Introduction
The breakpoint cluster region (BCR)-Abelson murine
leukemia viral oncogene homolog 1 (ABL) chimeric tyrosine kinase
has been mechanistically associated with in the pathogenesis of
Philadelphia chromosome positive malignancies (1,2).
Signaling initiated by BCR-ABL involves a number of pathways that
regulate the survival and proliferation of chronic myelogenous
leukemia (CML) cells (Fig. 1).
Inhibition of BCR-ABL with imatinib mesylate, also known as
Gleevec, has been reported to be notably efficient in patients with
CML. Indeed, the use of imatinib markedly improved clinical
outcome, with a 10 year survival of >80% of patients (3). Nevertheless, genetic and epigenetic
mechanisms within imatinib-resistant cells can lead to relapse;
thus, it is necessary to develop novel BCR-ABL inhibitors. A major
criterion to which these drugs must fit is the potency against both
wild-type (native) and mutated BCR-ABL with minimal interaction
with other protein kinases. Ponatinib, a Food and Drug
Administration (FDA) approved, third generation BCR-ABL inhibitor
for CML, is unique in its ability to downregulate native and all
known clinical mutations (including the most frequent T315I) in the
ABL kinase domain (4). However,
side effects, particularly cardiovascular toxicity, have been
associated with the limited target selectivity of ponatinib due to
the attenuation of unrelated protein kinases (4-7).
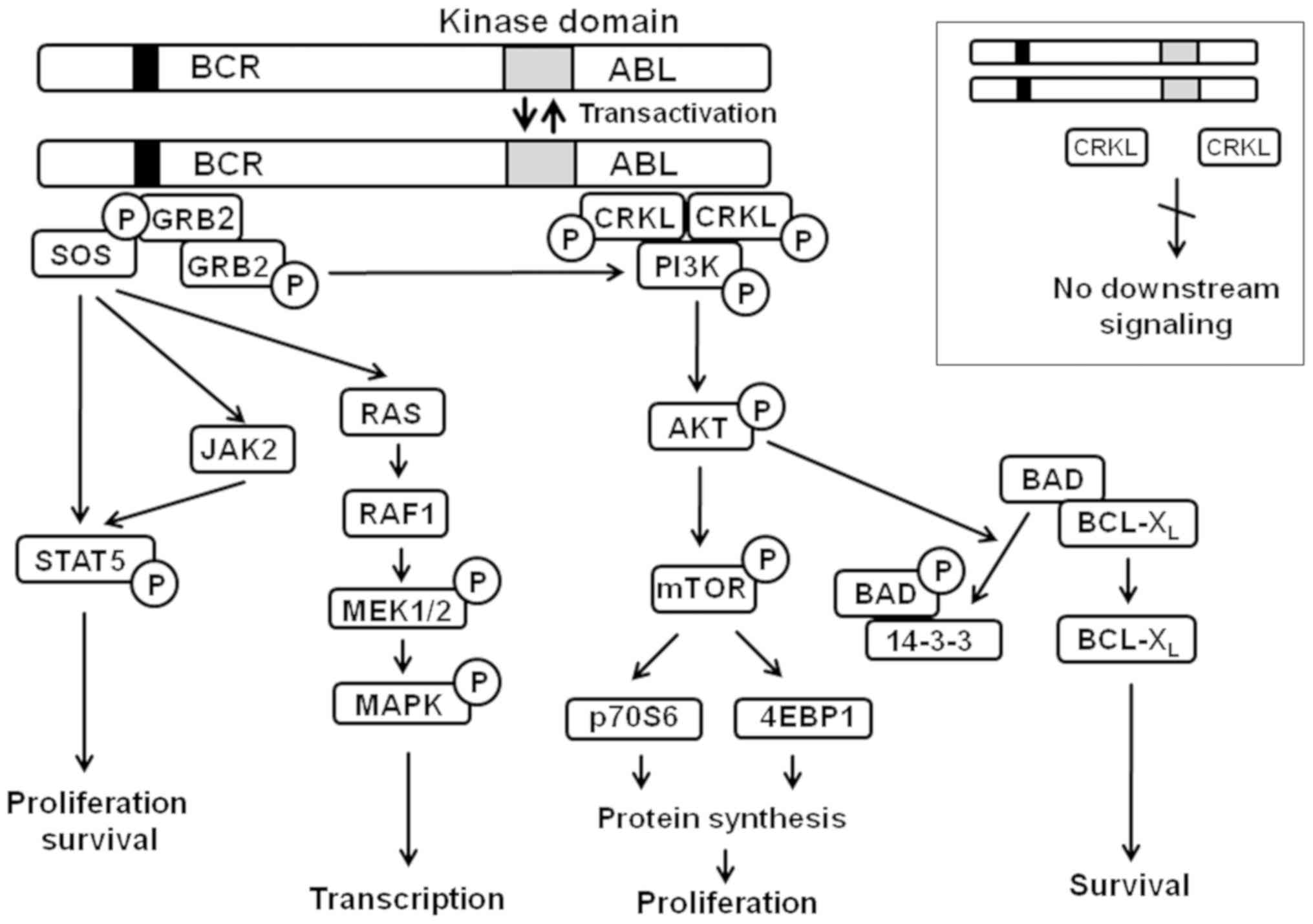 | Figure 1Signaling pathways regulated by
BCR-ABL chimeric tyrosine kinase. Adapted from (10). AKT, protein kinase B; ABL, Abelson
murine leukemia viral oncogene homolog 1; BCR, breakpoint cluster
region; BCL-XL, B-cell lymphoma 2-extra large; BAD,
BCL-2-associated death promoter; CRKL, Crk-like protein; MAPK,
mitogen-activated protein kinase; MEK, MAPK kinase; mTOR, mammalian
target of rapamycin; PI3K, phosphoinositide 3-kinase; STAT5, signal
transducer and activator of transcription 5. |
Aiming to design an optimized compound that combines
the potency of ponatinib (against the wild-type and mutant BCR-ABL
CML cells) with an improved kinase selectivity profile, Chilov and
Titov have developed 3-([1,2,4]triazolo[4,3-a]
pyridin-3-ylethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)
methyl)-3-(trifluoromethyl)phenyl)benzamide (PF-114) (8,9).
PF-114 demonstrated excellent inhibitory potency against the
wild-type and a number of clinically relevant ABL mutations,
including T315I, E255K, Y253F and others in in vitro kinase
assays; PF-114 exhibited notable selectivity in inhibiting ABL
compared with other kinases. The nanomolar kinase inhibitory
potency of PF-114 was paralleled by its efficacy against cultured
leukemia cells and in murine xenografts (8). Importantly, at tolerable doses of
orally administered PF-114, BCR-ABL positive human CML transplants
in immunocompromised mice were completely inhibited for ≤240 days
without tumor re-growth (8).
In the present study, we investigated the molecular
mechanisms of CML cell death induced by PF-114. It was demonstrated
that this compound is highly cytotoxic (at subnanomolar to low
nanomolar concentrations) against the CML derived K562 cell line
(native BCR-ABL), but not BCR-ABL negative leukemia cell lines,
indicating notable target selectivity. Dephosphorylation of
Crk-like protein (CrkL) adaptor protein (a BCR-ABL substrate) in
K562 cells preceded the inhibition of
pro-proliferative/pro-survival Akt-ERK1/2 signaling, and cell cycle
arrest in G1 phase. Apoptotic signaling involved mitochondrial
mechanisms, caspase activation, poly(ADP-ribose)polymerase (PARP)
cleavage and DNA fragmentation. These results indicated that PF-114
is a potent, CML cell specific inhibitor of BCR-ABL signaling.
Materials and methods
Cell lines, culture conditions and drug
treatment
Reagents were purchased from Sigma-Aldrich (Merck
KGaA) unless specified otherwise. Human leukemia cell lines K562
(CML, BCR-ABL-positive), HL60 [FAB-M2 acute myeloblastic leukemia
with maturation; (10)], U937
(monoblastic) and Jurkat (T-lymphocytic; all three harbor no
BCR-ABL fusion protein) obtained from the American Type Culture
Collection, were cultured in RPMI-1640 (PanEco) supplemented with
5% fetal calf serum (HyClone; GE Healthcare Life Sciences), 2 mM
L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin
at 37°C, 5% CO2 in a humidified atmosphere. Cells in the
logarithmic phase of growth were used in the experiments. The
synthesis of PF-114 was conducted as reported previously (8). This compound was dissolved in
dimethyl sulfoxide as 10 mM stock solution and stored at -20°C.
