6B-b). These results indicate that
VEGI may exert one aspect of its anti-angiogenic effects by
directly binding to VEGF-A and inhibiting neovascularization.
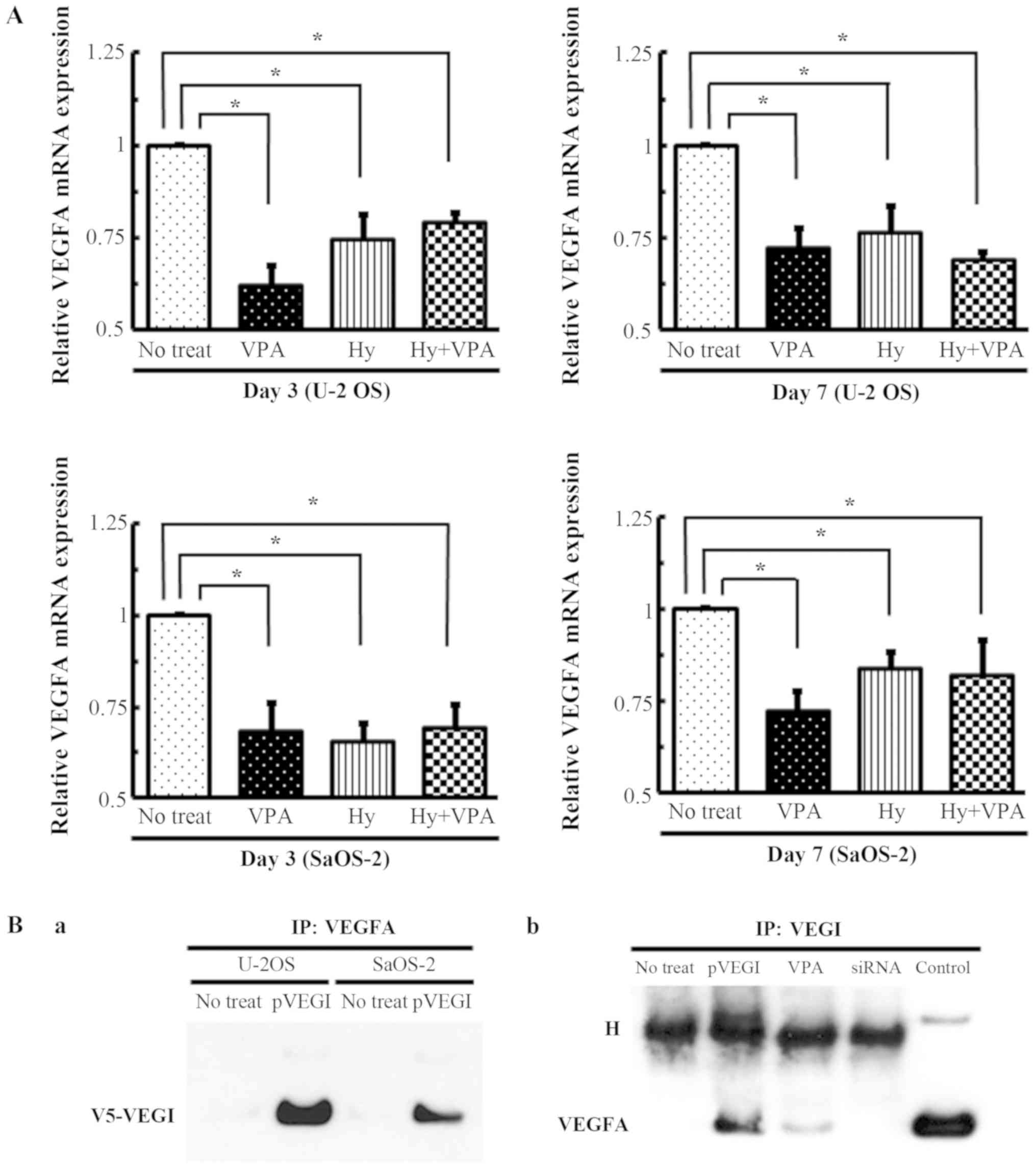 | Figure 6Anti-angiogenic effect of Hy and VPA
on VEGI. (A) U-2 OS and SaOS-2 cells were treated with 20 µM
Hy and/or 1.0 mM VPA for 3 (Day 3) or 7 days (Day 7) with medium
changed on day 3. The VEGF-A mRNAs was quantitated by qPCR. The
values are expressed as the ratio to the average value in no treat
as a control. Each bar indicates the mean ± SE of values obtained
from four independent experiments for each samples under three sets
of condition cultures. *P<0.05, significant
difference in comparison to no treatment. (B) An IP assay was
performed with OS cell lysis. (a) V5-His-tagged VEGI expression
vector and siRNA transfected into U-2 OS and SaOS-2 cells. IP with
VEGF-A was detected with V5-His-tag antibody by western blotting.