Aqueous dilutions in water were made immediately before
experiments.
CrkL phosphorylation assays
The total intracellular pool of CrkL protein
[phosphorylated (p) and non-phosphorylated forms) was determined by
flow cytometry (11). Cells were
incubated for 2 h at 37°С in the absence (control) or presence of
10, 30 and 100 nM PF-114, fixed with 2% buffered paraformaldehyde
(10 min, 37°С), then washed twice with saline and placed on ice.
Cells were permeabilized with methanol for 30 min, washed with
saline supplemented with 1% bovine serum albumin (BSA) and
resuspended in 100 μl of 10% human serum (30 min, 4°С) to
prevent non-specific antibody binding. Then, cells were pelleted
(2,700 × g, 5 min at room temperature), and the antibody to
non-phosphorylated CrkL (Table
S1; Santa Cruz Biotechnology, Inc.; 1:50) was added for 30 min
at room temperature. Secondary antibody conjugated with
AlexaFluor® 488 (Invitrogen; Thermo Fisher Scientific,
Inc., cat. no. A27034; 1:1,000) was added, cells were incubated for
30 min at room temperature and washed with saline. For the
detection of pCrkL, phycoerythrin (PE) conjugated antibody
(Table S1; BD Biosciences, 1:5)
was added followed by incubation for 30 min at room temperature and
washing in saline with 1% BSA. Samples were analyzed by flow
cytometry using FACSCanto II (BD Biosciences). Total CrkL was
determined based on AlexaFluor 488 fluorescence in a fluorescein
isothiocyanate-A channel whereas pCrkL positive cells were detected
in a PE channel. A total of 20,000 fluorescent 'events' were
collected per each sample. Data were analyzed with FACSDiva
software 6.0 (BD Biosciences).
Cell death assays
The cytotoxicity of PF-114 was determined via an MTT
assay (12). The assays were
performed in 96-well microtiter plates (SPL Life Sciences). To each
well, 5×103 cells and PF-114 (up to 50 μM, 2-fold
dilutions) were added. Cells were incubated for 72 h at 37°C in a
humidified CO2-controlled atmosphere. The half-maximal
inhibitory concentration (IC50) was determined. Analysis
of cell cycle distribution, as well as Annexin V-propidium iodide
(PI; Thermo Fisher Scientific, Inc.) staining were conducted
according to our previous study (12). The mitochondrial transmembrane
electric potential was determined by flow cytometry using
MitoTrackerRed (Thermo Fisher Scientific, Inc). Briefly, K562 cells
(5×104 in 2 ml of culture medium) were treated with 10
nM PF-114 for 24 or 48 h at 37°C, 5% CO2) MitoTracker
Red (1 μM) was added to the cells 2 h prior to the
completion of drug exposure. Cells were washed with ice cold
saline, resuspended in fresh saline and immediately analyzed by
flow cytometry in PE-A channel. At least 30,000 fluorescent events
were acquired per each sample. Data were processed using FACSDiva
software.
For the detection of DNA fragmentation (13), K562 cells (2×106 in 10
ml of culture medium) were treated with PF-114 and lysed for 30 min
on ice in buffer containing 0.35 M NaCl, 20 mM Tris-HCl pH 7.4, 2
mM MgCl2, 1 mM dithiothreitol and 0.5% NP-40. Buffered
phenol (pH 8.0, 1:1 v/v) and chloroform (1:5 v/v) were added to the
lysates. Samples were centrifuged for 10 min at 15,300 × g at 4°C.
Sodium acetate (3 M, pH 5.2; 0.1 v/v) and 1.5 v/v of ethanol were
added to the aqueous phase. Samples were incubated overnight at
-20°C and centrifuged at 15,300 × g, 4°C for 10 min. Pellets were
dissolved in TAE buffer containing 10 μg/ml RNase A. DNA
fragments were resolved by electrophoresis in 1% agarose gel,
stained with ethidium bromide and visualized under UV light.
Immunoblotting
K562 cells (1×105 in 2 ml of culture
medium) were treated with 0.1, 1, 10 and 100 nM PF-114 for 24 or 48
h, washed with ice cold saline and lysed for 30 min on ice in the
buffer containing 50 mM Tris-HCl pH 7.4, 1% NP-40, 0.1% sodium
dodecylsulfate, 150 mM NaCl, 1 mM EDTA, protein inhibitor cocktail
and 2 mM phenylmethylsulfonyl fluoride. Total protein concentration
in lysates was determined by the Bradford method. Proteins were
resolved in 10-15% SDS-PAGE (30 μg total protein per lane)
and transferred onto a 0.2 μm nitrocellulose membrane (GE
Healthcare). Non-specific protein-antibody interactions were
blocked with 5% skimmed milk for 30 min. at room temperature.
Primary antibodies (Table SI)
were diltuted in Tris buffered saline with Tween-20 (TBST; 1:1,000)
supplemented with 1% BSA. Membranes were incubated with primary
antibodies overnight at 4°C. Secondary antibodies conjugated with
horseradish peroxidase were obtained from Cell Signaling
Technology, Inc. (cat. nos. 7074 and 7076; Cell Signaling
Technology, Inc.) and diluted in 5% skimmed milk in TBST (1:1,000).
Membranes were incubated with secondary antibodies for 1 h at room
temperature. Proteins were detected via enhanced chemiluinescence
using the Image Quant LAS 4000 system (GE Healthcare).
Statistical analysis
Data are presented as the mean of four repeats ±
standard deviation. Comparisons were performed using one-way ANOVA
test with a Dunnett's post-hoc test (GraphPad Prism 6.0; GraphPad
Software, Inc.) Each experimental group was compared to control
cohort. P<0.05 was considered to indicate a statistically
significant difference.
Results
PF-114, a novel BCR-ABL inhibitor
A variety of chemotypes have been developed to
inactivate BCR-ABL chimera. The tyrosine kinase ABL can be
downregulated by targeting the ATP binding pocket and/or
allosterically via interactions with the myristoil binding site
[reviewed in (14)]. PF-114 is a
fourth generation inhibitor of the BCR-ABL protein kinase and a
close structural analog of the FDA approved drug ponatinib. The
molecular design of PF-114 was based on the ponatinib scaffold to
weaken the interactions of the inhibitor with off-target protein
kinases, a major reason for the insufficient selectivity of
ponatinib and a probable cause of its toxicity (7,15-17).
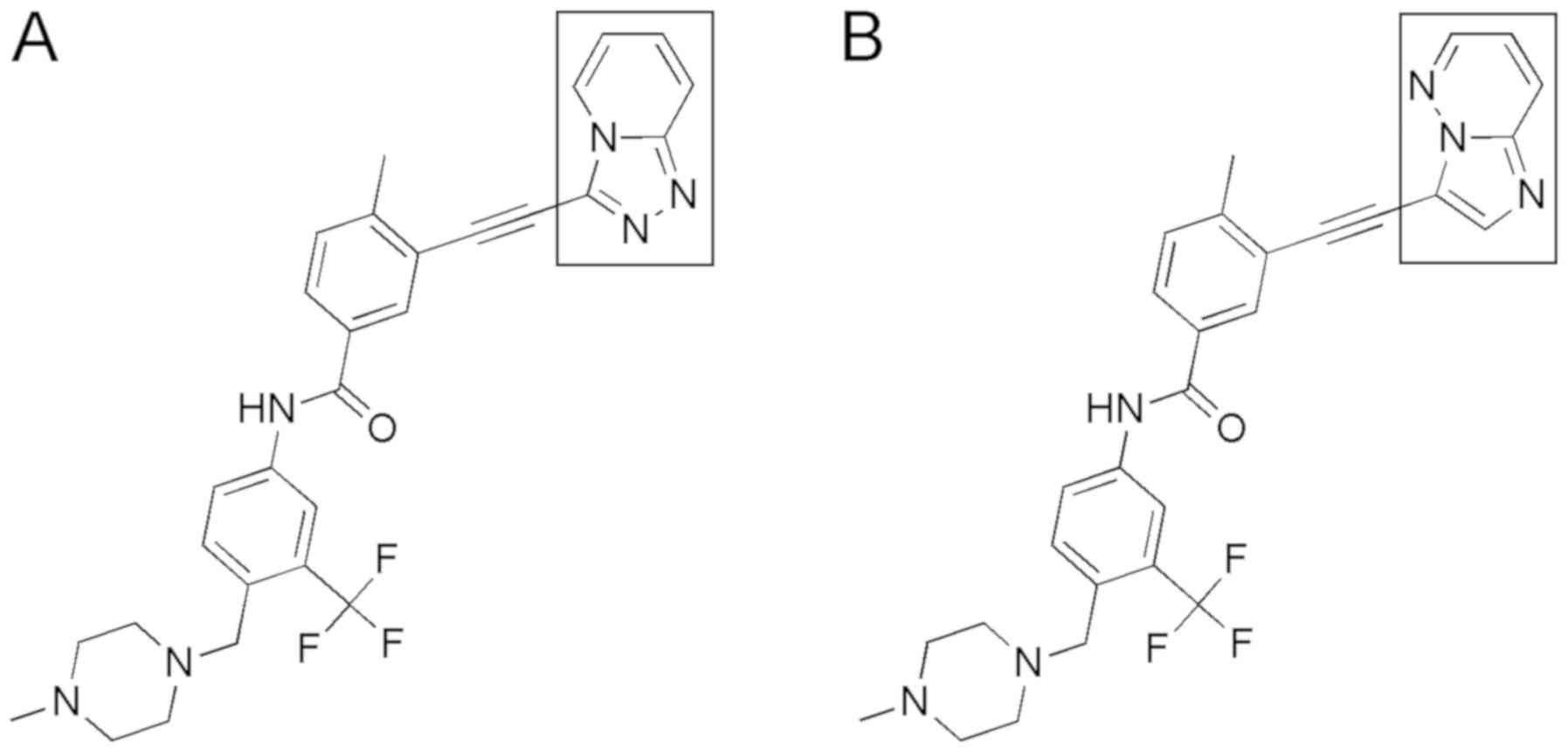
The triazolopyridine moiety of PF-114 (Fig. 2A) that replaced the bioisosteric
pyrazolopyrimidine moiety of ponatinib (Fig. 2B) has been designed to disfavor the
hydrogen bonds between the inhibitor and protein kinases other than
ABL. No hydrogen bonding with water molecules in the active site of
B-Raf and other enzymes (but not ABL) was possible after replacing
the nitrogen atom (an H bond acceptor) in ponatinib with the carbon
atom in PF-114. Additionally, the formation of the CH…O=C hydrogen
bond became unlikely due to the exchange of the CH moiety for the N
atom in PF-114 that is repulsed from the main chain O=C. These
modifications resulted in a more selective kinase inhibitory
profile and an improved in vivo safety of PF-114 (8).
Preferential cytotoxicity of PF-114 to
the BCR-ABL positive CML cell line
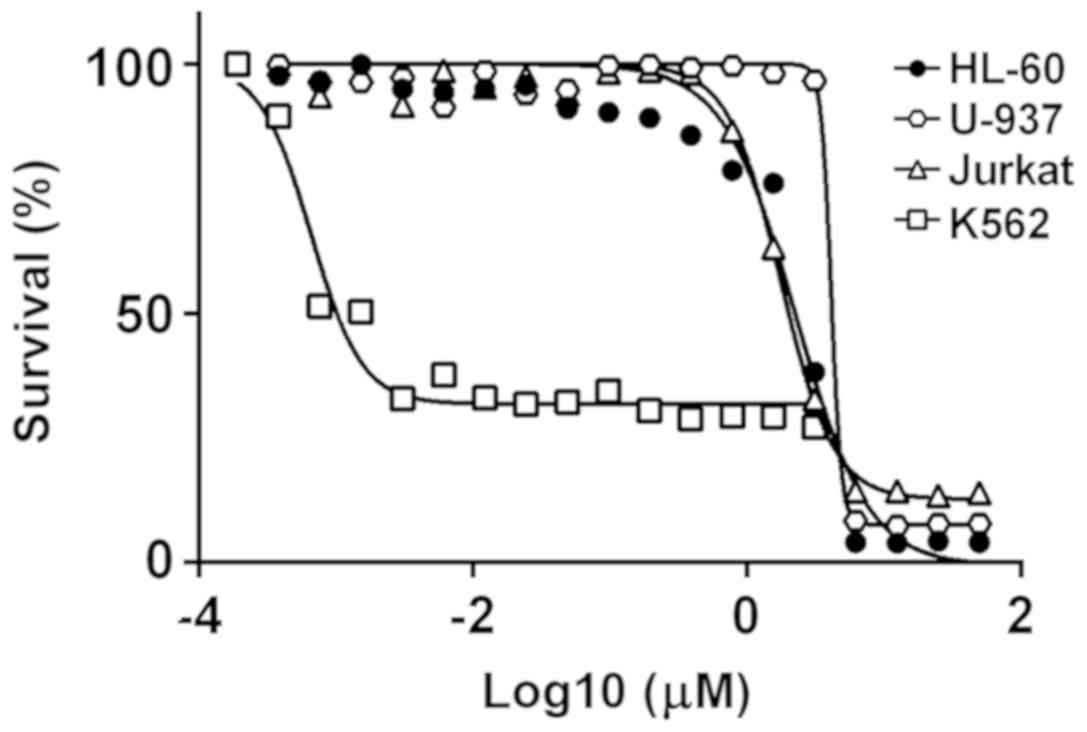
We compared the ability of PF-114 to induce the
death of leukemia cell lines depending on whether the cells carry
the chimeric BCR-ABL protein. As presented in Fig. 3, PF-114 was notably potent
(IC50=1-5 nM after 72 h exposure) for the
BCR-ABL-positive K562 cell line. The cytotoxic effect was time
dependent: By 24 h, the percentage of viable cells (by MTT data
conversion) remained high, whereas by 48 h the IC50
values peaked at their lowest levels in the low nanomolar range
(Fig. S1). Conversely, the HL60,
U-937 and Jurkat leukemia cell lines that lack BCR-ABL chimera were
significantly less sensitive to PF-114, with IC50 values
that were larger by three orders of magnitude. Furthermore, human
donor lymphocytes were resistant to concentrations of PF-114 >1
μM (data not shown). These results indicated that PF-114 was
preferentially cytotoxic against BCR-ABL positive cells.
PF-114 attenuates CrkL
phosphorylation
In addition, whether the cytotoxicity of PF-114
arises due to the prevention of BCR-ABL mediated phosphorylation of
its substrates was investigated. The CrkL adaptor protein is
phosphorylated at Tyr207 by BCR-ABL (18); this event generates a scaffold for
downstream proteins, thereby inducing BCR-ABL activated signaling.