(b) SaOS-2 cell lysate of VPA treatment after 7 days was performed
IP with VEGI-coated beads and was detected by western blotting
using VEGF-A antibody. H, heavy chain; Hy, hydralazine; VPA, sodium
valproate; VEGI, vascular endothelial growth inhibitor; VEGF,
vascular endothelial growth factor; qPCR, quantitative polymerase
chain reaction; SE, standard error; IP, immunoprecipitation; OS,
osteosarcoma. |
Discussion
Our previous study demonstrated that the histone
deacetylase inhibitors VPA and TSA increased the expression of VEGI
with little effect on its receptor (DR3), and sensitized both OS
and HMVE cells to apoptosis via the VEGI/DR3 autocrine and
paracrine pathways (18). In the
present study, the combination of the DNA methyltransferase
inhibitor Hy and the histone deacetylase inhibitor VPA induced the
expression of DR3 and VEGI more efficiently compared with either
monotherapy, without inducing DcR3 secretion. DNMT or HDAC
inhibitors usually activate gene expression within minutes/hours.
The expression of VEGI, DR3 and DcR3 were analyzed at 12, 24 and 48
h of Hy and/or VPA treatment. These genes displayed similar degrees
of fluctuation to that observed at 3 and 7 days (Fig. S1). It was also found that their
combination inhibited HMVE cell proliferation and, subsequently,
vascular tube formation by HMVE cells in comparison to Hy or VPA
alone. However, perhaps the most notable finding of the present
study was that VEGI bound directly to VEGF-A, and precipitated the
VEGI/VEGF-A immune complex as determined by to immunoprecipitation
studies.
In recent years, HDAC inhibitors have been tested
against various cancers in clinical trials (20). However, HDAC inhibitors alone are
not sufficiently effective, as several trials have demonstrated
increased histone acetylation in tumor samples, despite there being
only a slight clinical effect (21,22).
DNMT and HDAC inhibitors have been reported to act synergistically
against cancer development through regulating the expression of
tumor suppressor genes and oncogenes (23). Chavez-Blanco et al
investigated the growth inhibitory effect of VPA; however, Hy did
not exert a similar effect on cervical, colon and breast cancer,
sarcoma, glioma, or head and neck cancer cell lines. By contrast,
Hy in combination with VPA significantly inhibited cell growth in
all cell lines, and this combination improved the efficiency of
treatment with current anticancer agents (24). Bauman et al conducted a
phase I trial of the combination of Hy and VPA to determine the
maximum tolerated dose. The combination of Hy and VPA was found to
be non-toxic, and may be appropriate for patients with resistance
to anticancer drugs, or in combination with other cancer treatments
(25). Capobianco et al
suggested that 5-aza-dc and TSA inhibited cell growth and induced
reprogramming towards osteoblast differentiation in cases of OS
with multidrug resistance, through the induction of the
re-expression of several epigenetically silenced genes. These
agents exhibited greater efficacy in combination than either did as
monotherapy (26). In our previous
study, VPA and TSA alone were unable to increase the DR3 expression
in HMVE cells (18). Takami et
al reported that DR3 contained several CpG motifs in the
promoter region, and was hypermethylated in synovial cells in
patients with rheumatoid arthritis (RA). Thus, RA synovial cells
may become resistant to apoptosis as a result of DR3 downregulation
(27). However, in the present
study, we demonstrated that even Hy treatment alone was able to
enhance DR3 expression in HMVE cells, and its combination with VPA
further increased the expression of DR3 in both OS and HMVE cells.