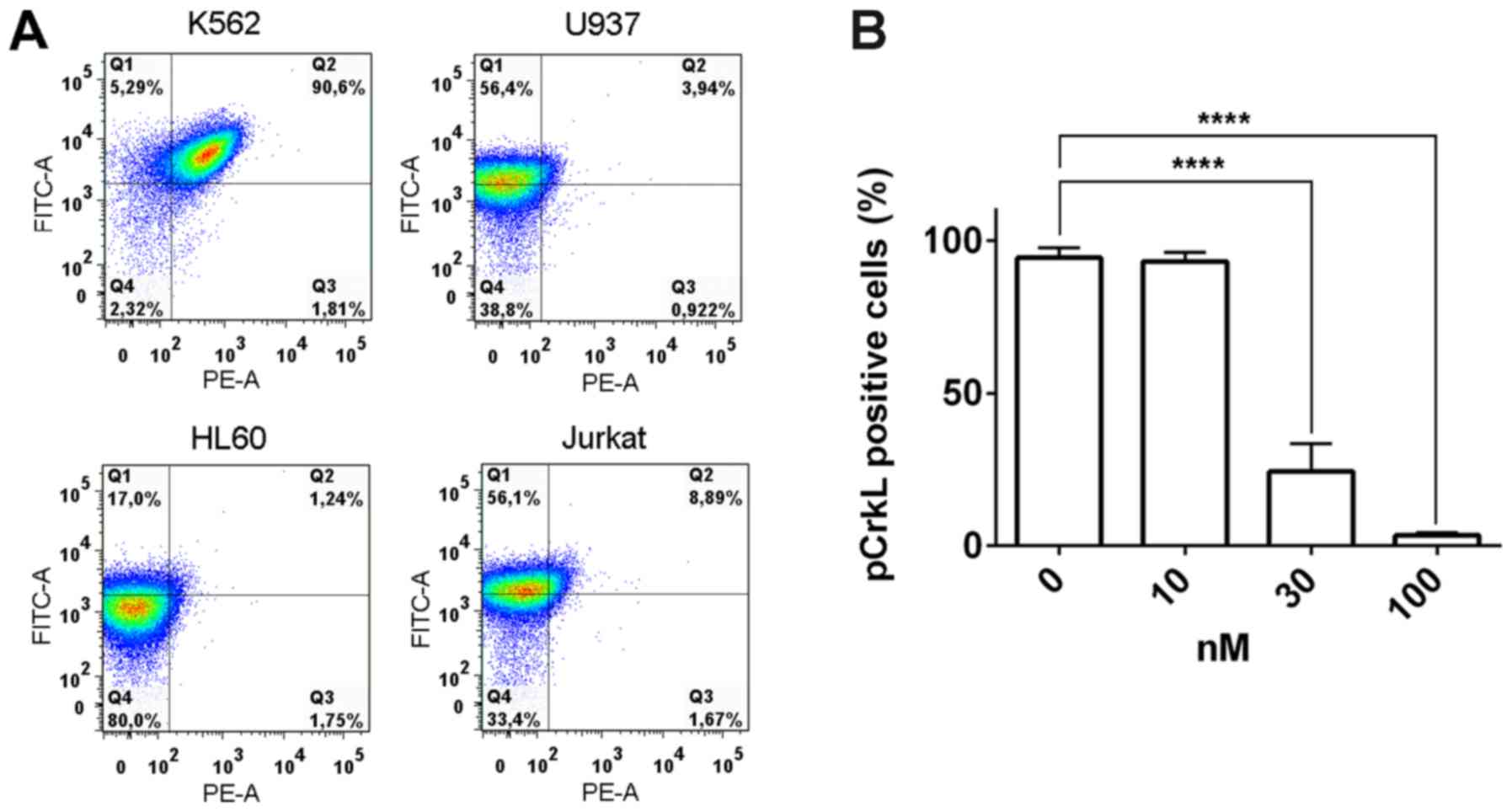
To determine the effects of PF-114 on CrkL phosphorylation, we
treated K562 cells with this compound; the intracellular amounts of
phosphorylated and non-phosphorylated CrkL pools were then
measured. Intact K562 cells contained pCrkL whereas in the BCR-ABL
negative leukemia cell lines (U937, HL-60 and Jurkat), only the
non-phosphorylated protein was detected (Fig. 4A). Within 2 h following treatment
with 30 nM PF-114 the amount of pCrkL in K562 cells decreased to
~25% of its initial level, indicating that PF-114 significantly
attenuated BCR-ABL mediated CrkL phosphorylation compared with the
control (Fig. 4B). Interestingly,
this effect was reversible as the percentage of pCrkL was gradually
restored following the removal of PF-114 and incubation of cells in
drug free medium for ≥20 h. By this time pCrkL was detectable in
~80% of cells (data not shown).
PF-114 attenuates Akt-ERK1/2 and induces
G1 arrest in BCR-ABL-positive CML cells
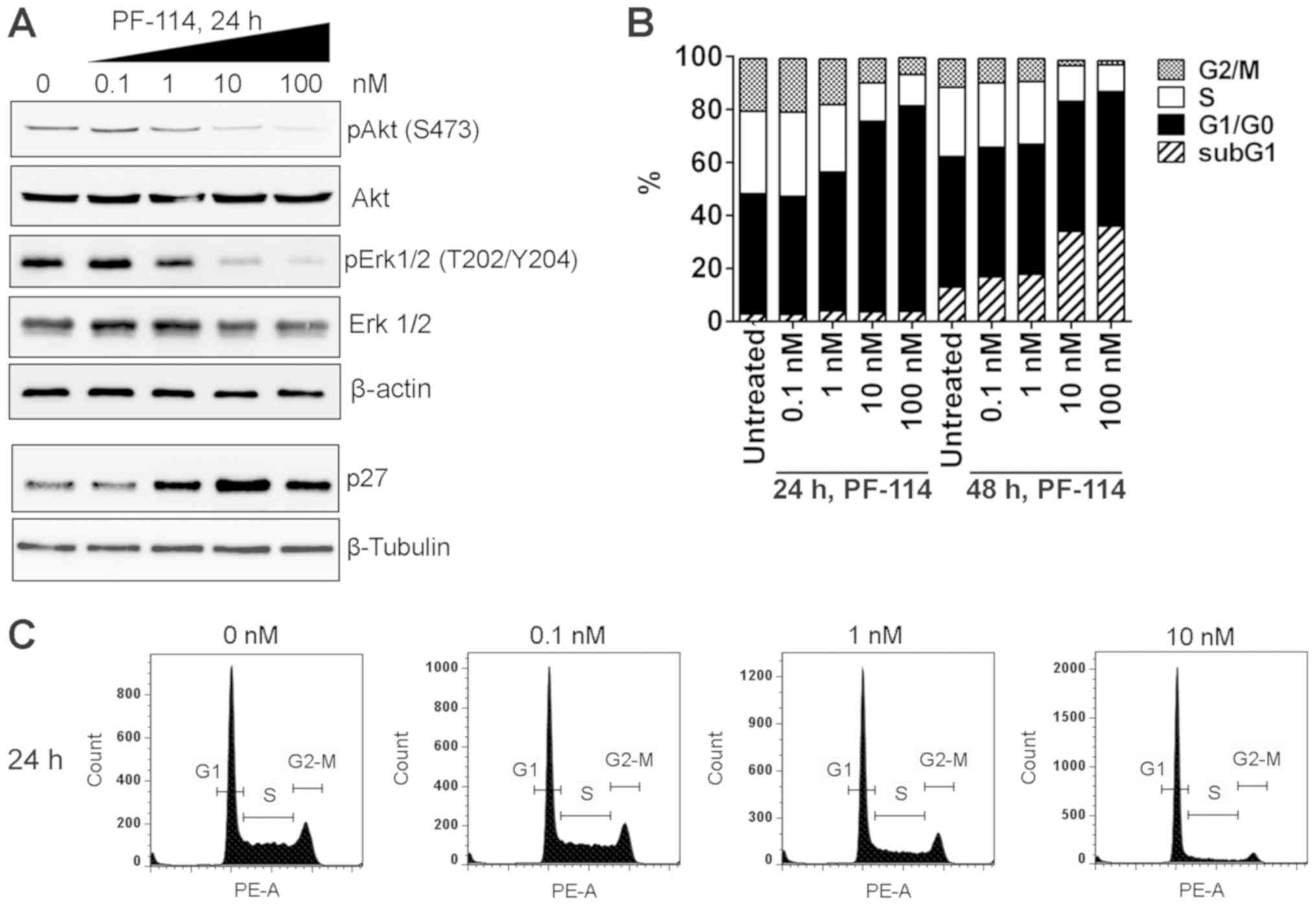
Non-phosphorylated CrkL fails to form dimers, which
prevents the induction of signaling cascades. Accordingly, the
phosphorylation of Akt and ERK1/2, the serine/threonine protein
kinases generally known for their pro-survival and
pro-proliferation activities (19-21),
was markedly attenuated by exposure of K562 cells to nanomolar
concentrations of PF-114 (Fig.
5A). Total amounts of Akt and ERK1/2 were markedly unchanged,
indicating that the attenuation of Akt and ERK1/2 phosphorylation
was a post-translational event. Importantly, the inhibition of Akt
and ERK1/2 phosphorylation was paralleled by an increase in p27
expression (Fig. 5A) and cell
arrest in G1 phase within 24 h exposure (Fig. 5B and C). These effects were
observed at 1, 10 and 100 nM PF-114.