These results suggest that the upregulation of the DR3 expression
after Hy treatment is mediated by promoter demethylation and is
associated with a decrease in the expression of DNMT. Hellebrekers
et al were the first to demonstrate that DNMT and HDAC
inhibitors directly inhibited endothelial cell growth and
angiogenesis by inducing the re-expression of growth suppression
genes in endothelial cells (7).
Our in vitro vascular tube formation assay also revealed
only a mild effect of VPA on HMVE cells, but the combination with
Hy synergistically prevented vascular tube formation.
As regards the clinicopathological study of
angiogenesis in OS, the expression of VEGF was detected in 63% of
27 primary OS biopsy samples. Patients with VEGF-positive tumors
exhibited higher rates of cancer recurrence and poorer survival in
comparison to those with VEGF-negative tumor (28). In fact, pulmonary metastasis was
confirmed in ~82% of the VEGF-positive samples (28). Qu et al demonstrated that a
notable reduction in the VEGF expression after chemotherapy was
correlated with a good prognosis (29). These reports suggest that VEGF may
exert a paradoxical effect in OS: It is associated with a poor
outcome, but can also contribute to a better response to
chemotherapy. The anti-angiogenic effects of HDAC inhibitors
downregulate VEGF expression via suppression of hypoxia-inducible
factor 1α activity (30,31). VPA inhibits angiogenesis by
inducing the expression of endogenous anti-angiogenic proteins,
such as thrombospondin-1, activin A and VEGI (18,32).
Thus, we assessed the effect of Hy and VPA on the VEGF-A gene
expression by qPCR analysis. Treatment of OS cells with Hy and/or
VPA significantly suppressed the VEGF-A gene expression. VEGI, also
referred to as TNFSF15, and VEGF interfere with each other in the
modulation of angiogenesis (33-36).
VEGI is capable of inhibiting the VEGFR1 and VEGFR2 activity of
vascular endothelial cells (33,34).
VEGI treatment of endothelial progenitor cells leads to the
accelerated degradation of membrane-bound VEGFR1 (mFlt1) and the
enhanced production of soluble VEGFR1 (sFlt1), thus inhibiting
VEGF-stimulated blood vessel growth in experimental animals
(34). Zhang et al
demonstrated that VEGI was able to suppress the VEGF gene
expression, thereby inducing miR-29b as result of JNK-GATA3
activation (35). We herein
assessed the effect of Hy and VPA on the miR-29b and GATA3
expression by qPCR. Treatment with VPA induced the expression of
the miR-29b and GATA3 genes in both OS and HMVE cells (data not
shown). Thus, epigenetic modification was able to control VEGF
expression and may have also occurred through the TNFSF15 gene
expression. These observations prompted us to investigate other
options by comparing VEGF to VEGI. Immunoprecipitation was
performed using either VEGF-A- or VEGI-coated beads. Surprisingly,
we observed that VEGI physically interacted with VEGF-A on pVEGI
transfected OS cell lysis and that VPA treatment induced VEGI
expression. This finding suggests that suppression of VEGF-A
production is controlled by the physical interaction of VEGF-A and
VEGI. While further studies are required, this finding may indicate
a novel mechanism for interfering with VEGF-driven
neovascularization.
In conclusion, the findings from our present and
previous studies suggest that the DNMT and HDAC inhibitors Hy and
VPA, respectively, not only induce the re-expression of tumor
suppressor genes in cancer cells, but also exert anti-angiogenic
effects, leading to the enhancement of the VEGI/DR3 pathway, and
that their combination synergistically enhances the VEGI and DR3
gene expression and promotes HMVE cell death. Moreover, the
precipitation of the VEGI/VEGF-A immune complex may constitute
evidence of an additional VEGI-mediated anti-angiogenic machinery.