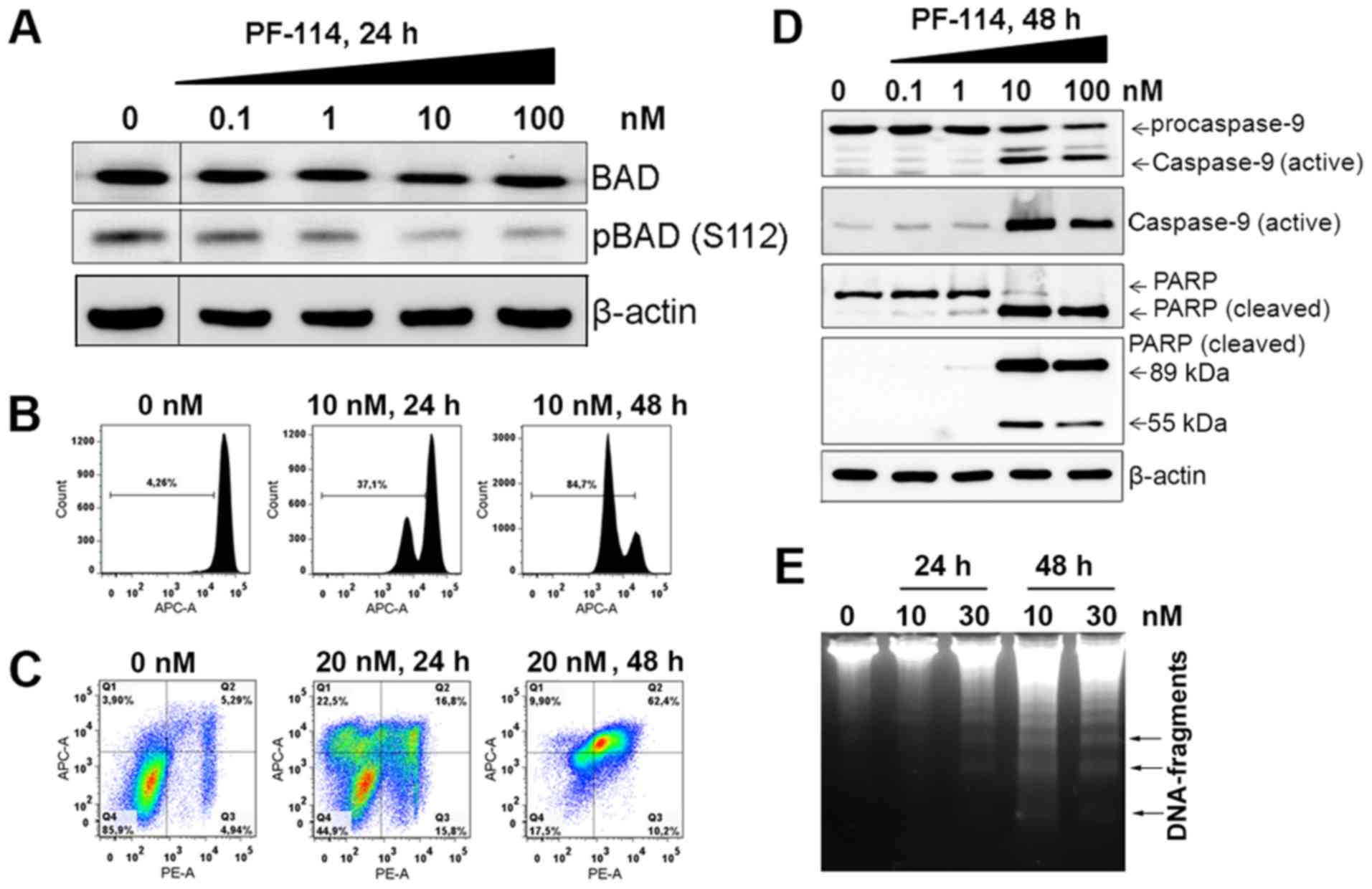
PF-114 induces apoptosis in K562
cells
Time course analysis of individual markers of
programmed cell death in PF-114-treated K562 cells demonstrated the
ordering of cell death-executing processes. By 24 h following
treatment, Bad dephosphorylation was observed (Fig. 6A), an event that allows this
protein to enter mitochondria, and interfere with the pro-survival
Bcl-2 family proteins (22). In
the same time period, changes in the mitochondrial transmembrane
potential were measurable, with a notable portion of cells
(>40%) exhibiting a decrease in this parameter (Fig. 6B). By 48 h, the decreased
mitochondrial transmembrane potential was detected in a predominant
portion of cell population. Concomitant with this phenomenon was
increased Annexin V staining (Fig.
6C), a marker of phosphatidylserine translocation across the
plasma membrane of apoptotic cells (23). The processes of caspase-9 and PARP
cleavage were revealed by 48 h; PARP cleavage generated a typical
89 kDa product and a 55 kDa band characteristic of later stages of
death cascades (24). The
proteolytic processing of caspase-3 was detectable by 24 h (data
not shown). These events culminated to late apoptotic DNA
degradation as indicated by the characteristic DNA fragments of
140-170 bp (Fig. 6E). The
activation of caspase cascades led to a decrease in the expression
of mitochondrial proteins BH3 interacting-domain death agonist
(Bid), Bcl-extra large (XL) and Bcl-2 family apoptosis regulator
(Mcl-1) achievable with notably high PF-114 concentrations
(Fig. 7A). Bak expression was
markedly unchanged, whereas that Bax levels markedly decreased at
100 nM PF-114 (Fig. S2).
Collectively, typical apoptotic mechanisms may be implicated in
PF-114-induced death of BCR-ABL positive, CML-derived K562
cells.
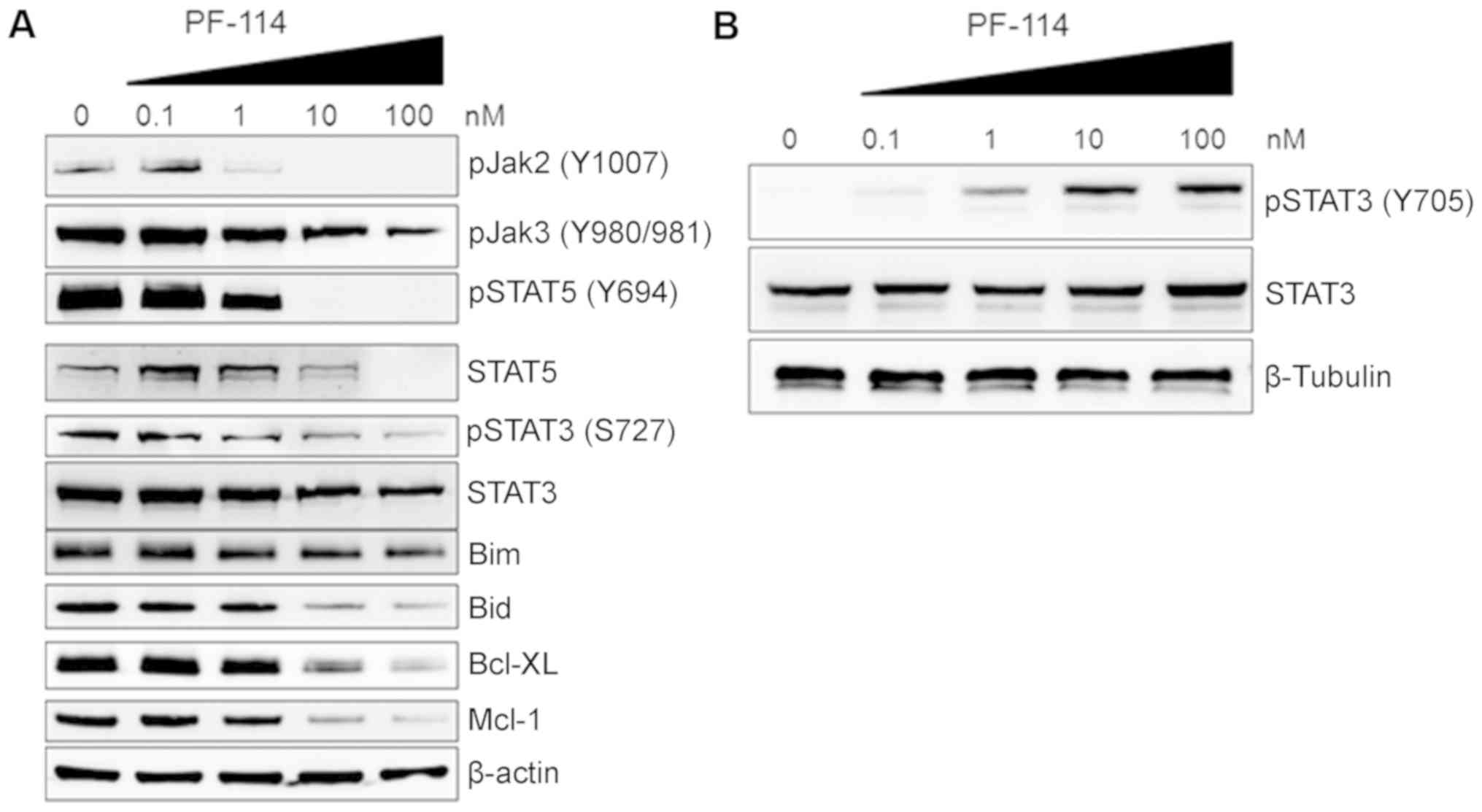
An imbalance between individual members of the
superfamily of Janus kinase-signal transducer and activator of
transcription proteins (JAK-STAT) has been reported to be
associated with cell responses to various cytotoxic stimuli
(25). As presented in Fig. 7A, pJAK2 at Tyr1007/1008 was
markedly downregulated with 1 nM PF-114. Downregulation of pJAK3
was observed following treatment with higher concentrations of
PF-114. Furthermore, individual amino acid residues within the
STAT3 phosphorylation sites were determined to be differentially
sensitive to PF-114. We observed a dose-dependent increase in
pSTAT3 Tyr705 with 1-100 nM PF-114 (Fig. 7B) whereas less pronounced Ser727
phosphorylation was detectable at 100 nM (Fig. 7A). Conversely, STAT5
phosphorylation appeared to be notably sensitive; low nanomolar
concentrations of PF-114 abrogated STAT5 phosphorylation at Tyr694
(Fig. 7A). The expression of total
STAT3 was markedly unchanged; total STAT5 protein expression
decreased with higher PF-114 concentrations (10 and 100 nM).
In the present study, the effects of PF-114 and
ponatinib were analyzed in K562 cells. The latter drug essentially
induced similar responses at similar concentrations and time
intervals as PF-114 (data not shown), which indicated the potential
of the novel compound as a therapeutic agent for treating CML.
Discussion
The present study revealed that blocking BCR-ABL
with the novel inhibitor PF-114 induced a tightly regulated
ensemble of molecular events in K562 cells. Cell responses were
manifested in a manner of cascades, suggesting a network involving
protein kinases, and mitochondrial and transcriptional processes.
Of note, dysregulation of signaling is not linear since several
pathways are affected. This multiplicity of death mechanisms
requires the inactivation of a key target downstream of BCR-ABL,
that is, the CrkL adaptor protein; this can impair a number of
kinase cascades. As a result, the divergent inactivation of
pro-survival signaling was detectable in PF-114-treated CML cells.
Downregulation of pAkt was concomitant with a decrease in pERK1/2,
caspase activation, downregulation of mitochondrial proteins (Bid,
Bcl-xL and Mcl-1) and reductions in the mitochondrial transmembrane
potential. Collectively, these events may contribute to cell growth
arrest and apoptosis. Therefore, targeting CrkL, an upstream (or
proximal) coordinator of various pro-proliferative and pro-survival
pathways, is therapeutically advantageous providing that certain
mechanisms of cytotoxicity are non-functional as a result of a
tumor genotype and/or preceding treatment.
Molecular ordering of signaling events in response
to BCR-ABL inhibition revealed that cell cycle arrest preceded cell
death. An elevation in p27 expression may be a candidate mechanism
of G1 arrest in PF-114 treated K562 cells. This pivotal finding
requires further investigation as p27 emerges as a therapeutic
target in CML. Matrine, a plant alkaloid, significantly inhibited
the proliferation of K562 cells, induced cell cycle arrest in G0/G1
and promoted apoptosis (26).
These effects were regulated by a number of mechanisms, including
p27 upregulation (27).
Importantly, Liu et al (28) have demonstrated that this protein
was critical for the maintenance of leukemia stem cells in
CML-bearing mice. Furthermore, a combination of p27 silencing and a
compound potent against imatinib-resistant K562 cells was
synergistic (28). The complex
regulation of p27 involves transcriptional and posttranscriptional
events, including its translocation between the nucleus and the
cytoplasm. The activity of Ca2+-calmodulin dependent
kinase IIγ reduces the amounts of nuclear p27, promoting the
accumulation of this protein in the cytoplasm (29). Thus, p27-mediated G1 arrest and
leukemia stem cell quiescence are alleviated, leading to blast
crisis and disease progression (29). The mechanisms underlying the
upregulation of p27 in response to PF-114 require further
investigation. The results of the present study may provide
suggests the necessity of drug combinations to increase the
efficacy of targeted CML treatments.
Downregulation of the individual members of the
JAK-STAT families by PF-114 requires further investigation. Lee
et al (30) demonstrated
that a variety of signaling cascades mediated by oncogenic protein
kinases converge to STAT3. This transcription factor has been
implicated in the protection of CML cells from apoptosis via
several mechanisms (31-33). STAT3 undergoes a plethora of
posttranslational modifications, in particular, phosphorylation,
which regulate its genomic and non-genomic functions (34,35).
STAT3 phosphorylation has been mechanistically associated with the
emergence of resistance to imatinib (36). Eiring et al (37) reported a potent and selective SH2
domain inhibitor capable of reducing STAT3 phosphorylation and
nuclear transactivation. This novel compound restored the
sensitivity of CML cells to imatinib, including those from
patients, otherwise resistant to tyrosine kinase inhibitors
(37). Ruxolitinib, an inhibitor
of JAK2, synergized with nilotinib in inducing CML cell death in a
Phase I clinical trial (38).
These findings favor for the combined use of BCR-ABL inhibitors
with antagonists of various signaling cascades (39).
In pancreatic adenocarcinoma cells, imatinib and
ponatinib exhibited no effect on pSTAT3 levels, but were elevated
in response to an ERK blocker (40); however, as to which site in the
STAT3 protein was phosphorylated remains unknown. In the present
study, we observed opposing effects of PF-114 on two forms of
pSTAT3; the levels of pSer727 decreased, whereas those of pTyr705
were elevated. The latter form was increased following treatment
with >1 nM PF-114; however, pSer727 was attenuated only at 100
nM. In contrast, Ser727 but not Tyr705 phosphorylation was proposed
as an important factor of glioblastoma radioresistance;
pharmacological downregulation of pSTAT3 Ser727 markedly sensitized
these cells to radiation (41).
The phosphorylation at these two sites in STAT3 signifies the
induction of differential epigenetic pathways in response to
BCR-ABL inhibitors, including PF-114. Therefore, further
investigation into the intracellular signaling mechanisms in
PF-114-treated cells is required. Our future studies of the
genome-wide transcriptional effects of PF-114 aim to identify genes
that may influence the responses of CML cells to BCR-ABL
inhibition.
Signaling events downstream of BCR-ABL inhibition
may include mechanisms other than the ones described above. The
present study reported that pAkt levels were decreased by PF-114;
however, the levels of total glycogen synthase kinase-3 (GSK-3β) or
its phosphorylated form were markedly unaltered in PF-114-treated
cells (Fig. S3). In K562 cells,
the role of GSK-3 as one of various substrates of Akt
phosphorylation has been addressed by Zhou et al (42). The pharmacological inhibition of
PI3K led to decreased pAkt levels, whereas the subsequent
downregulation of pGSK-3β was less pronounced (42). This observation suggests, at least
indirectly, that in K562 cells, GSK-3β could be phosphorylated by
other kinases in addition to Akt. This hypothesis is in line with
the synergistic effects of PI3K inhibitor and imatinib.
Furthermore, the PI3K-Akt-GSK-3β and the BCR-ABL-Akt pathways were
proposed to act in concert, which suggests the role of signaling
mechanisms that remain functional in cells treated with an BCR-ABL
inhibitor. This further supports the need for combinations of the
BCR-ABL inhibitor with other inhibitors of signal transduction.