Although further studies with in vivo assays and clinical
samples are required, these findings may indicate a novel mechanism
for interfering with VEGF-driven neovascularization. Hy and VPA are
administered long-term in anti-hypertensive therapy and for the
treatment of epilepsy and bipolar disorder, respectively.
Importantly, our results demonstrated that Hy and VPA exerted their
effects at concentrations that are attainable in the sera of
patients undergoing oral treatment with the respective agents,
without serious side effects (37,38).
Thus, Hy and VPA may achieve better results with regard to reducing
the population of vascular endothelial cells in OS, while also
reducing host toxicity. There is a possibility that the benefit is
due to an unknown activity of Hy and VPA (i.e., other than their
activity as DNMT and HDAC inhibitors). The targeting of epigenetic
modifications is currently being pursued in clinical studies. The
findings of the present study suggest that Hy and VPA may prevent
hematogenous pulmonary metastasis and support the need for further
investigation of epigenetic modifications as a novel therapeutic
approach to OS.
Supplementary Materials
Funding
The present study was supported in part by a
Grant-in-Aid for Scientific Research (C) (26462279), (15K11097) and
(18K09564) from the Ministry of Education, Culture, Sports, Science
and Technology of Japan, Grant-in-Aid for Researchers, Hyogo
College of Medicine, 2017.
Availability of data and materials
All the datasets generated and analyzed in the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
KY, HF and SY designed the study and participated in
the discussion during the preparation of the manuscript. SK, KK and
KY performed and contributed to all the experiments. YF and HF
performed OS cell experiments. HN and KN performed HMVE cell
experiments. SK, KK and KY performed IP experiments. SK and KY
analyzed the data and wrote the manuscript. All the authors have
read and approved the final version of this manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
Acknowledgments
Not applicable.
References
|
1
|
Dai Z, Tang H, Pan Y, Chen J, Li Y and Zhu
J: Gene expression profiles and pathway enrichment analysis of
human osteosarcoma cells exposed to sorafenib. FEBS Open Bio.
8:860–867. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qian M, Gong H, Yang X, Zhao J, Yan W, Lou
Y, Peng D, Li Z and Xiao J: MicroRNA-493 inhibits the proliferation
and invasion of osteosarcoma cells through directly targeting
specificity protein 1. Oncol Lett. 15:8149–8156. 2018.PubMed/NCBI
|
|
3
|
Jaenisch R and Bird A: Epigenetic
regulation of gene expression: How the genome integrates intrinsic
and environmental signals. Nat Genet. 33:245–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suzuki H, Gabrielson E, Chen W, Anbazhagan
R, van Engeland M, Weijenberg MP, Herman JG and Baylin SB: A
genomic screen for genes upregulated by demethylation and histone
deacetylase inhibition in human colorectal cancer. Nat Genet.
31:141–149. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feller L, Kramer B and Lemmer J:
Pathobiology of cancer metastasis: A short account. Cancer Cell
Int. 12:242012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yoo CB and Jones PA: Epigenetic therapy of
cancer: Past, present and future. Nat Rev Drug Discov. 5:37–50.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hellebrekers DM, Jair KW, Viré E, Eguchi
S, Hoebers NT, Fraga MF, Esteller M, Fuks F, Baylin SB, van
Engeland M, et al: Angiostatic activity of DNA methyltransferase
inhibitors. Mol Cancer Ther. 5:467–475. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deroanne CF, Bonjean K, Servotte S, Devy
L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens
BV, et al: Histone deacetylases inhibitors as anti-angiogenic
agents altering vascular endothelial growth factor signaling.
Oncogene. 21:427–436. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hsu YY, Shi GY, Wang KC, Ma CY, Cheng TL
and Wu HL: Thrombomodulin promotes focal adhesion kinase activation
and contributes to angiogenesis by binding to fibronectin.