In the present study, the effects of PF-114 on K562
cells that carry wild-type Abl in the context of BCR-ABL fusion
protein were analyzed. To further evaluate the potential of PF-114
the cellular effects of the novel compound should be investigated
against mutant forms of ABL kinase. As PF-114 potently inhibited
different ABL mutants (including T315I) in vitro, in cell
based assays and in animal models (8), the growth arrest/death mechanisms
associated may be similar or identical in CML cells carrying
wild-type or mutant Abl. However, the mutants may function beyond
CrkL/BCR-ABL interactions, thereby circumventing its downstream
cascades. Additionally, the T315I mutant is capable of restoring
ERK1/2 signaling, STAT5 phosphorylation, factor-independent CML
cell growth and leukemogenic potential in
tetramerization-incompetent and other loss-of-function BCR-ABL
mutants (43). These effects have
been attributed to transphosphorylation of BCR protein by T315I
mutants (44). These critical
findings highlight the interplay between serine-threonine and
tyrosine phosphorylation of BCR as an important mechanism of CML
recurrence. Providing that T315I mutants confer growth advantages
in the absence of the wild-type BCR-ABL variant that otherwise have
notably increased transformation potential, T315I-driven phenomena
may emerge after cells with wild-type BCR-ABL are eliminated by
treatment (44). It is therefore
critical to elucidate whether PF-114 can attenuate BCR
transphosphorylation.
Our findings require further investigation. The
present study aimed to identify the molecular mechanisms that
comprise the response of one CML cell line to PF-114; however,
which mechanism is closely associated with CML cell responses to
PF-114 remains unknown. Thus, experiments involving the
inactivation of individual pathways should be performed.
Furthermore, our future efforts will be focused on mechanistic
investigations using other BCR-ABL-positive cell lines and patient
samples. Although Mian et al (8) demonstrated a high potency of PF-114
against CML cells possessing wild-type and different mutant forms
of BCR-ABL, whether individual mechanisms of cell death are
supported by our findings with K562 cells remains unknown. Finally,
the mechanisms underlying cell death in cells with mutant BCR-ABL
should be determined.
PF-114 is structurally similar to the FDA approved
pan-BCR-ABL antagonist ponatinib. This compound is capable of
inhibiting the wild-type as well as various clinically relevant
mutant forms of chimeric BCR-ABL tyrosine kinase. We demonstrated
that the CML-derived human K562 cell line that carries wild-type
BCR-ABL was notably sensitive to PF-114. This agent at nanomolar
concentrations rapidly blocked the phosphorylation of CrkL adaptor
protein, thereby inhibiting a number of downstream signaling
pathways, inducing cell cycle arrest and subsequent apoptosis.
Together with favorable pharmacokinetics and good therapeutic
efficacy in a murine model (8),
this novel inhibitor may be considered as a potential therapeutic
drug candidate for the treatment of CML (45).
Supplementary Materials
Funding
This study was supported by the Megagrant (grant no.
14. W03.31.0020 between the Ministry of Science and Education of
the Russian Federation and Institute of Gene Biology, Russian
Academy of Sciences). VVT was supported by Ministry of Science and
Higher Education of the Russian Federation in the framework of
Increase Competitiveness Program of MISiS (grant no.
P02-2017-2-1).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ESI, VVT, GGC, YVD and AAS made substantial
contributions to the design of the study. AAZ, FNN and GGC
synthesized PF-114. ESI, VVT, MAY, AIK, AVS and AAK performed the
experiments and analyzed data. AAS wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interests.
Abbreviations:
|
BCR-ABL
|
breakpoint cluster region-Abelson
murine leukemia viral oncogene homolog 1
|
|
CML
|
chronic myelogenous leukemia
|
|
CrkL
|
Crk-like protein
|
|
GSK-3β
|
glycogen synthase kinase-3β
|
|
JAK
|
Janus kinase
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
Acknowledgments
We are grateful to Dr S. Kalinichenko and Dr Y.
Dobryakova for providing antibodies against GSK-3β and pGSK-3β.
References
|
1
|
Pasternak G, Hochhaus A, Schultheis B and
Hehlmann R: Chronic myelogenous leukemia: Molecular and cellular
aspects. J Cancer Res Clin Oncol. 124:643–660. 1998. View Article : Google Scholar
|
|
2
|
Lin X, Qureshi MZ, Attar R, Khalid S,
Tahir F, Yaqub A, Aslam A, Yaylim I, De Carlos Back LK, Farooqi AA,
et al: Targeting of BCR-ABL: Lessons learned from BCR-ABL
inhibition. Cell Mol Biol (Noisy-le-grand). 62:129–137. 2016.
|
|
3
|
Hochhaus A, Larson RA, Guilhot F, Radich
JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F,
Fujihara S, et al: IRIS Investigators: Long-term outcomes of
imatinib treatment for chronic myeloid leukemia. N Engl J Med.
376:917–927. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cortes JE, Kantarjian H, Shah NP, Bixby D,
Mauro MJ, Flinn I, O'Hare T, Hu S, Narasimhan NI, Rivera VM, et al:
Ponatinib in refractory Philadelphia chromosome-positive leukemias.
N Engl J Med. 367:2075–2088. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cortes JE, Kim DW, Pinilla-Ibarz J, le
Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ,
Talpaz M, et al: PACE Investigators: A phase 2 trial of ponatinib
in Philadelphia chromosome-positive leukemias. N Engl J Med.
369:1783–1796. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoy SM: Ponatinib: A review of its use in
adults with chronic myeloid leukaemia or Philadelphia
chromosome-positive acute lymphoblastic leukaemia. Drugs.
74:793–806. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moslehi JJ and Deininger M: Tyrosine
kinase inhibitor-associated cardiovascular toxicity in chronic
myeloid leukemia. J Clin Oncol. 33:4210–4218. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mian AA, Rafiei A, Haberbosch I, Zeifman
A, Titov I, Stroylov V, Metodieva A, Stroganov O, Novikov F, Brill
B, et al: PF-114, a potent and selective inhibitor of native and
mutated BCR/ABL is active against Philadelphia chromosome-positive
(Ph+) leukemias harboring the T315I mutation. Leukemia.
29:1104–1114. 2015. View Article : Google Scholar
|
|
9
|
Chilov GG and Titov IY: Protein kinase
inhibitor (versions), use thereof for treating oncological diseases
and based pharmaceutical composition. Patent RU2477723 (C2). Filed
July 10, 2017; issued August 31, 2017.
|
|
10
|
Cellosaurus - a knowledge resource on cell
lines. Version 29. https://web.expasy.org/cellosaurus/CVCL_0002.
|
|
11
|
Liu D: The adaptor protein Crk in immune
response. Immunol Cell Biol. 92:80–89. 2014. View Article : Google Scholar :
|
|
12
|
Shchekotikhin AE, Dezhenkova LG, Tsvetkov
VB, Luzikov YN, Volodina YL, Tatarskiy VV Jr, Kalinina AA,
Treshalin MI, Treshalina HM, Romanenko VI, et al: Discovery of
antitumor anthra[2,3-b]furan-3-carboxamides: Optimization of
synthesis and evaluation of antitumor properties. Eur J Med Chem.
112:114–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rahbar Saadat Y, Saeidi N, Zununi Vahed S,
Barzegari A and Barar J: An update to DNA ladder assay for
apoptosis detection. Bioimpacts. 5:25–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rossari F, Minutolo F and Orciuolo E:
Past, present, and future of Bcr-Abl inhibitors: From chemical
development to clinical efficacy. J Hematol Oncol. 11:842018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haguet H, Douxfils J, Mullier F, Chatelain
C, Graux C and Dogné JM: Risk of arterial and venous occlusive
events in chronic myeloid leukemia patients treated with new
generation BCR-ABL tyrosine kinase inhibitors: A systematic review
and meta-analysis. Expert Opin Drug Saf. 16:5–12. 2017. View Article : Google Scholar
|
|
16
|
Wehrle J and von Bubnoff N: Ponatinib: A
third-generation inhibitor for the treatment of CML. Recent Results
Cancer Res. 212:109–118. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zulbaran-Rojas A, Lin HK, Shi Q, Williams
LA, George B, Garcia-Manero G, Jabbour E, O'Brien S, Ravandi F,
Wierda W, et al: A prospective analysis of symptom burden for
patients with chronic myeloid leukemia in chronic phase treated
with frontline second- and third-generation tyrosine kinase
inhibitors. Cancer Med. 7:5457–5469. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aspinall-O'Dea M, Pierce A, Pellicano F,
Williamson AJ, Scott MT, Walker MJ, Holyoake TL and Whetton AD:
Antibody-based detection of protein phosphorylation status to track
the efficacy of novel therapies using nanogram protein quantities
from stem cells and cell lines. Nat Protoc. 10:149–168. 2015.
View Article : Google Scholar
|
|
19
|
Lux MP, Fasching PA, Schrauder MG, Hein A,
Jud SM, Rauh C and Beckmann MW: The PI3K pathway: Background and
treatment approaches. Breast Care (Basel). 11:398–404. 2016.
View Article : Google Scholar
|
|
20
|
Dey N, De P and Leyland-Jones B:
PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell
signaling to clinical trials. Pharmacol Ther. 175:91–106. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nussinov R, Tsai CJ and Jang H: A new view
of pathway-driven drug resistance in tumor proliferation. Trends
Pharmacol Sci. 38:427–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Correia C, Lee SH, Meng XW, Vincelette ND,
Knorr KL, Ding H, Nowakowski GS, Dai H and Kaufmann SH: Emerging
understanding of Bcl-2 biology: Implications for neoplastic
progression and treatment. Biochim Biophys Acta. 1853:1658–1671.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schutters K and Reutelingsperger C:
Phosphatidylserine targeting for diagnosis and treatment of human
diseases. Apoptosis. 15:1072–1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chaitanya GV, Steven AJ and Babu PP:
PARP-1 cleavage fragments: Signatures of cell-death proteases in
neurodegeneration. Cell Commun Signal. 8:312010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bousoik E and Montazeri Aliabadi H: 'Do We
Know Jack' about JAK? A closer look at JAK/STAT signaling pathway.
Front Oncol. 8:2872018. View Article : Google Scholar
|
|
26
|
Ma L, Zhu Z, Jiang L, Sun X, Lu X, Zhou M,
Qian S and Jianyong L: Matrine suppresses cell growth of human
chronic myeloid leukemia cells via its inhibition of the
interleukin-6/Janus activated kinase/signal transducer and
activator of transcription 3 signaling cohort. Leuk Lymphoma.
56:2923–2930. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma L, Xu Z, Wang J, Zhu Z, Lin G, Jiang L,
Lu X and Zou C: Matrine inhibits BCR/ABL mediated ERK/MAPK pathway
in human leukemia cells. Oncotarget. 8:108880–108889. 2017.
View Article : Google Scholar
|
|
28
|
Liu C, Nie D, Li J, Du X, Lu Y, Li Y, Zhou
J, Jin Y and Pan J: Antitumor effects of blocking protein
neddylation in T315I-BCR-ABL leukemia cells and leukemia stem
cells. Cancer Res. 78:1522–1536. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu Y, Zheng W, Zhang J, Gan X, Ma X, Meng
Z, Chen T, Lu X, Wu Z, Huang W, et al: Aberrant activation of
CaMKIIγ accelerates chronic myeloid leukemia blast crisis.
Leukemia. 30:1282–1289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and
Settleman J: Drug resistance via feedback activation of Stat3 in
oncogene-addicted cancer cells. Cancer Cell. 26:207–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mencalha AL, Corrêa S, Salles D, Du Rocher
B, Santiago MF and Abdelhay E: Inhibition of STAT3-interacting
protein 1 (STATIP1) promotes STAT3 transcriptional up-regulation
and imatinib mesylate resistance in the chronic myeloid leukemia.
BMC Cancer. 14:8662014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Xie B, Kong Y, Tao Y, Yang G, Gao
M, Xu H, Zhan F, Shi J, Zhang Y, et al: Overexpression of RPS27a
contributes to enhanced chemoresistance of CML cells to imatinib by
the transactivated STAT3. Oncotarget. 7:18638–18650.
2016.PubMed/NCBI
|
|
33
|
Shi Y, Zhang Z, Qu X, Zhu X, Zhao L, Wei
R, Guo Q, Sun L, Yin X, Zhang Y, et al: Roles of STAT3 in leukemia
(Review). Int J Oncol. 53:7–20. 2018.PubMed/NCBI
|
|
34
|
Guanizo AC, Fernando CD, Garama DJ and
Gough DJ: STAT3: A multifaceted oncoprotein. Growth Factors.
36:1–14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sgrignani J, Garofalo M, Matkovic M,
Merulla J, Catapano CV and Cavalli A: Structural biology of STAT3
and its implications for anticancer therapies development. Int J
Mol Sci. 19:E15912018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Al-Jamal HA, Jusoh SA, Yong AC, Asan JM,
Hassan R and Johan MF: Silencing of suppressor of cytokine
signaling-3 due to methylation results in phosphorylation of STAT3
in imatinib resistant BCR-ABL positive chronic myeloid leukemia
cells. Asian Pac J Cancer Prev. 15:4555–4561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Eiring AM, Page BDG, Kraft IL, Mason CC,
Vellore NA, Resetca D, Zabriskie MS, Zhang TY, Khorashad JS, Engar
AJ, et al: Combined STAT3 and BCR-ABL1 inhibition induces synthetic
lethality in therapy-resistant chronic myeloid leukemia. Leukemia.
31:1253–1254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sweet K, Hazlehurst L, Sahakian E, Powers
J, Nodzon L, Kayali F, Hyland K, Nelson A and Pinilla-Ibarz J: A
phase I clinical trial of ruxolitinib in combination with nilotinib
in chronic myeloid leukemia patients with molecular evidence of
disease. Leuk Res. 74:89–96. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh H, Williams RT and Guy KR: Methods
and compositions for the treatment of Bcr-Abl positive
lymphoblastic leukemias. Patent US20150133462 A1. Filed May 23,
2013; issued May 14, 2015.
|
|
40
|
Sahu N, Chan E, Chu F, Pham T, Koeppen H,
Forrest W, Merchant M and Settleman J: Cotargeting of MEK and
PDGFR/STAT3 pathways to treat pancreatic ductal adenocarcinoma. Mol
Cancer Ther. 16:1729–1738. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ouédraogo ZG, Müller-Barthélémy M, Kemeny
JL, Dedieu V, Biau J, Khalil T, Raoelfils LI, Granzotto A, Pereira
B, Beaudoin C, et al: STAT3 serine 727 phosphorylation: A relevant
target to radiosen-sitize human glioblastoma. Brain Pathol.
26:18–30. 2016. View Article : Google Scholar
|
|
42
|
Zhou Q, Chen Y, Chen X, Zhao W, Zhong Y,
Wang R, Jin M, Qiu Y and Kong D: In vitro antileukemia activity of
ZSTK474 on K562 and multidrug resistant K562/A02 cells. Int J Biol
Sci. 12:631–638. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Haberbosch I, Rafiei A, Oancea C, Ottmann
GO, Ruthardt M and Mian AA: BCR: A new target in resistance
mediated by BCR/ABL-315I? Genes Cancer. 7:36–46. 2016.PubMed/NCBI
|
|
44
|
Mian AA, Schüll M, Zhao Z, Oancea C,
Hundertmark A, Beissert T, Ottmann OG and Ruthardt M: The
gatekeeper mutation T315I confers resistance against small
molecules by increasing or restoring the ABL-kinase activity
accompanied by aberrant transphosphorylation of endogenous BCR,
even in loss-of-function mutants of BCR/ABL. Leukemia.
23:1614–1621. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
NIH: U.S. National Library of Medicine.
ClinicalTrials. Study to evaluate tolerability, safety,
pharmacokinetics and preliminary efficacy of PF-114 for oral
administration in adults with Ph+ chronic myeloid leukemia, which
is resistant to the 2-nd generation Bcr-Abl inhibitors or Has T315I
mutation in the BCR-ABL gene. (NCT02885766). https://clinicaltrials.gov/ct2/show/NCT02885766.
|