Oncotarget. 7:68122–68139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li T, Kang G, Wang T and Huang H: Tumor
angiogenesis and anti-angiogenic gene therapy for cancer. Oncol
Lett. 16:687–702. 2018.PubMed/NCBI
|
|
12
|
Tan KB, Harrop J, Reddy M, Young P,
Terrett J, Emery J, Moore G and Truneh A: Characterization of a
novel TNF-like ligand and recently described TNF ligand and TNF
receptor superfamily genes and their constitutive and inducible
expression in hematopoietic and non-hematopoietic cells. Gene.
204:35–46. 1997. View Article : Google Scholar
|
|
13
|
Zhai Y, Ni J, Jiang GW, Lu J, Xing L,
Lincoln C, Carter KC, Janat F, Kozak D, Xu S, et al: VEGI, a novel
cytokine of the tumor necrosis factor family, is an angiogenesis
inhibitor that suppresses the growth of colon carcinomas in vivo.
FASEB J. 13:181–189. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kitson J, Raven T, Jiang YP, Goeddel DV,
Giles KM, Pun KT, Grinham CJ, Brown R and Farrow SN: A
death-domain-containing receptor that mediates apoptosis. Nature.
384:372–375. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Migone TS, Zhang J, Luo X, Zhuang L, Chen
C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JXH, et al: TL1A is a
TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell
costimulator. Immunity. 16:479–492. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chinnaiyan AM, O'Rourke K, Yu GL, Lyons
RH, Garg M, Duan DR, Xing L, Gentz R, Ni J and Dixit VM: Signal
transduction by DR3, a death domain-containing receptor related to
TNFR-1 and CD95. Science. 274:990–992. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bai C, Connolly B, Metzker ML, Hilliard
CA, Liu X, Sandig V, Soderman A, Galloway SM, Liu Q, Austin CP, et
al: Overexpression of M68/DcR3 in human gastrointestinal tract
tumors independent of gene amplification and its location in a
four-gene cluster. Proc Natl Acad Sci USA. 97:1230–1235. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamanegi K, Kawabe M, Futani H, Nishiura
H, Yamada N, Kato-Kogoe N, Kishimoto H, Yoshiya S and Nakasho K:
Sodium valproate, a histone deacetylase inhibitor, modulates the
vascular endothelial growth inhibitor-mediated cell death in human
osteosarcoma and vascular endothelial cells. Int J Oncol.
46:1994–2002. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
20
|
Bolden JE, Peart MJ and Johnstone RW:
Anticancer activities of histone deacetylase inhibitors. Nat Rev
Drug Discov. 5:769–784. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Siu LL, Pili R, Duran I, Messersmith WA,
Chen EX, Sullivan R, MacLean M, King S, Brown S, Reid GK, et al:
Phase I study of MGCD0103 given as a three-times-per-week oral dose
in patients with advanced solid tumors. J Clin Oncol. 26:1940–1947.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schrump DS, Fischette MR, Nguyen DM, Zhao
M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Kruchin E, et al:
Clinical and molecular responses in lung cancer patients receiving
Romidepsin. Clin Cancer Res. 14:188–198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu S, Ren J, Chen HB, Wang Y, Liu Q, Zhang
R, Jiang SW and Li J: Cytostatic and apoptotic effects of DNMT and
HDAC inhibitors in endometrial cancer cells. Curr Pharm Des.
20:1881–1887. 2014. View Article : Google Scholar
|
|
24
|
Chavez-Blanco A, Perez-Plasencia C,
Perez-Cardenas E, Carrasco-Legleu C, Rangel-Lopez E, Segura-Pacheco
B, Taja-Chayeb L, Trejo-Becerril C, Gonzalez-Fierro A, Candelaria
M, et al: Antineoplastic effects of the DNA meth-ylation inhibitor
hydralazine and the histone deacetylase inhibitor valproic acid in
cancer cell lines. Cancer Cell Int. 6:22006. View Article : Google Scholar
|
|
25
|
Bauman J, Shaheen M, Verschraegen CF,
Belinsky SA, Houman Fekrazad M, Lee FC, Rabinowitz I,
Ravindranathan M and Jones DV Jr: A phase I protocol of hydralazine
and valproic acid in advanced, previously treated solid cancers.
Transl Oncol. 7:349–354. 2014. View Article : Google Scholar
|
|
26
|
Capobianco E, Mora A, La Sala D, Roberti
A, Zaki N, Badidi E, Taranta M and Cinti C: Separate and combined
effects of DNMT and HDAC inhibitors in treating human multi-drug
resistant osteosarcoma HosDXR150 cell line. PLoS One. 9:e955962014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takami N, Osawa K, Miura Y, Komai K,
Taniguchi M, Shiraishi M, Sato K, Iguchi T, Shiozawa K, Hashiramoto
A, et al: Hypermethylated promoter region of DR3, the death
receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis
Rheum. 54:779–787. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaya M, Wada T, Akatsuka T, Kawaguchi S,
Nagoya S, Shindoh M, Higashino F, Mezawa F, Okada F and Ishii S:
Vascular endothelial growth factor expression in untreated
osteosarcoma is predictive of pulmonary metastasis and poor
prognosis. Clin Cancer Res. 6:572–577. 2000.PubMed/NCBI
|
|
29
|
Qu Y, Xu J, Jiang T, Zhao H, Gao Y, Zheng
C and Shi X: Difference in pre- and postchemotherapy vascular
endothelial growth factor levels as a prognostic indicator in
osteosarcoma. J Int Med Res. 39:1474–1482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chelluri R, Caza T, Woodford MR, Reeder
JE, Bratslavsky G and Byler T: Valproic acid alters angiogenic and
trophic gene expression in human prostate cancer models. Anticancer
Res. 36:5079–5086. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang C, Yang C, Feldman MJ, Wang H, Pang
Y, Maggio DM, Zhu D, Nesvick CL, Dmitriev P, Bullova P, et al:
Vorinostat suppresses hypoxia signaling by modulating nuclear
translocation of hypoxia inducible factor 1 alpha. Oncotarget.
8:56110–56125. 2017.PubMed/NCBI
|
|
32
|
Cinatl J Jr, Kotchetkov R, Blaheta R,
Driever PH, Vogel JU and Cinatl J: Induction of differentiation and
suppression of malignant phenotype of human neuroblastoma BE(2)-C
cells by valproic acid: Enhancement by combination with
interferon-alpha. Int J Oncol. 20:97–106. 2002.
|
|
33
|
Yang GL, Zhao Z, Qin TT, Wang D, Chen L,
Xiang R, Xi Z, Jiang R, Zhang ZS, Zhang J, et al: TNFSF15 inhibits
VEGF-stimulated vascular hyperpermeability by inducing VEGFR2
dephosphorylation. FASEB J. 31:2001–2012. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qi JW, Qin TT, Xu LX, Zhang K, Yang GL, Li
J, Xiao HY, Zhang ZS and Li LY: TNFSF15 inhibits vasculogenesis by
regulating relative levels of membrane-bound and soluble isoforms
of VEGF receptor 1. Proc Natl Acad Sci USA. 110:13863–13868. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang K, Cai HX, Gao S, Yang GL, Deng HT,
Xu GC, Han J, Zhang QZ and Li LY: TNFSF15 suppresses VEGF
production in endothelial cells by stimulating miR-29b expression
via activation of JNK-GATA3 signals. Oncotarget. 7:69436–69449.
2016.PubMed/NCBI
|
|
36
|
Deng HT, Liu HL, Zhai BB, Zhang K, Xu GC,
Peng XM, Zhang QZ and Li LY: Vascular endothelial growth factor
suppresses TNFSF15 production in endothelial cells by stimulating
miR-31 and miR-20a expression via activation of Akt and Erk
signals. FEBS Open Bio. 7:108–117. 2016. View Article : Google Scholar
|
|
37
|
Spina E and Perugi G: Antiepileptic drugs:
Indications other than epilepsy. Epileptic Disord. 6:57–75.
2004.PubMed/NCBI
|
|
38
|
Kandler MR, Mah GT, Tejani AM and Stabler
SN: Hydralazine for essential hypertension. Cochrane Database Syst
Rev. 8:CD0049342010.
|